
Abstract
Objectives
This paper presents an Australian model that formed part of the health technology assessment for public investment in siltuximab for the rare condition of idiopathic Multicentric Castleman Disease (iMCD) in Australia.
Methods
Two literature reviews were conducted to identify the appropriate comparator and model structure. Survival gain based on available clinical trial data were modelled using an Excel-based model semi-Markov model including time-varying transition probabilities, an adjustment for trial crossover and long-term data. A 20-year horizon was taken, and an Australian healthcare system perspective was adopted, with both benefits and costs discounted at 5%. The model was informed with an inclusive stakeholder approach that included a review of the model by an independent economist, Australian clinical expert opinion and feedback from the Pharmaceutical Benefits Advisory Committee (PBAC). The price used in the economic evaluation reflects a confidential discounted price, which was agreed to with the PBAC.
Results
An incremental cost-effectiveness ratio of A$84,935 per quality-adjusted life-year (QALY) gained was estimated. At a willingness-to-pay threshold of A$100,000 per QALY, siltuximab has a 72.1% probability of being cost-effective compared with placebo and best supportive care. Sensitivity analyses results were most sensitive to the length of interval between administrations (from 3- to 6-weekly) and crossover adjustments.
Conclusion
Within a collaborative and inclusive stakeholder framework, the model submitted to the Australian PBAC found siltuximab to be cost-effective for the treatment of iMCD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
We describe accessing Australian public funding via a health technology assessment process, based on an acceptable incremental cost-effectiveness ration (ICER), for use of siltuximab in idiopathic Multicentric Castleman Disease, a rare condition. |
The proposed, semi-Markov framework captures the disease progression and the treatment failure via tunnel substates and reflects the long-term outcomes in terms of costs and disease progression. |
The result of this analysis suggests siltuximab is cost-effective for the treatment of idiopathic Multicentric Castleman Disease, based on the evaluation from the Australian Pharmaceutical Benefits Advisory Committee. |
1 Introduction
Castleman disease is a rare lymphoproliferative disorder characterised by systemic enlargement of the lymph nodes and related lymphatic tissues and can be classified as either Unicentric Castleman disease (UCD), where a slow-growing mass is localised to a single lymph node, or Multicentric Castleman disease (MCD), which affects multiple lymph node sites and often involves other lymphatic tissue organs, such as the spleen or liver [1,2,3]. MCD is further classified into two sub-types, primarily determined by human herpesvirus-8 (HHV-8) and human immunodeficiency virus (HIV) status [4]. In the population where the aetiology remains unknown (HHV-8 negative and HIV negative), the disease is classified as idiopathic MCD (iMCD) [4,5,6]. iMCD is a complex and rare condition, and its clinical course includes non-specific signs and symptoms, ranging from mild to progressive worsening of symptoms leading to life-threatening complications and episodic exacerbations accompanied by coma, seizures and cerebrovascular events [4, 5, 7,8,9,10,11]. Incidence and prevalence of iMCD in the USA has been reported, respectively, as 2.45 and 6.1 per million [12].
Currently, the most widely accepted epidemiological definition of a “rare disease” in the European Union and Australia is a condition that impacts less than 5 in 10,000 people per annum [13, 14]. Approval for public funding for a rare disease treatment can be challenging, given the often limited availability of high-quality comparative, randomised trials and long-term outcome data. These limitations impact the ability to demonstrate cost-effectiveness. For end-of-life treatments, reimbursement decisions may include a modifier effect [15], with increased thresholds set in some health technology assessment (HTA) environments such as National Institute for Clinical Excellence (NICE) with a severity modifier incremental cost-effectiveness ratio (ICER) threshold of approximately £50,000 per quality-adjusted life-years (QALY) [16] and, in Australia, a derived threshold between A$45,000 and A$75,000 per QALY, as the PBAC has not officially published any thresholds [17,18,19,20]. Where the primary outcome is overall survival (OS), if there are marginal improvements, the decision to fund may also consider whether the incremental survival is clinically meaningful to the patient and counterbalanced with the level of safety and toxicity, and impact on quality of life [21,22,23]; finally a determination is made by HTA Committees on whether the return on treatment investment maximised health returns to the health service and society.
This paper presents the model that formed part of an HTA that demonstrated that public investment in siltuximab for the treatment of iMCD in Australia would deliver positive outcomes to a small group of patients. We discuss the detailed, rigorous and collaborative stakeholder framework process undertaken, and highlight some of the challenges in the reimbursement of siltuximab for iMCD on the Australian Pharmaceutical Benefits Scheme (PBS). Stakeholder engagement has been identified as valuable in the model development process, making economic models more widely accepted by decision-makers [24]. The stakeholders included Australian clinical experts (clinical expert opinion), economic experts and the Pharmaceutical Benefits Advisory Committee (PBAC). Clinical Expert opinion consisted of a survey of 7 clinicians [25] and face-to-face discussions with a further 13 clinicians [26], all treating patients with iMCD. Clinical expert opinion was sought at differential time points during the process to determine the comparator, the treatment algorithm, confirmation of health states and the best fit extrapolated survival curves. Advice from economists was incorporated into the structure of the health states and model. Lastly, the PBAC was consulted after an initial HTA review of siltuximab [27] via the post-PBAC process. During this process, key issues that needed to be addressed were discussed, and a way forward was proposed. The results reported in this article incorporate the recommendations made during the post-PBAC process. Siltuximab was recommended for reimbursement by the PBAC via the expedited early re-entry pathway (providing entry at the earliest next PBAC cycle meeting; there are three key PBAC cycles per annum) [28] confirming the strength of the clinical data, the value of a collaborative approach and the acceptance of the validity of the model with only minor input adjustments proposed by the PBAC.
2 Methods
2.1 Intervention, Target Population and Comparator
Siltuximab (an anti-interleukin-6 monoclonal antibody treatment) is listed as a first-line treatment in the International iMCD treatment guidelines [6]. The population of interest for the intervention was based on the sole randomised controlled trial (RCT; double blind, placebo-controlled multicentre trial), MCD2001 [29], and its corresponding long-term follow-up study, MCD2002 [30]. The RCT MCD2001 contained the relevant population and intervention, with this trial design preferred by the PBAC. MCD2001, a regulatory focused study, compared siltuximab in addition to best supportive care (SIL/BSC) to placebo in addition to best supportive care (PLB/BSC) over a 48-week trial timeframe. MCD2001 included 79 symptomatic iMCD adult patients (HIV-negative and HHV-8 negative), who were either newly diagnosed or previously treated (most patients had prior corticosteroid treatment), from 38 hospitals across 19 countries [29, 30]. Overall survival (OS), an HTA-preferred outcome for modelling long-term effectiveness was reported but was not a suitable primary trial outcome, as few deaths would be expected in the trial population with patients’ level of functioning being high [Eastern Cooperative Oncology Group (ECOG): 0–2], indicating a population with good performance status within a 48-week period. Additionally, crossover of patients from the PLB/BSC arm to active SIL/BSC was allowed following treatment failure on PLB/BSC, thereby making meaningful within trial OS assessments subject to bias.
