
Abstract
Homeostasis of monoaminergic pathways is compromised in aging and neurodegenerative/neurological diseases such as Alzheimer’s disease and depression. On the other hand, their modulation has also been linked to the mechanism of action of several drugs. Therefore, monitoring the levels of noradrenaline (NA), adrenaline (AD), dopamine (DA), and serotonin (5-HT), as well as those of tryptophan (TRYP), the precursor of 5-HT, and DA metabolites, namely 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA), is fundamental for assessing disease severity and progression. This work aimed to develop and validate the first High-Performance Liquid Chromatography (HPLC) coupled with fluorescence detector (FLD) method that simultaneously and accurately quantifies NA, AD, DA, DOPAC, HVA, 5-HT and TRYP in mouse brain and prefrontal cortex (PFC) matrices. Previous sample preparation by protein precipitation was required to extract the compounds. Calibration curves were plotted using the background subtraction approach to reduce the interference of the endogenous analytes. Intra and inter-day accuracy and precision were within the ranges defined by ICH (The International Council for Harmonisation) guideline for bioanalytical method validation. Following validation, the impact of cannabidiol (CBD), cannabigerol (CBG), and cannabidivarin (CBDV) was explored in mice brains post-administration, revealing significant alterations in specific neurotransmitter levels upon cannabinoid exposure and shedding light on the complex modulation of neurochemical dynamics by cannabinoids. This research highlights the fit-for-purpose of the HPLC-FLD method and provides insights into potential mechanisms underlying phytocannabinoid actions in the central nervous system (CNS).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Neuronal plasticity is essential for healthy brain functions, requiring functional neurotransmission at the chemical synapses [1, 2]. Noradrenaline (NA), adrenaline (AD), dopamine (DA), and serotonin (5-HT) are the principal neurotransmitters in the monoamine pathways [2, 3] and regulate mood and behavior [4]. However, their homeostasis is compromised by aging as well as various psychiatric, neurological, and neurodegenerative diseases such as Major Depression Disorder, Autism Spectrum Disorder (ASD), Alzheimer's disease, and Parkinson’s disease (PD) [5,6,7,8]. For example, deficits in noradrenergic, serotoninergic, and dopaminergic levels have been observed in depression and Alzheimer’s disease, even though several other hypotheses have been proposed to explain their pathogenesis [5, 9, 10]. Interestingly, some specific brain regions are more affected than others [5, 11], such as the prefrontal cortex (PFC) in depression disease [10], Alzheimer’s disease [7], and ASD [12]. Nevertheless, PD is particularly characterized by extensive damage in the substantia nigra dopaminergic neural cells with decreased striatal levels of DA, which regulates cognition and motivation and controls motor activity and voluntary movements [5, 13]. On the other hand, it has been demonstrated that there is a deficit of 5-HT in the brain of ASD, even though it may be enhanced in the plasma, probably because the metabolic pathways of 5-HT and its precursor tryptophan (TRYP) are affected in ASD children [12, 14]. Indeed, the main source of 5-HT is TRYP, an amino acid that is provided from the diet and that crosses the blood–brain barrier (BBB) [15]. Its metabolism is mediated by the peripheral enzymes tryptophan hydroxylase 1 or by tryptophan hydroxylase 2, which is predominantly expressed in the central nervous system (CNS). Posteriorly, 5-hydroxytryptophan is decarboxylated by the aromatic acid decarboxylase to form 5-HT (Fig. 1) [16]. 5-HT cannot cross the BBB. Therefore, when peripherally synthesized in the enterochromaffin cells of the intestinal mucosa, 5-HT is stored in the platelets; when synthesized in the CNS, it is stored in neurons in the raphe nuclei [15].

Generic representation of monoamine pathways in the brain and the metabolism of the main neurotransmitters. NA is metabolized in adrenergic neurons into adrenaline (AD). Dopamine (DA) is metabolized in dopaminergic neurons forming the main metabolite 3,4-dihydroxyphenylacetic acid (DOPAC) and the final metabolite homovanillic acid (HVA). In adrenergic neurons, DA can be converted into NA. Serotonin (5-HT) is synthesized in serotoninergic neurons from tryptophan (TRYP). The bolded compounds are the ones that were herein quantified by the exposed bioanalytical method.ALDH: aldehyde dehydrogenase; COMT: catechol-O-methyltransferase; DOPAL: 3,4-Dihydroxyphenylacetaldehyde; HVA: homovanillic acid; L-DOPA: levodopa; MAO: monoamine oxidase
Considering these examples, it becomes evident why monoamines are used as biomarkers of disease progression and therapeutic response of a wide range of drugs, and to investigate the mechanisms of action of new drug candidates. Currently, phytocannabinoids have emerged as promising drug candidates to prevent and treat several neurological diseases. Despite the therapeutic potential of phytocannabinoids [10, 17], their main mechanisms of action remain unknown, including their influence on the monoamine pathways. Consequently, comprehensive studies are needed to unravel this modulation.
Monitoring monoamines in the brain is fundamental for diagnosing, treating, and understanding various neurological and psychiatric conditions. However, their interconversion and the change in the catalytic activity of their metabolic enzymes at pathophysiological states underscores the necessity for the simultaneous quantification of the monoamines and their main metabolites and precursors [3, 18]. For instance, in adrenergic neurons, phenylethanolamine N-methyltransferase converts NA into AD (Fig. 1) [13] while DA is converted into NA by dopamine-β-hydroxylase. However, in dopaminergic neurons, DA is metabolized into 3,4-dihydroxyphenylacetic acid (DOPAC) by monoamine oxidase (MAO) and aldehyde dehydrogenase (ALDH). The final metabolite, homovanillic acid (HVA), is converted by catechol-O-methyltransferase (COMT) (Fig. 1) [13]. PD patients have significantly higher urinary HVA/DA ratio than control subjects. Moreover, in the cerebrospinal fluid, untreated PD patients exhibit lower levels of DA and HVA compared with those patients treated with L-DOPA [19, 20].
