
Abstract
The emergence of all-solid-state Li batteries (ASSLBs) represents a promising avenue to address critical concerns like safety and energy density limitations inherent in current Li-ion batteries. Solid electrolytes (SEs) show significant potential in curtailing Li dendrite intrusion, acting as natural barriers against short circuits. However, the substantial challenges at the SEs−electrode interface, particularly concerning the anode, pose significant impediments to the practical implementation of ASSLBs. This review aims to delineate the most viable strategies for overcoming anode interfacial hurdles across four distinct categories of SEs: sulfide SEs, oxide SEs, polymer SEs, and halide SEs. Initially, pivotal issues such as anode interfacial side reactions, inadequate physical contact, and Li dendrite formation are comprehensively outlined. Furthermore, effective methodologies aimed at enhancing anode interfacial stability are expounded, encompassing approaches like solid electrolyte interface (SEI) interlayer insertion, SE optimization, and the adoption of Li alloy in lieu of Li metal, each tailored to specific SE categories. Moreover, this review presents novel insights into fostering interfaces between diverse SE types and Li anodes, while also advocating perspectives and recommendations for the future advancement of ASSLBs.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Since its inception in 1958, Li metal, renowned for its high energy density, has been a pivotal component in battery technology. The milestone moment arrived in 1976 when Whittingham introduced the first rechargeable Li-ion batteries (LIBs) featuring Li|Li2S, boasting an exceptional specific energy nearing 500 Wh kg−1 [1]. These groundbreaking LIBs, comprising the LiCoO2 cathode (crafted by Goodenough) and carbonaceous anode (engineered by Yoshino), emerged as the cornerstone of modern LIBs [2, 3]. Sony's development of the 18650-type LIB in 1991, which is post-acquisition of the aforementioned patent, marked a turning point, propelling the subsequent evolution and commercialization of LIBs through innovative electrode materials and electrolyte advancements [4]. The advent of LIBs heralded a new era, revolutionizing energy storage applications in automotive and portable electronics. However, inherent safety concerns, notably thermal runaway due to Li dendrites and the risk of short circuits, pose significant threats to LIB integrity. Furthermore, the limitation of current LIBs' energy density (capped at 300 Wh kg−1) contributes to the prevalent "range anxiety" in electric vehicles, compounding the paramount concerns.
Addressing these challenges necessitates a paradigm shift—replacing conventional flammable liquid electrolytes (LEs) with robust and secure solid electrolytes (SEs) to realize all-solid-state Li batteries (ASSLBs). ASSLBs offer a definitive solution to mitigate safety issues. While the cathode materials in ASSLBs remain akin to those in LIBs, the former possess the potential for a 50% or greater increase in energy density through stacking, obviating the need for individual cell packing, metal shell fixation, and allocated space for coolants—A distinctive advantage in practical applications [5]. Consequently, owing to their exceptional safety profile and heightened energy density, ASSLBs stand as the frontrunner in the quest for a new generation of energy storage devices.
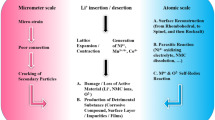
Typically, SEs need to satisfy several key criteria: (i) displaying appropriate ionic conductivity (> 0.1 mS cm−1); (ii) possessing a broad electrochemical stability window (ESW); (iii) showcasing robust mechanical properties to hinder Li dendrite infiltration; (iv) maintaining cost-effectiveness; and (v) offering ease of processing [6]. To date, four primary categories of SEs have emerged: sulfide SEs, oxide SEs, polymer SEs, and halide SEs. These distinct categories of SEs, when integrated into ASSLBs, present diverse advantages and challenges owing to the distinctive electrochemical and physical characteristics they exhibit. This differentiation is summarized in Fig. 1, while Table 1 delineates the ionic conductivities of typical SEs.

Holistic aspects of four main categories of SEs performance
The ionic conductivities of sulfide SEs rival those of traditional LEs and stand as one of the most promising directions in SE research. Notably, sulfide SEs showcase impressive mechanical properties, such as plastic deformation, and readily form densely packed interfaces [7]. A myriad of sulfide SE types have undergone exploration, including Li–P–S-based glasses, Li6PS5X (X = Cl, Br, or I), and Li10GeP2S12 (LGPS) varieties. Glassy sulfide SEs possess advantageous plastic properties, circumventing high-resistance grain boundaries and facilitating efficient ionic flux due to their composition. An exemplary instance lies in the binary xLi2S·(100 − x) P2S5 system, where x, ranging from 40 to 80 molar percentages, generates single-phase composition like 60Li2S·40P2S5, 75Li2S·25P2S5, among others [8]. The ionic conductivities of these glassy sulfide SEs typically fall within a moderate range of 10−2–10−1 mS cm−1 at room temperature (RT). Strategies enhancing charge carrier concentration and mobility in glasses, involving compounds like Li3PO4, Li4SiO4, LiBH4, and other Li salts, effectively bolster the ionic conductivity of glassy systems [9, 10]. Transitioning from glass to glassy ceramics via heat treatment influences their ionic conductivities, as observed in Li7P3S11 derived from 70Li2S·30P2S5 glass, exhibiting a high ionic conductivity of 1 mS cm−1 after heating within the range of 531–633 K. The substantial presence of interstitial sites and expansive open volumes between P2S74− di-tetrahedra and PS43− tetrahedra facilitates rapid Li+ transport. Notably, Li3PS4 demonstrates three crystal types—α, β, and γ phases—with β-phase PS43− tetrahedra presenting disorderly zig-zag structures and an ionic conductivity of approximately 3.2 mS cm−1 [11, 12]. Within the Li6PS5X argyrodite family, the anionic disorder plays a pivotal role in dictating differing ionic conductivities. Argyrodites like Li6PS5Cl (LPSC) and Li6PS5Br (LPSB) exhibit S2−/X− anion disorder due to partial exchange between normal X− and S2− sites, contributing to ionic conductivities of 1.9 and 0.68 mS cm−1 at RT, respectively. In contrast, Li6PS5I (LPSI) maintains an ordered anionic framework due to the larger non-interchangeable I−, resulting in a lower ionic conductivity of 4.6 × 10−4 mS cm−1 [13, 14]. Substituting elements in argyrodites not only stabilizes the structure but also enhances the ionic conductivity of sulfide SEs, occurring at halide sites, PS43− units, and free sulfur sites. Notably, Li5.3PS4.3Cl1.7 stands as a representative, boasting an ionic conductivity of up to 17 mS cm−1 [15, 16]. The LGPS family, described as Li11−xM2−xP1+xS12 (0 < x < 2) with M representing Si, Sn, or Ge, showcases remarkable potential. The groundbreaking LGPS superionic conductor reported in 2011 by Kanno’s group demonstrated an exceptional ionic conductivity of 12 mS cm−1, inspiring Japanese automakers like Toyota and Nissan to embrace sulfide SE systems [17]. Subsequent advancements led to the development of LGPS-type SEs, such as Li9.54Si1.74P1.44S11.7Cl0.3 (LSPS) in 2016, boasting an impressive ionic conductivity of 25 mS cm−1. The synthesis of LGPS with a single crystal structure reached the pinnacle, showcasing the highest ionic conductivity to date at 27 mS cm−1 [18, 19]. Despite LGPS achieving or surpassing LE-level ionic conductivities, researchers face lingering challenges, including interfacial properties between sulfide SEs and electrodes, a narrow ESW, Li dendrite formation, and the high cost associated with Ge. These hurdles warrant continued investigation and resolution in the pursuit of sulfide SEs' full potential in solid-state battery technology.
Oxide SEs have garnered significant attention from battery energy manufacturers like QuantumScape, WELION, Qingtao Development, and Prologium due to their superior safety compared to sulfide SEs and LEs. Typical oxide SEs, including garnets, NASICONs, and perovskites, offer advantages such as cost-effective processing and reduced sensitivity to air exposure. Among promising oxide SE candidates, garnet-type electrolytes stand out for their broad ESW, robust chemical stability at the Li − garnet interface, and high mechanical stiffness and hardness [20, 21]. Garnets are categorized based on their Li content into Li3, Li5, and Li7 types, corresponding to Li3Ln3Te2O12, Li5La3M2O12, and Li7La3M’2O12, respectively, where Ln includes Y, Pr, Nd, Sm–Lu, and M includes Nb, Ta, Sb, and M’ includes Zr, Sn, Hf [22,23,24,25,26,27,28,29,30]. The concentration of Li+ in garnet structures significantly influences the Li+ conduction pathway. Li7La3Zr2O12 (LLZO) garnet electrolyte has been identified as a potential catalyst for the commercialization of ASSLBs due to its moderate ionic conductivity (0.1–1 mS cm−1), robust mechanical strength, chemical stability, and wide ESW [31, 32]. To stabilize the cubic phase in LLZO, substituting Zr with metals like Ta or Nb or doping with elements like Ga or Al has proven effective [33]. For example, LLZTO garnets doped with Ta exhibit satisfactory stability with Li anodes [34]. NASICONs, originating from sodium super ion conductors, have intrigued researchers since their discovery in 1976, evolving into compounds like LiM2(PO)4, where M represents Ge, Ti, Hf. LiTi2(PO)4 displays superior ionic conductivity compared to others [35,36,37,38]. Additionally, Li1+xAlxTi2−x(PO4)3 (LATP) and Li1+xAlxGe2−x(PO4)3 (LAGP) achieve ionic conductivities exceeding 10−1 mS cm−1 at 25 °C by partially replacing trivalent ions [39]. However, LATP’s compatibility with anode materials possessing low potential properties like In and C6Li is unsatisfactory due to Ti4+ reduction at 2.5 V vs. Li/Li+ [34, 40]. Perovskite-type LixM1/3Nb1−xTixO3 (M = La, Nd) and Li0.5La0.5TiO3 were first reported in 1984, and the Li3xLa(2/3) (1/3)−2xTiO3 phase (LLTO) subsequently, where represents a structural vacancy and relative compounds, with 0 < x < 0.16 [41,42,43]. Comparing the heat generation rates of prevalent oxide SEs, the order of thermal stability toward Li anodes is LAGP < LATP < LLTO < LLZO [34]. These comparative insights into thermal stability and compatibility with anode materials highlight the nuanced strengths and limitations of different oxide SEs, informing ongoing research aimed at harnessing their full potential in advancing solid-state battery technology.
Polymer SEs possess remarkable flexibility, strong adhesion, and the capacity to form favorable membranes. These SEs typically comprise solid-state polymer hosts (PHs) with high dielectric properties and dissolved Li salts featuring low lattice energy. Among the PHs, those with functional groups play a pivotal role in dissociating and facilitating Li+ transport. Notably, poly(ethylene oxide) (PEO) stands out as one of the most promising PHs. Li salts like Li bis(trifluoromethanesulfonyl)imide (LiTFSI), LiBF4, and LiClO4 significantly contribute to maintaining high ionic conductivities in polymer SEs. The pioneering work in 1973 marked the inception of the typical PEO-based SE framework [44]. In PEO-based SEs, Li+ transport primarily occurs through ion migration and the strong complexation and dissociation effects of ether oxygen groups along the PEO chain segments within the polymer matrix [45]. Notably, due to its exceptional salt-solvating ability, low glass transition temperature (Tg), and compatibility with electrodes, PEO-based SEs have been the sole choice for large-scale EVs in the ASSLBs developed by Bolloré [6]. Further advancements have led to the development of other polymer hosts like polyvinylidene fluoride (PVDF), polyurethane (PU), polyimide (PI), and polyacrylonitrile (PAN). These hosts exhibit high ionic conductivities, good Li salt solubility, and continuous polar groups (such as –NH2, –O–, –P–, and –S–) within their molecular chain segments [46,47,48,49,50,51,52,53,54,55]. However, most polymer SEs available to date fail to meet practical requirements, primarily due to their low ionic conductivity (ranging from 10−5 to 10−3 mS cm−1) at RT and low Li+ transference number (ranging from 0.2 to 0.3). To enhance the Li+ conductivity of polymer SEs, researchers have explored various approaches, including incorporating inorganic fillers. However, due to space limitations, this review cannot delve deeper into additional relevant methods and findings in this field [56].
As a burgeoning family of SEs, halide SEs present compelling features akin to sulfide SEs, offering excellent machinability, a wide ESW spanning between 0.36 V and 6.71 V vs. Li/Li+, desirable ionic conductivity exceeding 1.0 mS cm−1, and robust chemical and electrochemical stability against oxide cathodes, as well as air/humidity tolerance [57]. Ternary compounds expressed as LiaMXb (where X = F, Cl, Br, I and M represents metal or non-metal elements) are the prevailing representatives of halide SEs. Significant breakthroughs in 2018 introduced halide SEs like Li3YCl6 and Li3YBr6, boasting ionic conductivities ranging from 0.51 to 1.70 mS cm−1 at RT [58]. Subsequent advancements led to the development of halide SEs like Li3InCl6, Li3InBr6, Li3Y1−xInxCl6, LixScCl3+x, Li2Sc2/3Cl4, showcasing ideal ionic conductivities exceeding 1.0 mS cm−1 at RT [59]. Notably, the ionic conductivity of Li3Y(Br3Cl3) has surged to an impressive 7.2 mS cm−1 at RT, signifying a substantial stride in this domain [60]. These impressive ionic conductivities in halide SEs hold promise for the design and advancement of next-generation ASSLBs. The choice of metal elements in halide SEs significantly influences their compatibility with Li anodes in ASSLB applications. Halide SEs featuring non-metal elements tend to demonstrate a narrower ESW, exemplified by Li3OCl (up to 2.55 V vs. Li/Li+). In contrast, those incorporating metal elements exhibit a broader ESW, such as Li3YCl6 (up to 4.21 V vs. Li/Li+) [61, 62]. Due to their advantageous properties—prominently, exceptional ionic conduction and wide ESWs—halide SEs with metal elements have garnered escalating interest. These halide SEs can be categorized based on the type of metal element involved: (i) LiaMiXb featuring group 3 elements like Sc, Y, La–Lu; (ii) LiaMiiXb involving group 13 elements such as Al, Ga, and In; and (iii) LiaMiiiXb encompassing divalent metal elements like Mg, Ti, Cu, Fe [59]. However, it's important to note that the reduction potential of halide SEs still surpasses 0.6 V (vs. Li/Li+), indicating that direct coupling with Li metal anodes remains a challenge.
While the performance of SEs remains a crucial factor for achieving efficient Li+ transport and high electrochemical performance in assembled ASSLBs, it's no longer the sole determinant. The establishment of a preferred interface, ensuring robust stability between SEs and electrodes, has emerged as a decisive element in meeting the commercial demands for ASSLBs. Most SEs encounter challenges in maintaining complete stability when faced with highly reductive Li anodes, creating bottlenecks for the practical application of ASSLBs. Recent papers have extensively covered protection methods specifically for the Li anode side [63,64,65,66], leaving a substantial gap in addressing interfacial issues across all SE categories. This discussion aims to delve into both the Li anode side and SEs side, highlighting the intricate interface stability challenges and proposing corresponding solutions. Our objective is to present the most promising outcomes in research. The exploration begins by uncovering the failure mechanisms within the interface between Li anodes and SEs, encompassing chemical side reactions, poor physical contact, and the formation of Li dendrites. Additionally, we emphasize design strategies aimed at stabilizing the interface between Li anodes and the four main types of SEs—sulfide, oxide, polymer, and halide SEs—pertaining to ASSLBs within the past decade. Lastly, we offer ideas and suggestions for interfacing different SE types with Li anodes and propose views and recommendations for the future development of ASSLBs. This comprehensive approach aims to address and bridge critical gaps in understanding and overcoming interface challenges, ultimately propelling advancements in ASSLB technology.
2 Issues of Li–Solid Electrolyte Interfaces/Interphases
In the realm of batteries, interfaces and interphases represent distinct yet crucial concepts. The interface serves as the primary site for electron/ion exchange in ASSLBs, where the anode/cathode meets the SEs, creating a sudden phase discontinuity experienced by both the bulk anode/cathode and bulk SEs. Conversely, the interphase emerges as a result of irreversible reactions between SEs and the anode/cathode, aiming for superior ionic conduction and electronic insulation [67]. The allure of Li metal, with its exceptionally high specific capacity of ~ 3 860 mAh g−1, has long been perceived as the holy grail in the landscape of commercial LIBs. Additionally, Li metal boasts a notably low potential of − 3.04 V vs. the standard hydrogen electrode (SHE) and a low density of 0.534 g cm−3, significantly contributing to the achievement of ultrahigh energy density in ASSLBs [68]. While incorporating Li anodes in ASSLBs is pivotal for enhancing energy density, the inherent low electrochemical potential of Li anodes leads to their reactivity with most SEs. This reactivity often triggers the nucleation and growth of Li dendrites, ultimately leading to cell short circuits. Unlike LEs, SEs lack the ability to infiltrate voids and gaps, resulting in undesirable physical contact between SEs and electrodes. Consequently, the primary challenge in ASSLBs lies in ensuring the stability of the interface between Li anodes and SEs. This challenge is directly linked to issues of side reactions, contact area, and the formation of Li dendrites, posing significant hurdles in advancing the performance and safety of these batteries.
Securing an ideal interphase that encompasses robust mechanical, chemical, and electrochemical properties holds immense significance. Such interphases are pivotal in establishing a stable interface that mitigates severe side reactions, maintains low interfacial impedance, and enables a high critical current density (CCD). These attributes play a crucial role in restraining side reactions, facilitating the swift transport of Li+, and suppressing the formation of Li dendrites at the interface. Addressing several challenges inherent in Li–SE interfaces/interphases is imperative before their practical implementation can be realized.
2.1 Side Reactions
The ESW of SEs delineates their capacity to resist oxidation or reduction by extracting or inserting ions and electrons. Meeting the escalating energy density demands of ASSLBs necessitates a high operating voltage, underscoring the need for SEs to retain stability across the widest possible ESW. Assessing the chemical stabilities of both LEs and SEs is often predicted by using an energy diagram, where the ESW is gauged by the relative energy of "Eg" [69]. Oxidation events occur when the top of the valence band (VB) surpasses the chemical potential of the cathode (μC), while reduction reactions are triggered when the bottom of the conduction band (CB) falls below the chemical potential of the anode (μA). The ESW, positioned between the bottom of CB and the top of VB in SEs, is determined by their oxidation and reduction (such as S and P redox for thiophosphate-based SEs, and O and Zr redox for garnet) [70]. A wide ESW is pivotal for achieving high-performance cells, often achieved by lowering the highest occupied molecular orbital (HOMO) and raising the lowest unoccupied molecular orbitals (LUMO) to counteract sluggish decomposition kinetics [71]. Regarding SEs and the cathode/anode interface, during the Li+ stripping/plating process, a cathode electrolyte interphase (CEI) interlayer and an anode solid electrolyte interphase (SEI) interlayer are formed. The stability issues at these interfaces significantly impact the state and energy density of ASSLBs, though beyond the review's scope. The formation of SEIs inevitably involves SE consumption and impedance increase, directly related to the reduction products of SEs. The ESW of commonly used SEs, along with their application prospects, is summarized in Fig. 2, providing a foundation for interpreting interface stability between Li and SEs. Notably, Li binary compounds merit a separate classification for summarization (Fig. 2, blue–green bars at the bottom). Extensive computational and experimental research into the ESW not only delineates the operating voltage range for various SE types but also elucidates oxidation and reduction processes. For example, the ESW of LGPS spans from 1.71 to 2.14 V vs. Li/Li+, with LGPS lithiated and reduced to Li4GeS4, P, and Li2S at 1.7 V, and oxidized to Li3PS3 and GeS2 at 2.14 V [32].

Electrochemical stability window of examples from various SEs (binary compounds, sulfide, oxide, polymer and halide)
According to Wenzel et al., the Li–SEs interface can be classified into three distinct categories: the thermodynamically stable interphase, the mixed ionic and electronic conducting (MIEC) interphase, and the metastable solid–electrolyte interphase (M–SEI), as illustrated in Fig. 3 [72].
-
(1)
Thermodynamically stable interphase. Figure 3a depicts a scenario where the Li anode and SEs exhibit stability with each other, forming a flat and clear 2D interface devoid of any side reactions. Li binary compounds stable with the Li anode undergo decomposition at 0 V vs. Li/Li+ (Fig. 2a, cyan bars), signaling a stable interphase [73]. Common binary SEs such as Li3N, Li2O, Li3P, and LiX (where X represents halide elements) are stable with the Li anode, although they typically exhibit low ionic conductivity, rendering them unsuitable as standalone SEs for ASSLBs [74].
-
(2)
MIEC interphase. Figure 3b illustrates the occurrence of chemical reactions between the Li anode and SEs, resulting in a thermodynamically unstable interface. This scenario often leads to the formation of an electronic conductive interphase, fostering an electronic pathway for reduction at the anode–SEs interface. This phenomenon contributes to gradual Li dendrite formation, ultimately leading to short circuits in ASSLBs, a significant impediment to practical use. However, in the case of PEO-based SEs, Wan et al. discovered that an in situ formed MIEC interphase, comprising the high ionic conductivity of Li3N and the electronic conductor Mg between PEO and Mg3N2 intermediary, effectively alleviated concentration gradients and enhanced uniform Li deposition [75]. In contrast, without the MIEC interlayer, anions tend to accumulate on the Li anode.
-
(3)
M–SEI. Figure 3c showcases chemical reaction products exhibiting low electronic conductivity or insulative properties, limiting the continuous growth of the M–SEI. This metastable interphase bears similarity to the SEI observed in LEs-based cells.