Seeking clinical expert opinion determined that Australian practice included treatments of last resort, such as chemotherapy combination treatment and unregistered treatments. Therefore, the advice provided considered the most appropriate comparator to be BSC, as a primary purpose of treatment was symptom control. BSC included a range of interventions such as management of effusions, use of antipyretic, antipruritic, antihistamine and pain drugs, management of infections and infusion-related adverse events, and transfusions. However, exploring the validity of including unregistered treatments as potential comparators was also important to address potential HTA concerns.
A literature search was conducted to identify publications relating to all available historical or clinical evidence in iMCD-treated patients. Online databases and the grey literature were searched with no restriction on the start date up to and including 1 November 2020 (Supplementary material). A total of 354 citations were identified and 134 articles were accessed and read independently by two reviewers, and of these, 20 citations were included in the analysis. The PBAC Guidelines Appendix 4 Table A4 “Example factors that might cause comparative treatment effect heterogeneity” [31] were critically and comprehensively applied in reviewing the comparator studies. A high level of heterogeneity was observed across these studies was found in most known factors identified as contributing to treatment effect, such as randomisation techniques, duration of follow-up, loss to follow-up, outcomes, median duration of disease since diagnosis and other factors in relation to participant population and circumstances. This high level of heterogeneity between the siltuximab and unregistered and/or last-resort treatment studies did not support the validity of indirectly comparing results between these studies. Consequently, the sole comparator in the model was PLB/BSC, which was that recommended by clinical opinion. The PBAC considered PLB/BSC to be an acceptable comparator, given that iMCD is a rare condition with an unmet need for effective treatment [27, 28].
2.2 Perspective
The base-case analysis was performed from an Australian healthcare perspective, which included direct costs (health and health-related resource use) and health-related outcomes within the Australian healthcare system.
2.3 Model Structure
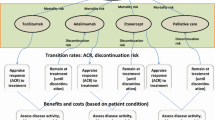
An Excel-based visual basic application (VBA) macros, health state semi-Markov model [32, 33] was constructed to estimate the costs and QALYs of the target population based on individual patient data from the MCD2001 study [29, 34]. Semi-Markov models are used, as they are a special case of the Markov chain where the time spent in the current state is dependent upon both the prior and future adjoining states [32]. This state-of-the-art model initially drew on experience from the 2014 model presented to the Canadian Agency for Drugs and Technologies in Health (CADTH), with improvement to adjust for trial crossover and utilisation of longer-term data [with up to 89 months (6.9 years) of follow-up] [34, 35]. Model validation was conducted at two stages (during and after model completion) and included comprehensive reviews by two experienced academic modellers and Australian clinical expert opinion [Figs. S1–S3 in the electronic supplementary material (ESM)]. The model consisted of four health states: (1) stable disease [SD; no treatment failure (TF) with no response (NR)], (2) responder (R; no TF with response), (3) post-treatment failure (pTF) and (4) death (D; Fig. 1). When TF occurred, patients moved to the pTF state and received subsequent therapies (e.g. rituximab, chemotherapy; Table 1) or BSC alone. Response to subsequent therapies was not assessed. The model projected that SIL/BSC treatment resulted in a longer time to TF. This effect was observed due to more patients in the R state and a lower rate of TF from the NR state versus PLB/BSC. Costs and quality-adjusted life-years (QALYs) for each treatment were accumulated based on the mean time spent in each health state, from which the incremental cost-effective ratio (cost/QALY) was determined, which is the primary economic consideration for the PBAC.

Model states and transitions. NR no response, R responder, pTF post-treatment failure, D death
2.4 Time Horizon, Cycle Length and Discounting
The base-case model was initially run over a lifetime horizon but was reduced to a 20-year time horizon at the request of the PBAC to decrease uncertainty in the analysis. To reflect survival observed in patients enrolled in MDC2001 [36], the model was divided into two stages. The first stage included the last event data from the MCD2001 study using Kaplan–Meier estimates. The second stage included information beyond the last event observed during the MCD2001 and MCD2002 trials and was informed by standard parametric survival models fitted to data from the clinical trials for SIL/BSC (for time to treatment failure (TTF) and OS or to external data to assess patients’ movement between states. Akaike information criteria (AIC) and Bayesian information criteria (BIC) were employed with clinical verification from Australian clinical expert opinion to assess OS curves of best fit (refer to Table S1 and Figs. S4–S6 in ESM). The model cycle length was 21 days to reflect the siltuximab administration protocol. The state occupancy at the start of a given cycle was informed by the clinical data. No half-cycle correction was implemented due to the short cycle length. Costs and health outcomes were both discounted at a rate of 5.0% in line with the Australian PBAC Guidelines [31].
2.5 Transition Probabilities
The semi-Markov structure allowed for some transition probabilities to vary with respect to time since entering the current state and for observance of the time since TF, via tunnel substates. The probability of transition between pTF and D states depended on ‘time since TF’ in one of the scenarios tested, partially circumventing the memoryless assumption of the Markov process.
2.6 Response Rates
The independently reviewed MCD2001 primary efficacy endpoint of durable tumour and symptomatic response were used in the base case [29] with 34% (18 of 53) of SIL/BSC patients responding completely or partially, compared with 0% (0 of 26) of PLB/BSC patients. Conservatively, response could only be achieved during stage 1 of the model; no further durable responses from the longer follow-up stage were used. Patients discontinued treatment from the NR state after 2 years.
2.7 Time To Treatment Failure Extrapolation
The base-case model adopted the definition of TF from MCD2001. Two scenarios of TTF in the SIL/BSC arm were considered: a core analysis of MCD2001 and an extended analysis of MCD2001 and MCD2002. Kaplan–Meier estimates of TTF with SIL/BSC and PLB/BSC treatment were presented.