In this context, the present work was carried out to develop and validate a new bioanalytical method that accurately and simultaneously quantifies not only NA, AD, DA, and 5-HT but also their metabolites DOPAC and HVA, and the precursor TRYP in homogenate samples of mouse brain, employing the direct fluorescence of the analytes.
Until nowadays, high-performance liquid chromatography (HPLC) methods that quantify neurotransmitters have been validated using different detection methods [21], such as electrochemical [22, 23], chemiluminescence [24,25,26], mass spectrometry (MS or MS/MS) [27, 28], or fluorescence detection (FLD) [1, 3, 18, 29]. Electrochemical detection exposes a lack of reproducibility due to the hysteretic degradation of the electrode. On the other hand, chemiluminescence detection presents higher sensitivity and specificity but requires a post-column system for the initiation and detection of chemiluminescence while MS or MS/MS detection allows the quantification and identification of the compounds but depends on compound ionization and requires complex and expensive instrumentation [21]. These drawbacks prompted us to take advantage of the native fluorescence of the monoamines and use the FLD. Although other HPLC–FLD methods have already been developed for measuring neurotransmitters, they do not include all the monoamines and the metabolites/precursors herein proposed. Moreover, the majority of them requires derivatization, hampering the sample preparation process [1, 29], or they did not employ an internal standard (IS), which is important to improve the precision and accuracy of the results, particularly in bioanalytical processes that involve complex sample preparation procedures (e.g., analyte derivatization, extraction, concentration).
Therefore, to the best of our knowledge, this is the first HPLC-FLD assay reporting the simultaneous quantitative determination of NA, AD, DA, 5-HT, DOPAC, HVA, and TRYP in mice brain homogenate, requiring one simple step for sample preparation and employing an appropriate IS [4-(Aminomethyl)benzene-1,2-diol]. Moreover, an experimental preclinical study was performed to investigate the impact of phytocannabinoids on noradrenergic, serotoninergic, and dopaminergic pathways in the PFC and to demonstrate the method is fit for purpose.
2 Experimental
2.1 Chemicals and Reagents
NA, AD, DOPAC, HVA, TRYP, 5-HT, L-Cysteine, Cannabinol (CBN), Cannabidiol (CBD), Cannabichromene (CBC), Cannabigerol (CBG) and Cannabidivarin (CBDV) were acquired from Sigma Aldrich (St. Louis, MO, USA), DA from Fluka (Buchs, Switzerland), and 4-(Aminomethyl)benzene-1,2-diol, the IS, was acquired from BLDpharm (Kaiserslautern, Germany). The perchloric acid (60%, V/V), used to prepare the work solutions of neurotransmitters and brain homogenates, was acquired from Panreac Quimica (Barcelona, Spain).
To prepare the acetate buffer, anhydrous sodium acetate was acquired from Carlo Erba (Val de Reuil, France), octane sulfonic acid sodium salt from Thermo Fisher Scientific (Karlsruhe, Germany), acetic acid glacial from Honeywell Fluka (Seelze, Germany) and ethylenediaminetetraacetic acid (EDTA) from Sigma Aldrich (St. Louis, MO, USA).
Gradient grade methanol was acquired from Honeywell (Saint-Germain-en-Laye, France), and ultrapure water (HPLC grade; 18.2 MΩ·cm) was obtained using a Milli-Q water purification apparatus, Sartoriusarium® pro from Sartorius (Goettingen, Germany).
2.2 Reference Standards Solution
Stock solutions (SS) of NA, AD, DA, DOPAC, HVA, TRYP, and 5-HT were prepared independently in 4% (V/V) perchloric acid at a concentration of 0.5 mg/mL. Posteriorly, SS was diluted with 4% (V/V) perchloric acid 4% to prepare intermediate work solutions at a concentration of 0.05 mg/mL and 0.005 mg/mL. The storage temperature of the SS and intermediate work solutions was −20 °C. Calibration standards and quality control (QC) solutions were prepared from the respective intermediate work solutions by dilution with 0.2% (V/V) perchloric acid and stored at 4 °C. The IS selected was 4-(Aminomethyl)benzene-1,2-diol, prepared in 4% (V/V) perchloric acid at a concentration of 0.5 mg/mL with posterior dilution in 0.2% (V/V) perchloric acid to a final concentration of 0.005 mg/mL.
2.3 Calibration Curve and Quality Controls
Six calibration standard solutions were prepared by blending the respective quantity of each neurotransmitter. According to the defined calibration curve range, it was possible to quantify NA between 10 and 400 ng/mL, AD from 10 to 200 ng/mL, DA from 10 to 800 ng/mL, DOPAC from 50 to 300 ng/mL, HVA from 55 to 300 ng/mL, TRYP from 35 to 1000 ng/mL and 5-HT from 35 to 300 ng/mL.
QC solutions were prepared according to the ICH guideline M10 to Bioanalytical Method Validation [30]. Lower limit of quantification (LLOQ) was prepared at a final concentration of 10 ng/mL to NA, AD, and DA, 50 ng/mL to DOPAC, 55 ng/mL to HVA, and 35 ng/mL to TRYP and 5-HT. QC1, the lower quality control solution, inferior to three times LLOQ, was prepared at a final concentration of 25 ng/mL to NA, AD, and DA, 100 ng/mL to DOPAC, 110 ng/mL to HVA, and 87.5 ng/mL to TRYP and 5-HT. QC2, the medium quality control solution incorporating 30% to 50% of the calibration curve range, was prepared at a final concentration of 180 ng/mL to NA, 90 ng/mL to AD, 360 ng/mL to DA, 150 ng/mL to DOPAC and HVA, 450 ng/mL to TRYP and 135 ng/mL to 5-HT. QC3, the high-quality control solution, superior to 75% of the calibration curve range, was prepared at a final concentration of 320 ng/mL to NA, 160 ng/mL to AD, 640 ng/mL to DA, 255 ng/mL to DOPAC and HVA, 800 ng/mL to TRYP and 240 ng/mL to 5-HT.