Copyright © 2015, Elsevier
Interfaces between the Li anode and SEs can be categorized into three key types: a thermodynamically stable interface; b mixed ionic and electronic conducting (MIEC) interphase; c metastable solid–electrolyte interphase (M–SEI). Reprinted with permission from Ref. [72].
In terms of sulfide SEs, Li3PS4-based glasses, glass ceramics, and Li argyrodites demonstrate greater stability than highly conducting sulfide SEs like LGPS and LSPS when exposed to the Li anode in ASSLBs. When Li metal interacts with Li3PS4-based glasses, glass ceramics, and argyrodites, the decomposition products are Li2S and Li3P. The former compound is insulative, while the latter is ionic conductive, indicating the formation of an M–SEI. On the other hand, LGPS shows a favorable decomposition energy (− 1.2 eV atom−1), resulting in decomposed products of insulative Li2S, ionic conductive Li3P, and electronic conductive Li–Ge alloy after contact with the Li anode, indicating an MIEC interphase [74, 76]. The accumulation of these products increases interfacial impedance and encourages continuous Li dendrite growth, leading to eventual cell failure. This behavior is similar in sulfide SEs containing Si, Sn, and Sb. Li3PS4-based glasses, glass ceramics, and argyrodites can serve as protective layers between the Li anode and highly conducting sulfide SEs like LGPS and LSPS. LLZO, known as an ionic conductor, shows a decomposition close to 0 V, suggesting greater stability with Li metal compared to sulfide SEs. However, slight reduction of Zr4+ in LLZO by Li can elevate electronic conductivity in the interphase, leading to the formation of an MIEC interphase [77]. LPON reduced by Li metal produces Li3P, Li2O, and Li3N, indicating an M–SEI favorable for Li+ transport without blocking electron conduction, described as a metastable interphase, enabling a long cycle life of around ≈10 000 cycles [78, 79]. NASICON-type oxide SEs containing Ti, Ge, and Al, such as Ti4+ in LLTO, Ti4+ and Al3+ in LATP, Ti4+, Ge4+, and Al3+ in LAGP, are prone to forming MIEC interphases with Li metal via reduction reactions, making them unsuitable for use with the Li anode [80]. Most polymer SEs are stable with the Li anode, except for poly(N-methyl-malonic amide) (PMA) and polyacrylonitrile (PAN) [81, 82]. Halide SEs, due to high valence metal elements, exhibit high reactivity with the Li anode [83, 84]. For instance, LiaMiXb exhibits a reduction onset of 0.41–0.92 V, while LiaMiiXb demonstrates a higher value of 1.06–0.92 V [62]. When Li3YCl6 contacts Li metal, it decomposes into Li3Y (an electronic conductor) and LiCl (an ionic conductor), indicating an MIEC interphase [85]. The metal component is crucial in determining the reduction onset of halide SEs.
2.2 Poor Physical Contact
Compared to the effective contact between sufficient LEs and electrodes, establishing a solid connection between SEs and electrodes is notably more intricate. Inadequate contact between the Li anode and SEs results in interfacial issues like the formation of Li dendrites, elevated charge transfer impedance, and uneven current distribution. Even with sulfide SEs known for their pliability, which aids in compacting the Li anode and SEs, they still struggle to entirely fill gaps, leading to subpar physical contact. Applying moderate stack pressure during cycling becomes essential to enable cycling in ASSLBs by preventing void formation [86]. Most electrochemical tests in ASSLBs employ external pressure to counteract the adverse effects of insufficient solid–solid contact and high interfacial impedance. However, the specific parameters of applied external pressure vary based on the properties of sulfide SEs and electrodes and lack uniformity.
Evaluating the contact performance between solid-state electrodes and SEs involves assessing wettability and cyclic stress. Inadequate physical contact between the Li anode and SEs often arises from poor wettability, which can be gauged by the contact angle between molten Li and SEs. Contact angles below 90° signify good wettability, representing a lithiophilic interface, while angles larger than 90° denote poor wettability, indicating a lithiophobic interface [87]. Oxide SEs, typically brittle when assembled, exhibit weak interfacial compatibility. Within the garnet family, while they demonstrate remarkable electrochemical stability with the Li anode, the metallic Li anode showcases poor wettability against garnets, hindering further applications in ASSLBs [88]. LLZO, boasting an ionic conductivity of 6.46 × 10−1 mS cm−1, tends to develop impurities (like LiOH and Li2CO3) in air, leading to a significant reduction in conductivity to 3.62 × 10−1 mS cm−1, adversely affecting wettability. Surface impurity removal as a pretreatment is necessary to enhance LLZO's wettability with Li metal [74].
Furthermore, during the repeated plating and stripping processes of the Li anode or Li alloy anode (e.g., Li–Si or Li–Sn), cyclic stress occurs due to volume expansion and compression, resulting in void formation and contact loss. This insufficient contact can create uneven Li+ flux distribution, facilitating Li dendrite formation. These dendrites may penetrate SEs through microdefects like pores or grain boundaries, leading to short circuits, especially at relatively low current densities [89]. Therefore, high-quality SEs should be capable of reversible deformation to accommodate anode volume changes, maintaining close contact for sustaining capacity and cycling performance in ASSLBs [90].
The mechanical properties of SEs significantly influence the physical contact between electrodes and SEs. For instance, sulfide SEs lacking metal additives, like LPSC, exhibit better physical contact with Li compared to LGPS due to increased hardness from metal additives. Polymer electrolytes outperform other SE categories, establishing an intimate interface with enhanced flexibility [91].
2.3 Li Dendrites
Li dendrites pose a significant challenge in addressing anode interfacial issues within secondary batteries, capable of causing severe short circuits. ASSLBs were initially devised to couple high-energy Li metal with SEs, aiming to leverage a robust mechanical barrier to prevent the infiltration of problematic Li dendrites through the SEs. However, despite this design, Li dendrite growth remains a critical factor in short circuits within ASSLBs, sometimes occurring even more rapidly than in cells using LEs. The exact mechanisms driving Li dendrite formation are multifaceted and not yet fully understood, as they can either grow from the Li anode toward the SEs or originate directly within the SEs. Previous studies suggest that the primary causes of Li dendrites in ASSLBs encompass interface defects, the inherent mechanical properties of SEs, and the critical current for stripping (CCS).
Anode interfacial defects, such as voids and grain boundaries, typically arise due to inadequate physical contact, volume fluctuations, and low compacting density. When the applied current density exceeds a moderate range, deposited Li tends to nucleate and propagate primarily within the voids and grain boundaries within the SEs. This infiltration can extend throughout the entire SE, culminating in cell failure. The formation and growth of Li dendrites are not confined solely to grain boundaries and cracks but can also occur within amorphous SEs lacking distinct grain boundaries [92, 93].
The well-known Monroe–Newman model outlines that inhibiting Li dendrites becomes feasible when the shear modulus of SEs surpasses twice that of Li metal, which is around 4.8 GPa at RT [94]. As a result, SEs generally need to have a shear modulus higher than 9.6 GPa at RT. However, even inorganic SEs like LLZO (100 GPa) and β-Li3PS4 (12 GPa) possess shear moduli exceeding 9.6 GPa at RT, yet Li dendrite growth remains prevalent [90]. For garnets, an excessively high shear modulus can lead to mechanical flaws and fractures due to strain experienced during cycling [68]. It's important to note that the Monroe–Newman model is only applicable to interfaces devoid of defects or irregularities in SEs [66]. Consequently, Li dendrite growth is notably pronounced in the inherently softer nature of polymer SEs compared to other inorganic SEs, despite the positive traits such as flexibility and excellent processability they offer. Despite extensive efforts to enhance the elastic modulus, it still remains considerably lower than that of the Li anode for all PEO-based SEs, presenting a major challenge for the utilization of polymer SEs [95].
The CCD, which denotes the maximum permissible current density without leading to a short circuit, can be further categorized into CCD for plating (CCP) and CCS. Among these, CCS holds greater significance as it is a pivotal factor in causing ASSLBs failure due to the emergence of Li dendrites, superseding the importance of CCP [96]. Exceeding the CCS threshold results in uneven Li stripping, leading to various drawbacks including void formation on the Li anode surface, diminished solid–solid contact area, escalated local current density at contact points, and ultimately, the short circuit of ASSLBs. External pressure tests reveal that moderate external pressure enhances the CCD. At the Li–SEs interface, it's not diffusion but rather creep that acts as the actual mechanism for Li+ migration. Li metal possesses a yield strength of ≈0.8 MPa, which surpasses the threshold at which Li starts to creep within SEs' pores, eventually fostering Li dendrite formation and culminating in a short circuit [86]. The U.S. Department of Energy aims for an ASSBs power density exceeding 33 kWh L−1, necessitating a CCD of more than 10 mA cm−2 to attain this benchmark [97]. However, currently employed ASSLBs typically operate within a limited CCD range of 1–2 mA cm−2, presenting a challenging path to achieving the desired high power density and expanded CCD. Addressing anode–SEs interfacial issues, curbing Li dendrite formation, boosting CCD, and reducing interfacial resistance require urgent attention through numerous dedicated studies.
On the whole, Li dendrite formation and growth, SEI properties, and volume changes within the Li anode pose the primary bottlenecks at the Li anode–SEs interface, as depicted in Fig. 4. Consequently, key strategies revolve around interlayer insertion to segregate the Li anode and SEs, modifications to the Li anode, and SEs optimization to prevent side reactions and Li dendrite growth while preserving SEs' ionic conduction. Here's a summary of recent strategies aimed at fostering efficient and secure ASSLBs based on the preliminary analysis of these challenges.

Illustrative representation of key interfacial challenges between the Li anode and SEs, along with effective solutions
3 Interfacial Engineering Toward Li and Sulfide SEs
Based on recent studies and earlier discussions, sulfide SEs showcase exceptional ionic conductivity, yet their high reactivity and limited ESW pose significant challenges for sulfide-based ASSLBs at the anode interface. These encompass parasitic reactions, subpar contact, low CCD, and the growth of Li dendrites. A variety of successful strategies have emerged to combat these persistent interfacial concerns between Li and sulfide SEs. These strategies involve incorporating stable interlayers, fine-tuning SE properties, and alloying the Li anode, all aimed at resolving the intricate interface issues between Li and sulfide SEs.
3.1 Insertion of Interlayer
Certainly, one prevalent approach involves incorporating an artificial interlayer to mitigate parasitic reactions and prevent direct contact between the Li anode and sulfide SEs, all while minimizing any significant increase in cell impedance. To effectively serve this purpose, these interlayers must fulfill specific criteria: (i) exhibiting thermodynamic stability with the Li anode; (ii) demonstrating ionic conductivity while being electronically insulative (or exhibiting low electronic conductivity); and (iii) possessing good mechanical ductility. Based on their preparation methods, these artificial interlayers can be categorized into two types: the in situ SEI interlayer and the ex situ buffer interlayer.
3.1.1 In Situ SEI Interlayer
The incorporation of an in situ SEI interlayer serves to prevent parasitic reactions and the formation of mixed ionic and electronic conducting interphases while preserving the ionic conductivity of sulfide SEs. Addressing the challenges associated with the narrow ESW and contact stability with both ambient environment and Li metal is crucial for highly active sulfide SEs in practical applications. Researchers have dedicated significant efforts toward achieving stability at the anode interface, aiming to establish a uniform and robust in situ layer. For instance, Sun et al. demonstrated an in situ generated, air-stable, and highly ionic conductive LixSiSy interphase layer on Li metal, enhancing the stability of Li metal against both air exposure and sulfide SEs. This LixSiSy interphase comprises Li2SiS3/Li4SiS4 and Li2S; the former facilitates rapid Li+ migration through the protective layer, while the latter ensures direct contact with Li metal without triggering parasitic reactions (Fig. 5A) [98].

Insertion of stable in situ SEIs between Li and sulfide SEs. A An air stable SEI formed at the anode interface. a Schematic illustration of formation processes for the in situ LixSiSy interlayer. b Cycling performance of Li–LixSiSy|Li3PS4|LiCoO2 cells. Reprinted with permission from Ref. [98]. Copyright © 2019, John Wiley and Sons. B A stable in situ SEI composed of binary materials formed at the anode interface. a Schematic illustration of LiF-rich in situ interlayer’s formation process between Li3PS4 SEs and the Li anode. b Cycling performance of Li|LiFSI@Li3PS4|LiCoO2 at 0.3 mA cm−2 at RT. Reprinted with permission from Ref. [99]. Copyright © 2018, Chunsheng Wang. c Schematic illustration of a stable SEI LiF interlayer formation process. d Cycling performance of assembled ASSLBs including Li|Li7P3S11|LiCoO2, Li|Li7P3S11(I)|LiCoO2 and Li/Li7P3S11(HFE)|LiCoO2 at 0.1 mA cm−2. Reprinted with permission from Ref. [100]. Copyright © 2018, Elsevier. C In situ SEI interlayer formed at the Li anode interface. a Schematic diagram of the modified interface with a Li3PO4–Li3N composite SEI. b Cycling performance of NPO–Li|LPSC1.59|LiCoO2 batteries and Li|LPSC1.59|LiCoO2 batteries. Reprinted with permission from Ref. [102]. Copyright © 2022, Elsevier
The chemical composition of the interphase formed at the anode–SE interface profoundly influences both the CCD value and the long-term cycling performance. Binary SEI interlayers like Li2S, Li3P, Li3N, LiF, LiCl, LiBr, and LiI demonstrate high interfacial energy, negative decomposition energy, and acceptable electronic conduction, making them favorable for establishing a thermodynamically stable interphase. For instance, when the Li anode is coated with Li bis(fluorosulfonyl)imide (LiFSI) and brought into contact with Li3PS4, an in situ formation of a LiF-rich SEI layer effectively inhibits Li dendrites, resulting in an increased CCD exceeding > 2 mA cm−2 at RT, as showcased in Fig. 5B(a) and Fig. 5B(b) [99]. Another method employs the in situ liquid phase infiltration of methoxyperfluorobutane (HFE) into sulfide SEs, generating a stable interphase comprising LiF at the Li anode–sulfide SE interface, effectively suppressing Li dendrites, as depicted in Fig. 5B(c) and Fig. 5B(d) [100]. Sun et al. synthesized LiPSCl0.3F0.7 as an SE via fluorination, yielding a stable LiF interphase toward Li metal, enabling ultra-stable Li+ transport even under high current densities [101]. Moreover, in Fig. 5C, a Li3PO4–Li3N hybrid interphase, exhibiting desirable ionic conduction and sufficiently low electronic conduction, was constructed on the Li surface through a chemical reaction involving LiNO3/H3PO4 (NPO). The resulting cell displayed commendable cycling performance at 1 mA cm−2 [102]. Density function theory (DFT) calculations revealed that Li3PO4 possesses the highest interfacial energy against Li metal, while Li3N exhibits the highest adhesion energy. Wang et al. introduced trace amounts of propylene carbonate (PC) to form a stable and robust SEI with LiCl-rich LPSC, not only enhancing the Li–SE interface for Li+ transport but also impeding undesirable parasitic reactions and Li dendrite growth [103]. The assembled ASSLBs with LiCoO2|Li6.25PS4O1.25Cl0.75|Li showcased a specific capacity of 116 mAh g−1 under a 1 C rate for 200 cycles.
As a remarkable milestone in ASSLB research, LGPS boasts ultrahigh ionic conduction comparable, and sometimes superior, to LEs at RT [17]. Yet, the reduction of Ge4+ in LGPS against the Li anode can accelerate LGPS decomposition. However, recent research has employed an in situ SEI interlayer to address this concern, yielding promising outcomes. Several effective strategies have emerged to foster a stable and high Li+ transport SEI between LGPS and Li metal. One of these approaches involves leveraging in situ gelation, electrochemical deposition of LEs on the Li anode, and chemical modification of the Li anode. In the Li–LGPS interface realm, substantial efforts have been dedicated to crafting an in situ gel polymer interlayer (GPI) using in situ polymerization technology. This elastic GPI exhibits desirable ionic conduction, enhancing physical contact, promoting uniform Li+ transport, and curbing parasitic reactions and Li dendrite formation [104]. Similarly, in Fig. 6A, a dual-layered structure (GI-P) was created via ring-opening polymerization on the Li metal surface. This structure consists of an inorganic species (the bottom layer) and a polymer (the upper layer), effectively restraining Li dendrites and fostering a deformable SEI [105]. Electrochemically reducing LEs at the Li–sulfide SE interface can generate a nanocomposite interlayer, as depicted in Fig. 6B. Gao et al. demonstrated an in situ preparation of a nano-level SEI composed of organic and inorganic Li salts on Li metal, effectively curbing LGPS reduction reactions during extensive cycling [106]. The stable composite interlayer enabled electrodeposition of the Li anode for over 3 000 h. Another ingenious strategy involves employing an interlayer with high ionic conductivity on the Li anode through chemical modification. Zhou et al. employed a smart chemical iodine vapor deposition method to create an in situ densely interweaving structural LiI interphase. This LiI SEI serves as a bridge between Li and LGPS, exhibiting negligible electronic conductivity yet high ionic conductivity and mechanical strength. This approach showcased effective Li dendrite suppression and outstanding cycling performance even under challenging conditions [107]. Further exploration led to the investigation of a manipulated LiH2PO4 interphase via the chemical reaction of H3PO4 in tetrahydrofuran solvents with Li metal. The resulting LiH2PO4 interlayer on the Li surface demonstrated enhanced protection and intimate physical contact, indicating improved stability between the Li anode and LGPS, as illustrated in Fig. 6C [108].

Insertion of a stable in situ SEI between Li and LGPS. a Schematic illustration of the Li–sulfide SE interface with GI-P@Li and with bare Li, where PR and IR represent polymer-rich and inorganic-rich, respectively. Reprinted with permission from Ref. [105]. Copyright © 2022, Elsevier. b Schematic diagram of composite Li salts as an in situ interphase for improving the anode interface: (i) undesirable interface between Li and LGPS without a composite interlayer; (ii) desirable interface between Li and LGPS with composite Li salts as a protective interlayer. Reprinted with permission from Ref. [106]. Copyright © 2018, John Wiley and Sons. c LiCoO2|LGPS|Li cells with the LiH2PO4 interlayer between the anode and SE. Reprinted with permission from Ref. [108]. Copyright © 2018, ACS publications
3.1.2 Ex Situ Buffer Interlayer
In terms of ex situ buffer interlayers, the integration of a protective layer has emerged as a common approach to stabilize both Li metal and sulfide SEs. However, considering the discussed mechanism of Li+ transport primarily via creep, substantial pressure is required [96, 109]. Nonetheless, extremely high stack pressure can promote Li dendrite formation and lead to short circuits in ASSLBs. To address this challenge, the mechanical constriction mechanism has introduced a graphite-protected Li metal (Li/G) setup (Fig. 7A) that accommodates high external stack pressures [110,111,112]. The Li/G composite anode has demonstrated exceptional performance across different sulfide SE systems, showcasing an 82% capacity retention for an SE system composed of LPSC–LSPS–LPSC after 10 000 cycles at a high rate of 20 C. Additionally, a multi-LPSC composite system exhibited an ultra-high capacity retention of 95% over 700 cycles at 55 °C. Moreover, solid-state plastic crystal electrolytes, known for their high ionic conductivity at RT, have been identified as promising interlayers to address interfacial challenges [113, 114]. For instance, the succinonitrile (SN)-based plastic crystal electrolyte (PCE) interlayer (Fig. 7B) offers good thermal stability, nonflammability, and chemical compatibility with the Li anode [115]. Assembled Li-S batteries with a PCE interlayer showcased outstanding high initial capacity and extremely high capacity retention after 100 cycles. Introducing Al2O3 as an interlayer via atomic layer deposition (ALD) has been another successful strategy to prevent parasitic reactions by forming a thermodynamically stable Li2O–Al2O3 phase for various SE types [116,117,118]. Yao et al. suppressed short circuits in Li–Al2O3|Li5.4PS4.4Cl1.6|Al2O3–Li symmetric cells by sputtering a 400 nm-thick Al2O3 interlayer at the interface between the Li anode and the Li5.4PS4.4Cl1.6 thin membrane electrolyte [119]. Molecular layer deposition (MLD) has also proven effective; for instance, an alucone layer was created via MLD on the anode interface to block electron transfer, suppressing side reactions and Li dendrites at the anode interface (Fig. 7C) [120]. Additionally, binary composite compounds with acceptable ionic conduction and high interfacial energy have served as effective layers to ensure stable interphases for Li+ transport and Li dendrite suppression during cycling. A Li3N–LiF composite designed for a dendrite-free Li–Li3PS4 interface exhibited exceptional performance with an ultra-high value of CCD, even at an outstanding area capacity (6.0 mAh cm−2) (Fig. 7D) [121]. Furthermore, the insertion of an Au thin film by Masahiro Tatsumisago proved advantageous in inhibiting side reactions within the Li anode and Li3PS4, exhibiting excellent rate performance at elevated temperatures [122]. Other types of SEs have also been explored as interlayers. For instance, a LiPON thin amorphous interlayer introduced via radio frequency sputtering by Grovenor et al. improved the wettability between molten Li and LPSC and reduced interfacial resistance, increasing CCD for Li/LiPON|LPSC|LiPON/Li symmetric cells to 4.1 mA cm−2, a promising value for practical applications of ASSLBs [123].

Insertion of a stable ex situ buffer interlayer between Li and SEs. A a The illustration of Li/G structure design. b Scanning electron microscopy of the cross-section for Li/G electrodes. Reprinted with permission from Ref. [110]. Copyright © 2020, Royal Society of Chemistry. c Electrochemical performance of ASSLBs for 10 000 cycles with Li/G electrodes at a high rate of 20 C. Reprinted with permission from Ref. [111]. Copyright © 2021, Springer Nature. B: a Schematic illustration of ASSLBs with the PCE interlayer. b Photographs of the PCE at different temperature. c Cycling performance of ASSLBs with and without the PCE interlayer at 0.13 mA cm−2 for 100 cycles. Reprinted with permission from Ref. [115]. Copyright © 2019, John Wiley and Sons. C a Schematic illustration of the alucone layer on the Li surface through MLD. b Scanning electron microscopy of the Li surface after MLD treatment. c Cycling performance of ASSLBs with and without the alucone layer at 55 °C. Reprinted with permission from Ref. [120]. Copyright © 2018, Elsevier. D a Schematic illustration of the interfacial activation process. b Scanning electron microscopy of structure of the Li3N–LiF interlayer and Li3PS4 SEs. c Voltage profile for the symmetric cell at 1.0 mA cm−2 for 220 h. d Voltage profile of the symmetric cell at high current density of 6.0 mA cm−2. Reprinted with permission from Ref. [121]. Copyright © 2020, John Wiley and Sons
3.2 Optimization of Sulfide SEs
Optimizing sulfide SEs involves modifications and structural designs aimed at reducing residual electronic conductivity and broadening the ESW. This process holds significant importance in restraining the formation of lithium dendrites.
3.2.1 Doping of Exotic Elements
Strengthening the weak P–S bonds is crucial to enhancing the stability of sulfide SEs. Modifying the composition of these SEs offers a straightforward method to boost their thermodynamic stability and widen the ESW while preserving their ionic conduction properties. The β-Li3PS4, a low-symmetry crystalline material, boasts high ionic conductivity but faces instability at RT, limiting its practicality. An effective approach involves anion doping to establish a stable interface between sulfide SEs and Li anodes [31, 124]. The substitution of sulfur with oxygen in β-Li3PS4, suggested by Chen’s group based on bond valence and DFT, strengthens the P–O bonds, offering increased stability without compromising ionic conductivity. This strategy not only stabilizes the medium-temperature crystal phase but also facilitates smoother ion transport pathways, transitioning from a 2D to 3D transport behavior. This advancement preserves the wide ESW of β-Li3PS4. This substitution technique extends to argyrodites as well [125, 126]. Shao et al. [103] explored Li6.25PS4O1.25Cl0.75, demonstrating exceptional cycling performance when paired with Li anodes and LiCoO2, retaining the pristine argyrodite's exceptional ionic conduction. This modification led to the in situ formation of a Li3PO4 interphase, serving as an SEI interlayer at the anode interface, as illustrated in Fig. 8A. This O-doped argyrodite showcased superior capabilities in suppressing Li dendrites and exhibiting resilience against air and humidity. Beyond single atom doping or substitution, various oxides like ZnO[127, 128], Li2O[129, 130], Sb2O5[131], P2O5[132] and Nb2O5[133] have been employed to reinforce the stability of sulfide SEs against both air and Li anodes, as depicted in Fig. 8B. Notably, incorporating ZnO atoms onto the sulfur site not only enhances the SEs' stability but also leads to the formation of a highly conductive LiZn alloy, effectively curbing the nucleation and growth of Li dendrites without sacrificing ionic conductivity.