The approach to extrapolate time-to-event data (during stage 2 only) included an assessment of log-cumulative hazard and residual plots to assess whether proportional hazards (or accelerated failure time) could be assumed. If plots were not parallel, then independent functions were fitted to each arm. Alternatively, if plots showed non-straight lines, consideration was given to other flexible modelling techniques, including fitting standard parametric models such as exponential, Weibull, lognormal, log-logistic, Gompertz and generalised gamma.
Though several extrapolation methodologies were explored, in the base case an exponential model of TTF was used among patients who responded in the SIL/BSC arm [providing the lowest AICs, and best visual fit (refer to Table S2 and Figs. S7–S10 in ESM)]. A lognormal model was used among patients without response in the SIL/BSC arm (core analysis, the lowest AICs, visual inspection: best fit). A generalised gamma model was used among patients without response in the PLB/BSC arm (the lowest AICs, visual inspection: best fit). The selected models are shown in Fig. 2.

a Proportion of patients with response on SIL/BSC treatment who were alive without TF during first 160 cycles: Kaplan–Meier estimates versus selected PSM. PSM parametric survival model, TF treatment failure, TTF time to treatment failure. b Proportion of patients without response on SIL/BSC treatment who were alive without TF during first 160 cycles: Kaplan–Meier estimates versus selected PSM. NR no response, PSM parametric survival model, TF treatment failure, TTF time to treatment failure. c Proportion of patients without response on PLB/BSC who were alive without TF during first 160 cycles: Kaplan–Meier estimates versus selected PSM. PSM parametric survival model, TF treatment failure, TTF time to treatment failure
2.8 Overall Survival
For HTA purposes, the OS data (not a primary outcome) from the MCD2001 clinical trial were relatively immature (6-year follow-up, with few events for this rare disease) and impacted by the crossover effect since patients in the PLB/BSC arm could crossover to SIL/BSC following protocol-defined TF; a total of 13 of the 26 PLB/BSC patients switched to SIL/BSC [29, 30]. The 1-year survival rate was 100% in the siltuximab arm and 92% in the placebo arm [36]. Therefore, improved outcomes after patients crossed over to SIL/BSC treatment may have increased the Kaplan–Meier estimates of OS among patients randomised to the PLB/BSC arm, thus underestimating the SIL/BSC treatment effect (Fig. 3) [37].

OS: follow-up analysis with adjustment for crossover (proportion of patients alive as a function of time from randomisation. PLB/BSC placebo in addition to best supportive care, SIL/BSC siltuximab in addition to best supportive care. Source: [73]
Different statistical methods have been proposed to adjust OS for crossover, i.e., censoring at the time of crossover, inverse probability of censoring weighting, rank preserving structural failure time models, or ‘two-stage’ methods [38]. All these methods were attempted, but most failed to adjust the observed OS for the PLB/BSC arm in MCD2001 due to a small sample size and few events (including none among patients after crossover). Censoring as a crossover adjustment method is usually considered to be a limited, and unreliable, method due to concerns for selection bias. However, in this instance, the potential for selection bias could only be assigned to one patient (≤ 3.8%) who withdrew consent to participate in the study a month after randomisation. The risk is further decreased considering the rarity of the disease and the problem with finding study participants. In addition, there is a low risk of selection bias indicated by similar baseline characteristics of patients from the PLB/BSC (n = 13) arm who did not switch to SIL/BSC treatment. Therefore, despite 13 out of 13 (100%) of the switching patients experiencing prior treatment failure (a condition of switching), the majority (11 of 13: 84.6%) responded to SIL/BSC treatment in MCD2001 after switching from PLB/BSC. This suggests that patients may respond to SIL/BSC even though initially they had disease progression, potentially translating into improved OS if treated with SIL/BSC.
Crossover allowance is important in trials of diseases that impact mortality (and indeed crossover mirrors clinical practice), allowing greater likelihood of patient recruitment and addressing ethical considerations [39]. If a drug has a true mortality benefit, the intention to treat (ITT) analysis likely underestimates OS in the presence of crossover (Fig. 3), and a cost utility analysis (CUA) based on these data will likely overestimate the ICER of the new therapy [37]. Thus, censoring as a method to adjust OS for crossover effect was considered a plausible adjustment method. In the survival analysis, general Australian mortality rates were applied (refer to Table S5 and Figs. S4–S6 in ESM).
Additionally, to stress test the overall survival scenarios, due to limited survival data for SIL/BSC and PLB/BSC and crossover adjustments, clinical expert opinion was utilised to determine the most clinically appropriate OS extrapolation curves. Three alternative scenarios (Fig. 4) were presented, and Scenario #2 was selected as representing the most plausible long-term extrapolation of the available clinical data (using an exponentially fitted parametric survival model). Published case series or cohort studies confirmed the base-case analysis in the placebo arm of the model (Fig. 4). The projections with censoring of 13 patients in placebo arm reflected all available OS data (Fig. 3 and Fig. S6 in ESM).

Extrapolation OS curves validated by Australian clinical expert opinion. OS overall survival, HR hazard ratio, pTF post-treatment failure, TF treatment failure, TTF time to treatment failure
2.9 Adverse Events
Grade 3 or higher adverse events (AEs) that were reported in more than 5% of patients and that incurred a financial cost in either treatment arm were included in the model. Therefore, anaemia, hypertension, fatigue, nausea and neutropenia were included in the model [30, 36]. The average incidence rate for each AE in the SIL/BSC arm was identified and transformed into a cycle probability, in the NR and R states. The probability of AEs with PLB/BSC was obtained using the odds ratio (OR) of AEs (SIL/BSC versus PLB/BSC) from the double-blind phase of MCD2001 [29] (refer to Table S3 and S4 in ESM).
2.10 QALYs
Health outcomes were measured in QALYs, with utilities assigned for each of the health states. The comprehensive literature review identified two sources of health-related quality of life (HRQoL) utilities for this population [34, 35, 40]. HRQoL derived from the ACCELERATE [40] registry were initially utilised in the base case with clinical-trial-derived utilities applied in the sensitivity analysis. Following discussions with the PBAC during the post-PBAC process, the base-case utilities were changed to include the clinical trial EQ-5D-derived utilities (based on EQ-5D-3L) [34], which were also the source for utilities in the CADTH model [35]. The utilities were based directly on MCD2001 trial data. The quality of life (QoL) of patients in MCD2001 was measured using the 36-Item Short Form Survey (SF-36) questionnaire at cycles 1, 3 and 6; every third cycle thereafter; and at the end-of-treatment visit. To derive these utilities, patients with iMCD were stratified by treatment (either SIL/BSC or PLB/BSC) and classified as either responders to treatment [complete response (CR)/partial response (PR) as defined by the trial primary efficacy endpoint] or non-responders (including patients with SD). Mixed-effects models were applied to accommodate imbalances and missing data (e.g. repeated measures with data missing in a single response variable, under the assumption of missing at random) [41,42,43]. The translation algorithm from SF-36 to EQ-5D was based on published methodology [44]. The PBAC preference for the CADTH EQ-5D-3L utility estimates are that they are derived directly from the trial patients, ensuring that the outcomes are appropriately incorporated into the model.