2.4 Brain Tissue Preparation
Healthy adult male CD-1 mice were sacrificed by cervical dislocation and decapitation after anesthesia with isoflurane, and the total brain or PFC was quickly collected, weighed, and placed on ice. Tissues were homogenized using a Teflon® pestle from Thomas Scientific (Swedesboro, NJ, United States) for total brain samples or an Ultra-Turrax X 10/25 (Ystral GmbH, Gottingen, Germany) for PFC samples. Tissues were homogenized in ice-cold perchloric acid (0.2 mol/L) supplemented with 6 mmol/L of cysteine (4 mL per g of tissue). Then, the homogenized tissue was centrifugated at 4800 r/min at 4 °C for 15 min and the supernatant was collected (100 µL), spiked with 10 µL of IS, 4-(Aminomethyl)benzene-1,2-diol, and 10 µL of calibration standard solution to construct the calibration curve. This mixture was subjected to protein precipitation (PP) with 10 µL of perchloric acid (2.0 mol/L). Then, the sample was vortexed for 1 min, centrifuged at 14,000 r/min for 4 min, the supernatant was collected and 10 µL was injected into the HPLC apparatus. All procedures were carried out on ice.
2.5 Apparatus and Chromatographic Conditions
Chromatographic analysis was performed on a Shimadzu Prominence HPLC system (Shimadzu Corporation, Kyoto, Japan) constituted by a solvent delivery unit (LC-20A), a degasser system (DGU-20A5), an autosampler (SIL-20AHT), a column oven (CTO-10ASVP), a diode-array detection (DAD) (SPD-M20A) and a fluorescence detector (RF-20AXS). The HPLC system and data acquisition were controlled by LCsolution software (Shimadzu Corporation, Kyoto, Japan). Chromatographic separation was achieved on a Grace Apollo C18 chromatographic column (250 × 4.6 mm, 5 µm).
The chromatographic conditions were optimized and the best conditions that ensured the guideline parameters were a gradient elution (Fig. 2) with a mobile phase constituted by acetate buffer (anhydrous sodium acetate (0.1 mol/L), EDTA (0.3 mmol/L), octane sulfonic acid sodium salt (0.5 mmol/L) and acetic acid (0.05 mol/L), final pH of 4.40) and methanol, pumped at 1.0 mL/min. The chromatographic column was maintained at 27 °C and the total run time was 23 min. The fluorescence detector was set at 279 nm (excitation) and 320 nm (emission).

Representation of the HPLC apparatus and the chromatographic conditions herein used. λEm: wavelength of emission; λExc: wavelength of excitation
2.6 Method Validation
The present HPLC method was fully validated in mice brain and partially validated in PFC taking into account the ICH M10 guideline for bioanalytical method validation [30]. Accordingly, selectivity, carry-over, specificity, calibration curve and respective range, linearity, accuracy, precision, and stability were assessed in mice brain (n = 5), while selectivity, carry-over, calibration curve range, linearity, accuracy, and precision were investigated in triplicate (n = 3) in PFC.
Demonstrating method selectivity guarantees that it properly differentiates and measures the analytes without interferents. However, the analytes under investigation are endogenous compounds present in the biological matrices. To circumvent this, the peak purity was analyzed, recurring to a double detection with DAD and FLD. Hence, following ICH guideline, six blank samples were collected from different animal donors and were subjected to the optimized HPLC method to assess the presence of interferents at the retention time of the analytes. Moreover, the carry-over effect was also tested by running a mobile phase after the highest calibration standard to ensure that the signals of the analytes do not interfere with the next analytical run.
Specificity ensures that the method detects and differentiates the analytes from other substances that potentially will interfere with the analyte quantification, such as substances structurally similar to the analytes, fluorescent compounds, metabolites, isomers, impurities, and degradation products, as well as other substances that may be administered in future preclinical studies. To assess specificity, direct injections of several fluorescent compounds (30 µg/mL each) were prepared by spiking 100 µL of mobile phase acetate buffer: methanol (90:10, V:V) with 10 µL of the respective concentrated solution of the test compound. Moreover, direct injections of CBD, CBN, CBG, CBC, and CBDV (6.5 µg/mL each) were tested to check future possible interferents in vivo preclinical studies.
Calibration curves and their respective ranges were defined based on the endogenous basal concentrations. This approach not only ensured a statistical difference between the endogenous concentrations and the spiked samples at the LLOQ and QC1, but also guaranteed that endogenous concentrations observed in the in vivo studies were within the concentration ranges. Six calibration standards and QCs were defined as described in Sect. 2.3, and at least 75% of calibration standards must present accuracy and precision within ± 15% of the nominal concentration, except LLOQ, which must be within ± 20%. Linearity was confirmed for each compound by five consecutive and individual calibration curves when considering the total validation performed in brain tissue. For that, the homoscedasticity test was performed at linear regression, and F-value, which expresses the ratio between the variances obtained at the lowest and highest concentration levels of the calibration range, was calculated. Regarding partial validation in PFC tissue, three calibration curves were considered in accordance with the ICH guideline.