Substitution or doping for sulfide SEs. A O substitution of S for sulfide SEs. a Process diagram of dendrites growth without O doping for the Li–LPSC interface. b Schematic illustration of inhibition for Li dendrite with a stable interface. Reprinted with permission from Ref. [103]. Copyright © 2022, John Wiley and Sons. B ZnO co-doping in argyrodite. a Schematic illustration of advantages of ZnO-doped argyrodite sulfide SEs. b Elemental mapping on the Li–Li5.7Zn0.15PS5.85O0.15Br interface. c Galvanostatic intermittent cycling of Li|Li5.7Zn0.15PS5.85O0.15Br|Li symmetric cells at RT for 20 h. Reprinted with permission from Ref. [127]. Copyright © 2019, ACS Publications. C Halide doping in sulfide SEs. a Arrhenius plot of these two sulfide SEs of Li3PS4 and Li7P2S8I. Reprinted with permission from Ref. [135]. Copyright © 2015, ACS Publications. b Cycling performance of Li|LPS30I|Li cells at 100 °C [136] Copyright © 2018, John Wiley and Sons. D Sn substitution of P in argyrodite sulfide LPSI SEs. a Schematic illustration of Li|LPSI–20Sn/LGPS|LCO@LNO/LGPS ASSLBs. b Rate performance of Li|LPSI–20Sn/LGPS|LCO@LNO/LGPS ASSLBs at RT. Reprinted with permission from Ref. [137]. Copyright © 2020, John Wiley and Sons
LiX, encompassing various halide elements, stands out as an ideal dopant for enhancing sulfide SEs. This group of dopants contributes significantly to widening the ESW, refining compatibility with the Li anode, and augmenting ionic conductivity. Among these halides, the ionic conductivity follows the order: LiI > LiBr > LiCl > LiF [134]. Liang et al. introduced LiI into Li3PS4, resulting in the creation of a Li7PS8I interphase, exhibiting remarkable electrochemical stability reaching up to 10 V vs. Li/Li+ [135]. This innovative interphase not only demonstrates significantly higher Li+ conductivity (6.3 × 10−1 mS cm–1) compared to β-Li3PS4 and LiI but also bolsters stability against Li metal while reducing charge transfer resistance. Wang et al. showcased the effectiveness of LiI incorporation into Li2S–P2S5 glass electrolytes in suppressing Li dendrite formation, facilitating favorable Li deposition at the anode interface, and enabling a CCD of up to 3.9 mA cm−2 at 100 °C, as illustrated in Fig. 8C [136]. An alternative strategy involves substituting Sn for P in sulfide SEs, yielding stable SEs not only against Li anodes but also in moist air, thanks to the robust Sn–S bonding and high ionic conductivity of Sn-doped SEs [137, 138]. As depicted in Fig. 8D, the LPSI–20Sn electrolyte displayed exceptional plating/stripping behavior over 200 h in symmetric cells, serving as a stabilizing interlayer for Li anodes and exhibiting excellent performance and rate capability [137].
3.2.2 Design of Electrolyte structures
Vertical multilayered sulfide SEs represent a promising strategy, leveraging the best of single SE layers to enhance compatibility and stability at the SE–electrode interface. Li's group introduced a novel sandwiched electrolyte structure (Fig. 9A) to achieve exceptional rate capability and cycling performance [111]. This design concept, likened to an expansion screw effect, employs the newly formed Li dendrites as screws, the inner sulfide SEs like LGPS as anchors, and the surrounding SEs such as LPSC as drywalls. Similarly, a similar structural design involving Cl substitution in argyrodite SEs not only widens the ESW but also suppresses Li dendrite nucleation [112]. Addressing the challenge of poor interfacial stability between superionic conductor LGPS and Li anodes, bi-layer structural designs like LGPS/75%Li2S–24%P2S5–1%P2O5 and LGPS/Li3PS4 have been developed [139, 140]. Additionally, for improved anode stability, Sb2O5 is incorporated into Li3PS4 to create a bi-layer structure of LGPS/Li3P0.98Sb0.02S3.95O0.05. This combination of LGPS with high ionic conductivity and Li3P0.98Sb0.02S3.95O0.05 boasting exceptional stability enhances Li+ flux even at − 10 °C while improving anode interfacial compatibility [131]. However, solely protecting the Li anode doesn't entirely prevent side reactions and Li dendrites. Ci's group devised a robust layer coating on SEs, such as graphene oxide (GO) and Li2S, to mitigate interfacial reactions between Li and Li7P3S11 particles, as well as to guide or induce homogeneous Li deposition (Fig. 9B) [141, 142]. In the Li2S@Li7P3S11 system, the resulting ASSLB exhibited exceptional cycling performance over 150 cycles, serving as an effective means to inhibit Li dendrite formation.

Structural design for sulfide SEs. A Vertically multilayered structured sulfide SEs. a Schematic illustration of Li/G|LPSC–LGPS–LPSC|G/Li. b Cycling performance of sandwiched structural sulfide SEs at a rate of 1 C with different central electrolytes at RT coupled with Li/G anodes and NCM811. c The Ragone plot for typical ASSLBs’ performance. Reprinted with permission from Ref. [111]. Copyright © 2021, Springer Nature. B In situ coated on sulfide SEs. a Schematic illustration of synthesis procedures of Li2S@Li7P3S11 composite sulfide SEs. b The voltage profiles of Li/Li symmetric batteries with and without Li2S coating for Li7P3S11 SEs at 0.1 mA cm−2. The expression “wt%” means “% by weight”. c Bulk transition energy barrier for Li2S and Li7P3S11. Reprinted with permission from Ref. [142]. Copyright © 2022, Elsevier. C Ion-insulation wood template coupled with LPSC structural design. a Schematic illustration of synthesis procedures of LPSC@wood SEs. b Li deposition morphology and distribution within LPSC@wood SEs. c Electrochemical performance of symmetric cells for LPSC (green) and LPSC@wood (orange) SEs at 0.2 mA cm−2. Reprinted with permission from Ref. [143]. Copyright © 2020, Elsevier
In the realm of sulfide SEs-based ASSLBs systems, the strategies mentioned earlier—SEI fabrication and SEs modification—have shown significant promise in improving anode interfacial stability and restraining Li dendrite growth to varying degrees. However, the fundamental issue persists. Completely eliminating the possibility of Li dendrite formation demands a design that facilitates the uniform deposition and growth of Li. In an innovative approach illustrated in Fig. 9C, Wang et al. explored a strategy involving a wood template LPSC to manipulate a consistent Li+ flux. Electrolyte cells assembled with such modified systems exhibited exceptional stability for 1 000 h at 0.2 mA cm−2 (in symmetric cells) and maintained excellent battery performance over 100 cycles (Li4Ti5O12|LPSC@wood|Li cells) [143]. Another intriguing approach was introduced by Liang et al., who engineered an adhesive sulfide SEs by employing hot melting adhesive porous membranes made of ethyl vinyl acetate porous (EVAP) on LGPS. The adhesive force played a pivotal role in enhancing the contact area at the anode interface and improving interfacial stability, offering a fresh perspective on interfacial engineering in ASSLB applications [144].
3.3 Alloying of Li Anode
Li alloys present an enticing alternative to pure Li anodes in ASSLBs, offering enhanced solid–solid contact wettability, promoting uniform Li deposition, and curbing Li dendrite formation during plating/stripping. Several high-capacity Li alloys—like Li–In, Li–Si, Li–Al, and Li–Sn alloys—serve as effective Li dendrite inhibitors, elevating charge–discharge reversibility and long-term cyclability for ASSLBs [145]. Moreover, these alloys offer superior mechanical properties compared to pure Li metal [145]. The newly formed alloyed interphase during cycling also holds promise for enabling high capacity and stable long-term performance in ASSLBs. Among these alloys, Li–In alloys (with a capacity of 220 mAh g–1 for LiIn) are frequently used in lab-scale experiments due to their thermodynamic and kinetic stability [146,147,148]. The Li–In alloy exhibits higher Li+ diffusivity compared to Li metal owing to lower energy barriers for bulk Li diffusion. However, the high voltage difference of ≈ 0.6 V between In and Li leads to a significantly reduced discharging platform, resulting in decreased energy density in ASSLBs. Despite this, most ASSLBs using Li–In anodes only cycle at low current density, insufficient to effectively demonstrate Li–In alloy's ability to suppress dendrite formation. Zhang et al. reported the short-circuiting of a Li–In|LPSC|NCM622 full cell due to Li–In dendrite generation after long-term cycling at high current density and cathode loading (Fig. 10A) [149]. Analysis via scanning electron microscope (SEM), X-ray photoelectron spectroscopy (XPS), and Ab initio molecular dynamics (AIMD) indicated that metallic In lacks absolute thermodynamic and kinetic stability with sulfide SEs, causing volume expansion and interfacial reactions, leading to Li–In dendrite formation and penetration under high current density until cell failure. Unlike vertically grown Li dendrites, the laterally striped morphology of Li–In dendrites exhibits reduced growth rates, minimizing electrolyte structure damage—an important guideline for alloy anode research and development [150]. Li–In alloys can also be obtained from Li-free In foil anodes during Li plating, showcasing improved interfacial contact due to volume expansion without dendrites and exhibiting excellent cycling stability for over 160 cycles. Beyond mechanical and physical mixing methods, Li–In alloy incorporation on sulfide SEs stabilizes the anode surface. Lee et al. devised a unique In–I–Li–P–S layer inserted between the Li anode and the Li3PS4 electrolyte. Composed of Li2S, P2S5, and InI3, this layer forms a Li–In alloy and a stable LiI interphase with chemical stability toward the Li anode [151]. This In–I–Li–P–S protector effectively separates the Li anode and the Li3PS4 electrolyte without causing side reactions, exhibiting outstanding electrochemical performance (cycling over 500 h) in assembled ASSLBs.

Alloyed for Li anodes to improve interfacial properties with sulfide SEs. A Li–In alloy anode used in LPSC. a Schematic diagram of anode interface evolution with the Li anode and the Li–In anode for LPSC before and after cycling. b Long-term cycling performance of Li–In|LPSC |LNO@NCM622 at 3.8 mA cm−2. Reprinted with permission from Ref. [149]. Copyright © 2021, Springer Nature. B: Pure silicon anode enabled by sulfide SEs. a Schematic illustration of interfacial change during lithiation processes between the Si anode and LPSC. b Si|LPSC|NCM811 cycling performance at RT. Reprinted with permission from Ref. [155]. Copyright © 2021, Science. C Carbon-free and binder-free Li–Al alloys used in LGPS. a Schematic illustration of charging and discharging of Li–Al alloy in ASSLBs. b Cycling stability of ASSLBs using Li0.8Al alloy anodes for 200 cycles at 0.2 C. Reprinted with permission from Ref. [162]. Copyright © 2022, Science family of journals/AAAS
Li–Si alloys, offering a voltage plateau of < 0.3 V vs. Li/Li+, boast high theoretical specific capacities of 3 572 mAh g−1 (Li15Si4). Earlier studies using Li4.4Si and Li4.4GexSi1–x as anodes for sulfide-based cells yielded unsatisfactory results [152, 153]. However, recent research utilizing pure Si anodes in sulfide-based cells has shown exceptional performance. Stefan Kaskel et al. investigated a columnar Si anode employing a 1D breathing mechanism via a scalable physical vapor deposition (PVD) process [154]. This 1D Si anode design maintains the interface, reduces the active contact area, and minimizes side reactions during cycling. The assembled Si|LPSC|NCM full cell demonstrated an outstanding area capacity of 3.5 mAh cm−2 and stable cycling performance with an excellent Coulombic efficiency (CE) of 99.7% − 99.9% over 100 cycles. In cyclic voltammetry (CV) measurements of cathodic scans for Si|SE|Li half cells, Si exhibited three phase transformations: a-Si to PI phases (Li–50% by atom number (hereinafter the expression “% by atom number” is referred to as “at%”) Si; LiSi), the PI phase to the PII phase (Li–30 at% Si; Li7Si3) at 0.21 V vs. Li/Li+, and the PII phase to the PIII phase (Li–24 at% Si; Li3.16Si) at 0.05 V vs. Li/Li+. The discharge voltage is influenced by the Si anode's voltage plateau, which extends to 1.0 V instead of providing a constant voltage plateau (as seen in Li or In anodes). Consequently, the cut-off voltage for Si anode-based ASSLBs is reduced to 2.0 V for complete anode utilization. Meng et al. reported a 99.9% by weight (hereinafter the expression “% by weight” is referred to as “wt%”) μSi anode used in μSi|LPSC|NCM811 full cells, achieving remarkable interfacial stability and cycling performance (Fig. 10B) [155]. This μSi anode, devoid of carbon additives, prevents sulfide SE decomposition while forming a Li–Si interphase between Si and sulfide SEs, facilitating both electronic and ionic transport. Operating at wide temperatures ranging from − 20 to 80 °C, the critical current density (CCD) reaches 5 mA cm−2, and for the first time, the area capacity reaches up to 11 mAh cm−2, retaining an outstanding capacity of 80% over long-term cycling. This provides compelling evidence for the viability of Si anodes in ASSLBs. Zhu et al. investigated ASSLBs using nano-Si anodes, achieving an excellent capacity of 145 mAh g−1 over 1 000 cycles [156]. Unlike Meng’s research [155], carbon additives in nano-Si did not result in sulfide SE decomposition, possibly due to the lower electronic conductivity of nano-Si, three orders of magnitude lower than μSi, attributed to its larger surface area and SiO coating. Furthermore, in the same system, Zhu’s group designed bipolar stacking ASSLBs with a high voltage of 8.2 V and an energy density of 204 Wh kg−1 [157]The ionic conductivities o The advancements in Si anode research are poised to drive large-scale commercialization and progression in ASSLBs.
Kim et al. introduced the Ag–Li anode to establish a stable interface between the anode and LPSC by forming an in situ intermetallic layer composed of Ag–Li [158]. Their assembled Ag–Li|LPSC|NCM full cell exhibited superior cycling performance even under a high rate of 12 C. Utilizing a thin Ag–C nanocomposite layer as the anode effectively regulated Li deposition and improved stable cycling performance [159]. The solubility of Ag in Li forms a stable Li–Ag alloy that maintains an interface, while carbon acts as a separator, preventing SEs from direct contact with the Li anode. This assembled pouch cell demonstrated an outstanding energy density of > 900 Wh L−1 and a CE of > 99.8% over 1 000 cycles. Building on the insights gained from the Ag–C layer [159], Yang et al. employed a clever design by incorporating a Li-based Li–B alloy within a 3D skeleton and an Ag@C layer. The Li–B alloy countered the volume changes in the anode during cycling, while the Ag@C layer maintained a stable interphase [160]. Choi chose an Ag–C composite anode with an elastic spandex binder, resulting in the formation of Ag–Li alloys at the anode interface [161]. Zhou et al. designed a reliable anode using a Li0.8Al alloy without carbon and binder additives for ASSLBs, as depicted in Fig. 10C [162]. The Li0.8Al alloy exhibited excellent performance toward LGPS, retaining an outstanding capacity of 93.29% for 200 cycles and an energy density of 541 Wh kg−1. These various strategies employing Li alloying provide promising options for anodes and significantly expedite the practical implementation of ASSLBs.
4 Interfacial Engineering Toward Li and Oxide SEs
Numerous oxide SEs, such as Li phosphorus oxynitride (LiPON), despite being relatively inexpensive, exhibit undesirable ionic conduction of 2 × 10−3 mS cm−1, making them unsuitable for practical applications—an aspect extensively covered in previous reviews and thus won't be discussed here [163]. Instead, the focus lies on the most promising oxide SEs, particularly garnets and NASICONs. In the case of garnets, while the interface stability between oxide SEs and the Li anode is commendable, garnets possess a lithiophobic nature that hampers their contact with the Li anode, leading to undesirable interfacial resistance reaching several or even hundreds of kΩ cm2. Additionally, this lithiophobic characteristic restricts effective wettability at the anode interface, resulting in uneven current distribution and the formation of Li dendrites, which significantly hinders the practical application of garnets. On the other hand, NASICONs demonstrate high Li+ conductivity and exceptional air stability. However, their development faces hurdles due to interfacial challenges, including high interfacial impedance and severe side reactions with Li metal, impeding their progress in practical applications. Researchers have explored various approaches to address these issues in oxide SEs, encompassing electrolyte modification, the creation of artificial solid-state electrolyte interfaces, and composite Li anodes. In the subsequent sections, we will consolidate the recent strides made in advancing strategies to enhance the interfaces of oxide SEs-based ASSLBs.
4.1 Insertion of Interlayer
Introducing an ideal SEI stands as a potent strategy to resolve the interfacial challenges encountered between the Li anode and oxide SEs. This approach not only enhances the wettability of Li on oxide SEs but also reduces interfacial impedance significantly. This section will focus on summarizing the SEI interlayer insertion strategy for two primary types of oxide SEs—garnets and NASICONs.
4.1.1 Interlayer for Li–Garnets
The interfacial resistance between garnets and the Li anode is heavily influenced by the presence of impurities like Li2CO3 and LiOH. Addressing these issues involves introducing an artificial SEI to reduce interfacial impedance, enhance wettability, and improve the lithiophilic affinity between the Li anode and garnets. Additionally, the insertion of an SEI interlayer proves beneficial in curbing cracks and defects within oxide SEs. To meet the necessary criteria for an effective artificial SEI, several key requirements must be fulfilled: (1) stability with garnets; (2) high Li+ diffusivity; (3) minimal volume change during cycling; (4) minimal thickness to not compromise the energy density of ASSLBs. Various types of SEI interlayers exist, including liquid metals, metal oxides, conductive binary compounds, Li alloys, electrically conductive materials, covalent organic frameworks (COFs), polymers, and others. Liquid metals can infiltrate garnet grain boundaries to create additional ionic transport paths. Li's group proposed using liquid Ga as a lithiophilic layer on garnets, avoiding the need to remove Li2CO3. This method significantly reduces Li–garnet interfacial resistance, ensuring high battery reversibility [164]. Oxide interlayers enhance the wettability of oxide SEs for Li metal. For instance, an ultrathin Al2O3 film deposited on Li7La2.75Ca0.25Zr1.75Nb0.25O12 using the ALD method has shown promising results. This lithiated Al2O3 interface drastically reduces interfacial impedance and improves wettability, albeit with some complexity and cost limitations in the ALD method (Fig. 11A) [88]. Another approach involves coating a Ta2O5 nanofilm on LLZTO, significantly decreasing interfacial impedance and resulting in high-performance full cells with impressive endurance [165].

For optimizing the interface between Li anodes and garnet SEs, a stable SEI interlayer insertion strategy is applied. A Al2O3 interlayer for improving interface between Li and Li7La2.75Ca0.25Zr1.75Nb0.25O12. a Scanning electron microscopy of the Li–garnet interface without and with Al2O3 interlayers. b Corresponding EIS’s comparison results. Reprinted with permission from Ref. [88]. Copyright © 2017, Springer Nature. B Schematic illustration of the desired Li–garnet interface. a Schematic illustration of the Li–garnet interface with and without surface modification. b The first charge–discharge curve of Li|Sn–LLZNO|LiFePO4 ASSLBs. Reprinted with permission from Ref. [167]. Copyright © 2018, The Royal Society of Chemistry. C Binary materials interlayer for improving the interface between garnet–type oxide SEs and electrodes. a Schematic diagram and advantages of the interfacial stability strategies of garnet–type oxide SE’s ASSLBs toward electrodes. b Capacity and CE at 0.2 C under 60 °C for NCM622|modified–LLZTO|Li cells. c CCD test results of symmetric cells with and without garnet surface modification. Reprinted with permission from Ref. [177]. Copyright © 2021, Elsevier. D Combing alloy phases and binary materials as the interlayer for garnet–type oxide SEs. a Diagram of preparation processes of the Li–LLZTO interface through chemical reaction between Zn(NO3)2 and Li. b Long-term cycling tests results with improved interfaces at 1 − 2 mA cm−2 for Li symmetric cells. Reprinted with permission from Ref. [183]. Copyright © 2020, John Wiley and Sons
The right interlayer can establish an alloy interphase, stabilizing the Li–garnet interface, reducing interfacial impedance, and enhancing garnet surface wettability. Zhang et al. introduced In2(1–x)Sn2xO3 (ITO) as an interlayer, forming a LixIn/LixSn interphase that effectively bonds the Li anode onto the LLZTO surface through conversion and alloying reactions [166]. Guo et al. addressed LLZTO–Li anode interfacial issues by modifying LLZTO with a Sn film, reducing interfacial resistance by approximately 20 times through Li–Sn alloy formation (Fig. 11B) [167]. Liu et al. applied a SnS2 film on LLZTO, significantly lowering interfacial impedance to ~ 17 Ω cm2, enhancing wettability, and improving CCD for ASSLBs [168]. Hu et al. in situ coated a Si film on garnets, creating a super lithiophilic SE that displayed low impedance and excellent cycling stability [169]. Alloy formations like Li–Ag [170, 171], Li–Zn [172, 173] and Li–Al [174] alloys have also proven effective in adjusting garnet wettability. However, there's a continual need for more appropriate alloy interphases to meet evolving requirements.
Interphases consisting of conductive binary materials like Li3N, Li3P, Li2O, and LiF offer strong capabilities in impeding Li penetration in garnets [175,176,177]. Guo et al. introduced a practical SEI insertion strategy involving inorganic and organic compounds (Fig. 11C) [177]. This SEI interlayer, fostering high Li+ and electronic conductivities between LLZTO and the Li anode, enhances wettability without impeding charge transfer. Sun et al. implemented an MIEC interlayer with Li3N/Cu nanoparticles at the anode–garnet interface via magnetic sputtering technology [89]. This interlayer aids Li+ transport, regulates electric field distribution, suppresses Li dendrite generation, and optimizes anode interface contact. A chemical reaction between Li and SnF2 forms an in-situ mosaic structural Li7Sn3/LiF interphase at the Li–garnet interface [178]. Li7Sn3 acts as an ionic conductor, relieving mechanical stress during cycling, while LiF expedites Li+ transport through Li7Sn3.
Combining alloy phase construction with conductive binary materials, a mixed conductive layer showcasing robust electronic and ionic conductivity effectively diminishes interfacial impedance, improves Li–garnet anode physical contact, and suppresses Li dendrite formation. An introduced Si3N4 layer on garnets forms a stable interphase of Li–Si alloy and Li3N through a reduction reaction with Li metal, ensuring tight Li anode contact, homogeneous electric field distribution, and a significant drop in interfacial resistance [179, 180]. A garnet-based ASSLB incorporating a Li2S/LixSn interphase exhibited an interfacial impedance of 47 Ω cm2 and sustained a long-term cycling life over 1 000 h [181]. A homogenous lithiophilic interphase comprising InLix and LiCl was crafted between the Li anode and LLZTO by using a straightforward, cost-effective wet chemistry method [182]. This approach showcased a reduction in interface impedance from 189 to 1 Ω, an increase in CCD from 0.2 to 0.7 mA cm−2, and exceptional electrochemical performance over 475 cycles. Wang et al. proposed an in situ method to generate a ZnLix and Li3N interlayer between Li and LLZTO via an energetic chemical reaction between Zn(NO3)2 and Li [183]. This approach demonstrated a CE of > 99.5% without Li dendrite generation over thousands of hours of cycling, as depicted in Fig. 11D.
The cycling-induced localized stress often triggers the formation of Li dendrites. Carbon materials, known for their high ionic and electronic conductivity, serve as excellent soft buffer layers, ensuring electro-chemo-mechanical stability. Li et al. devised a lithiophilic candle soot (CS) shield atop LLZTO using flame vapor deposition, which displayed a polycrystalline structure with graphitic domains (Fig. 12a, b) that mitigated the carbonate layer [184]. The Li|CS-LLZTO|FeF3 cell exhibited an exceptional capacity of 500 mAh g−1 over 1 500 cycles, showcasing remarkable long-term cyclability. Similarly, a graphite-based soft interface was created as a buffer layer via pencil drawing, demonstrating outstanding ionic and electronic conductivities (Fig. 12c, e). This straightforward method ensures uniform Li+ distribution, resulting in ASSLBs with superior rate capacity and long-term cyclability [185]. Hu et al. utilized amorphous carbon between Li and garnets to uphold electronic conductivity and regulate Li+ flux, addressing electro-chemo-mechanical stability concerns (Fig. 12f, g) [186]. Li et al. introduced a novel strategy involving the one-step fabrication of LLZTO with a lithiophilic graphite (iGr@LLZTO) interface. This approach reduced resistance from 4 351.6 to 26.2 Ω cm2, decreased overpotential from 500 to 30 mV, and improved contact between iGr@LLZTO and the Li anode while ensuring compatibility and efficient Li+ transport [187].