As shown in Table 1, the baseline utility score was 0.7034 for all iMCD trial patients [34]. A key observation made in the derivation of the trial-based utilities was that SIL/BSC improved HRQoL not only through the achievement of durable response (i.e. separate variable for SIL/BSC treatment and the response/no response disease in linear mixed models were statistically significant) but also through other benefits, including alleviation of iMCD symptoms [34]. Consequently, patients not responding to SIL/BSC had higher HRQoL than non-responding patients treated with PLB/BSC. This is reflected in the additional incremental utility attributed to SIL/BSC of 0.0819 per year, regardless of response status, with responders also experiencing another 0.0352 utility increase directly attributable to response. Post-treatment failure utility decrement (0.1801) was identical for all patients independent of prior treatment.
2.11 Costs and Resource Use Inputs
Direct healthcare costs at 2020–2021 A$ prices were used, including the cost of drug acquisition (SIL/BSC) treatments, subsequent treatments, concomitant therapies, cost of drug administration, tests before administration, monitoring the drug’s effectiveness (Medical Benefits Schedule items), cost of management of AEs and health-state-dependent cost–standard of care: routine surveillance costs (consultations, diagnostic tests and procedures) and end-of-life costs (Table 1).
The cost for siltuximab was based on a dose of 11 mg/kg per cycle based on a discounted price, assuming 90% public hospital use. In the base case, wastage is incorporated by assuming full vials are used. BSC was based on treatment used in MCD2001 to manage iMCD symptoms; corticosteroids were predominantly utilised (with the dose capped at 1 mg/kg/day prednisone or equivalent, therefore not considered a disease-modifying dose). Post-treatment failure regimen costs are based on an average of 3.875 cycles derived from the average cycles, predominantly chemotherapies, observed in a retrospective study of two large medical centres : cyclophosphamide was used 62.5% of the time and rituximab for the remaining 37.5% [45].
Hospitalisation costs were computed by model health state based on input from an Australian clinical survey [25] using the AR-DRG R61B LYMPHOMA&N-ACUTE LEUK, INTC (cost A$5265) [46], as there is no iMCD-specific National Hospital Cost Data Collection cost weight available (due to the small number of patients, such data are not collected). End-of-life cost is based on the 2009/2010 cost of the last 6 months of life of the Australian cancer population, inflated to 2021 using the Australian Bureaus of Statistics Health Price Index – A$44,040 [47].
2.12 One-Way Sensitivity Analyses
Scenario analyses were conducted to assess alternate model scenarios. One-way (deterministic) sensitivity analyses were performed to assess the impact of individual parameters of the model on a variety of variables, assuming different discount rates, body weight, mean age, crossover effect, utilities and effect on mortality (refer to Table S5 in ESM).
2.13 Probabilistic Sensitivity Analyses
Probabilistic sensitivity analyses were conducted using assigned distributions (refer to Table S5–S6 in ESM); 10,000 iterations were performed to obtain a stable estimate. Mean probabilistic sensitivity analyses results were illustrated through an incremental cost-effectiveness plane.
2.14 Model Validation
Economic modelling in such a rare disease is complicated by the inherent limitations of the clinical trial data including patient heterogeneity. The model was validated considering the Assessment of the Validation Status of Health-Economic decision models (AdViSHE) tool [48], and underwent cross-validity. Initially during the development of the model, a UK economist (Keith R Abrahams) was consulted to ensure that the appropriate health states, model inputs and outcomes had been identified and that the methodology was sound. Moreover, several publications reporting on historical OS for iMCD patients were used to validate OS projections in the PLB/BSC arm [49,50,51,52,53,54,55,56]. The next steps included the validation of the comparator, health states and selection of extrapolated survival cures for best fit by Australian clinical experts [22, 26]. Following completion, the model was reviewed by an Australian economist (Zanfina Ademi) for structural validity and internal consistency. Part of this review included extreme value testing for values, including discounting, time horizon, model transition probabilities and survival (as shown in Figs. S1–S3 in the ESM), concluding that the model functioned as expected.
3 Results
The model calculated total (discounted) costs and QALYs for SIL/BSC and PLB/BSC. Results of the cost–utility analysis were expressed as ICERs. Life-years with the tumour and a symptomatic response or life-years without treatment failure are key drivers of the incremental life-years gained, which is also reflected in the incremental QALY gain. Siltuximab (84.7%), BSC, visit, test and hospitalisation (13.5%) costs were key drivers of the incremental total costs.
3.1 Base-Case Results
Administration of siltuximab was every 21 days; however by the end of MCD2001 the mean cycle length was 24.2 days, and this formed the base-case assumption. The clinical trial demonstrated greater durable tumour and symptomatic response for siltuximab versus placebo (34% versus 0%; p = 0.0012) [36] which leads to improved survival (the key model driver) with the model demonstrating siltuximab’s cost-effectiveness with an ICER of A$84,935 per QALY gained (Table 2).
3.2 One-Way Sensitivity Analyses
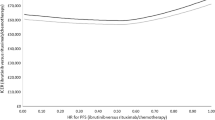
Overall, the results show that the ICER was most sensitive to the length of interval between administrations (from 3- to 6-weekly; ICER A$52,736 for every 6 weeks [57] versus ICER A$179,986 every 3 weeks [58]) followed by the type of censoring for crossover effect (censoring of 18 patients ICER A$70,650 versus no censoring of patients ICER A$145,867). Other variables had a limited impact, such as using a different source of utilities (ACCELERATE [40] ICER A$77,971 versus Vernon [34] ICER A$98,301). The results of deterministic sensitivity analyses are presented as a Tornado Diagram in Fig. 5.