Accuracy and precision were evaluated by spiking blank samples with the QC standard solutions. Accuracy is represented as bias percentage and expresses the difference between the estimated and nominal concentration. Precision, expressed as the coefficient of variation (CV), demonstrates the nearness of repeated individual measures. The accuracy was calculated using the following formula:
On the other hand, precision was assessed by determining the CV applying the following equation:
Considering the total validation performed in mice brain, intra-day accuracy and precision were verified by five replicates of each QC on the same day, while inter-day accuracy and precision were obtained by five replicates on independent days. For partial validation in the PFC, inter-day accuracy and precision were analyzed, recurring to three replicates.
Sample recovery reflects the efficiency of the extraction process, and it is calculated by comparing non-extracted working solutions of QC1, 2, and 3 with spiked samples with the same QC1, 2, and 3 standard solutions submitted to the extraction process.
Stability conditions were tested in work standard solution and blank samples spiked with QC1 or QC3 standard solutions at different experimental conditions to ensure that, during sample handling, analysis, and storage. Stability is given in % according to the equation bellow and it must be within \(\pm\) 15%.
where t represents the stability condition.
Thus, to investigate analyte stability during sample preparation, unprocessed brain homogenate samples were remained for 2 h at 4 °C and then, prepared and analyzed. In addition, brain homogenate samples were prepared and remained for 3, 6, and 9 h at room temperature (RT), and 24 h at 4 °C to assess the stability of the processed samples until analysis. Additionally, endogenous stability was also assessed in the frozen brain at −20 °C for 3 d and in homogenized brain tissue after freezing it for 3 d at −20 °C.
2.7 Method Application
HPLC method was applied in a preclinic in vivo study to investigate the impact of BD, CBG, and CBDV on monoaminergic pathways. Healthy adult male CD-1 mice were acquired from certified animal suppliers (Harlan Laboratories, Milan, Italy). Animals had ad libitum access to food and tap water and they were accommodated in a relative humidity of (55 ± 5) %, a controlled temperature of (20 ± 2) °C, and reversed 12 h light–dark cycles. All in vivo experiments fulfilled the European Directive (2010/63/EU) regarding the protection of laboratory animals used for scientific purposes and the Portuguese law on animal welfare (Decree-law 113/2013). Moreover, the project was first reviewed and approved by the Animal Welfare Board (ORBEA, 01–2020—Órgão Responsável pelo Bem-Estar Animal) at the Faculty of Pharmacy of the University of Coimbra and the Direção-Geral de Alimentação e Veterinária (DGAV, 0421/000/000/2020, Lisbon, Portugal). Reducing the number of used animals and their suffering was a priority during all the experiments.
Briefly, animals were divided into four groups: the control group (n = 5) and three treated groups (n = 4). The control group received the vehicle composed of polyethylene glycol (PEG) 400 and 0.9% sodium chloride (NaCl) solution (50:50, V/V) [31], and it was administered by intraperitoneal (ip) route (5.8 mL/kg). Treated groups received CBD, CBG, or CBDV at an equimolar dose of 55 µmol/kg (ip) and were sacrificed 30 min post-administration. PFC was quickly removed to quantify NA, AD, DA, DOPAC, HVA, TRYP, and 5-HT. Brain tissue was collected and prepared as described in Sect. 2.4, replacing the 10 µL of calibration standard solution with the 10 µL of 0.2% (V/V) perchloric acid.
GraphPad Prism® 8 software (San Diego, CA, USA) was used for statistical analyses in quantified concentration results for each compound. Kruskal–Wallis test with multiple comparisons using the Uncorrected Dunn’s test was applied, and the results were represented as mean ± Standard Error of Mean (SEM). Statistical significance was considered when p < 0.05.
3 Results and Discussion
3.1 Method Development
Several chromatographic conditions regarding mobile phase composition, excitation and emission wavelengths, and column temperature, were tested to acquire and separate the signal of each analyte. Then, the compounds were singly injected to acknowledge their retention time.
Since this method was applied to biological samples, hence requiring analyte extraction and spiking with a predefined concentration of an IS avoid possible sample preparation errors and guarantee a reproducible and accurate quantification. Potential intrinsic fluorescent compounds, such as norfloxacin, ofloxacin, prulifloxacin, gatifloxacin, pazufloxacin, clinofloxacin, lacosamide, levetiracetam, zonisamide, perampanel, amitriptyline, antipyrine, pitavastatin, atorvastatin and 4-(Aminomethyl)benzene-1,2-diol were tested as IS. The last one was selected because it was the only one with a measurable peak, while none of the others were detected.
The mobile phase was composed of acetate buffer supplemented with EDTA and octane sulfonic acid. While this buffer shows good compatibility with fluorescence detection, EDTA improved peak shapes, and octane sulfonic acid, an ion-pairing reagent, increased the retention time of the compounds.
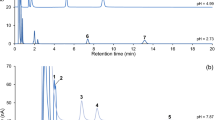
The pH of the acetate buffer was readjusted to 4.40 with acetic acid because it allowed the complete separation and resolution of the peaks. It is noteworthy that slight variations in pH strongly influence the resolution of DA, DOPAC, and HVA, as specified in Fig. 3.