Insertion of the carbon buffer layer between Li anodes and garnet-type SEs. a Illustration of CS preparation processes. b Cycling tests results of the Li|CS–LLZTO|FeF3 ASSLBs under different current densities from 100 to 400 μA cm−2. Reprinted with permission from Ref. [184]. Copyright © 2020, ACS Publications. c Schematic illustration of preparation processes for graphite-based interlayers through pencil drawing. d Comparison of wettability for molten Li metal on Li5.9Al0.2La3Zr1.75W0.25O12 (LLZWO) with and without interlayers. e Cycling performance of ASSLBs at 0.5 C. Reprinted with permission from Ref. [185]. Copyright © 2018, ACS Publications. f and g Schematics illustration of Li+ transport behaviors without and with carbon interlayers during plating/stripping process. Reprinted with permission from Ref. [186]. Copyright © 2021, ACS Publications
To maintain electro-chemo-mechanical stability, a stress-adaptive interlayer proves effective. Luo et al. demonstrated a soft hyperelastic polydimethylsiloxane (PDMS) substrate (Fig. 13A) that effectively suppressed Li dendrite formation and cracks in garnets during cycling [188]. In garnet-based ASSLBs, the rigid contact between oxide SEs and Li demands a softer polymer interlayer to reduce anode interfacial impedance and enhance contact with the Li anode. An in situ solidified gel polymer electrolyte (GPE) buffered the interface between the electrode and garnets, fortifying the garnet blocks and enabling flexible solid batteries [189]. Additionally, Fan et al. devised a polymer SE and 3D Li metal structure specifically for suppressing Li dendrites using 3D frameworks [190]. COFs, with their regular open channels, facilitate improved wetting of garnets by the Li anode and swift Li+ conduction. COF-based layers serve as interlayers to enhance garnet lithiophilicity and diminish interfacial impedance. For instance, sulfonated COFs were prepared to boost the lithiophilicity of LLZTO against the Li anode and establish a pathway for Li+ diffusion at the anode interface (Fig. 13B) [191]. The resulting symmetric solid-state Li cells demonstrated lower interfacial impedance and a high CCD of 3 mA cm−2.

Organic materials interlayer between Li and garnets. A Integration of the Li anode with the PDMS substrate. a Optical illustration of Li foil. b Optical illustration of compressible PDMS/the Li soft anode. c Cycling performance of ASSLBs with and without PDMS with LiCoO2 at 1 C for 100 cycles. d and e Electrochemical profiles of ASSLBs with and without PDMS after different cycles. Reprinted with permission from Ref. [188]. Copyright © 2020, ACS Publications. B Constructed COF interlayer between the Li anode and LLZTO. a Preparation process for building the COF interlayer on the garnet surface. b Comparison of wettability for molten Li on garnetw with and without COF interlayers. c Cross-section scanning electron microscopy of the interface between LLZTO and Li metal with COF interlayers. d EIS results of the symmetric cells with and without COF interlayers. Reprinted with permission from Ref. [191]. Copyright © 2020, John Wiley and Sons
4.1.2 Interlayer for Li–NASICONs
The reduction of NASICONs by Li metal can be controlled by incorporating an SEI interlayer. In the case of LATP, various artificial buffer interlayers have been explored to tackle interfacial issues, spanning from inorganic to organic solutions. For instance, the use of ultrathin ZnO helped create a multifunctional SEI with low electronic conductivity on LATP [192]. Similarly, like LLZTO, an Al2O3 protective interlayer applied via ALD technology can safeguard the Li–LATP interface [193]. Employing organic LEs on the Li anode allows the formation of an artificial SEI at the Li–LATP interface. A stable interphase containing LiF, MgF2, and B2O3 was achieved by using a DME solution containing LiBF4 and Mg(ClO4)2, showcasing excellent capacity retention (84.6%) over 500 cycles [194, 195]. Polymer SEs, known for their viscoelastic and flexible properties, particularly PEO-based polymers, have been utilized, although PEO's efficiency tends to decrease below 60 °C [196,197,198]. Additionally, a biomaterial-derived interlayer made of a conformal sericin protein film (SPF) has emerged as a novel prospect for NASICONs-based ASSLBs, effectively preventing LATP reduction and Li dendrite formation (Fig. 14A) [199].

Insertion of the SEI for NASICONs. A Schematic illustration of interface environment of Li/IL–LATP (left) and Li/IL@SPF–LATP (right) during cycling processes. Reprinted with permission from Ref. [199]. Copyright © 2022, Elsevier. B GPE layer for stabilizing the interface between LAGP and the Li anode. a Diagram of processes for preparing the GPE modified at the interface between LAGP and Li. b The crystal structure of LAGP. c The cross-linked PMMA’s chemical structure. d Schematic illustration of ASSLBs with the pristine LAGP. e Charge/discharge profiles of ASSLBs at different rates at RT. Reprinted with permission from Ref. [204]. Copyright © 2022, Elsevier
Various interlayers have been employed to enhance surface stability and reduce impedance for LAGP. Strategies like the implementation of a LiF-Li3N layer as an SEI or using a metallic Bi thin film have been proposed to boost compatibility and inhibit side reactions at the Li–LAGP interface [200, 201]. Additionally, the application of LiPON thin films as separators on B2O3-added LAGP, generated via RF-sputtering technology, has shown promise in mitigating side reactions [202]. Efforts have also focused on creating PEO-based buffer interlayers to stabilize the anode interface for LAGP. For instance, a poly(vinylene carbonate) (PVCA)-based buffer interlayer successfully improved interfacial compatibility, achieving a full cell with exceptional cycling performance, retaining 96% capacity over 200 cycles [203]. Similarly to garnets, the GPE employing an 8.85 μm thickness PMMA as an SEI exhibited efficient suppression of side reactions and Li dendrite formation, showcasing good cycling stability for 200 cycles at 25 °C (Fig. 14B) [204].
4.2 Optimization of Oxide SEs
The diversity in ionic conductivities and chemical properties across various types of oxide SEs results in distinct advantages and disadvantages. For example, garnets demonstrate acceptable high ionic conduction properties ranging from 0.1 to 1 mS cm−1. They exhibit relatively high stability but are plagued by issues such as large impedance and limited wettability toward the Li anode. On the other hand, NASICONs showcase remarkable ionic conduction properties within the range of 1 to 10 mS cm−1. However, they also suffer from significant side reactions and poor contact with Li. Consequently, modifications in various approaches are essential for optimizing oxide SEs to achieve ASSLBs with high capacity and stable long-term cycling performance.
4.2.1 Conditioning of Electrolyte Surface
The primary challenge with garnets lies in their inadequate physical contact, leading to substantial interfacial impedance. Moreover, garnets exhibit a propensity to spontaneously exchange Li+ and H+, making them susceptible to water and air, resulting in impurities like LiOH and Li2CO3 on their surface. These impurities hinder effective contact with Li metal and diminish ionic conductivity, posing significant barriers to the commercial use of garnets. Surface conditioning emerges as a promising method to address these issues through physical or chemical means [205]. Physical methods like polishing or heat treatment have been employed to eliminate the Li2CO3/LiOH passivation layer, although complete impurity removal via polishing alone remains challenging. Elevated temperature treatment aids in breaking down Li2CO3 to form garnets without Li2CO3, as depicted in Fig. 15A. Chemical treatments, such as using acids like HCl or H3PO4, have proven effective in removing the passivation layer [206]. For instance, Wen et al. utilized H3PO4 to eliminate the passivation layer, forming desirable Li3PO4 with enhanced wettability and ionic conductivity, illustrated in Fig. 15B [164]. An alternative surface chemistry method involves creating a fluorinated interface to facilitate ionic diffusion. However, precise control over the acid treatment duration is crucial to preserve the garnet's volume and prevent further corrosion [207].

Physic and chemical method for garnet surface conditioning. A Contact angle measurements for oxide SEs with different physical treatments for molten metallic Li. a Molten metallic Li on Li2CO3 without any treatment. b Molten metallic Li on dry polishing (DP) LLZO. c Molten metallic Li on wet polishing (WP) LLZO. d Molten metallic Li on wet-polishing–heat-treatment (DP–HT) LLZO. Reprinted with permission from Ref. [205]. Copyright © 2017, ACS Publications. B Diagram of processes for the Li3PO4 modified interlayer for improving the interface between garnet SEs and molten metallic Li. Reprinted with permission from Ref. [164]. Copyright © 2019, The Royal Society of Chemistry
4.2.2 Doping of Exotic Elements
High grain boundary resistance is a critical factor leading to cell failure, especially in garnets. Two effective approaches involve doping exotic elements or inorganics into oxide SEs and optimizing the preparation processes. The former method is a compelling strategy to enhance the relative density of oxide SEs, reducing microdefects like voids and pores while eliminating grain boundaries. For instance, Murugan et al. explored Al-doped Li7La3Zr2O12 with 1 wt% Li4SiO4 additives, achieving an impressive relative density of 98% [208]. Similarly, Ti-doping, Ta-substitution, and Al2O3-additives for garnets have shown promise in enhancing density and improving interfacial wettability [209,210,211]. These modifications result in garnets with a relative density exceeding 99%, significantly reducing grain boundary effects. Regarding the optimization of the preparation process, various strategies have been employed to enhance the densification of oxide SEs. Techniques such as sintering processes, rapid induction hot pressing, and cold isostatic pressing have been pivotal in this regard [212,213,214]. Guo et al., utilizing flowing oxygen sintering technology, achieved garnets with an exceptional relative density of up to 96% and an impressive ionic conduction rate of 0.74 mS cm−1 [215]. Another innovative approach by Wen et al. involved a Li2TiO3-assisted sintering strategy for modifying grain boundaries in Li6.5La3Zr1.5Ta0.5O12. The presence of Li2TiO3 at the grain boundary reduced electronic conductivity while enhancing mechanical properties and bonding strength in the modified oxide SEs [216].
4.2.3 Design of Electrolyte Structures
Due to the inherent challenge of establishing a strong connection between oxide SEs and the Li anode, applying high stack pressure has been a reliable method to eliminate pores and foster their contact. However, excessive pressure, acting as a driving force, can lead to mechanical short circuits in ASSLBs if it surpasses a critical threshold [86]. To address this issue, Cui et al. introduced an innovative three-dimensional (3D) micropatterned approach, ensuring a stable morphology at the Li metal interface even under limited stack pressure and high current density [217]. The adapted ASSLBs displayed exceptional performance, sustaining stable cycling for over 500 h at 0.5 mA cm−2. This innovation mitigated local current density and reduced mechanical stress at the anode interface. The 3D framework, facilitating robust ionic conduction, played a pivotal role in suppressing Li dendrites [218,219,220]. Additionally, a multilayer design for LLZO has emerged as another viable strategy to address interfacial challenges, notably through bilayer and trilayer garnet frameworks [221]. Hu et al. introduced a 3D garnet framework, maintaining a porous-dense-porous (PDP) trilayer structure that concurrently supported both ionic and electronic conduction [220]. During controlled electrochemical deposition, this structure facilitated the formation of a 3D Li anode with increased contact area and reduced interfacial resistance. Similar PDP designs have been employed in Li-S batteries, resulting in outstanding specific capacities exceeding 1 200 mAh g−1 and nearly 100% Coulombic efficiency [219].
4.3 Composition of Li Anode
The optimal Li anode–garnets interface requires expansive contact, minimal interfacial resistance (< 100 Ω cm2), and a dendrite-free environment. Creating composite Li anodes serves a dual purpose: reducing interfacial impedance and enhancing garnets' wettability. For example, casting 0.5 wt% Na-doped Li melt (Li☆) on LLZTO surfaces successfully transferred impurities from the oxide SE's grain boundaries to the Li☆ top layer (Fig. 16A) [222]. This innovative approach resolves wettability issues, enabling high-performance full cells with interfacial resistance < 1 Ω cm2, a high current density of 1.5 mA cm2, and a lifespan exceeding 1 250 cycles. A design strategy introduces a 3D LiCux nanowire network infiltrated with Li metal at the Li–garnets interface, facilitating high-performance cells with a reversible capacity of 147 mAh g−1 and long-term cycling capabilities [223]. Efforts to improve Li's viscosity and enhance the anode interface wettability remain paramount. While a low-fluidity, high-viscosity Li–C composite anode improves cell performance (Fig. 16B), lithiated Li–C's high electronic conductivity poses risks of MIEC interphase formation, leading to Li dendrite formation [224]. A solution arrives in the form of a high-viscosity Li-BN nanosheets (Li-BNNS) composite anode adhering to garnets, resulting in an in situ formed Li3N interphase that effectively inhibits Li dendrite generation (Fig. 16C) [225]. Introducing Li0.3La0.5TiO3 into the Li anode resolves the physical contact issue at the Li–garnets interface. This composite anode enhances anode interface wettability, decreases interfacial impedance, and offers additional Li storage owing to Li0.3La0.5TiO3’s inherent storage capacity [226]. Similarly, the Li–Mo composite anode reduces cohesive energy and improves interfacial binding energy with garnets [227].

Li metal modification for improving compatibility with oxide SEs. A The wettability performance of garnets toward different liquid metals. a The digital photos of Li on different garnet surfaces; b The digital photos of Li☆ on different garnet surfaces; c The digital photos of Na on different garnet surfaces. d Cross-section of scanning electron microscopy for Li☆–LLZTO interfaces. Reprinted with permission from Ref. [222]. Copyright © 2020, John Wiley and Sons. B Interfacial performance for the LLZTO/pure Li or Li–C composite anode. a The photos of the LLZTO surface. b Digital photos of molten Li–C. c Digital photos of molten Li–C on LLZTO. d Scanning electron microscopy images of the garnet/Li–C interface. e Scanning electron microscopy images of the garnet–pure Li interface. Reprinted with permission from Ref. [224]. Copyright © 2019, John Wiley and Sons. C Li-BNNS composite strategy for improving the garnet/Li interface. a Contact angle measurements of pure Li on garnets. b Contact angle measurements of Li-BNNS composites (1%). c Contact angle measurements of Li-BNNS composites (2%). d Contact angle measurements of Li-BNNS composites (5%). e Scanning electron microscopy images of pure Li–garnets. f Scanning electron microscopy images of pure Li-BNNS–garnets. Reprinted with permission from Ref. [225]. Copyright © 2019, ACS Publications
The pursuit of reducing interfacial impedance between garnets and Li anodes has been a central focus. Achieving a lithiophilic interphase to enhance wetting while simultaneously managing electronic conductivity to prevent Li dendrite formation poses a significant challenge. Balancing these factors in interface design has been a persistent challenge for researchers. Taking cues from biological functional gradient materials, Goodenough et al. devised a functional gradient Li anode (FGLA), comprising Li–LiAl–LiF with substantial interfacial energy differences, illustrated in Fig. 17A [228]. This FGLA not only lowers interfacial resistance but also supports a CCD exceeding 3.0 mA cm−2. In a divergence from the lithiophilic interlayer strategy, Huang et al. introduced Sr doping into the Li anode to create a lithiophilic/lithiophobic bifunctional layer, encompassing Li–Sr/SrO-doped Li2O, detailed in Fig. 17B. This bifunctional layer aims to inhibit reduction reactions between Li and oxide SEs by positioning the lithiophilic layer toward the Li anode and the lithiophobic layer toward the garnets [229].

Composite Li anode to balance the low impedance and suppress Li dendrites. A The functional gradient Li anode strategy for improving anode–garnet interfaces. a The atomic structures and interfacial energies for different materials of Li, LiF, LLZTO and Li9Al4. b Schematic illustration of the FGLA strategy for improving the LLZTO–anode interface. c Scanning electron microscopy images of the FGLA–LLZTO interface. d Energy dispersive X-ray spectroscopy results of the FGLA–LLZTO interface. e CCD profiles of FGLA|LLZTO|FGLA symmetrical cells. Reprinted with permission from Ref. [228]. Copyright © 2022, John Wiley and Sons. B Lithiophilic/lithiophobic bifunctional layers for improving the Li–garnet interface. a Scanning electron microscopy images of the cross-section for the Li–Sr-1%|LLZTO interface. b TOF–SIMS profiles for the Li–Sr-1%|LLZTO interface. c XPS results for the Li–Sr-1%|LLZTO interface. d Atomic structures of the Li (110)–Li2O (111) interface, the Li2O (111)–garnet (100) interface, the Li (110)–SrO (100) interface, and the SrO (100)–garnet (100) interface, with the corresponding calculated interface energy γ by DFT. e Diagram of the Li–Sr alloy|SrO-doped Li2O|garnet. Reprinted with permission from Ref. [229]. Copyright © 2022, ACS Publications
5 Interfacial Engineering Toward Li and Polymer SEs
Unlike sulfide and oxide SEs, polymer-based SEs present advantages in adhesion to Li–polymer interfaces. However, practical applications still face various unresolved challenges. These include low Li+ conductivity, elevated interfacial resistance, limited chemical and mechanical stability against Li metal, potential side reactions, and the formation and growth of Li dendrites. Moreover, the persistent hurdle of the shuttle effect leads to internal short circuits, self-discharge, and compromises in cycle efficiency for polymer SEs. Interactions between the polymer SEs, the lithium salt system, and the Li anode are pronounced, making the formation of a stable SEI difficult. This results in substantial interfacial resistance, even during cell inactivity. Addressing the interfacial issues within Li–polymer SEs necessitates enhancements in Li+ conductivity and SEI stability, alongside bolstering the mechanical properties of polymer SEs to surpass a shear modulus of > 9.6 GPa [94]. Various design strategies, predominantly interlayer construction and electrolyte optimization, have been introduced to tackle these challenges.
5.1 Insertion of Interlayer
The formation of a stable SEI between the Li anode and polymer SEs stands as a pivotal factor in suppressing Li dendrite formation, ultimately ensuring exceptional cycle efficiency and longevity for the cells [230]. Zhou et al. delved into a series of polyoxalate (POE) structural polymer SEs terminating with trifluoroacetate (POE–F) units to lower the HOMO value and enhance antioxidative capacity [231]. Consequently, this approach facilitated the creation of a LiF-based interphase, optimizing the compatibility between Li and POE–F at the interface. Goodenough et al. proposed an in situ Li3P interphase formation at the Li–polymer SEs interface using black phosphorus additives to reduce interfacial impedance and enhance the wettability of polymer SEs [232]. This interlayer exhibited robust adhesion to the Li anode, accommodating volume changes and mitigating internal stress during the cycling of assembled ASSLBs. Cui's group developed an ultrathin polymer SE comprising polyacrylonitrile (PAN)–PEO/LiTFSI with a PAN membrane matrix, displaying remarkable mechanical strength and effective suppression of Li dendrite formation. The resulting stable interphase, consisting of LiF and Li3N (Fig. 18), was attributed to PEO for the LiF interphase and PAN for generating the Li3N interphase [233]. PAN's robustness also enhances the safety and stability of assembled ASSLBs, ensuring improved performance even at higher temperatures. Compared to PEO-based counterparts, PVDF-based polymer SEs present a wider ESW, heightened mechanical strength, and superior ionic conductivity [234]. An intricately structured interphase with a mere 20 nm thickness emerged between the PVDF–LiFSI electrolyte and the Li anode, encompassing LiF, Li2CO3, Li2O, and sulfur compounds [235]. This high ionic conductivity, low electronic conductivity, and impressive mechanical strength in the interphase promote uniform Li deposition and stripping during cycling.

a Diagram of preparation processes for PAN membranes. b Digital photo for PAN membranes. c Structure of PAN membranes. d SEM images of PAN membranes. e SEM images for the cross-section of PAN membranes. f Diagram of PAN–PEO/LiTFSI SEs. g SEM images of PAN–PEO/LiTFSI SEs. h SEM images for the cross-section of PAN–PEO/LiTFSI SEs. i EIS results for different times for symmetrical cells. j Diagram of the SE–Li interface, where the SE is with and without PAN. Reprinted with permission from Ref. [233]. Copyright © 2022, John Wiley and Sons.
Furthermore, ALD-prepared Al2O3 finds application in stabilizing the interface between the Li anode and PEO–LiTFSI polymer SEs, effectively curbing the shuttle effect caused by lithium polysulfides. This ALD–Al2O3 layer enhances the electrochemical properties of Li–S batteries, manifesting in heightened areal capacity, improved CE, and reduced self-discharge rates [236]. Addressing the compatibility concerns between the Li anode and polymer SEs, Shen et al. utilized a GO coating on PPC-based SE membranes, leading to the formation of reduced GO at the interface with the Li anode. This modification resulted in an enhanced ion conduction property (0.222 mS cm−1), a widened ESW reaching up to 4.8 V, and improved Li+ migration capability [237]. Similarly, a graphite layer serves as a protective element to enhance the compatibility between polymer SEs and Li, effectively suppressing Li dendrite formation [238].
In the realm of LEs-based LIBs, the integration of 3D structural Li anodes is considered a strategy to inhibit Li dendrites while providing an enlarged surface area with lithiophilic sites. Inspired by this concept, 3D structural Li/Cu mesh anodes [239] and 3D Li/Ni anodes [240] have been employed in assembling polymer SEs-based ASSLBs, showcasing exceptional cycling performance. However, relying solely on 3D Li anodes might not suffice in reducing local current density and optimizing physical contact with polymer SEs. Hence, it is imperative to complement these approaches with additional strategies for anode interfacial modification [56].
5.2 Optimization of Polymer SEs
In the domain of soft polymer SEs, enhancing the mechanical properties can effectively inhibit Li dendrite growth to a certain extent. Strategies to optimize polymer SEs encompass various approaches: incorporating fillers like inorganic materials, choosing high modulus polymer hosts, creating composite electrolytes, initiating cross-link networks through in situ lighting or heating, and engineering molecular channels. These techniques aim to augment ionic conductivity, bolster mechanical strength, and improve the interfacial stability of polymer SEs.
5.2.1 Addition of Fillers
Fillers integrated into polymer SEs facilitate the creation of composite polymer electrolytes (CPEs), employing a rigid-flexible coupling design that enhances Li+ transference and interfacial stability, extends the ESW, and fortifies mechanical properties. These fillers encompass Li+-insulating agents like Al2O3[241], SiO2[242] and TiO2[243], alongside Li+-conductive substances such as LLZTO [244], LATP [197, 245, 246] and LAGP [247]. Using an in situ sol–gel method, Chen et al. developed a dual-network structure, combining 1 mol L−1 LiTFSI/[Py13][TFSI], PEO, and Al2O3 [248]. This design broadened the ESW, enhanced nonflammability, and improved Li compatibility in PEO-based CPEs. The addition of Al2O3 aimed at augmenting the ionic conductivity of the CPE. Palygorskite ((Mg,Al)2Si4O10(OH)) fibers as inorganic fillers create an interconnecting network within the polymer matrix, significantly enhancing mechanical properties. Yang et al. engineered CPEs using a PVDF polymer matrix and Palygorskite fillers, enhancing nonflammability, mechanical strength, ionic conduction, and the Li+ transference number of the SEs (Fig. 19A) [249].