Tornado diagram for SIL/BSC versus PLB/BSC. A$ Australian dollars 2019, BSC best supportive care, CI confidence interval, LY life-years, NR no response, PLB/BSC placebo in addition to best supportive care, PSM parametric survival model, pTF post-treatment failure, QALY quality-adjusted life-years, R responder, SIL siltuximab, SIL/BSC siltuximab in addition to best supportive care, TF treatment failure, TTF time to treatment failure
3.3 Probablistic Sensitivity Analyses
A probabilistic sensitivity analysis (PSA) was conducted with 10,000 iterations for the base-case analysis (Suppl. Table S6). The mean probabilistic ICER was A$85,094/QALY (95% CI A$83,926, A$86,294) using parametric methodology. For a willingness-to-pay threshold of A$100,000/QALY, SIL/BSC had a 72.1% probability of being cost-effective compared with PLB/BSC. The derived policy threshold for siltuximab carries a higher derived threshold than non-rare diseases (estimated to be A$45,000–A$75,000 [17,18,19,20]). In recommending public funding for siltuximab, the PBAC considered the resulting ICER to be acceptable in the context of iMCD being such a rare disease with an unmet need for effective treatment [28]. The cost-effectiveness plane for SIL/BSC versus PLB/BSC is presented in Fig. 6. The ellipse contains 95% of the modelled analyses. It is not known what the true opportunity cost is [59, 60], as consideration of broader health benefits foregone (in non-pharmaceutical expenditure) are not part of Australian PBAC consideration.

Cost-effectiveness plane for SIL/BSC versus PLB/BSC. PLB/BSC placebo in addition to best supportive care, PSA probabilistic sensitivity analysis, SIL/BSC siltuximab in addition to best supportive care
4 Discussion
This model, and its journey as part of the Australian HTA process, provides comprehensive insight into the process undertaken to obtain public funding for siltuximab for the treatment of iMCD in Australia. The research framework and the process undertaken included a collaborative stakeholder involvement consisting of health economists, clinical expert opinion and feedback from the PBAC during the post-PBAC process. The extensive collaboration between key stakeholders that was part of the process eventuated in developing a model and an HTA within the well-acknowledged constraints of research in rare diseases while still considering ethical elements of distributive justice such as longer survival, reduced health system costs, demonstrated efficacy and safety.
A critical step in this evaluation was identifying and validating the key inputs for the cost-effective analysis (CEA) model. As with many rare diseases, this was complicated by the limited evidence for the intervention (SIL/BSC), limited published data on patients’ survival and understanding of the disease in clinical practice. Although the comparator (BSC) was identified based on Australian clinical expert opinion, it was appropriate to explore all evidence available for treatments used for iMCD. A comprehensive literature review of iMCD treatments was conducted to identify relevant clinical evidence for siltuximab and other treatments used for iMCD. A key approach was the critical appraisal/assessment for risk of bias commonly evident in the use of all unlicensed therapies in clinical practice for the treatment of iMCD based on the PBAC bias assessment criteria [31], and the exclusion of these therapies as comparators. This provided clarity that the heterogeneity seen in these studies, including in trial design, population characteristics and diagnosis of iMCD [45, 49, 50, 61,62,63,64,65,66,67], confirmed their unsuitability for consideration in an HTA process that has a preference for data based on randomised controlled trials (where this is available). Consequently, the MCD2001 RCT provided the efficacy and safety data for the appropriate population, iMCD patients and both the licensed intervention (SIL/BSC) and comparator (PLB/BSC – where the comparator arm was subsequently adjusted for historical control [45]) in the CEA model [29, 30]. These data, where appropriate, were supplemented with clinical expert opinion in the absence of robust real-world evidence of iMCD treatment with siltuximab. The CEA also considered an equity-centred clinical aspect, allowing patients to choose no treatment or treatment every 3 or 6 weeks, making the model flexible and adaptive to individual patients and their clinical needs in real-world practice.
The second literature review identified that a Markov model had been submitted to the CADTH [35]. However, the Pan-Canadian Oncology Drug Review approved funding for siltuximab despite concluding that siltuximab was not cost-effective for the treatment of iMCD [35]. A critical review of the CADTH model found it incorporated less mature data from the clinical trial and follow-up data of 1 year [35]. The current analysis used individual patient data, incorporated more recent and longer-term data, an additional 4.91 years of follow-up data, and adapted inputs to reflect Australian clinical practice and costs, adding to the rigour of the model. For example, the CADTH model assumed a high risk of TF among patients with durable response. Due to the low number of events in this group of patients (1 TF in MCD2001), TTF of all patients using SIL/BSC was used as a proxy of TTF among patients with response. However, updated data from MCD2001 and MCD2002, which included 83 months of follow-up data, indicated that the average rate of TF among patients with response was almost 20 times lower than that of patients without response in the SIL/BSC arm [30, 36]. Therefore, the assumption implemented in the previous model appears overly conservative.
In addition, the model submitted to CADTH used 1-year Kaplan–Meier estimators for OS among patients from MCD2001 without adjusting for the crossover of patients in the PLB/BSC after disease progression to open-label SIL/BSC. In doing so, it also made assumptions regarding the proportion of patients who were alive in years 2–5 due to lack of long-term follow-up data. In the current model, MCD2002 provided additional longer-term survival data, and an adjustment for crossover was included. With rigorous model testing [48], it was determined that not censoring for crossover had the single greatest impact upon the ICER; therefore, in the cost-effectiveness modelling of SIL/BSC, adjustment for crossover was included, as it was considered clinically and ethically important and was also accepted by the PBAC, though they noted that there remained limitations with this type of analysis [27] .
A limitation was the need to adjust for the PLB/BSC patients being allowed to cross over into SIL/BSC. Patients were allowed to cross over to alleviate ethical issues of patients experiencing treatment failure in the placebo arm; the MCD2001 study allowed patients to switch to open-label siltuximab. This accurately reflected patient experience and the non-linear, unpredictable changes in siltuximab disease progression. However, small patient numbers and the need to adjust for crossover are limitations in the analysis that are not uncommon in rare diseases. Not adjusting for the crossover would have delivered results that were not reflective of the true incremental benefit of SIL/BSC.
A strength of the model was that the structure allowed for the varying the transition probabilities between post-treatment failure and death health states, based on the time since treatment failure, thereby partially overcoming the memoryless Markov assumption.
Overall, the PBAC-recommended changes in the model produced an ICER of A$84,935 per QALYs gained, which was determined to be cost-effective. However, it is worth noting that the model was heavily informed with MCD2001 [36], MCD2002 [30] and several other non-randomised studies for the PLB/BSC patient population [45, 49,50,51,52,53,54,55,56, 68]. This raises some universal limitations that are applicable to most HTA economic evaluations, including the lack of longer-term data and the number of OS years extrapolated in rare diseases. The 2017 NICE analysis of single technology appraisals for 22 cancer therapies from 28 appraisals [69] found that only a median of 6.7% (mean 6.5%) of the modelled time duration was observed, with a duration of observed follow-up across the assessments of only a median of 1.3 years (mean 2.0 years) out of a total median model duration of 25 years (mean 31.4 years). While the duration of SIL/BSC observed trial data is comparatively extensive, the median model time duration is shorter when compared with empirical evidence of observed versus modelled time frames in NICE cancer therapy assessments. Hence, the siltuximab 20-year modelled duration encompasses a much greater than usual 30% period of observed data, providing a much higher level of certainty than that usually observed in modelled evaluations to key HTA bodies such as NICE.