Influence of the mobile phase pH in peak resolution for each compound. The COOH/COO− group of DOPAC (pKa = 4.53) and HVA (pKa = 4.35) leads to variations in retention time and, consequently, peak resolution. TRYP also has a COOH/COO− group. However, TRYP is ionized at pH 4.30 and 4.40 (pKa = 2.36). The remaining compounds do not undergo variations at this pH because their NH2/NH3+ group grants a pKa > 8 (all non-ionized). The variation in DA resolution is related to the variation in retention time of HVA
Preparation and pretreatment of brain and PFC tissue samples were optimized to attain the best compound recuperation without interferences, initiated by tissue homogenization with ice-cold perchloric acid 0.2 mol/L in the proportion of 4 mL per g tissue. Perchloric acid solubilizes the compounds allowing their remotion from the tissue. Solid phase extraction was tested on Oasis HLB 1 mL 30 mg extraction cartridges (Waters™), however, the recoveries of compounds were meager (< 5%) with changes in the retention time. Liquid–liquid extraction (LLE) involves solvent evaporation under high temperatures. Since the neurotransmitters are temperature-sensitive, requiring sample manipulation on ice, LLE was not herein feasible. Therefore, PP extraction was optimized by testing different solvents. To 100 µL of brain homogenized, perchloric acid (0.2 mol/L, 1:2, V/V), perchloric acid (0.7 mol/L, 1:1, V/V), and acetonitrile (1:1, V/V) were tested resulting in a high sample dilution with consequent lower recoveries. To avoid sample dilution, perchloric acid (2 mol/L, 1:0.2, V/V) and trichloroacetic acid (20%, V/V) (1:0.2, V/V) were tested with recoveries ranging from 60 to 73%. The highest and most precise recuperation procedure was achieved with perchloric acid at 2 mol/L (1:0.1, V/V), with recovery values equal to or exceed 67%. Distinct times of vortex and centrifugation conditions, such as acceleration and time, were also investigated. Higher vortex and centrifugation time did not increase the recovery percentage leading to the final conditions, which are described in Sect. 2.4. However, under these experimental conditions, the stability of 5-HT was compromised, requiring the addition of cysteine to the ice-cold perchloric acid (0.2 mol/L) during homogenization. Different concentrations of cysteine (3, 6, and 9 mmol/L) were tested, and 6 mmol/L was selected as it ensured higher stability.
An isocratic elution, where the same proportion of acetate buffer and methanol is constant during all the running time, was the first approach. Moreover, there was an endogenous interferent at the retention time of NA, which demanded to optimize the chromatographic conditions. To a complete separation of the interferent from NA, it was necessary to decrease de proportion of methanol in the mobile phase. Nevertheless, TRYP, HVA, and 5-HT were eluted at higher retention times and with a broad peak shape. The complete separation was achieved recurring to a gradient elution as in Sect. 2.5.
The oven temperature was defined at 27 °C since it was the optimal condition for the most symmetric peak and best resolution without compromising the compounds stability.
The wavelengths for excitation (279 nm) and emission (320 nm) were defined based on the fluorescence spectrum of each compound and a diluted homogenized brain sample.
3.2 Full Method Validation in Brain
For total method validation in brain tissue, ICH Guideline M10 to Bioanalytical methods validation requirements must be fulfilled, namely selectivity, carry-over, specificity, calibration curve range and linearity, accuracy and precision, recovery, and stability [30].
Selectivity was evaluated by running six blank samples from independent donors, obtaining similar chromatograms. Nonetheless, neurotransmitters are endogenous compounds constitutively present in the brain, existing in the blank samples. According to the ICH Guideline M10 for Bioanalytical methods validation, since the interference of the endogenous compounds does not avoid an adequate signal-to-noise ratio, the background subtraction approach should be chosen for method validation [30]. This approach comprises pooling and mixing different brain homogenates to obtain a representative matrix, which will be used to construct the calibration curve. Blank samples (pool) are analyzed, and the endogenous concentrations measured will be subtracted from the areas observed on the calibration standard solutions. The obtained difference regards the concentration of the sample. Thus, the calibration curve was constructed resorting to the difference between the spiked matrix sample and the blank sample. The background subtraction approach allows an equal matrix effect because the study samples matrix is the same biological matrix used to construct the calibration curve [30].
An additional step to confirm selectivity was implemented using a double detection system encompassing DAD and FLD to compare the chromatographic runs of mobile phase spiked with one calibration standard, blank samples, and blank samples spiked with the same calibration standard (Fig. 4). The retention time of the compound was the same. The areas of the blank spiked sample would be the sum of the areas of the spiked mobile phase with the blank sample. Furthermore, overlapping the spiked mobile phase with the blank sample and the spiked sample confirmed the addition of areas without other interferences, supporting selectivity and peak purity (Fig. 4).

Chromatogram of a a mobile phase spiked with the calibration standard P6, b a blank sample, and c a blank sample spiked with the fortification solution P6. IS (0.5 µg/mL) presents a retention time of 7.7 min and it is present in all spiked samples
The carry-over effect was also analyzed by running the highest QC sample. No peaks were observed in successive analytical runs at the retention times of the analytes.
Specificity was also performed particularly bearing in mind the in vivo studies herein were developed with cannabinoids (Sect. 2.7). Indeed, cannabinoids emit fluorescence and could interfere with the analytes under investigation [32]. We demonstrated that CBG, CBC, CBN, CBD, and CBDV did not interfere with the neurotransmitters, their metabolites, or precursors since their elution occurs with the mobile phase at void time.
Calibration curves and QC samples were defined according to the endogenous levels quantified in blank samples of brain tissue. During full validation, five calibration curves (n = 5) were independently tested for each neurotransmitter in brain-homogenized tissue. The calibration curves ranges were defined as follows: NA (10–400 ng/mL); AD (10–200 ng/mL); DA (10–800 ng/mL); DOPAC (50–300 ng/mL); HVA (55–300 ng/mL), TRYP (35–1000 ng/mL), and 5-HT (35–300 ng/mL). Since heteroscedasticity was confirmed for all the analytes except DOPAC, several weighting factors were tested and 1/x2 was selected as it ensured the smallest sum of absolute percentage relative errors for intra-day and the best weighted linear regression factor (r). The calibration parameters attained for each compound, including slope, interception, and regression coefficient (r2) are shown in Table 1. Good linearity was found for all analytes (r2 ≥ 0.988).