Schematic illustration of fillers for polymer SEs to enhance properties. A PVDF/palygorskite nanowires CPE with passive fillers. a Schematic diagram of the PVDF/palygorskite CPE. b Voltage profile of NCM|PVDF/palygorskite nanowires|Li cells at 1 C. Reprinted with permission from Ref. [249]. Copyright © 2018, ACS Publications. B Polymer SEs with active fillers to form the PEO–Li@LLZTO composite electrolyte (PL@LCSE). a The diagram of PL@LCSE synthesis processes. Schematics of the difference on the surface of Li electrodes with b PLCSE and c PL@LCSE during Li plating/stripping processes. Reprinted with permission from Ref. [250]. Copyright © 2022, Elsevier
Li+-conductive fillers are pivotal in Li+ transport and suppressing Li dendrites compared to their Li+-insulating counterparts. Zhang et al. coupled Li single-ion polymer (Li (4-styrenesulfonyl) (trifluoromethanesulfonyl) imide (LiSTFSI)) onto LLZTO surfaces via silane coupling to create PEO–Li@LLZTO composite electrolytes (PL@LCSE) [250]. This design effectively reduced PEO crystallinity and dendrite generation, showcasing exceptional cycling performance in the assembled full cell (Fig. 19B). PEO integrated with LLZTO, fluorinated carbonate (FEC), and SN additives produced a CPE with improved Li+ transfer efficiency, exhibiting superior cycling performance (over 700 cycles at 1 C and RT) [251].
Furthermore, the addition of Li salts and plasticizers into polymer SEs enhances Li+ conductivity and suppresses Li dendrites. For example, the introduction of Li2S into PEO tackles its relatively low Li+ conductivity, promoting the formation of a LiF-rich interphase. This effectively augments Li+ diffusion capacity and curtails side reactions by inhibiting C–O bond breakage [196]. PVDF-based polymer SEs show reduced flammability compared to PEO-based SEs, presenting a promising avenue for solid-state electrolytes in ASSLBs [249].
5.2.2 Utilization of High Modulus Host
Overcoming the intrinsic softness and low ionic conductivity of polymer SEs involves utilizing a high modulus polymer host to optimize interfacial stability, augment ionic conduction, and bolster the hardness of polymer SEs for suppressing Li dendrite formation. In a study by Cui et al., a CPE was developed by embedding PEO/LiTFSI into a nano-porous polyimide (PI) film (8.6 μm thick), as depicted in Fig. 20A. The vertically aligned nanochannels within the PI film enhance ionic conductivity and Li.+ transport rates, while the high modulus of the PI host effectively suppresses Li dendrite formation [54]. In another approach, Goodenough et al. highlighted the significance of a single polymer host's energy gap concerning Li plating and stripping processes under high voltage. Their work introduced a bilayer electrolyte comprising PEO interfacing with the Li anode and poly(N-methyl-malonic amide) interfacing with the cathode. This innovative electrolyte-based full cell showcased remarkable performance, notably Li dendrite-free operation and low impedance [81]. Research attention has been drawn toward CPEs employing diverse polymer matrices, showcasing higher ionic conductivity and mechanical strength at RT. Fan et al., for instance, utilized porous PI film and PVDF as hosts to create a CPE by incorporating LiTFSI salts and LLZTO filler. This CPE demonstrated improved mechanical properties and efficient suppression of Li dendrite formation [252].

A Design of ultrathin, lightweight and flexible composite polymer SEs. a Illustration of design principles for composite polymer SEs. b SEM images of the cross-section for the PI film. c Digital photo for the PI film. d Diagram of energy density with different electrolytes. Reprinted with permission from Ref. [54]. Copyright © 2019, Springer Nature. B An intercalated PEC–LiMNT CSE. a Digital images of the Ni foam. b Digital images of the Li–Ni composite. c SEM images of the Ni foam. d SEM images of the Li–Ni composite. e Long-term cycling performance for different systems for polymer SEs-based ASSLBs at 25 °C. f–h Principles of ion-transfer-number enhancement composite solid electrolytes. Reprinted with permission from Ref. [240]. Copyright © 2019, John Wiley and Sons
5.2.3 Fabrication of Composite Electrolytes
Zhang et al. illustrated in Fig. 20B their development of a poly(ethylene carbonates)–Li montmorillonite CPE exhibiting desirable ionic conduction properties and a high Li+ transference number of 0.83. The incorporation of Li montmorillonite improved electrochemical and thermal stability, while additives such as FEC and poly(tetrafluoroethylene) binders optimized ion conduction and film-forming properties. When coupled with a 3D Li anode, the assembled ASSLBs displayed exceptional performance with a Li dendrite-free characteristic [240]. Similarly, various CPEs utilizing different polymer hosts, including triblock copolymer polystyrene–poly(ethylene glycol)–polystyrene (PS–PEG–PS) [253], sandwich-like poly(propylene carbonate)/polypropylene/poly(propylene carbonate) (PPC/PP/PPC) [254], and PEO–LiTFSI–Ni phosphate [255], exhibited high ionic conductivity and favorable compatibility with the Li anode. Elastomers, renowned for their mechanical elasticity and dispersion capabilities, have been recently introduced as polymer SE hosts for metal-based batteries (e.g., Na, K, Mg, Zn) via the polymerization-induced phase separation method [256]. Lee et al. innovated a plastic crystal-embedded elastomer electrolyte (PCEE) featuring mechanical robustness, acceptable Li+ conductivity (1.1 mS cm−1), and a high Li+ transference number (0.75). The symmetric Li cell based on PCEE showed uniform morphologies without Li dendrite formation after 100 cycles at 10 mA cm−1, owing to the formation of an inorganic–organic hybrid SEI layer. Furthermore, the asymmetric Li|PCEE|Cu cell demonstrated outstanding cycling performance with low voltage hysteresis below 26 mV and 100% Coulombic efficiency, achieving an exceptional energy density of 410 Wh kg−1. Dimethyl sulfoxide (DMSO), known for its high flash point of 95 °C and a high dielectric permittivity of 47.2, is desirable for dissolving Li salts, although free DMSO molecules are unstable with a Li anode. To address this, Li et al. utilized DMSO as a Li+ carrier and PVDF–HFP as a polymer matrix to create a solidified localized high-concentration electrolyte (S–LHCE) through a freeze-drying method [257]. This strategy enhanced compatibility with the Li anode, broadened the ESW, and decoupled electrolytes with a non-solvating solid framework. The stable interphase formed during cycling, composed of LiF at the anode interface, facilitated uniform Li deposition. Consequently, the assembled solid-state batteries exhibited exceptional electrochemical performance across a wide temperature range of − 10 to 100 °C.
5.2.4 Design of Cross-Link Networks
Polymer SEs featuring 3D cross-linked amorphous polymer hosts demonstrate exceptional mechanical robustness and ionic conduction, crucial for enhancing Li deposition uniformity on the anode and suppressing Li dendrite growth. In their work, Nanda et al. developed a cross-linked polymer SE derived from PEO and woven glass fiber matrices (Fig. 21a–e) [95]. This SE displayed a robust elastic modulus surpassing 1 GPa, effectively curbing Li dendrite formation, and showcased noteworthy ionic conductivity, moderate mechanical strength, and high tolerance to stretching during the manufacturing process. Li et al. introduced polymer SEs by cross-linking hierarchical structured g-C3N4, PEO, and LiTFSI, yielding SEs that offer excellent robustness and mechanical properties to inhibit Li dendrite growth even under substantial bending and prolonged cycling [258]. Consequently, the assembled cells demonstrated longevity with over 1 200 cycles and maintained an outstanding capacity (200 mAh g−1) at a high rate of 5 C. Another compelling development involved a well-cross-linked polymer SE comprised of LiSTFSI and PEGDA with PC plasticizer on PAN nanofibers (SIPE–PAN–NFs), showcasing remarkable ionic conduction (0.809 mS cm−1 at RT), improved mechanical strength, flexibility, and excellent cycling performance [259]. Furthermore, an ultrathin cross-linked polymer SE, just 12 μm thick, was devised by integrating 1,3-dioxolane and trimethylolpropane triglycidyl ether (TTE) into a mesoporous polymer host releasing Li nitrate. This SE exhibited favorable ionic conductivity, high mechanical strength, a preferred SEI composed of Li3N and LiF, and remarkable durability even with 2000 cycles of bending (Fig. 20f–h) [260]. Additionally, the cross-linking of polysiloxane-based polymer SEs with natural terpenes emerges as a promising avenue for ASSLBs, particularly in mitigating Li dendrite formation [261].

Cross-linked network polymer electrolytes. a Digital photo of cross-linked membranes with and without glass fibers. b Geometric construction and diameter distribution of glass fibers. c SEM images of the cross-section for the CPE. d Magnified view detailing of (c). Reprinted with permission from Ref. [95]. Copyright © 2021, Elsevier. e Optical photos of the ultrathin cross-linked polymer SE and SE thickness. f Specific capacity of the pouch cells under different bending tests. Reprinted with permission from Ref. [260]. Copyright © 2022, Elsevier
5.2.5 Engineering of Molecular Channels
Numerous promising polymer SEs have emerged from these methodologies. Yet, advancing novel strategies for improved polymer SEs remains imperative. Hu et al. pioneered a groundbreaking approach utilizing molecular channel engineering to craft a novel polymer SE by coordinating Cu2+ with cellulose nanofibrils (CNFs), resulting in an SE boasting outstanding attributes (Fig. 22) [262]. This innovation showcased exceptional ionic conduction (1.5 mS cm−1), a wide ESW (0 − 4.5 V), strong compatibility with both the Li anode and cathode, and a high transference number (0.78). The one-dimensional CNFs, embedded with oxygen-containing polar functional groups derived from wood, play a pivotal role in facilitating Li+ solvation and efficient Li+ movement within the SE. Furthermore, the Li–Cu–CNFs composite exhibited versatility across various metals and polymers, setting a precedent for the development of other high-performance SEs.

Copyright © 2021, Springer Nature
Design concept of Li–Cu–CNF SEs. a Origins, structures and Li+ transport pathways for Li–Cu–CNF. b Digital photo for the Li–Cu–CNF film. c Illustration of ASSLBs structure, including the Li anode, the Li–Cu–CNF SEs and the cathode with Li–Cu–CNF binders. d SEM image of the Li surface after long cycling for Li–Cu–CNF–based SEs ASSLBs. Reprinted with permission from Ref. [262].
6 Interfacial Engineering Toward Li and Halide SEs
Numerous studies have explored functional interlayers and electrolyte modifications to stabilize the interface between Li and halide SEs. While considerable efforts aim to establish a direct coupling between halide SEs and Li anodes, utilizing Li–In alloy or In foil [58, 60, 263,264,265,266,267] and Li11Sn6[268] remains an effective strategy to circumvent interfacial side reactions. Despite the limitation of direct contact between halide SEs and Li anode, they present promising potential as protective layers for cathodes due to their remarkable oxidation stability [269].
6.1 Insertion of Interlayer
Due to the inherent instability of halide electrolytes toward Li anodes, the use of an LPSC layer has been a common approach to stabilize the interface between halide SEs and Li. This strategy leverages the minimal interfacial ion transport resistance between sulfide SEs and halide SEs [111]. Among the halide SEs employed, Li3YCl6, Li3ScCl6, Li3HoCl6, and Li3InCl6 have been investigated [85, 269,270,271]. Sun et al. demonstrated that despite the thermodynamic instability of sulfide SEs toward Li anodes, the interaction between LPSC and Li generates a stable and conductive interface comprising Li2S, Li3P, and LiCl (depicted in Fig. 23A). This Li+ conductive interface mitigates the compatibility issues between Li and Li3YCl6, showcasing a commendable initial CE of 98.1% [85].