The model structure presented can generally be applied to other countries. However, the comparator, clinical practice, cost inputs, burden of disease and epidemiological inputs are Australia-specific, and these need to be adapted for other countries, as all models need to be jurisdiction-specific. The decision to fund is specific to Australia.
The ability to treat rare diseases through both new and existing therapies provides treatment alternatives. However, new therapies are often expensive or difficult to obtain. Consequently, many health jurisdictions around the world face immense challenges providing equitable and ethical healthcare. This HTA shows that, by following a rigorous, logical, detailed, collaborative and inclusive stakeholder framework, siltuximab for the treatment of iMCD was recommended for public subsidy by the Australian PBAC. It acknowledges the universal problems of conducting economic modelling for rare disease treatments, with limited prior research, complicated clinical trials with small number of participants, geographical spread and, at times, heterogeneity in disease characteristics. A rigorous and stakeholder-inclusive process that demonstrates the importance of conducting the most appropriate comprehensive literature reviews using detailed logic and reasoning for the CEA model inputs and analyses is described.
5 Conclusion
Within a collaborative, inclusive stakeholder framework, the PBAC-accepted model assessed siltuximab as being cost-effective in the very rare disease iMCD. This approach produced an incremental cost-effectiveness ratio of A$84,935 per QALY. This was a collaborative approach that enabled assessment of cost-effectiveness to allow people with a rare disease to have access to a well-researched, effective treatment within an HTA environment.
References
El-Osta HE, Kurzrock R. Castleman’s disease: from basic mechanisms to molecular therapeutics. Oncologist. 2011;16(4):497–511.
Roca B. Castleman’s disease. A review. AIDS Rev. 2009;11(1):3–7.
van Rhee F, Stone K, Szmania S, Barlogie B, Singh Z. Castleman disease in the 21st century: an update on diagnosis, assessment, and therapy. Clin Adv Hematol Oncol. 2010;8(7):486–98.
Fajgenbaum DC, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646–57.
van Rhee F, Greenway A, Stone K. Treatment of idiopathic Castleman disease. Hematology/Oncol Clinics. 2018;32(1):89–106.
Van Rhee F, Voorhees P, Dispenzieri A, Fossa A, Srkalovic G, Ide M, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018;132(20):2115–24.
Fajgenbaum DC. HHV-8-negative, idiopathic multicentric Castleman disease. Wolters Kluwer. UptoDate Web site. http://cdcn.org/wp-content/uploads/2019/08/HHV-8-negative_idiopathic-multicentric-Castleman-disease-UpToDate.pdf. 2019. Accessed 1 Oct 2022.
Fajgenbaum DC, Liu A, Ruth J, Nabel C, Finkelman B, Kurzrock R, et al. HHV-8-negative, idiopathic multicentric Castleman disease (IMCD): a description of clinical features and therapeutic options through a systematic literature review. Blood. 2014;124(21):4861.
Muskardin TW, Peterson BA, Molitor JA. Castleman disease and associated autoimmune disease. Curr Opin Rheumatol. 2012;24(1):76–83.
Oksenhendler E, Boutboul D, Fajgenbaum D, Mirouse A, Fieschi C, Malphettes M, et al. The full spectrum of Castleman disease: 273 patients studied over 20 years. Br J Haematol. 2018;180(2):206–16.
Vernon M, Teschendorf B, van Rhee F, editors. Qualitative research in Castleman’s disease: exploring patients’ perspectives of symptoms through qualitative interviews. In: Poster Presented at the 16th annual conference of the International Society for Quality of Life Research (ISOQOL); 2009 October 28-31, 2009; New Orleans, USA.
Mukherjee S, Martin R, Sande B, Paige JS, Fajgenbaum DC. Epidemiology and treatment patterns of idiopathic multicentric Castleman disease in the era of IL-6–directed therapy. Blood Adv. 2022;6(2):359–67.
Nguengang Wakap S, Lambert DM, Olry A, Rodwell C, Gueydan C, Lanneau V, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet. 2020;28(2):165–73.
TGA. Orphan drug designation eligibility criteria 2021. https://www.tga.gov.au/publication/orphan-drug-designation-eligibility-criteria. Accessed 10 Aug 2022.
Zhang K, Garau M. International cost-effectiveness thresholds and modifiers for HTA decision making. OHE; 2020.
NICE. Task and finish group report: CHTE methods review – modifiers. National Institute for Health and Care Excellence; 2020.
Griffiths E, Hendrich J, Stoddart S, Walsh S. Acceptance of health technology assessment submissions with incremental cost-effectiveness ratios above the cost-effectiveness threshold. ClinicoEcon Outcomes Res CEOR. 2015;7:463–76.
Makarounas-Kirchmann K. Review of PBAC decisions on rare diseases: pricing implications. 2020.
PBAC. 6.01 Everolimus, dispersible tablets, 2mg, 3mg and 5mg, Afinitor®, Novartis : December 2017. Canberra: Department of Health; 2017.
Lybrand S, Wonder M. Analysis of PBAC submissions and outcomes for medicines (2010–2018). Int J Technol Assess Health Care. 2020;36(3):224–31.
Vitry A, Mintzes B, Lipworth W. Access to new cancer medicines in Australia: dispelling the myths and informing a public debate. J Pharm Policy Pract. 2016;9(1):13.
Ellis LM, Bernstein DS, Voest EE, Berlin JD, Sargent D, Cortazar P, et al. American Society of Clinical Oncology perspective: raising the bar for clinical trials by defining clinically meaningful outcomes. J Clin Oncol. 2014;32(12):1277–80.
Niraula S, Seruga B, Ocana A, Shao T, Goldstein R, Tannock IF, et al. The price we pay for progress: a meta-analysis of harms of newly approved anticancer drugs. J Clin Oncol. 2012;30(24):3012–9.
Xie RZ, Malik ED, Linthicum MT, Bright JL. Putting stakeholder engagement at the center of health economic modeling for health technology assessment in the United States. Pharmacoeconomics. 2021;39(6):631–8.