To perform accuracy and precision, inter and intra-day tests were conducted using the QCLLOQ, QC1, QC2, and QC3. Precision and accuracy were recorded during five independent days (n = 5) for total validation, intra-day analysis was performed by the repetition of each QC five times on the same day (n = 5), while inter-day analysis was performed comparing each QC on five different days. The results are presented in Table 2, and all of them are within the required ranges admitted by the ICH Guideline M10 on Bioanalytical Method Validation defined, i.e. ± 20% in the QCLLOQ and ± 15% in the QC1, QC2, and QC3 [30]. Recovery of each neurotransmitter was investigated recurring to QC1, QC2, and QC3 by comparing the values obtained in the spiked samples subjected to the extraction process with spiked samples non-subjected to the extraction process for each compound. The recoveries of each neurotransmitter are presented in Table 3. The CV values were lower than 13.87%, demonstrating the precision of the sample preparation procedure.
The absolute recovery of the IS (500 ng/mL) was (108.01 \(\pm\) 2.72) %, expressed as (mean \(\pm\) SD) %, which also proves precision in the sample preparation process.
Stability conditions were defined based on the sample preparation, storage, and the time to their analyses. The stability of unprocessed brain samples revealed that all the compounds were stable at 4 °C for 2 h (89.30%—109.47%, which is in the defined range by the ICH Guideline). Regarding processed samples, all compounds were stable for 24 h at 4 °C and 6 h at RT (the stability percentage was between 86.89% and 111.67% and in accordance with ICH). NA, AD, DA, and HVA were stable for 9 h at RT (the stability percentage was between 91.65% and 103.90%) (Table 4).
Since the monoamines are endogenous compounds, according to the ICH Guideline, stability experiments should mimic as much as possible the study samples and should be analyzed in the authentic biological matrix [30]. In this way, the endogenous stability in the brain homogenates and brain tissue was also investigated, without spiking with any QC standard solution. Hence, at −20 °C for 3 d, only DOPAC and 5-HT were not stable in brain homogenate, while the remaining compounds were within 93.89% to 99.74%. All compounds were stable after one cycle of freezing and thawing of the brain tissue (94.1–111.74%, Table 5). Note that, the endogenous stability of AD was not possible to determine since the endogenous concentration was inferior to the LLOQ.
The stability of the combined calibration standards and SS was demonstrated during 7 d at 4 °C (Table 6) as well as the IS standard solution (102.44%). However, the stability of the IS and SS was guaranteed during 7 d at −20 °C (100.43%).
3.3 Partial Method Validation in PFC
Established the best experimental conditions and once validated, the HPLC method was partially validated in mice PFC matrix to quantify the analytes in vivo preclinical studies. According to ICH guideline, partial validation of the method ideally requires selectivity, carry-over effect, calibration curve range, linearity, as well as inter-day accuracy and precision [30].
Herein, selectivity was evaluated by running 6 different PFC homogenates obtained from independent animals. The chromatograms of 6 blank PFC homogenates were very similar to those observed for the total brain homogenates, revealing no additional peaks or statistical differences at the areas of the identified peaks. The carry-over effect was absent and the interferents was not observed from previous runs.
Considering the similarity between PFC and total brain homogenates, the calibration curve range applied in the PFC matrix was the same as the total brain matrix. Linearity was investigated through the construction of three calibration curves on independent days (n = 3) and a good linearity was observed for all analytes (r2 ≥ 0.986). The calibration parameters collected for each compound, including slope, interception, and regression coefficient (r2) are shown in Table 7.
Inter-day accuracy and precision were achieved recurring to the QCLLOQ, QC1, QC2, and QC3 and were recorded during three independent days (n = 3). The results are presented in Table 8, and all of them are within the required ranges admitted by the ICH Guideline M10 on Bioanalytical Method Validation defined, i.e. ± 20% in the QCLLOQ and ± 15% in the QC1, QC2, and QC3 [30]. The partial validation in PFC homogenates demonstrated reproducibility and can be applied in in vivo preclinical studies.
3.4 Method Application
To investigate the impact of CBD, CBG, and CBDV on monoaminergic pathways, healthy male mice received one single dose of CBD, CBG, or CBDV (55 µmol/kg, ip, n = 4). The control group of healthy CD-1 male mice (n = 5) was administered with the vehicle. Animals were sacrificed 30 min post-administration to quantify the neurotransmitters, metabolites, and precursors at the PFC (Fig. 5).

Representative chromatogram of a PFC sample collected from mice treated with 55 µmol/kg of CBD intraperitoneally
Except for AD, all neurotransmitters, their metabolites, and precursors were quantified in PFC matrices and were within the calibration curve ranges herein defined. In fact, AD has been also reported as unmeasurable with other analytical methods. For instance, Benedetto et al. stated AD concentrations were lower than LLOQ (6.0 ng/mL) in healthy mice [3]. Importantly, non-treated animals evidenced concentration ranges of NA, 5-HT, and DA in PFC similar to those previously reported in the literatures [3, 18, 24, 33]. Only two methods currently quantify neurotransmitters by HPLC-FLD without derivatization, however, they require higher injection volumes [3, 18]. Benedetto et al. only quantified four neurotransmitters (NA, DA, DOPAC, and 5- HT) in mice brain homogenates, with higher LLOQ for NA and DA, whereas the LLOQ of 5-HT was very similar to that reported herein (31 ng/mL vs. 35 ng/mL) [3]. The technique of Fonseca et al. also quantified DA, NA, AD and HVA but lacked neurotransmitters from the serotoninergic pathway. Moreover, it was developed in rat matrices and required gradient elution using two different buffers [18].