Insertion of LPSC as an interlayer to stabilize the surface between the Li anode and Li3YCl6. A a The symmetric cell performance comparison with and without LPSC interlayers. b Schematic diagram of Li3YCl6-based ASSLBs. c The initial charge/discharge profiles for halide SEs-based ASSLBs. d The MIEC for the Li–Li3YCl6 interface. e LPSC protector for the Li–Li3YCl6 interface. f Rate performance of Li3ScCl6-based ASSLBs coupled with Li metal and NMC622, as well as the LPSC protector for the anode interface. Reprinted with permission from Ref. [85]. Copyright © 2021, Science family of journals/AAAS. B Development of new halide SEs with good compatibility toward Li metal. a Advantages and voltage of new halide SEs (Li2ZrCl6 and Li2HfCl6). Reprinted with permission from Ref. [272]. Copyright © 2022, ACS Publications. b CV graph of various metal-doped LPSBrI SEs. c DC charge–discharge graph of various metal-doped LPSBrI SEs. Reprinted with permission from Ref. [273]. Copyright © 2022, Elsevier
6.2 Optimization of Halide SEs
6.2.1 Development of New Electrolytes
Yao et al. pioneered an innovative approach by employing stochastic surface walking global optimization in tandem with global neural network potential methods to strategically design halide SEs. Through this method, they identified two unique layered halide SE candidates: Li2ZrCl6 and Li2HfCl6, exhibiting exceptional stability (as depicted in Fig. 23B) [272]. The synthesized Li2ZrCl6 and Li2HfCl6 demonstrated unprecedented endurance against the Li anode, setting a remarkable record with a cycling time reaching 4 000 h. This groundbreaking utilization of machine learning strategies not only enhances the development of halide SEs but also presents a promising avenue for advancing SEs in ASSLBs.
6.2.2 Doping of Exotic Elements
The strategic doping of elements in SEs requires a delicate balance between enhancing ionic conductivity and widening the ESW. Pan et al. conducted an insightful study by doping varying amounts of Zr into Li3ErCl6, revealing that Zr doping improved the ionic conductivity of halide SEs but concurrently narrowed the ESW [267]. Employing a fluorine doping strategy optimized the stability of the Li–Li3YBr6 interface. The resultant Li3YBr5.7F0.3 exhibited a wider ESW compared to Li3YBr6, attributed to the in situ formation of a fluorine-rich interlayer during the cycling process [274].
7 Summary and Outlook
In this review, recent progress in addressing critical interfacial challenges between Li anodes and various SEs—including sulfide, oxide, polymer, and halide SEs—has been discussed. These SEs present diverse physical, chemical, electrochemical, and mechanical properties, necessitating distinct strategies to mitigate their respective interfacial problems.
Sulfide SEs are regarded as promising options for commercial ASSLBs due to their high RT ionic conductivities, akin to liquid electrolytes, and good ductility. However, their limitations such as a narrow ESW, sensitivity to air and humidity, and high cost pose significant challenges. The main solutions involve integrating stable SEI layers—either in situ or ex situ—to curb parasitic reactions, optimizing electrolytes to minimize residual electron conductivity, and alloying the Li anode to prevent dendrite formation and undesirable interactions with sulfide SEs.
Oxide SEs encompass a broad spectrum, notably garnets and NASICONs, exhibiting excellent Li+ conduction at room temperature (~ 1 mS cm−1), mechanical robustness, and wider ESWs. While garnets' mechanical rigidity inhibits Li dendrite penetration, it also leads to less dense interfaces and higher resistance. Strategies include surface conditioning to remove impurities, doping to enhance density, and structural designs to reduce resistance. For NASICONs like LAGP and LATP, stable SEI interlayers effectively mitigate their poor stability toward Li anodes.
Polymer SEs boast flexibility, good adhesion, and membrane-forming capabilities, but suffer from low RT ionic conductivity, poor mechanical properties, narrow ESWs, and weak interfacial compatibility with Li anodes. Improving their intrinsic properties and integrating protective interlayers are crucial strategies to enhance ionic conductivity, mechanical strength, and compatibility with Li anodes, addressing interfacial impedance.
Halide SEs, lauded for their desirable RT ionic conductivity, wide ESWs, and excellent compatibility with oxide cathodes, yet face stability issues with Li anodes due to the high valence states of metal elements. Approaches often include utilizing LPSC interlayers or Li–In alloys instead of Li metal. Strategies involve optimizing halide SEs' stability, such as fluorine doping and exploring unique layered structures like Li2ZrCl6 and Li2HfCl6.
ASSLBs aim for a delicate balance between exceptional safety and robust electrochemical performance. Despite extensive strategies for anode interfacial engineering, there are still notable areas for improvement in SEs, Li anodes, and their interfaces in ASSLBs.
-
I.
Multilayer SEs Exploration. Exploring multilayer SEs offers an opportunity to leverage the distinct advantages of individual SE layers while addressing stability issues at the electrode–SE interface. This approach allows the design of SEs structures that can withstand concurrent reduction and oxidation at the anode and cathode. Luo et al. extensively summarize recent progress on multilayer SEs, emphasizing the potential of vertically hetero-structured designs [71]. However, additional interfacial engineering remains crucial to stabilize the anode interface, recognizing that no SEs are entirely immune to instability with Li metal.
-
II.
SEI Interlayer Optimization. While SEI interlayers have proven successful in enhancing anode interfacial properties, practical ASSLBs still face challenges in accommodating high current density and real capacity. Designing an optimized 3D porous scaffold anode with superior ionic and electronic transport properties is crucial to achieving higher current density and area capacity.
-
III.
Li Alloy Anode Consideration. Li alloy anodes, compared to Li anodes, offer enhanced stability to SEs. Notably, the volume change effect of Li alloy anodes in ASSLBs is more manageable than in LIBs. Various representative Li alloys, such as Li–In, Li–Al, Li–Ag, Ag–C and Li–Sn alloys, and pure Si anodes (forming Li–Si alloys during cycling), show promise. However, these alloys often have higher density and potential, thereby potentially reducing the overall energy density of ASSLBs. Therefore, the ideal alloy anode should exhibit lower density and potential while maintaining high compactness. Li–Si alloys, especially, emerge as a promising candidate for new commercial ASSLBs due to their properties.
References
Whittingham, M.S.: Electrical energy storage and intercalation chemistry. Science 192, 1126–1127 (1976). https://doi.org/10.1126/science.192.4244.1126
Mizushima, K., Jones, P.C., Wiseman, P.J., et al.: LixCoO2 (0<x<1): a new cathode material for batteries of high energy density. Mater. Res. Bull. 15, 783–789 (1980). https://doi.org/10.1016/0025-5408(80)90012-4
Yoshino, A.: The birth of the lithium-ion battery. Angew. Chem. Int. Ed. 51, 5798–5800 (2012). https://doi.org/10.1002/anie.201105006
Song, R.S., Ge, Y.Q., Wang, B., et al.: A new reflowing strategy based on lithiophilic substrates towards smooth and stable lithium metal anodes. J. Mater. Chem. A 7, 18126–18134 (2019). https://doi.org/10.1039/c9ta05503g
Tan, D.H.S., Banerjee, A., Chen, Z., et al.: From nanoscale interface characterization to sustainable energy storage using all-solid-state batteries. Nat. Nanotechnol. 15, 170–180 (2020). https://doi.org/10.1038/s41565-020-0657-x
Al-Salih, H., Houache, M.S.E., Baranova, E.A., et al.: Composite cathodes for solid-state lithium batteries: “catholytes” the underrated giants. Adv. Energy Sustain. Res. 3, 2200032 (2022). https://doi.org/10.1002/aesr.202200032
Wu, J.H., Liu, S.F., Han, F.D., et al.: Lithium/sulfide all-solid-state batteries using sulfide electrolytes. Adv. Mater. 33, 2000751 (2021). https://doi.org/10.1002/adma.202000751
Dietrich, C., Weber, D.A., Sedlmaier, S.J., et al.: Lithium ion conductivity in Li2S–P2S5 glasses-building units and local structure evolution during the crystallization of superionic conductors Li3PS4, Li7P3S11 and Li4P2S7. J. Mater. Chem. A 5, 18111–18119 (2017). https://doi.org/10.1039/C7TA06067J
Tatsumisago, M., Hayashi, A.: Superionic glasses and glass–ceramics in the Li2S–P2S5 system for all-solid-state lithium secondary batteries. Solid State Ion. 225, 342–345 (2012). https://doi.org/10.1016/j.ssi.2012.03.013
Zhang, Q., Cao, D.X., Ma, Y., et al.: Sulfide-based solid-state electrolytes: synthesis, stability, and potential for all-solid-state batteries. Adv. Mater. 31, 1901131 (2019). https://doi.org/10.1002/adma.201901131
Homma, K., Yonemura, M., Nagao, M., et al.: Crystal structure of high-temperature phase of lithium ionic conductor, Li3PS4. J. Phys. Soc. Jpn. 79, 90–93 (2010). https://doi.org/10.1143/jpsjs.79sa.90
Mizuno, F., Hayashi, A., Tadanaga, K., et al.: New, highly ion-conductive crystals precipitated from Li2S–P2S5 glasses. Adv. Mater. 17, 918–921 (2005). https://doi.org/10.1002/adma.200401286
Deiseroth, H.J., Kong, S.T., Eckert, H., et al.: Li6PS5X: a class of crystalline Li-rich solids with an unusually high Li+ mobility. Angew. Chem. Int. Ed. Engl. 47, 755–758 (2008). https://doi.org/10.1002/anie.200703900
Rao, R.P., Adams, S.: Studies of lithium argyrodite solid electrolytes for all-solid-state batteries. Phys. Status Solidi A 208, 1804–1807 (2011). https://doi.org/10.1002/pssa.201001117
Bai, X.T., Duan, Y., Zhuang, W.D., et al.: Research progress in Li–argyrodite-based solid-state electrolytes. J. Mater. Chem. A 8, 25663–25686 (2020). https://doi.org/10.1039/d0ta08472g
Feng, X.Y., Chien, P.H., Wang, Y., et al.: Enhanced ion conduction by enforcing structural disorder in Li-deficient argyrodites Li6−xPS5−xCl1+x. Energy Storage Mater. 30, 67–73 (2020). https://doi.org/10.1016/j.ensm.2020.04.042
Kamaya, N., Homma, K., Yamakawa, Y., et al.: A lithium superionic conductor. Nat. Mater. 10, 682–686 (2011). https://doi.org/10.1038/nmat3066
Kato, Y., Hori, S., Saito, T., et al.: High-power all-solid-state batteries using sulfide superionic conductors. Nat. Energy 1, 16030 (2016). https://doi.org/10.1038/nenergy.2016.30
Iwasaki, R., Hori, S., Kanno, R., et al.: Weak anisotropic lithium-ion conductivity in single crystals of Li10GeP2S12. Chem. Mater. 31, 3694–3699 (2019). https://doi.org/10.1021/acs.chemmater.9b00420
Luo, W., Gong, Y.H., Zhu, Y.Z., et al.: Reducing interfacial resistance between garnet-structured solid-state electrolyte and Li-metal anode by a germanium layer. Adv. Mater. 29, 1606042 (2017). https://doi.org/10.1002/adma.201606042
Hu, S.Q., Li, Y.F., Yang, R., et al.: Structure and ionic conductivity of Li7La3Zr2−xGexO12 garnet-like solid electrolyte for all solid state lithium ion batteries. Ceram. Int. 44, 6614–6618 (2018). https://doi.org/10.1016/j.ceramint.2018.01.065
Cussen, E.J.: Structure and ionic conductivity in lithium garnets. J. Mater. Chem. 20, 5167–5173 (2010). https://doi.org/10.1039/B925553B
O’Callaghan, M.P., Lynham, D.R., Cussen, E.J., et al.: Structure and ionic-transport properties of lithium-containing garnets Li3Ln3Te2O12 (Ln = Y, Pr, Nd, Sm–Lu). Chem. Mater. 18, 4681–4689 (2006). https://doi.org/10.1021/cm060992t
Cussen, E.J., Yip, T.W.S., O’Neill, G., et al.: A comparison of the transport properties of lithium-stuffed garnets and the conventional phases Li3Ln3Te2O12. J. Solid State Chem. 184, 470–475 (2011). https://doi.org/10.1016/j.jssc.2010.12.021
Murugan, R., Thangadurai, V., Weppner, W.: Lattice parameter and sintering temperature dependence of bulk and grain-boundary conduction of garnet-like solid Li–electrolytes. J. Electrochem. Soc. 155, A90 (2008). https://doi.org/10.1149/1.2800764
Murugan, R., Thangadurai, V., Weppner, W.: Fast lithium ion conduction in garnet-type Li7La3Zr2O12. Angew. Chem. Int. Ed. 46, 7778–7781 (2007). https://doi.org/10.1002/anie.200701144
Percival, J., Kendrick, E., Smith, R.I., et al.: Cation ordering in Li containing garnets: synthesis and structural characterisation of the tetragonal system, Li7La3Sn2O12. Dalton Trans (2009). https://doi.org/10.1039/B907331K
Gupta, A., Murugan, R., Paranthaman, M.P., et al.: Optimum lithium-ion conductivity in cubic Li7−xLa3Hf2−xTaxO12. J. Power. Sources 209, 184–188 (2012). https://doi.org/10.1016/j.jpowsour.2012.02.099
Huang, X., Liu, C., Lu, Y., et al.: A Li-Garnet composite ceramic electrolyte and its solid-state Li–S battery. J. Power. Sources 382, 190–197 (2018). https://doi.org/10.1016/j.jpowsour.2017.11.074
Wang, D.W., Zhu, C.B., Fu, Y.P., et al.: Interfaces in garnet-based all-solid-state lithium batteries. Adv. Energy Mater. 10, 2001318 (2020). https://doi.org/10.1002/aenm.202001318
Zhu, Y.Z., He, X.F., Mo, Y.F.: Origin of outstanding stability in the lithium solid electrolyte materials: insights from thermodynamic analyses based on first-principles calculations. ACS Appl. Mater. Interfaces 7, 23685–23693 (2015). https://doi.org/10.1021/acsami.5b07517
Han, F.D., Zhu, Y.Z., He, X.F., et al.: Electrochemical stability of Li10GeP2S12 and Li7La3Zr2O12 solid electrolytes. Adv. Energy Mater. 6, 1501590 (2016). https://doi.org/10.1002/aenm.201501590
Campanella, D., Belanger, D., Paolella, A.: Beyond garnets, phosphates and phosphosulfides solid electrolytes: new ceramic perspectives for all solid lithium metal batteries. J. Power. Sources 482, 228949 (2021). https://doi.org/10.1016/j.jpowsour.2020.228949
Reddy, M.V., Julien, C.M., Mauger, A., et al.: Sulfide and oxide inorganic solid electrolytes for all-solid-state Li batteries: a review. Nanomaterials 10, 1606 (2020). https://doi.org/10.3390/nano10081606
Goodenough, J.B., Hong, H.Y.P., Kafalas, J.A.: Fast Na+-ion transport in skeleton structures. Mater. Res. Bull. 11, 203–220 (1976). https://doi.org/10.1016/0025-5408(76)90077-5
Hong, H.Y.P.: Crystal structures and crystal chemistry in the system Na1+xZr2SixP3−xO12. Mater. Res. Bull. 11, 173–182 (1976). https://doi.org/10.1016/0025-5408(76)90073-8
Kohler, H., Schulz, H.: NASICON solid electrolytes part I: the Na+-diffusion path and its relation to the structure. Mater. Res. Bull. 20, 1461–1471 (1985). https://doi.org/10.1016/0025-5408(85)90164-3
Aono, H., Imanaka, N., Adachi, G.Y.: High Li+ conducting ceramics. Acc. Chem. Res. 27, 265–270 (1994). https://doi.org/10.1021/ar00045a002
Zheng, F., Kotobuki, M., Song, S.F., et al.: Review on solid electrolytes for all-solid-state lithium-ion batteries. J. Power. Sources 389, 198–213 (2018). https://doi.org/10.1016/j.jpowsour.2018.04.022
Feng, J.K., Lu, L., Lai, M.O.: Lithium storage capability of lithium ion conductor Li1.5Al0.5Ge1.5(PO4)3. J. Alloys Compd. 501, 255–258 (2010). https://doi.org/10.1016/j.jallcom.2010.04.084
Itoh, M., Inaguma, Y., Jung, W.H., et al.: High lithium ion conductivity in the perovskite-type compounds Ln12Li12TiO3(Ln=La, Pr, Nd, Sm). Solid State Ion. 70(71), 203–207 (1994). https://doi.org/10.1016/0167-2738(94)90310-7
Inaguma, Y., Yu, J.D., Shan, Y.J., et al.: The effect of the hydrostatic pressure on the ionic conductivity in a perovskite lanthanum lithium titanate. J. Electrochem. Soc. 142, L8–L11 (1995). https://doi.org/10.1149/1.2043988
Inaguma, Y., Itoh, M.: Influences of carrier concentration and site percolation on lithium ion conductivity in perovskite-type oxides. Solid State Ion. 86(87/88), 257–260 (1996). https://doi.org/10.1016/0167-2738(96)00100-2
Fenton, D.E., Parker, J.M., Wright, P.V.: Complexes of alkali metal ions with poly(ethylene oxide). Polymer 14, 589 (1973). https://doi.org/10.1016/0032-3861(73)90146-8
Xue, Z.G., He, D., Xie, X.L.: Poly(ethylene oxide)-based electrolytes for lithium-ion batteries. J. Mater. Chem. A 3, 19218–19253 (2015). https://doi.org/10.1039/C5TA03471J
Wu, Y.X., Li, Y., Wang, Y., et al.: Advances and prospects of PVDF based polymer electrolytes. J. Energy Chem. 64, 62–84 (2022). https://doi.org/10.1016/j.jechem.2021.04.007
Kim, J.U., Sung, C.H., Moon, S.I., et al.: Conductivity and transference number of poly(ethylene oxide)/poly(vinylidene fluoride) blend plasticized polymer electrolytes. In: Proceedings of 5th International Conference on Properties and Applications of Dielectric Materials. IEEE, pp. 646–649, Seoul, (1997). https://ieeexplore.ieee.org/document/616518
Kim, H.S., Paik, C.H., Cho, B.W., et al.: Discharge characteristics of an Li/LiCoO2 cell with poly(acrylonitirile)-based polymer electrolyte. J. Power. Sources 68, 361–363 (1997). https://doi.org/10.1016/S0378-7753(97)02654-2
Zhang, Z., Zhang, G.Z., Chao, L.: Three-dimensional fiber network reinforced polymer electrolyte for dendrite-free all-solid-state lithium metal batteries. Energy Storage Mater. 41, 631–641 (2021). https://doi.org/10.1016/j.ensm.2021.06.030
Such, K., Stevens, J.R., Wieczorek, W., et al.: Polymer solid electrolytes from the PEG–PMMA–LiCF3SO3 system. J. Polym. Sci. B Polym. Phys. 32, 2221–2233 (1994). https://doi.org/10.1002/polb.1994.090321309
Borah, S., Guha, A.K., Saikia, L., et al.: Nanofiber induced enhancement of electrical and electrochemical properties in polymer gel electrolytes for application in energy storage devices. J. Alloys Compd. 886, 161173 (2021). https://doi.org/10.1016/j.jallcom.2021.161173
Lv, Z.L., Tang, Y., Dong, S.M., et al.: Polyurethane-based polymer electrolytes for lithium batteries: advances and perspectives. Chem. Eng. J. 430, 132659 (2022). https://doi.org/10.1016/j.cej.2021.132659
Cong, B., Song, Y.X., Ren, N.Q., et al.: Polyethylene glycol-based waterborne polyurethane as solid polymer electrolyte for all-solid-state lithium ion batteries. Mater. Des. 142, 221–228 (2018). https://doi.org/10.1016/j.matdes.2018.01.039
Wan, J.Y., Xie, J., Kong, X., et al.: Ultrathin, flexible, solid polymer composite electrolyte enabled with aligned nanoporous host for lithium batteries. Nat. Nanotechnol. 14, 705–711 (2019). https://doi.org/10.1038/s41565-019-0465-3
Wang, B.Y., Wang, G.X., He, P.G., et al.: Rational design of ultrathin composite solid-state electrolyte for high-performance lithium metal batteries. J. Membr. Sci. 642, 119952 (2022). https://doi.org/10.1016/j.memsci.2021.119952
Ding, P.P., Lin, Z.Y., Guo, X.W., et al.: Polymer electrolytes and interfaces in solid-state lithium metal batteries. Mater. Today 51, 449–474 (2021). https://doi.org/10.1016/j.mattod.2021.08.005
Liang, J.W., Li, X.N., Adair, K.R., et al.: Metal halide superionic conductors for all-solid-state batteries. Acc. Chem. Res. 54, 1023–1033 (2021). https://doi.org/10.1021/acs.accounts.0c00762
Asano, T., Sakai, A., Ouchi, S., et al.: Solid halide electrolytes with high lithium-ion conductivity for application in 4 V class bulk-type all-solid-state batteries. Adv. Mater. 30, 1803075 (2018). https://doi.org/10.1002/adma.201803075
Li, X.N., Liang, J.W., Yang, X.F., et al.: Progress and perspectives on halide lithium conductors for all-solid-state lithium batteries. Energy Environ. Sci. 13, 1429–1461 (2020). https://doi.org/10.1039/C9EE03828K
Liu, Z.T., Ma, S., Liu, J., et al.: High ionic conductivity achieved in Li3Y(Br 3Cl3) mixed halide solid electrolyte via promoted diffusion pathways and enhanced grain boundary. ACS Energy Lett. 6, 298–304 (2021). https://doi.org/10.1021/acsenergylett.0c01690
Emly, A., Kioupakis, E., Van der Ven, A.: Phase stability and transport mechanisms in antiperovskite Li3OCl and Li3OBr superionic conductors. Chem. Mater. 25, 4663–4670 (2013). https://doi.org/10.1021/cm4016222
Wang, S., Bai, Q., Nolan, A.M., et al.: Lithium chlorides and bromides as promising solid-state chemistries for fast ion conductors with good electrochemical stability. Angew. Chem. Int. Ed. Engl. 58, 8039–8043 (2019). https://doi.org/10.1002/anie.201901938
Wang, X.Y., He, K., Li, S.Y., et al.: Realizing high-performance all-solid-state batteries with sulfide solid electrolyte and silicon anode: a review. Nano Res. 16, 3741–3765 (2023). https://doi.org/10.1007/s12274-022-4526-9
Lv, Q., Jiang, Y.P., Wang, B., et al.: Suppressing lithium dendrites within inorganic solid-state electrolytes. Cell Rep. Phys. Sci. 3, 100706 (2022). https://doi.org/10.1016/j.xcrp.2021.100706
Paul, P.P., Chen, B.R., Langevin, S.A., et al.: Interfaces in all solid state Li-metal batteries: a review on instabilities, stabilization strategies, and scalability. Energy Storage Mater. 45, 969–1001 (2022). https://doi.org/10.1016/j.ensm.2021.12.021
Liang, Y.H., Liu, H., Wang, G.X., et al.: Challenges, interface engineering, and processing strategies toward practical sulfide-based all-solid-state lithium batteries. InfoMat 4, e12292 (2022). https://doi.org/10.1002/inf2.12292
Xu, K.: Interfaces and interphases in batteries. J. Power. Sources 559, 232652 (2023). https://doi.org/10.1016/j.jpowsour.2023.232652
Lee, D., Lee, H., Song, T., et al.: Toward high rate performance solid-state batteries. Adv. Energy Mater. 12, 2200948 (2022). https://doi.org/10.1002/aenm.202200948
Goodenough, J.B., Park, K.S.: The Li-ion rechargeable battery: a perspective. J. Am. Chem. Soc. 135, 1167–1176 (2013). https://doi.org/10.1021/ja3091438
Schwietert, T.K., Arszelewska, V.A., Wang, C., et al.: Clarifying the relationship between redox activity and electrochemical stability in solid electrolytes. Nat. Mater. 19, 428–435 (2020). https://doi.org/10.1038/s41563-019-0576-0
He, F., Hu, Z.L., Tang, W.J., et al.: Vertically heterostructured solid electrolytes for lithium metal batteries. Adv. Funct. Mater. 32, 2201465 (2022). https://doi.org/10.1002/adfm.202201465
Wenzel, S., Leichtweiss, T., Krüger, D., et al.: Interphase formation on lithium solid electrolytes: an in situ approach to study interfacial reactions by photoelectron spectroscopy. Solid State Ion. 278, 98–105 (2015). https://doi.org/10.1016/j.ssi.2015.06.001
Nolan, A.M., Zhu, Y.Z., He, X.F., et al.: Computation-accelerated design of materials and interfaces for all-solid-state lithium-ion batteries. Joule 2, 2016–2046 (2018). https://doi.org/10.1016/j.joule.2018.08.017
Banerjee, A., Wang, X.F., Fang, C.C., et al.: Interfaces and interphases in all-solid-state batteries with inorganic solid electrolytes. Chem. Rev. 120, 6878–6933 (2020). https://doi.org/10.1021/acs.chemrev.0c00101
Yan, M., Liang, J.Y., Zuo, T.T., et al.: Stabilizing polymer–lithium interface in a rechargeable solid battery. Adv. Funct. Mater. 30, 1908047 (2020). https://doi.org/10.1002/adfm.201908047
Wenzel, S., Randau, S., Leichtweiß, T., et al.: Direct observation of the interfacial instability of the fast ionic conductor Li10GeP2S12 at the lithium metal anode. Chem. Mater. 28, 2400–2407 (2016). https://doi.org/10.1021/acs.chemmater.6b00610
Xiao, Y.H., Wang, Y., Bo, S.H., et al.: Understanding interface stability in solid-state batteries. Nat. Rev. Mater. 5, 105–126 (2019). https://doi.org/10.1038/s41578-019-0157-5
Richards, W.D., Miara, L.J., Wang, Y., et al.: Interface stability in solid-state batteries. Chem. Mater. 28, 266–273 (2016). https://doi.org/10.1021/acs.chemmater.5b04082
Li, J.C., Ma, C., Chi, M.F., et al.: Lithium-ion batteries: solid electrolyte: the key for high-voltage lithium batteries (adv. energy mater. 4/2015). Adv. Energy Mater. 5, 1401408 (2015). https://doi.org/10.1002/aenm.201570018
Hartmann, P., Leichtweiss, T., Busche, M.R., et al.: Degradation of NASICON-type materials in contact with lithium metal: formation of mixed conducting interphases (MCI) on solid electrolytes. J. Phys. Chem. C 117, 21064–21074 (2013). https://doi.org/10.1021/jp4051275
Zhou, W.D., Wang, Z.X., Pu, Y., et al.: Double-layer polymer electrolyte for high-voltage all-solid-state rechargeable batteries. Adv. Mater. 31, 1805574 (2019). https://doi.org/10.1002/adma.201805574