Clinical Expert Opinion Survey. iMCD Survery: Clinical Expert Opinion Responses 2020.
Clinical Expert Opinion. Clinical Expert Opinion on the clinical treatment of patients with iMCD in Australia. 2021.
PBAC. 5.10 Siltuximab, Powder for injection 100 mg, Powder for injection 400 mg, Sylvant®, EUSA Pharma (UK) Ltd. Canberra: Department of Health; 2021.
PBAC. 7.12 Siltuximab, Powder for injection 100 mg, Powder for injection 400 mg, Sylvant®, EUSA Pharma (UK) Ltd. Canberra: Department of Health; 2021.
van Rhee F, Munshi NC, Wong R, Ke X, Fossa A, Simpson D, et al. Efficacy of siltuximab in patients with previously treated multicentric Castleman’s disease (MCD). J Clin Oncol. 2014;32(15_suppl):8514.
van Rhee F, Casper C, Voorhees PM, Fayad LE, van de Velde H, Vermeulen J, et al. A phase 2, open-label, multicenter study of the long-term safety of siltuximab (an anti-interleukin-6 monoclonal antibody) in patients with multicentric Castleman disease. Oncotarget. 2015;6(30):30408–19.
PBAC. Guidelines for preparing submissions to the Pharmaceutical Benefits Advisory Committee (PBAC) (version 5). Commonwealth of Australia as represented by the Department of Health Care DofaA; 2016. https://pbac.pbs.gov.au/information/about-the-guidelines.html. Accessed 1 Oct 2022.
Abner EL, Charnigo RJ, Kryscio RJ. Markov chains and semi-Markov models in time-to-event analysis. J Biom Biostat. 2013;Suppl 1(e001):19522.
Woods BS, Sideris E, Palmer S, Latimer N, Soares M. Partitioned survival and state transition models for healthcare decision making in oncology: where are we now? Value Health. 2020;23(12):1613–21.
Vernon M, Robinson D Jr, Trundell D, Ishak J, Jen MH, Brazier J. Deriving health utility values from a randomized, double-blind, placebo-controlled trial of siltuximab in subjects with multicentric Castleman’s disease. Curr Med Res Opin. 2016;32(7):1193–200.
Pan-Canadian Oncology Drug Review. Siltuximab (Sylvant) for Multicentric Castleman’s Disease – Final Economic Guidance Report. 2015. https://www.cadth.ca/sites/default/files/pcodr/pcodr_siltuximab_sylvant_mcd_fn_egr.pdf. Accessed 1 Oct 2022.
van Rhee F, Wong RS, Munshi N, Rossi JF, Ke XY, Fossa A, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2014;15(9):966–74.
Jönsson L, Sandin R, Ekman M, Ramsberg J, Charbonneau C, Huang X, et al. Analyzing overall survival in randomized controlled trials with crossover and implications for economic evaluation. Value Health. 2014;17(6):707–13.
Latimer NR, Abrams KR. Technical Support Document 16: Adjusting survival time estimates in the presence of treatment switching 2014. http://www.nicedsu.org.uk. Accessed 1 Oct 2022.
Yeh J, Gupta S, Patel SJ, Kota V, Guddati AK. Trends in the crossover of patients in phase III oncology clinical trials in the USA. Ecancer. 2020;14:1142.
ACCELERATE. Data from the ACCELERATE registry database (data on file). 2021.
Howell DC. The treatment of missing data reprinted in Babones S. Fundamentals of Regression Modeling. Sage Publications; 2013.
Molenberghs G, Kenward MG. Missing data in clinical studies. Chichester: Wiley; 2007.
Schafer JL, Yucel RM. Computational strategies for multivariate linear mixed-effects models with missing values. J Comput Graph Stat. 2002;11(2):437–57.
Rowen D, Brazier J, Roberts J. Mapping SF-36 onto the EQ-5D index: how reliable is the relationship? Health Qual Life Outcomes. 2009;7(1):27.
Dong Y, Zhang L, Nong L, Wang L, Liang Z, Zhou D, et al. Effectiveness of rituximab-containing treatment regimens in idiopathic multicentric Castleman disease. Ann Hematol. 2018;97(9):1641–7.
Independent Hospital Pricing Authority. National Hospital Cost Data Collection Report: Public Sector, Round 22 (Financial Year 2017-18). Independent Hospital Pricing Authority; 2020. https://www.ihacpa.gov.au/resources/national-hospital-cost-data-collection-nhcdc-public-hospitals-report-round-22-financial-year-2017-18. Accessed 1 Oct 2022.
Reeve R, Srasuebkul P, Langton JM, Haas M, Viney R, Pearson S-A, et al. Health care use and costs at the end of life: a comparison of elderly Australian decedents with and without a cancer history. BMC Palliat Care. 2017;17(1):1.
Vemer P, Corro Ramos I, van Voorn GA, Al MJ, Feenstra TL. AdViSHE: a validation-assessment tool of health-economic models for decision makers and model users. Pharmacoeconomics. 2016;34(4):349–61.
Dispenzieri A, Armitage JO, Loe MJ, Geyer SM, Allred J, Camoriano JK et al. The clinical spectrum of Castleman’s disease. Am J Hematol. 2012;87(11):997–1002.
Liu AY, Nabel CS, Finkelman BS, Ruth JR, Kurzrock R, van Rhee F, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol. 2016;3(4):e163–75.
Seo S, Yoo C, Yoon DH, Kim S, Park JS, Park C-S, et al. Clinical features and outcomes in patients with human immunodeficiency virus-negative, multicentric Castleman’s disease: a single medical center experience. Blood Res. 2014;49(4):253–8.
Shin DY, Jeon YK, Hong Y-s, Kim TM, Lee S-h, Kim D-w, et al. Clinical dissection of multicentric Castleman disease. Leuk Lymphoma. 2011;52(8):1517–22.
Szturz P, Adam Z, Rehák Z, Koukalová R, Sprláková-Puková A, Michalka J, et al. Castleman disease: retrospective single-center study of therapeutic results in 10 patients. Klin Onkol. 2013;26(2):124–34.
Talat N, Belgaumkar AP, Schulte KM. Surgery in Castleman’s disease: a systematic review of 404 published cases. Ann Surg. 2012;255(4):677–84.
Wojtys M, Piekarska A, Kunc M, Ptaszynski K, Biernat W, Zaucha JM, et al. Clinicopathological comparison and therapeutic approach to Castleman disease – a case-based review. J Thorac Dis. 2019;11(11):4859–74.