The levels of NA in the PFC of healthy mice 30 min following the administration of the cannabinoids were lower than those observed in the control group. Specifically, animals treated with CBDV presented statistical differences (p < 0.05, Fig. 6a) relative to the control group. In opposition, no statistical differences were found between the 5-HT concentrations in the PFC of treated and untreated animal groups. However, animals treated with CBD or CBDV presented 5-HT concentrations lower than those observed in CBG-treated animals (p < 0.05, Fig. 6b), suggesting that CBD and CBDV modulate the serotoninergic pathway in healthy animals. In addition, cannabinoids revealed a tendency to increase TRYP levels, particularly CBG (p < 0.05 compared to the control group, Fig. 6c), suggesting the potential of CBG to restore TRYP levels in CNS diseases in which it is decreased, such as major depressive disorder [34, 35] and bipolar disorder [36]. These results are innovative and only Jenny, M. et al. have already demonstrated that CBD modulates serotoninergic signaling increasing the availability of TRYP in mitogen-stimulated peripheral blood mononuclear cells [37].

Results of neurotransmitters quantification. Concentrations of a noradrenaline, b serotonin, c tryptophan, d dopamine and dopamine metabolites, e DOPAC and f HVA in PFC 30 min after an acute intraperitoneal administration of CBD, CBG or CBDV (55 µmol/kg) to mice (n = 4). Results are represented as mean ± Standard Error of Mean (SEM). *p < 0.05 and **p < 0.01 as extracted from Kruskal–Wallis test, with multiple comparison Uncorrected Dunn’s test
CBD and CBDV also significantly decreased the levels of DA in PFC compared to the control group and the animals treated with CBG (p < 0.05, Fig. 6d). Complementarily, DOPAC and HVA, which are the main metabolites of DA, are enhanced after cannabinoid administration (Fig. 6e and f), even though statistical differences were found only between CBD and the control group for DOPAC. These findings suggest that CBD and CBDV can increase the metabolism of DA in PFC. In rats, Rossignoli, M. T. et al. reported a decrease in the DOPAC/DA ratio 5 d after intra-PFC microinjection of CBD [11].
These results evidence that this bioanalytical method could be accurately and precisely applied to demonstrate that cannabinoids modulate the monoaminergic pathways in healthy mice with a single intraperitoneal administration. Since this method has revealed its translational potential from brain tissue to PFC, it is expected to also be well succeeded if applied in other brain regions and after administration of other drugs or xenobiotics besides cannabinoids.
4 Conclusions
The simultaneous quantification of NA, AD, DA, and 5-HT, as well as the metabolites DOPAC and HVA, and the 5-HT precursor, TRYP, in mice brains make this HPLC-FLD method a pioneering approach for neurotransmitter quantification. It was fully validated according to the ICH M10 guideline for bioanalytical method validation using the authentic biological matrix and the background subtraction approach, ensuring reliability, accuracy, and precision of sample preparation and reduction of the matrix effect and interferences.
A major benefit of this HPLC method is the use of native fluorescence of analytes, avoiding complex sample derivatization procedures, saving time and resources, and reducing technical errors. The high recovery and good CV values corroborate the quality of the sample preparation procedure. In addition, the method demonstrated to be selective, specific, accurate, precise, and fit-to-purpose, allowing the simultaneous measurement of neurotransmitters, metabolites, and precursors, which are interconverted in vivo and have distinct neurochemical activities in diverse regions of the brain. Despite these advantageous, the present method does not differentiate between intra and extracellular neurotransmitter levels as it could be attained, for example, with brain microdialysis.
Indeed, the results obtained in the preclinic in vivo studies highlight the ability of CBG to increase TRYP levels while CBD enhanced the metabolism of DA and CBDV reduced NA and DA levels in PFC from mice. To our knowledge, this is probably the first scientific evidence that NA, DA, DOPAC, HVA, 5-HT, and TRYP levels are modulated by CBG, CBD, and CBDV.
This bioanalytical method is expected to support the design of targeted therapies and medications that address specific neurotransmitter imbalances, ultimately improving the quality of life for individuals with neurological dysfunctions.
Abbreviations
- 5-HT:
-
Serotonin
- AD:
-
Adrenaline
- ALDH:
-
Aldehyde dehydrogenase
- COMT:
-
Catechol-O-methyltransferase
- ASD:
-
Autism spectrum disorders
- CBC:
-
Cannabichromene
- CBD:
-
Cannabidiol
- CBDV:
-
Cannabidivarin
- CBG:
-
Cannabigerol
- CBN:
-
Cannabinol
- CNS:
-
Central nervous system
- DA:
-
Dopamine
- DAD:
-
Diode-array detection
- DOPAC:
-
3,4-Dihydroxyphenylacetic acid
- FLD:
-
Fluorescence detection
- HPLC:
-
High-performance liquid chromatography
- HVA:
-
Homovanillic acid
- IS:
-
Internal standard
- LLOQ:
-
Lower limit of quantification
- NA:
-
Noradrenaline
- PD:
-
Parkinson’s disease
- PFC:
-
Prefrontal cortex
- QC:
-
Quality control
- SS:
-
Stock solution
- TRYP:
-
Tryptophan
- MAO:
-
Monoamine oxidase
References
Fonseca BM, Cristóvão AC, Alves G. An easy-to-use liquid chromatography method with fluorescence detection for the simultaneous determination of five neuroactive amino acids in different regions of rat brain. J Pharmacol Toxicol Method. 2018;91:72–9.
Da Y, Luo S, Tian Y. Real-time monitoring of neurotransmitters in the brain of living animals. ACS Appl Mater Interfaces. 2023;15:138–57.