Duan, H., Fan, M., Chen, W.P., et al.: Extended electrochemical window of solid electrolytes via heterogeneous multilayered structure for high-voltage lithium metal batteries. Adv. Mater. 31, 1807789 (2019). https://doi.org/10.1002/adma.201807789
Khurana, R., Schaefer, J.L., Archer, L.A., et al.: Suppression of lithium dendrite growth using cross-linked polyethylene/poly(ethylene oxide) electrolytes: a new approach for practical lithium–metal polymer batteries. J. Am. Chem. Soc. 136, 7395–7402 (2014). https://doi.org/10.1021/ja502133j
Lu, Q.W., Fang, J.H., Yang, J., et al.: A novel solid composite polymer electrolyte based on poly(ethylene oxide) segmented polysulfone copolymers for rechargeable lithium batteries. J. Membr. Sci. 425(426), 105–112 (2013). https://doi.org/10.1016/j.memsci.2012.09.038
Wang, C.H., Liang, J.W., Luo, J., et al.: A universal wet-chemistry synthesis of solid-state halide electrolytes for all-solid-state lithium-metal batteries. Sci. Adv. 7, eabh1896 (2021). https://doi.org/10.1126/sciadv.abh1896
Doux, J.M., Nguyen, H., Tan, D.H.S., et al.: Stack pressure considerations for room-temperature all-solid-state lithium metal batteries. Adv. Energy Mater. 10, 1903253 (2020). https://doi.org/10.1002/aenm.201903253
Pang, Y.P., Pan, J.Y., Yang, J.H., et al.: Electrolyte/electrode interfaces in all-solid-state lithium batteries: a review. Electrochem. Energy Rev. 4, 169–193 (2021). https://doi.org/10.1007/s41918-020-00092-1
Han, X.G., Gong, Y.H., Fu, K., et al.: Negating interfacial impedance in garnet-based solid-state Li metal batteries. Nat. Mater. 16, 572–579 (2017). https://doi.org/10.1038/nmat4821
Huo, H.Y., Chen, Y., Li, R.Y., et al.: Design of a mixed conductive garnet/Li interface for dendrite-free solid lithium metal batteries. Energy Environ. Sci. 13, 127–134 (2020). https://doi.org/10.1039/C9EE01903K
Feng, X.Y., Fang, H., Wu, N., et al.: Review of modification strategies in emerging inorganic solid-state electrolytes for lithium, sodium, and potassium batteries. Joule 6, 543–587 (2022). https://doi.org/10.1016/j.joule.2022.01.015
Ha, H.J., Kil, E.H., Kwon, Y.H., et al.: UV-curable semi-interpenetrating polymer network-integrated, highly bendable plastic crystal composite electrolytes for shape-conformable all-solid-state lithium ion batteries. Energy Environ. Sci. 5, 6491–6499 (2012). https://doi.org/10.1039/C2EE03025J
Nagao, M., Hayashi, A., Tatsumisago, M., et al.: In situ SEM study of a lithium deposition and dissolution mechanism in a bulk-type solid-state cell with a Li2S–P2S5 solid electrolyte. Phys. Chem. Chem. Phys. 15, 18600–18606 (2013). https://doi.org/10.1039/C3CP51059J
Porz, L., Swamy, T., Sheldon, B.W., et al.: Mechanism of lithium metal penetration through inorganic solid electrolytes. Adv. Energy Mater. 7, 1701003 (2017). https://doi.org/10.1002/aenm.201701003
Monroe, C., Newman, J.: The impact of elastic deformation on deposition kinetics at lithium/polymer interfaces. J. Electrochem. Soc. 152, A396 (2005). https://doi.org/10.1149/1.1850854
Yang, G., Lehmann, M.L., Zhao, S., et al.: Anomalously high elastic modulus of a poly(ethylene oxide)-based composite electrolyte. Energy Storage Mater. 35, 431–442 (2021). https://doi.org/10.1016/j.ensm.2020.11.031
Kasemchainan, J., Zekoll, S., Spencer Jolly, D., et al.: Critical stripping current leads to dendrite formation on plating in lithium anode solid electrolyte cells. Nat. Mater. 18, 1105–1111 (2019). https://doi.org/10.1038/s41563-019-0438-9
Sarkar, S., Thangadurai, V.: Critical current densities for high-performance all-solid-state Li–metal batteries: fundamentals, mechanisms, interfaces, materials, and applications. ACS Energy Lett. 7, 1492–1527 (2022). https://doi.org/10.1021/acsenergylett.2c00003
Liang, J.W., Li, X.N., Zhao, Y., et al.: An air-stable and dendrite-free Li anode for highly stable all-solid-state sulfide-based Li batteries. Adv. Energy Mater. 9, 1902125 (2019). https://doi.org/10.1002/aenm.201902125
Fan, X.L., Ji, X., Han, F.D., et al.: Fluorinated solid electrolyte interphase enables highly reversible solid-state Li metal battery. Sci. Adv. 4, eaau9245 (2018). https://doi.org/10.1126/sciadv.aau9245
Xu, R.C., Han, F.D., Ji, X., et al.: Interface engineering of sulfide electrolytes for all-solid-state lithium batteries. Nano Energy 53, 958–966 (2018). https://doi.org/10.1016/j.nanoen.2018.09.061
Zhao, F.P., Sun, Q., Yu, C., et al.: Ultrastable anode interface achieved by fluorinating electrolytes for all-solid-state Li metal batteries. ACS Energy Lett. 5, 1035–1043 (2020). https://doi.org/10.1021/acsenergylett.0c00207
Su, H., Liu, Y., Zhong, Y., et al.: Stabilizing the interphase between Li and argyrodite electrolyte through synergistic phosphating process for all-solid-state lithium batteries. Nano Energy 96, 107104 (2022). https://doi.org/10.1016/j.nanoen.2022.107104
Xu, H.J., Cao, G.Q., Shen, Y.L., et al.: Enabling argyrodite sulfides as superb solid-state electrolyte with remarkable interfacial stability against electrodes. Energy Environ. Mater. 5, 852–864 (2022). https://doi.org/10.1002/eem2.12282
Wang, Y.H., Yue, J.P., Wang, W.P., et al.: Constructing a stable interface between the sulfide electrolyte and the Li metal anode via a Li+-conductive gel polymer interlayer. Mater. Chem. Front. 5, 5328–5335 (2021). https://doi.org/10.1039/D1QM00395J
Li, J.R., Su, H., Li, M., et al.: A deformable dual-layer interphase for high-performance Li10GeP2S12-based solid-state Li metal batteries. Chem. Eng. J. 431, 134019 (2022). https://doi.org/10.1016/j.cej.2021.134019
Gao, Y., Wang, D.W., Li, Y.C., et al.: Salt-based organic–inorganic nanocomposites: towards a stable lithium metal/Li10GeP2S12 solid electrolyte interface. Angew. Chem. Int. Ed. 57, 13608–13612 (2018). https://doi.org/10.1002/anie.201807304
Duan, C., Cheng, Z., Li, W., et al.: Realizing the compatibility of a Li metal anode in an all-solid-state Li−S battery by chemical iodine–vapor deposition. Energy Environ. Sci. 15, 3236–3245 (2022). https://doi.org/10.1039/D2EE01358D
Zhang, Z.H., Chen, S.J., Yang, J., et al.: Interface re-engineering of Li10GeP2S12 electrolyte and lithium anode for all-solid-state lithium batteries with ultralong cycle life. ACS Appl. Mater. Interfaces 10, 2556–2565 (2018). https://doi.org/10.1021/acsami.7b16176
Chen, Y.M., Wang, Z.Q., Li, X.Y., et al.: Li metal deposition and stripping in a solid-state battery via Coble creep. Nature 578, 251–255 (2020). https://doi.org/10.1038/s41586-020-1972-y
Su, Y.B., Ye, L.H., Fitzhugh, W., et al.: A more stable lithium anode by mechanical constriction for solid state batteries. Energy Environ. Sci. 13, 908–916 (2020). https://doi.org/10.1039/C9EE04007B
Ye, L.H., Li, X.: A dynamic stability design strategy for lithium metal solid state batteries. Nature 593, 218–222 (2021). https://doi.org/10.1038/s41586-021-03486-3
Gil-González, E., Ye, L.H., Wang, Y.C., et al.: Synergistic effects of chlorine substitution in sulfide electrolyte solid state batteries. Energy Storage Mater. 45, 484–493 (2022). https://doi.org/10.1016/j.ensm.2021.12.008
Alarco, P.J., Abu-Lebdeh, Y., Abouimrane, A., et al.: The plastic-crystalline phase of succinonitrile as a universal matrix for solid-state ionic conductors. Nat. Mater. 3, 476–481 (2004). https://doi.org/10.1038/nmat1158
MacFarlane, D.R., Forsyth, M.: Plastic crystal electrolyte materials: new perspectives on solid state ionics. Adv. Mater. 13, 957–966 (2001). https://doi.org/10.1002/1521-4095(200107)13:12/13957:aid-adma957%3e3.0.co;2-#
Wang, C.H., Adair, K.R., Liang, J.W., et al.: Solid-state plastic crystal electrolytes: effective protection interlayers for sulfide-based all-solid-state lithium metal batteries. Adv. Funct. Mater. 29, 1900392 (2019). https://doi.org/10.1002/adfm.201900392
Kazyak, E., Wood, K.N., Dasgupta, N.P.: Improved cycle life and stability of lithium metal anodes through ultrathin atomic layer deposition surface treatments. Chem. Mater. 27, 6457–6462 (2015). https://doi.org/10.1021/acs.chemmater.5b02789
Jung, S.C., Han, Y.K.: How do Li atoms pass through the Al2O3 coating layer during lithiation in Li-ion batteries? J. Phys. Chem. Lett. 4, 2681–2685 (2013). https://doi.org/10.1021/jz401231e
Kozen, A.C., Lin, C.F., Pearse, A.J., et al.: Next-generation lithium metal anode engineering via atomic layer deposition. ACS Nano 9, 5884–5892 (2015). https://doi.org/10.1021/acsnano.5b02166
Zhang, Z.H., Wu, L.P., Zhou, D., et al.: Flexible sulfide electrolyte thin membrane with ultrahigh ionic conductivity for all-solid-state lithium batteries. Nano Lett. 21, 5233–5239 (2021). https://doi.org/10.1021/acs.nanolett.1c01344
Wang, C.H., Zhao, Y., Sun, Q., et al.: Stabilizing interface between Li10SnP2S12 and Li metal by molecular layer deposition. Nano Energy 53, 168–174 (2018). https://doi.org/10.1016/j.nanoen.2018.08.030
Ji, X., Hou, S., Wang, P.F., et al.: Solid-state electrolyte design for lithium dendrite suppression. Adv. Mater. 32, 2002741 (2020). https://doi.org/10.1002/adma.202002741
Kato, A., Suyama, M., Hotehama, C., et al.: High-temperature performance of all-solid-state lithium-metal batteries having Li/Li3PS4 interfaces modified with Au thin films. J. Electrochem. Soc. 165, A1950–A1954 (2018). https://doi.org/10.1149/2.1451809jes
Su, J., Pasta, M., Ning, Z.Y., et al.: Interfacial modification between argyrodite-type solid-state electrolytes and Li metal anodes using LiPON interlayers. Energy Environ. Sci. 15, 3805–3814 (2022). https://doi.org/10.1039/D2EE01390H
Zhu, Y.Z., He, X.F., Mo, Y.F.: First principles study on electrochemical and chemical stability of solid electrolyte-electrode interfaces in all-solid-state Li-ion batteries. J. Mater. Chem. A 4, 3253–3266 (2016). https://doi.org/10.1039/C5TA08574H
Xiao, R.J., Li, H., Chen, L.Q.: High-throughput design and optimization of fast lithium ion conductors by the combination of bond-valence method and density functional theory. Sci. Rep. 5, 14227 (2015). https://doi.org/10.1038/srep14227
Wang, X.L., Xiao, R.J., Li, H., et al.: Oxygen-driven transition from two-dimensional to three-dimensional transport behaviour in β-Li3PS4 electrolyte. Phys. Chem. Chem. Phys. 18, 21269–21277 (2016). https://doi.org/10.1039/C6CP03179J
Chen, T., Zhang, L., Zhang, Z.X., et al.: Argyrodite solid electrolyte with a stable interface and superior dendrite suppression capability realized by ZnO co-doping. ACS Appl. Mater. Interfaces 11, 40808–40816 (2019). https://doi.org/10.1021/acsami.9b13313
Liu, G.Z., Xie, D.J., Wang, X.L., et al.: High air-stability and superior lithium ion conduction of Li3+3xP1-xZnxS4-xOx by aliovalent substitution of ZnO for all-solid-state lithium batteries. Energy Storage Mater. 17, 266–274 (2019). https://doi.org/10.1016/j.ensm.2018.07.008
Trevey, J.E., Gilsdorf, J.R., Miller, S.W., et al.: Li2S–Li2O–P2S5 solid electrolyte for all-solid-state lithium batteries. Solid State Ion. 214, 25–30 (2012). https://doi.org/10.1016/j.ssi.2012.02.034
Ohtomo, T., Hayashi, A., Tatsumisago, M., et al.: Characteristics of the Li2O–Li2S–P2S5 glasses synthesized by the two-step mechanical milling. J. Non Cryst. Solids 364, 57–61 (2013). https://doi.org/10.1016/j.jnoncrysol.2012.12.044
Xie, D.J., Chen, S.J., Zhang, Z.H., et al.: High ion conductive Sb2O5-doped β-Li3PS4 with excellent stability against Li for all-solid-state lithium batteries. J. Power. Sources 389, 140–147 (2018). https://doi.org/10.1016/j.jpowsour.2018.04.021
Cengiz, M., Oh, H., Lee, S.H.: Lithium dendrite growth suppression and ionic conductivity of Li2S–P2S5–P2O5 Glass solid electrolytes prepared by mechanical milling. J. Electrochem. Soc. 166, A3997–A4004 (2019). https://doi.org/10.1149/2.0311916jes
Jiang, Z., Liang, T.B., Liu, Y., et al.: Improved ionic conductivity and Li dendrite suppression capability toward Li7P3S11-based solid electrolytes triggered by Nb and O cosubstitution. ACS Appl. Mater. Interfaces 12, 54662–54670 (2020). https://doi.org/10.1021/acsami.0c15903
Mercier, R., Malugani, J.P., Fahys, B., et al.: Superionic conduction in Li2S-P2S5-LiI - glasses. Solid State Ion. 5, 663–666 (1981). https://doi.org/10.1016/0167-2738(81)90341-6
Rangasamy, E., Liu, Z.C., Gobet, M., et al.: An iodide-based Li7P2S8I superionic conductor. J. Am. Chem. Soc. 137, 1384–1387 (2015). https://doi.org/10.1021/ja508723m
Han, F.D., Yue, J., Zhu, X.Y., et al.: Suppressing Li dendrite formation in Li2S–P2S5 solid electrolyte by LiI incorporation. Adv. Energy Mater. 8, 1703644 (2018). https://doi.org/10.1002/aenm.201703644
Zhao, F.P., Liang, J.W., Yu, C., et al.: A versatile Sn-substituted argyrodite sulfide electrolyte for all-solid-state Li metal batteries. Adv. Energy Mater. 10, 1903422 (2020). https://doi.org/10.1002/aenm.201903422
Park, C., Lee, S., Kim, M., et al.: Li metal stability enhancement of Sn-doped Li2S–P2S5 glass-ceramics electrolyte. Electrochim. Acta 390, 138808 (2021). https://doi.org/10.1016/j.electacta.2021.138808
Yao, X.Y., Huang, N., Han, F.D., et al.: High-performance all-solid-state lithium–sulfur batteries enabled by amorphous sulfur-coated reduced graphene oxide cathodes. Adv. Energy Mater. 7, 1602923 (2017). https://doi.org/10.1002/aenm.201602923
Shin, B.R., Nam, Y.J., Oh, D.Y., et al.: Comparative study of TiS2/Li–In all-solid-state lithium batteries using glass-ceramic Li3PS4 and Li10GeP2S12 solid electrolytes. Electrochim. Acta 146, 395–402 (2014). https://doi.org/10.1016/j.electacta.2014.08.139
Li, J.W., Li, Y.Y., Cheng, J., et al.: A graphene oxide coated sulfide-based solid electrolyte for dendrite-free lithium metal batteries. Carbon 177, 52–59 (2021). https://doi.org/10.1016/j.carbon.2021.01.159
Li, J.W., Li, Y.Y., Cheng, J., et al.: In situ modified sulfide solid electrolyte enabling stable lithium metal batteries. J. Power. Sources 518, 230739 (2022). https://doi.org/10.1016/j.jpowsour.2021.230739
Li, Y., Cao, D.X., Arnold, W., et al.: Regulated lithium ionic flux through well-aligned channels for lithium dendrite inhibition in solid-state batteries. Energy Storage Mater. 31, 344–351 (2020). https://doi.org/10.1016/j.ensm.2020.06.029
Jiang, W., Yan, L.J., Zeng, X.M., et al.: Adhesive sulfide solid electrolyte interface for lithium metal batteries. ACS Appl. Mater. Interfaces 12, 54876–54883 (2020). https://doi.org/10.1021/acsami.0c17828
Lewis, J.A., Cavallaro, K.A., Liu, Y., et al.: The promise of alloy anodes for solid-state batteries. Joule 6, 1418–1430 (2022). https://doi.org/10.1016/j.joule.2022.05.016
Hou, L.P., Yuan, H., Zhao, C.Z., et al.: Improved interfacial electronic contacts powering high sulfur utilization in all-solid-state lithium–sulfur batteries. Energy Storage Mater. 25, 436–442 (2020). https://doi.org/10.1016/j.ensm.2019.09.037
Nagao, M., Hayashi, A., Tatsumisago, M.: Bulk-type lithium metal secondary battery with indium thin layer at interface between Li electrode and Li2S–P2S5 solid electrolyte. Electrochemistry 80, 734–736 (2012). https://doi.org/10.5796/electrochemistry.80.734
Santhosha, A.L., Medenbach, L., Buchheim, J.R., et al.: The indium–lithium electrode in solid-state lithium-ion batteries: phase formation, redox potentials, and interface stability. Batter. Supercaps 2, 524–529 (2019). https://doi.org/10.1002/batt.201800149
Luo, S.T., Wang, Z.Y., Li, X.L., et al.: Growth of lithium-indium dendrites in all-solid-state lithium-based batteries with sulfide electrolytes. Nat. Commun. 12, 6968 (2021). https://doi.org/10.1038/s41467-021-27311-7
Park, S.W., Choi, H.J., Yoo, Y., et al.: Stable cycling of all-solid-state batteries with sacrificial cathode and lithium-free indium layer (adv. funct. mater. 5/2022). Adv. Funct. Mater. 32, 2108203 (2022). https://doi.org/10.1002/adfm.202270030
Lee, G.H., Lee, S.G., Park, S.H., et al.: Interface engineering on a Li metal anode for an electro-chemo-mechanically stable anodic interface in all-solid-state batteries. J. Mater. Chem. A 10, 10662–10671 (2022). https://doi.org/10.1039/D1TA11066G
Sakuma, M., Suzuki, K., Hirayama, M., et al.: Reactions at the electrode/electrolyte interface of all-solid-state lithium batteries incorporating Li–M (M=Sn, Si) alloy electrodes and sulfide-based solid electrolytes. Solid State Ion. 285, 101–105 (2016). https://doi.org/10.1016/j.ssi.2015.07.010
Hashimoto, Y., Machida, N., Shigematsu, T.: Preparation of Li4.4GexSi1−x alloys by mechanical milling process and their properties as anode materials in all-solid-state lithium batteries. Solid State Ion. 175, 177–180 (2004). https://doi.org/10.1016/j.ssi.2004.08.022
Cangaz, S., Hippauf, F., Reuter, F.S., et al.: Enabling high-energy solid-state batteries with stable anode interphase by the use of columnar silicon anodes. Adv. Energy Mater. 10, 2001320 (2020). https://doi.org/10.1002/aenm.202001320
Tan, D.H.S., Chen, Y.T., Yang, H.D., et al.: Carbon-free high-loading silicon anodes enabled by sulfide solid electrolytes. Science 373, 1494–1499 (2021). https://doi.org/10.1126/science.abg7217
Cao, D.X., Sun, X., Li, Y.J., et al.: Long-cycling sulfide-based all-solid-state batteries enabled by electrochemo-mechanically stable electrodes. Adv. Mater. 34, 2200401 (2022). https://doi.org/10.1002/adma.202200401
Cao, D.X., Sun, X., Wang, Y., et al.: Bipolar stackings high voltage and high cell level energy density sulfide based all-solid-state batteries. Energy Storage Mater. 48, 458–465 (2022). https://doi.org/10.1016/j.ensm.2022.03.012
Choi, H.J., Kang, D.W., Park, J.W., et al.: In situ formed Ag–Li intermetallic layer for stable cycling of all-solid-state lithium batteries (adv. sci. 1/2022). Adv. Sci. 9, 2103826 (2022). https://doi.org/10.1002/advs.202270001
Lee, Y.G., Fujiki, S., Jung, C., et al.: High-energy long-cycling all-solid-state lithium metal batteries enabled by silver–carbon composite anodes. Nat. Energy 5, 299–308 (2020). https://doi.org/10.1038/s41560-020-0575-z
Chen, Z.R., Liang, Z.T., Zhong, H.Y., et al.: Bulk/interfacial synergetic approaches enable the stable anode for high energy density all-solid-state lithium–sulfur batteries. ACS Energy Lett. 7, 2761–2770 (2022). https://doi.org/10.1021/acsenergylett.2c01334
Oh, J., Choi, S.H., Chang, B., et al.: Elastic binder for high-performance sulfide-based all-solid-state batteries. ACS Energy Lett. 7, 1374–1382 (2022). https://doi.org/10.1021/acsenergylett.2c00461
Pan, H., Zhang, M.H., Cheng, Z., et al.: Carbon-free and binder-free Li–Al alloy anode enabling an all-solid-state Li–S battery with high energy and stability. Sci. Adv. 8, eabn4372 (2022). https://doi.org/10.1126/sciadv.abn4372
Bates, J.B., Dudney, N.J., Gruzalski, G.R., et al.: Fabrication and characterization of amorphous lithium electrolyte thin films and rechargeable thin-film batteries. J. Power. Sources 43, 103–110 (1993). https://doi.org/10.1016/0378-7753(93)80106-Y
Ruan, Y.D., Lu, Y., Huang, X., et al.: Acid induced conversion towards a robust and lithiophilic interface for Li–Li7La3Zr2O12 solid-state batteries. J. Mater. Chem. A 7, 14565–14574 (2019). https://doi.org/10.1039/C9TA01911A
Guo, S.J., Wu, T.T., Sun, Y.G., et al.: Interface engineering of a ceramic electrolyte by Ta2O5 nanofilms for ultrastable lithium metal batteries. Adv. Funct. Mater. 32, 2201498 (2022). https://doi.org/10.1002/adfm.202201498
Lou, J.T., Wang, G.G., Xia, Y., et al.: Achieving efficient and stable interface between metallic lithium and garnet-type solid electrolyte through a thin indium tin oxide interlayer. J. Power. Sources 448, 227440 (2020). https://doi.org/10.1016/j.jpowsour.2019.227440
He, M.H., Cui, Z.H., Chen, C., et al.: Formation of self-limited, stable and conductive interfaces between garnet electrolytes and lithium anodes for reversible lithium cycling in solid-state batteries. J. Mater. Chem. A 6, 11463–11470 (2018). https://doi.org/10.1039/C8TA02276C
Zhou, D., Ren, G.X., Zhang, N., et al.: Garnet electrolytes with ultralow interfacial resistance by SnS2 coating for dendrite-free all-solid-state batteries. ACS Appl. Energy Mater. 4, 2873–2880 (2021). https://doi.org/10.1021/acsaem.1c00193
Luo, W., Gong, Y.H., Zhu, Y.Z., et al.: Transition from superlithiophobicity to superlithiophilicity of garnet solid-state electrolyte. J. Am. Chem. Soc. 138, 12258–12262 (2016). https://doi.org/10.1021/jacs.6b06777
Luo, Y.L., Feng, W.W., Meng, Z.J., et al.: Interface modification in solid-state lithium batteries based on garnet-type electrolytes with high ionic conductivity. Electrochim. Acta 397, 139285 (2021). https://doi.org/10.1016/j.electacta.2021.139285
Feng, W.L., Dong, X.L., Li, P.L., et al.: Interfacial modification of Li/Garnet electrolyte by a lithiophilic and breathing interlayer. J. Power. Sources 419, 91–98 (2019). https://doi.org/10.1016/j.jpowsour.2019.02.066
Alexander, G.V., Sreejith, O.V., Indu, M.S., et al.: Interface-compatible and high-cyclability lithiophilic lithium–zinc alloy anodes for garnet-structured solid electrolytes. ACS Appl. Energy Mater. 3, 9010–9017 (2020). https://doi.org/10.1021/acsaem.0c01430
Wan, Z.P., Shi, K., Huang, Y.F., et al.: Three-dimensional alloy interface between Li6.4La3Zr1.4Ta0.6O1.2 and Li metal to achieve excellent cycling stability of all-solid-state battery. J. Power Sources 505, 230062 (2021). https://doi.org/10.1016/j.jpowsour.2021.230062
Fu, K.K., Gong, Y.H., Liu, B.Y., et al.: Toward garnet electrolyte-based Li metal batteries: an ultrathin, highly effective, artificial solid-state electrolyte/metallic Li interface. Sci. Adv. 3, e1601659 (2017). https://doi.org/10.1126/sciadv.1601659
Huang, Y., Chen, B., Duan, J., et al.: Graphitic carbon nitride (g-C3N4): an interface enabler for solid-state lithium metal batteries. Angew. Chem. Int. Ed. 59, 3699–3704 (2020). https://doi.org/10.1002/anie.201914417
Lu, G.J., Dong, Z.C., Liu, W., et al.: Universal lithiophilic interfacial layers towards dendrite-free lithium anodes for solid-state lithium-metal batteries. Sci. Bull. 66, 1746–1753 (2021). https://doi.org/10.1016/j.scib.2021.04.034
Huang, W.L., Bi, Z.J., Zhao, N., et al.: Chemical interface engineering of solid garnet batteries for long-life and high-rate performance. Chem. Eng. J. 424, 130423 (2021). https://doi.org/10.1016/j.cej.2021.130423
Hu, B.K., Yu, W., Xu, B.Q., et al.: An in situ-formed mosaic Li7Sn3/LiF interface layer for high-rate and long-life garnet-based lithium metal batteries. ACS Appl. Mater. Interfaces 11, 34939–34947 (2019). https://doi.org/10.1021/acsami.9b10534
Baniya, A., Gurung, A., Pokharel, J., et al.: Mitigating interfacial mismatch between lithium metal and garnet-type solid electrolyte by depositing metal nitride lithiophilic interlayer. ACS Appl. Energy Mater. 5, 648–657 (2022). https://doi.org/10.1021/acsaem.1c03157
Du, M.J., Sun, Y., Liu, B., et al.: Smart construction of an intimate lithium|garnet interface for all-solid-state batteries by tuning the tension of molten lithium. Adv. Funct. Mater. 31, 2101556 (2021). https://doi.org/10.1002/adfm.202101556
Zhao, B., Ma, W.C., Li, B.B., et al.: A fast and low-cost interface modification method to achieve high-performance garnet-based solid-state lithium metal batteries. Nano Energy 91, 106643 (2022). https://doi.org/10.1016/j.nanoen.2021.106643
Leng, J., Liang, H.M., Wang, H.Y., et al.: A facile and low-cost wet-chemistry artificial interface engineering for garnet-based solid-state Li metal batteries. Nano Energy 101, 107603 (2022). https://doi.org/10.1016/j.nanoen.2022.107603
Zhong, Y.R., Xie, Y.J., Hwang, S., et al.: A highly efficient all-solid-state lithium/electrolyte interface induced by an energetic reaction. Angew. Chem. Int. Ed. 59, 14003–14008 (2020). https://doi.org/10.1002/anie.202004477
Zhang, Y., Meng, J.W., Chen, K.Y., et al.: Behind the candelabra: a facile flame vapor deposition method for interfacial engineering of garnet electrolyte to enable ultralong cycling solid-state Li–FeF3 conversion batteries. ACS Appl. Mater. Interfaces 12, 33729–33739 (2020). https://doi.org/10.1021/acsami.0c08203