Zhang L, Zhao AL, Feng J, Cao X, Zhang Y, Zhu T, et al. A prospective phase II study of thalidomide, cyclophosphamide and prednisone in newly diagnosed idiopathic multicentric Castleman’s disease. Blood. 2018;132(Suppl. 1):4142.
Janssen Research & Development. Synoptic Clinical Study Report (Interim Analysis): An Open-label, Multicenter Study to Evaluate the Safety of Long-term Treatment with Siltuximab in Subjects with Multicentric Castleman’s Disease – Clinical study report (CNTO328MCD2002). 2013.
Recordati Rare Diseases Australia Pty Ltd. Sylvant (siltuximab) Product Information: TGA; 2022. https://www.ebs.tga.gov.au/ebs/picmi/picmirepository.nsf/pdf?OpenAgent&id=CP-2022-PI-02499-1&d=20230201172310101. Accessed 1 Oct 2022.
Cubi-Molla P, Buxton M, Devlin N. Allocating public spending efficiently: is there a need for a better mechanism to inform decisions in the UK and elsewhere? Appl Health Econ Health Policy. 2021;19(5):635–44.
Lomas J, Ochalek J, Faria R. Avoiding opportunity cost neglect in cost-effectiveness analysis for health technology assessment. Appl Health Econ Health Policy. 2022;20(1):13–8.
Fujimoto S, Koga T, Kawakami A, Kawabata H, Okamoto S, Mizuki M, et al. Tentative diagnostic criteria and disease severity classification for Castleman disease: a report of the research group on Castleman disease in Japan. Mod Rheumatol. 2018;28(1):161–7.
Nishimoto N, Honda O, Sumikawa H, Johkoh T, Aozasa K, Kanakura Y. A long-term (5-Year) sustained efficacy of tocilizumab for multicentric Castleman’s disease and the effect on pulmonary complications. Am Soc Hematol. 2007;110:646.
Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106(8):2627–32.
Yu L, Tu M, Cortes J, Xu-Monette ZY, Miranda RN, Zhang J, et al. Clinical and pathological characteristics of HIV-and HHV-8–negative Castleman disease. Blood. 2017;129(12):1658–68.
Ebisawa K, Shimura A, Honda A, Masamoto Y, Nakahara F, Kurokawa M. Hemoglobin and C-reactive protein levels as predictive factors for long-term successful glucocorticoid treatment for multicentric Castleman’s disease. Leuk Lymphoma. 2020;62(3):614–19.
Frizzera G, Peterson BA, Bayrd ED, Goldman A. A systemic lymphoproliferative disorder with morphologic features of Castleman’s disease: clinical findings and clinicopathologic correlations in 15 patients. J Clin Oncol. 1985;3(9):1202–16.
Hasija N, Bustamante L, Jaglal MV, Sokol L. Human immunodeficiency virus-negative Castleman’s disease: clinicopathological and laboratory characteristics and outcomes in patients treated in a single institution. Blood. 2015;126(23):5027.
Melikyan AL, Egorova EK, Kovrigina A, Subortseva I, Gilyazitdinova E, Karagyulyan S, et al. Clinical and morphological features of different types of Castleman’s disease. Ter Arkh. 2015;87(7):64–71.
Gallacher D, Auguste P, Connock M. How do pharmaceutical companies model survival of cancer patients? A review of NICE Single Technology Appraisals in 2017. Int J Technol Assess Health Care. 2019;35(2):160–7.
MBS. MBS Online: Australian Government, Department of Health and Aged Care; 2020. http://www9.health.gov.au/mbs/search.cfm. Accessed 1 Oct 2022.
Pharmaceutical Benefits Scheme. The Pharmaceutical Benefits Schedule: Australian Government, Department of Health and Aged Care; 2020. https://www.pbs.gov.au/pbs/home. Accessed 1 Oct 2022.
Chemist Warehouse. Chemist Warehouse Prices 2020. https://www.chemistwarehouse.com.au/?gclid=Cj0KCQjwrs2XBhDjARIsAHVymmR7q2J-BsU9GP8Cq9WC5EfjHoaRh1EL1j5hb7Z8Fb7IwqDKyODpITEaAvRwEALw_wcB&gclsrc=aw.ds. Accessed 1 Oct 2022.
EUSA Pharma. Additional analyses MCD2001–for Australia (data on file). 2020.
Acknowledgements
Pawel Kawalec and Przemyslaw Holko were involved in the building of the model on behalf of EUSA Pharma UK (LTD.) and had received funding for this project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
EUSA Pharma UK (LTD.) funded the work taken to write this manuscript.
Conflict of Interest
Francis Shupo, Grace Wayi-wayi and Natasa Zibelnik were employees of EUSA Pharma UK (LTD.) at the time of conducting this study. EUSA Pharma UK (LTD.) provided funding to KMC Health Care to support the work of Matt Kirchmann (Director), Carolyn Rutherford (Senior Health Economist) and Kelly Makarounas-Kirchmann (Principal) in undertaking the health technology assessment of Siltuximab. Zanfina Ademi reviewed the model and received no payment for this. EUSA Pharma UK (LTD.) also provided funding to Visible Analytics Limited for the work of Keith R Abrams (Director and Partner) in undertaking the Health Technology Assessment of Siltuximab.
Data Availability Statements
The datasets and code generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics Approval
Studies MCD2001 (NCT01024036) and MCD2002 (NCT01400503) had received relevant ethics approvals. Ethics approval was not needed for the cost-effectiveness analysis, as it was based on existing and published data.
Consent to Participate
The cost-effectiveness data were based on existing published data, and there was no need for consent.
Consent for Publications
The cost-effectiveness data were based on existing published data, and there was no need for consent.
Author Contributions
All authors contributed to the study conception and design. Zanfina Ademi and Keith R Abrams independently assessed the structural validity and internal consistency of the model. Material preparation, data collection and analysis were performed by Kelly Makarounas-Kirchmann, Matt Kirchmann, Carolyn Rutherford and Przemyslaw Holko. The first draft of the manuscript was primarily written by Francis Shup, Grace Wayi-Wayi and Kelly Makarounas-Kirchmann, and all authors provided comment and revision on subsequent versions of the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Shupo, F., Abrams, K.R., Ademi, Z. et al. Cost-Effectiveness Analysis of Siltuximab for Australian Public Investment in the Rare Condition Idiopathic Multicentric Castleman Disease. PharmacoEconomics Open 7, 777–792 (2023). https://doi.org/10.1007/s41669-023-00426-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41669-023-00426-x