Shao, Y.J., Wang, H.C., Gong, Z.L., et al.: Drawing a soft interface: an effective interfacial modification strategy for garnet-type solid-state Li batteries. ACS Energy Lett. 3, 1212–1218 (2018). https://doi.org/10.1021/acsenergylett.8b00453
Xie, H., Yang, C.P., Ren, Y.Y., et al.: Amorphous-carbon-coated 3D solid electrolyte for an electro-chemomechanically stable lithium metal anode in solid-state batteries. Nano Lett. 21, 6163–6170 (2021). https://doi.org/10.1021/acs.nanolett.1c01748
Cui, C., Ye, Q., Zeng, C., et al.: One-step fabrication of garnet solid electrolyte with integrated lithiophilic surface. Energy Storage Mater. 45, 814–820 (2022). https://doi.org/10.1016/j.ensm.2021.12.027
Zhang, X.Y., Xiang, Q., Tang, S., et al.: Long cycling life solid-state Li metal batteries with stress self-adapted Li/garnet interface. Nano Lett. 20, 2871–2878 (2020). https://doi.org/10.1021/acs.nanolett.0c00693
Bi, Z.J., Huang, W.L., Mu, S., et al.: Dual-interface reinforced flexible solid garnet batteries enabled by in situ solidified gel polymer electrolytes. Nano Energy 90, 106498 (2021). https://doi.org/10.1016/j.nanoen.2021.106498
Chi, S.S., Liu, Y.C., Zhao, N., et al.: Solid polymer electrolyte soft interface layer with 3D lithium anode for all-solid-state lithium batteries. Energy Storage Mater. 17, 309–316 (2019). https://doi.org/10.1016/j.ensm.2018.07.004
Cheng, Z.Y., Xie, M.L., Mao, Y.Y., et al.: Building lithiophilic ion-conduction highways on garnet-type solid-state Li+ conductors. Adv. Energy Mater. 10, 1904230 (2020). https://doi.org/10.1002/aenm.201904230
Hao, X.G., Zhao, Q., Su, S.M., et al.: Constructing multifunctional interphase between Li1.4Al0.4Ti1.6(PO4)3 and Li metal by magnetron sputtering for highly stable solid-state lithium metal batteries. Adv. Energy Mater. 9, 1901604 (2019). https://doi.org/10.1002/aenm.201901604
Liu, Y.L., Sun, Q., Zhao, Y., et al.: Stabilizing the interface of NASICON solid electrolyte against Li metal with atomic layer deposition. ACS Appl. Mater. Interfaces 10, 31240–31248 (2018). https://doi.org/10.1021/acsami.8b06366
Yang, K., Chen, L.K., Ma, J.B., et al.: Progress and perspective of Li1+xAlxTi2−x(PO4)3 ceramic electrolyte in lithium batteries. Infomat. 3, 1195–1217 (2021). https://doi.org/10.1002/inf2.12222
Yang, L.Y., Song, Y.L., Liu, H., et al.: Nanocomposite coatings: stable interface between lithium and electrolyte facilitated by a nanocomposite protective layer (small methods 3/2020). Small Methods 4, 1900751 (2020). https://doi.org/10.1002/smtd.202070014
Sheng, O.W., Zheng, J.H., Ju, Z.J., et al.: In situ construction of a LiF-enriched interface for stable all-solid-state batteries and its origin revealed by cryo-TEM. Adv. Mater. 32, 2000223 (2020). https://doi.org/10.1002/adma.202000223
Wang, Y., Wang, G.X., He, P.G., et al.: Sandwich structured NASICON-type electrolyte matched with sulfurized polyacrylonitrile cathode for high performance solid-state lithium–sulfur batteries. Chem. Eng. J. 393, 124705 (2020). https://doi.org/10.1016/j.cej.2020.124705
Yang, L.Y., Wang, Z.J., Feng, Y.C., et al.: Lithium-ion batteries: flexible composite solid electrolyte facilitating highly stable “soft contacting” Li–electrolyte interface for solid state lithium-ion batteries (adv. energy. mater 22/2017). Adv. Energy Mater. 7, 1701437 (2017). https://doi.org/10.1002/aenm.201770130
Lei, M., Fan, S.S., Yu, Y.F., et al.: NASICON-based solid state Li–Fe–F conversion batteries enabled by multi-interface-compatible sericin protein buffer layer. Energy Storage Mater. 47, 551–560 (2022). https://doi.org/10.1016/j.ensm.2022.02.031
Hou, G.M., Ma, X.X., Sun, Q.D., et al.: Lithium dendrite suppression and enhanced interfacial compatibility enabled by an ex situ SEI on Li anode for LAGP-based all-solid-state batteries. ACS Appl. Mater. Interfaces 10, 18610–18618 (2018). https://doi.org/10.1021/acsami.8b01003
Hu, F., Li, Y.Y., Wei, Y., et al.: Construct an ultrathin bismuth buffer for stable solid-state lithium metal batteries. ACS Appl. Mater. Interfaces 12, 12793–12800 (2020). https://doi.org/10.1021/acsami.9b21717
Jadhav, H.S., Kalubarme, R.S., Jadhav, A.H., et al.: Highly stable bilayer of LiPON and B2O3 added Li1.5Al0.5Ge1.5(PO4) solid electrolytes for non-aqueous rechargeable Li–O2 batteries. Electrochim. Acta 199, 126–132 (2016). https://doi.org/10.1016/j.electacta.2016.03.143
Zhang, S.N., Zeng, Z., Zhai, W., et al.: Bifunctional in situ polymerized interface for stable LAGP-based lithium metal batteries. Adv. Mater. Interfaces 8, 2100072 (2021). https://doi.org/10.1002/admi.202100072
Wang, Q.C., Ding, X.Y., Li, J.B., et al.: Minimizing the interfacial resistance for a solid-state lithium battery running at room temperature. Chem. Eng. J. 448, 137740 (2022). https://doi.org/10.1016/j.cej.2022.137740
Sharafi, A., Kazyak, E., Davis, A.L., et al.: Surface chemistry mechanism of ultra-low interfacial resistance in the solid-state electrolyte Li7La3Zr2O12. Chem. Mater. 29, 7961–7968 (2017). https://doi.org/10.1021/acs.chemmater.7b03002
Huo, H.Y., Chen, Y., Zhao, N., et al.: In situ formed Li2CO3-free garnet/Li interface by rapid acid treatment for dendrite-free solid-state batteries. Nano Energy 61, 119–125 (2019). https://doi.org/10.1016/j.nanoen.2019.04.058
Duan, H., Chen, W.P., Fan, M., et al.: Building an air stable and lithium deposition regulable garnet interface from moderate-temperature conversion chemistry. Angew. Chem. Int. Ed. Engl. 59, 12069–12075 (2020). https://doi.org/10.1002/anie.202003177
Patra, S., Krupa, B.R., Chakravarty, V., et al.: Microstructural engineering in lithium garnets by hot isostatic press to cordon lithium dendrite growth and negate interfacial resistance for all solid state battery applications. Electrochim Acta 312, 320–328 (2019). https://doi.org/10.1016/j.electacta.2019.05.003
Tsai, C.L., Roddatis, V., Chandran, C.V., et al.: Li7La3Zr2O12 interface modification for Li dendrite prevention. ACS Appl. Mater. Interfaces 8, 10617–10626 (2016). https://doi.org/10.1021/acsami.6b00831
Suzuki, Y., Kami, K., Watanabe, K., et al.: Transparent cubic garnet-type solid electrolyte of Al2O3-doped Li7La3Zr2O12. Solid State Ion. 278, 172–176 (2015). https://doi.org/10.1016/j.ssi.2015.06.009
Zhu, J.X., Li, X.L., Wu, C.W., et al.: A multilayer ceramic electrolyte for all-solid-state Li batteries. Angew. Chem. Int. Ed. 60, 3781–3790 (2021). https://doi.org/10.1002/anie.202014265
Rangasamy, E., Wolfenstine, J., Sakamoto, J.: The role of Al and Li concentration on the formation of cubic garnet solid electrolyte of nominal composition Li7La3Zr2O12. Solid State Ion. 206, 28–32 (2012). https://doi.org/10.1016/j.ssi.2011.10.022
Sharafi, A., Meyer, H.M., Nanda, J., et al.: Characterizing the Li–Li7La3Zr2O12 interface stability and kinetics as a function of temperature and current density. J. Power. Sources 302, 135–139 (2016). https://doi.org/10.1016/j.jpowsour.2015.10.053
Kumar, P.J., Nishimura, K., Senna, M., et al.: A novel low-temperature solid-state route for nanostructured cubic garnet Li7La3Zr2O12 and its application to Li-ion battery. RSC Adv. 6, 62656–62667 (2016). https://doi.org/10.1039/C6RA09695F
Li, Y.Q., Wang, Z., Li, C.L., et al.: Densification and ionic-conduction improvement of lithium garnet solid electrolytes by flowing oxygen sintering. J. Power. Sources 248, 642–646 (2014). https://doi.org/10.1016/j.jpowsour.2013.09.140
Zheng, C.J., Lu, Y., Su, J.M., et al.: Grain boundary engineering enabled high-performance garnet-type electrolyte for lithium dendrite free lithium metal batteries. Small Methods 6, 2200667 (2022). https://doi.org/10.1002/smtd.202200667
Xu, R., Liu, F., Ye, Y.S., et al.: A morphologically stable Li/electrolyte interface for all-solid-state batteries enabled by 3D-micropatterned garnet. Adv. Mater. 33, 2104009 (2021). https://doi.org/10.1002/adma.202104009
Xu, S.M., McOwen, D.W., Wang, C.W., et al.: Three-dimensional, solid-state mixed electron-ion conductive framework for lithium metal anode. Nano Lett. 18, 3926–3933 (2018). https://doi.org/10.1021/acs.nanolett.8b01295
Xu, S.M., McOwen, D.W., Zhang, L., et al.: All-in-one lithium-sulfur battery enabled by a porous-dense-porous garnet architecture. Energy Storage Mater. 15, 458–464 (2018). https://doi.org/10.1016/j.ensm.2018.08.009
Yang, C.P., Zhang, L., Liu, B.Y., et al.: Continuous plating/stripping behavior of solid-state lithium metal anode in a 3D ion-conductive framework. Proc. Natl. Acad. Sci. U. S. A. 115, 3770–3775 (2018). https://doi.org/10.1073/pnas.1719758115
Yi, E., Shen, H., Heywood, S., et al.: All-solid-state batteries using rationally designed garnet electrolyte frameworks. ACS Appl. Energy Mater. 3, 170–175 (2020). https://doi.org/10.1021/acsaem.9b02101
Fu, X.J., Wang, T.T., Shen, W.Z., et al.: A high-performance carbonate-free lithium|garnet interface enabled by a trace amount of sodium. Adv. Mater. 32, 2000575 (2020). https://doi.org/10.1002/adma.202000575
Dai, Q.S., Zhao, J., Ye, H.J., et al.: Ultrastable anode/electrolyte interface in solid-state lithium–metal batteries using LiCux nanowire network host. ACS Appl. Mater. Interfaces 13, 42822–42831 (2021). https://doi.org/10.1021/acsami.1c11613
Duan, J., Wu, W.Y., Nolan, A.M., et al.: Solid-state batteries: lithium–graphite paste: an interface compatible anode for solid-state batteries (adv. mater. 10/2019). Adv. Mater. 31, 1807243 (2019). https://doi.org/10.1002/adma.201970068
Wen, J.Y., Huang, Y., Duan, J., et al.: Highly adhesive Li-BN nanosheet composite anode with excellent interfacial compatibility for solid-state Li metal batteries. ACS Nano 13, 14549–14556 (2019). https://doi.org/10.1021/acsnano.9b08803
Cao, C.C., Zhong, Y.J., Wasalathilake, K.C., et al.: A low resistance and stable lithium-garnet electrolyte interface enabled by a multifunctional anode additive for solid-state lithium batteries. J. Mater. Chem. A 10, 2519–2527 (2022). https://doi.org/10.1039/d1ta07804f
Liu, B., Du, M.J., Chen, B.B., et al.: A simple strategy that may effectively tackle the anode-electrolyte interface issues in solid-state lithium metal batteries. Chem. Eng. J. 427, 131001 (2022). https://doi.org/10.1016/j.cej.2021.131001
Wang, T.R., Duan, J., Zhang, B., et al.: A self-regulated gradient interphase for dendrite-free solid-state Li batteries. Energy Environ. Sci. 15, 1325–1333 (2022). https://doi.org/10.1039/d1ee03604a
He, X.Z., Ji, X., Zhang, B., et al.: Tuning interface lithiophobicity for lithium metal solid-state batteries. ACS Energy Lett. 7, 131–139 (2022). https://doi.org/10.1021/acsenergylett.1c02122
Lu, Q.W., He, Y.B., Yu, Q.P., et al.: Dendrite-free, high-rate, long-life lithium metal batteries with a 3D cross-linked network polymer electrolyte. Adv. Mater. 29, 1604460 (2017). https://doi.org/10.1002/adma.201604460
Sun, H., Xie, X.X., Huang, Q., et al.: Fluorinated poly-oxalate electrolytes stabilizing both anode and cathode interfaces for all-solid-state Li/NMC811 batteries. Angew. Chem. Int. Ed. Engl. 60, 18335–18343 (2021). https://doi.org/10.1002/anie.202107667
Wu, N., Li, Y.T., Dolocan, A., et al.: In situ formation of Li3P layer enables fast Li+ conduction across Li/solid polymer electrolyte interface. Adv. Funct. Mater. 30, 2000831 (2020). https://doi.org/10.1002/adfm.202000831
Ma, Y.X., Wan, J.Y., Yang, Y.F., et al.: Scalable, ultrathin, and high-temperature-resistant solid polymer electrolytes for energy-dense lithium metal batteries. Adv. Energy Mater. 12, 2103720 (2022). https://doi.org/10.1002/aenm.202103720
Zhang, X., Liu, T., Zhang, S.F., et al.: Synergistic coupling between Li6.75La3Zr1.75Ta0.25O12 and poly(vinylidene fluoride) induces high ionic conductivity, mechanical strength, and thermal stability of solid composite electrolytes. J. Am. Chem. Soc. 139, 13779–13785 (2017). https://doi.org/10.1021/jacs.7b06364
Zhang, X., Wang, S., Xue, C.J., et al.: Self-suppression of lithium dendrite in all-solid-state lithium metal batteries with poly(vinylidene difluoride)-based solid electrolytes. Adv. Mater. 31, 1806082 (2019). https://doi.org/10.1002/adma.201806082
Fan, Z.J., Ding, B., Zhang, T.F., et al.: Solid/solid interfacial architecturing of solid polymer electrolyte-based all-solid-state lithium–sulfur batteries by atomic layer deposition. Small 15, 1903952 (2019). https://doi.org/10.1002/smll.201903952
Zhang, L.K., Xu, H.M., Jing, M.X., et al.: Solid electrolyte/lithium anodes stabilized by reduced graphene oxide interlayers: implications for solid-state lithium batteries. ACS Appl. Nano Mater. 4, 9471–9478 (2021). https://doi.org/10.1021/acsanm.1c01932
Zhang, L.K., Jing, M.X., Yang, H., et al.: Highly efficient interface modification between poly(propylene carbonate)-based solid electrolytes and a lithium anode by facile graphite coating. ACS Sustain. Chem. Eng. 8, 17106–17115 (2020). https://doi.org/10.1021/acssuschemeng.0c05153
Ye, Y., Deng, Z., Gao, L., et al.: Lithium-rich anti-perovskite Li2OHBr-based polymer electrolytes enabling an improved interfacial stability with a three-dimensional-structured lithium metal anode in all-solid-state batteries. ACS Appl. Mater. Interfaces 13, 28108–28117 (2021). https://doi.org/10.1021/acsami.1c04514
Chen, L., Li, W.X., Fan, L.Z., et al.: Solid-state lithium batteries: intercalated electrolyte with high transference number for dendrite-free solid-state lithium batteries (adv. funct. Mater. 28/2019). Adv. Funct. Mater. 29, 1901047 (2019). https://doi.org/10.1002/adfm.201970196
Croce, F., Appetecchi, G.B., Persi, L., et al.: Nanocomposite polymer electrolytes for lithium batteries. Nature 394, 456–458 (1998). https://doi.org/10.1038/28818
Zhou, D., Liu, R.L., He, Y.B., et al.: SiO2 hollow nanosphere-based composite solid electrolyte for lithium metal batteries to suppress lithium dendrite growth and enhance cycle life. Adv. Energy Mater. 6, 1502214 (2016). https://doi.org/10.1002/aenm.201502214
Gurevitch, I., Buonsanti, R., Teran, A.A., et al.: Nanocomposites of titanium dioxide and polystyrene-poly(ethylene oxide) block copolymer as solid-state electrolytes for lithium metal batteries. J. Electrochem. Soc. 160, A1611–A1617 (2013). https://doi.org/10.1149/2.117309jes
Zhao, C.Z., Zhang, X.Q., Cheng, X.B., et al.: An anion-immobilized composite electrolyte for dendrite-free lithium metal anodes. Proc. Natl. Acad. Sci. U. S. A. 114, 11069–11074 (2017). https://doi.org/10.1073/pnas.1708489114
Yu, X.W., Manthiram, A.: A long cycle life, all-solid-state lithium battery with a ceramic–polymer composite electrolyte. ACS Appl. Energy Mater. 3, 2916–2924 (2020). https://doi.org/10.1021/acsaem.9b02547
Yu, X.W., Manthiram, A.: Enhanced interfacial stability of hybrid-electrolyte lithium–sulfur batteries with a layer of multifunctional polymer with intrinsic nanoporosity. Adv. Funct. Mater. 29, 1805996 (2019). https://doi.org/10.1002/adfm.201805996
Wang, C.H., Yang, Y.F., Liu, X.J., et al.: Suppression of lithium dendrite formation by using LAGP–PEO (LiTFSI) composite solid electrolyte and lithium metal anode modified by PEO (LiTFSI) in all-solid-state lithium batteries. ACS Appl. Mater. Interfaces 9, 13694–13702 (2017). https://doi.org/10.1021/acsami.7b00336
Wu, F., Wen, Z.Y., Zhao, Z.K., et al.: Double-network composite solid electrolyte with stable interface for dendrite-free Li metal anode. Energy Storage Mater. 38, 447–453 (2021). https://doi.org/10.1016/j.ensm.2021.03.020
Yao, P.C., Zhu, B., Zhai, H.W., et al.: PVDF/palygorskite nanowire composite electrolyte for 4 V rechargeable lithium batteries with high energy density. Nano Lett. 18, 6113–6120 (2018). https://doi.org/10.1021/acs.nanolett.8b01421
Liu, M., Guan, X., Liu, H.M., et al.: Composite solid electrolytes containing single-ion lithium polymer grafted garnet for dendrite-free, long-life all-solid-state lithium metal batteries. Chem. Eng. J. 445, 136436 (2022). https://doi.org/10.1016/j.cej.2022.136436
Song, C., Li, Z.G., Peng, J., et al.: Enhancing Li ion transfer efficacy in PEO-based solid polymer electrolytes to promote cycling stability of Li–metal batteries. J. Mater. Chem. A 10, 16087–16094 (2022). https://doi.org/10.1039/D2TA03283J
Hu, J.K., He, P.G., Zhang, B.C., et al.: Porous film host-derived 3D composite polymer electrolyte for high-voltage solid state lithium batteries. Energy Storage Mater. 26, 283–289 (2020). https://doi.org/10.1016/j.ensm.2020.01.006
Zhang, B.H., Zhang, Y.H., Zhang, N., et al.: Synthesis and interface stability of polystyrene-poly(ethylene glycol)-polystyrene triblock copolymer as solid-state electrolyte for lithium-metal batteries. J. Power. Sources 428, 93–104 (2019). https://doi.org/10.1016/j.jpowsour.2019.04.033
Yue, H.Y., Li, J.X., Wang, Q.X., et al.: Sandwich-like poly(propylene carbonate)-based electrolyte for ambient-temperature solid-state lithium ion batteries. ACS Sustain. Chem. Eng. 6, 268–274 (2018). https://doi.org/10.1021/acssuschemeng.7b02401
Wu, Z.J., Xie, Z.K., Yoshida, A., et al.: Nickel phosphate nanorod-enhanced polyethylene oxide-based composite polymer electrolytes for solid-state lithium batteries. J. Colloid Interface Sci. 565, 110–118 (2020). https://doi.org/10.1016/j.jcis.2020.01.005
Lee, M.J., Han, J., Lee, K., et al.: Elastomeric electrolytes for high-energy solid-state lithium batteries. Nature 601, 217–222 (2022). https://doi.org/10.1038/s41586-021-04209-4
Xu, S.J., Xu, R.G., Yu, T., et al.: Decoupling of ion pairing and ion conduction in ultrahigh-concentration electrolytes enables wide-temperature solid-state batteries. Energy Environ. Sci. 15, 3379–3387 (2022). https://doi.org/10.1039/D2EE01053D
Hu, J.L., Chen, K.Y., Yao, Z.G., et al.: Unlocking solid-state conversion batteries reinforced by hierarchical microsphere stacked polymer electrolyte. Sci. Bull. 66, 694–707 (2021). https://doi.org/10.1016/j.scib.2020.11.017
Cheng, H., Yan, C.Y., Orenstein, R., et al.: Polyacrylonitrile nanofiber-reinforced flexible single-ion conducting polymer electrolyte for high-performance, room-temperature all-solid-state Li–metal batteries. Adv. Fiber Mater. 4, 532–546 (2022). https://doi.org/10.1007/s42765-021-00128-1
Wen, S.J., Luo, C., Wang, Q.R., et al.: Integrated design of ultrathin crosslinked network polymer electrolytes for flexible and stable all-solid-state lithium batteries. Energy Storage Mater. 47, 453–461 (2022). https://doi.org/10.1016/j.ensm.2022.02.035
Shim, J., Lee, J.W., Bae, K.Y., et al.: Dendrite suppression by synergistic combination of solid polymer electrolyte crosslinked with natural terpenes and lithium-powder anode for lithium–metal batteries. Chemsuschem 10, 2274–2283 (2017). https://doi.org/10.1002/cssc.201700408
Yang, C.P., Wu, Q.S., Xie, W.Q., et al.: Copper-coordinated cellulose ion conductors for solid-state batteries. Nature 598, 590–596 (2021). https://doi.org/10.1038/s41586-021-03885-6
Xu, G.F., Luo, L., Liang, J.W., et al.: Origin of high electrochemical stability of multi-metal chloride solid electrolytes for high energy all-solid-state lithium-ion batteries. Nano Energy 92, 106674 (2022). https://doi.org/10.1016/j.nanoen.2021.106674
Liang, J.W., Li, X.N., Wang, S., et al.: Site-occupation-tuned superionic LixScCl3+x halide solid electrolytes for all-solid-state batteries. J. Am. Chem. Soc. 142, 7012–7022 (2020). https://doi.org/10.1021/jacs.0c00134
Li, X.N., Liang, J.W., Luo, J., et al.: Air-stable Li3InCl6 electrolyte with high voltage compatibility for all-solid-state batteries. Energy Environ. Sci. 12, 2665–2671 (2019). https://doi.org/10.1039/C9EE02311A
Kwak, H., Han, D., Lyoo, J., et al.: All-solid-state batteries: new cost-effective halide solid electrolytes for all-solid-state batteries: mechanochemically prepared Fe3+-substituted Li2ZrCl6 (adv. energy mater. 12/2021). Adv. Energy Mater. 11, 2003190 (2021). https://doi.org/10.1002/aenm.202170045
Shao, Q.N., Yan, C.H., Gao, M.X., et al.: New insights into the effects of Zr substitution and carbon additive on Li3–xEr1–xZrxCl6 halide solid electrolytes. ACS Appl. Mater. Interfaces 14, 8095–8105 (2022). https://doi.org/10.1021/acsami.1c25087
Park, K.H., Kaup, K., Assoud, A., et al.: High-voltage superionic halide solid electrolytes for all-solid-state Li-ion batteries. ACS Energy Lett. 5, 533–539 (2020). https://doi.org/10.1021/acsenergylett.9b02599
Riegger, L.M., Schlem, R., Sann, J., et al.: Lithium–metal anode instability of the superionic halide solid electrolytes and the implications for solid-state batteries. Angew. Chem. Int. Ed. 60, 6718–6723 (2021). https://doi.org/10.1002/anie.202015238
Li, X.N., Liang, J.W., Kim, J.T., et al.: Highly stable halide-electrolyte-based all-solid-state Li–Se batteries. Adv. Mater. 34, 2200856 (2022). https://doi.org/10.1002/adma.202200856
Ji, W.X., Zheng, D., Zhang, X.X., et al.: A kinetically stable anode interface for Li3YCl6-based all-solid-state lithium batteries. J. Mater. Chem. A 9, 15012–15018 (2021). https://doi.org/10.1039/D1TA03042F
Li, F., Cheng, X.B., Lu, L.L., et al.: Stable all-solid-state lithium metal batteries enabled by machine learning simulation designed halide electrolytes. Nano Lett. 22, 2461–2469 (2022). https://doi.org/10.1021/acs.nanolett.2c00187
Rajagopal, R., Cho, J.U., Subramanian, Y., et al.: Preparation of highly conductive metal doped/substituted Li7P2S8Br(1–x)Ix type lithium superionic conductor for all-solid-state lithium battery applications. Chem. Eng. J. 428, 132155 (2022). https://doi.org/10.1016/j.cej.2021.132155
Yu, T.W., Liang, J.W., Luo, L., et al.: Superionic fluorinated halide solid electrolytes for highly stable Li–metal in all-solid-state Li batteries. Adv. Energy Mater. 11, 2101915 (2021). https://doi.org/10.1002/aenm.202101915
Tachez, M., Malugani, J.P., Mercier, R., et al.: Ionic conductivity of and phase transition in lithium thiophosphate Li3PS4. Solid State Ion. 14, 181–185 (1984). https://doi.org/10.1016/0167-2738(84)90097-3
Liu, Z.C., Fu, W.J., Payzant, E.A., et al.: Anomalous high ionic conductivity of nanoporous β-Li3PS4. J. Am. Chem. Soc. 135, 975–978 (2013). https://doi.org/10.1021/ja3110895
Bron, P., Johansson, S., Zick, K., et al.: Li10SnP2S12: an affordable lithium superionic conductor. J. Am. Chem. Soc. 135, 15694–15697 (2013). https://doi.org/10.1021/ja407393y
Adeli, P., Bazak, J.D., Park, K.H., et al.: Boosting solid-state diffusivity and conductivity in lithium superionic argyrodites by halide substitution. Angew. Chem. Int. Ed. 58, 8681–8686 (2019). https://doi.org/10.1002/anie.201814222
Xiao, W., Wang, J.Y., Fan, L.L., et al.: Recent advances in Li1+xAlxTi2−x(PO4)3 solid-state electrolyte for safe lithium batteries. Energy Storage Mater. 19, 379–400 (2019). https://doi.org/10.1016/j.ensm.2018.10.012
Fu, J.: Fast Li+ ion conducting glass-ceramics in the system Li2O–Al2O3–GeO2–P2O5. Solid State Ion. 104, 191–194 (1997). https://doi.org/10.1016/S0167-2738(97)00434-7
Ohta, S., Kobayashi, T., Asaoka, T.: High lithium ionic conductivity in the garnet-type oxide Li7−XLa3(Zr2−X, NbX)O12 (X=0–2). J. Power. Sources 196, 3342–3345 (2011). https://doi.org/10.1016/j.jpowsour.2010.11.089
Inaguma, Y., Chen, L.Q., Itoh, M., et al.: High ionic conductivity in lithium lanthanum titanate. Solid State Commun. 86, 689–693 (1993). https://doi.org/10.1016/0038-1098(93)90841-A
Jiang, Y., Yan, X.M., Ma, Z.F., et al.: Development of the PEO based solid polymer electrolytes for all-solid-state lithium ion batteries. Polymers 10, 1237 (2018). https://doi.org/10.3390/polym10111237
Acknowledgements
This work was financially sponsored by National Natural Science Foundation of China (Grant No. 52177218), the National Key Special Projects for International Cooperation in Science and Technology Innovation (2019YFE0100200) and Shanghai Pujiang Program (22PJ1408700).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jia, L., Zhu, J., Zhang, X. et al. Li–Solid Electrolyte Interfaces/Interphases in All-Solid-State Li Batteries. Electrochem. Energy Rev. 7, 12 (2024). https://doi.org/10.1007/s41918-024-00212-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41918-024-00212-1