De Benedetto GE, Fico D, Pennetta A, Malitesta C, Nicolardi G, Lofrumento DD, De Nuccio F, La Pesa V. A rapid and simple method for the determination of 3,4-dihydroxyphenylacetic acid, norepinephrine, dopamine, and serotonin in mouse brain homogenate by HPLC with fluorimetric detection. J Pharm Biomed Anal. 2014;98:266–70.
Nutt DJ. Relationship of neurotransmitters to the symptoms of major depressive disorder. J Clin Phychiatric. 2008;69:1–7.
Teleanu RI, Niculescu AG, Roza E, Vladâcenco O, Grumezescu AM, Teleanu DM. Neurotransmitters—key factors in neurological and neurodegenerative disorders of the central nervous system. Int J Mol Sci. 2022;23(11):1–24.
Kraus C, Castrén E, Kasper S, Lanzenberger R. Serotonin and neuroplasticity —links between molecular, functional and structural pathophysiology in depression. Neurosci Biobehav Rev. 2017;77:317–26.
Mather M. Noradrenaline in the aging brain: Promoting cognitive reserve or accelerating Alzheimer’s disease? Semin Cell Dev Biol Elsevier Ltd. 2021;116:108–24.
Davidson M, Rashidi N, Nurgali K, Apostolopoulos V. The role of tryptophan metabolites in neuropsychiatric disorders. Int J Mol Sci. 2022;23(17):1–18.
Ju Y, Tam KY. Pathological mechanisms and therapeutic strategies for Alzheimer’s disease. Neural Regen Res. 2022;17(3):543–9.
Fonseca C, Ettcheto M, Bicker J, Fernandes MJ, Falcão A, Camins A, Fortuna A. Under the umbrella of depression and Alzheimer’s disease physiopathology: can cannabinoids be a dual-pleiotropic therapy? Ageing Res Rev. 2023;90:1–26.
Rossignoli MT, Lopes-Aguiar C, Ruggiero RN, Do Val da Silva RA, Bueno-Junior LS, Kandratavicius L, Peixoto-Santos JE, Crippa JA, Cecilio Hallak JE, Zuardi AW, Szawka RE, Anselmo-Franci J, Leite JP, Romcy-Pereira RN. Selective post-training time window for memory consolidation interference of cannabidiol into the prefrontal cortex: reduced dopaminergic modulation and immediate gene expression in limbic circuits. Neuroscience. 2017;350:85–93.
Xiao L, Yan J, Yang T, Zhu J, Li T, Wei H, Chen J. Fecal microbiome transplantation from children with autism spectrum disorder modulates tryptophan and serotonergic synapse metabolism and induces altered behaviors in germ-free mice. M Systems. 2021;6(2):1–17.
Channer B, Matt SM, Nickoloff-bybel EA, Pappa V, Agarwal Y, Wickman J, Gaskill PJ. Dopamine, immunity, and disease. Pharmacol Rev. 2023;75:62–158.
Pais ML, Martins J, Castelo-Branco M, Gonçalves J. Sex differences in tryptophan metabolism: a systematic review focused on neuropsychiatric disorders. Int J Mol Sci. 2023;24(6):1–25.
Kanova M, Kohout P. Serotonin—Its synthesis and roles in the healthy and the critically Ill. Int J Mol Sci. 2021;22(9):1–12.
Correia AS, Vale N. Tryptophan metabolism in depression: a narrative review with a focus on serotonin and kynurenine pathways. Int J Mol Sci. 2022;23(15):1–17.
Stone NL, Murphy AJ, England TJ, O’Sullivan SE. A systematic review of minor phytocannabinoids with promising neuroprotective potential. Br J Pharmacol. 2020;177(19):4330–52.
Fonseca BM, Rodrigues M, Cristóvão AC, Gonçalves D, Fortuna A, Bernardino L, Falcão A, Alves G. Determination of catecholamines and endogenous related compounds in rat brain tissue exploring their native fluorescence and liquid chromatography. J Chromatogr B Anal Technol Biomed Life Sci. 2017;1049–1050:51–9.
Andersen AD, Blaabjerg M, Binzer M, Kamal A, Thagesen H, Kjaer TW, Stenager E, Gramsbergen JBP. Cerebrospinal fluid levels of catecholamines and its metabolites in Parkinson’s disease: effect of l-DOPA treatment and changes in levodopa-induced dyskinesia. J Neurochem. 2017;141(4):614–25.
Wichit P, Thanprasertsuk S, Phokaewvarangkul O, Bhidayasiri R, Bongsebandhu-phubhakdi S. Monoamine levels and Parkinson’s disease progression: evidence from a high-performance liquid chromatography study. Front Neurosci. 2021;15:1–8.
Bicker J, Fortuna A, Alves G, Falcão A. Liquid chromatographic methods for the quantification of catecholamines and their metabolites in several biological samples—a review. Anal Chim Acta. 2013;768(1):12–34.
Gunaratna PC, Cadle KK, Kissinger CB. An improved liquid chromatographic method with electrochemical detection for direct determination of serotonin in microdialysates from Caudate-putamen and pineal gland regions of rat brain. J Neurosci Methods. 2006;155(1):143–8.
Monge-Acuña AA, Fornaguera-Trías J. A high performance liquid chromatography method with electrochemical detection of gamma-aminobutyric acid, glutamate and glutamine in rat brain homogenates. J Neurosci Methods. 2009;183(2):176–81.
Tsunoda M, Funatsu T. Catecholamine analysis with strong cation exchange column liquid chromatography-peroxyoxalate chemiluminescence reaction detection. Anal Bioanal Chem. 2012;402(3):1393–7.
Ragab GH, Nohta H, Zaitsu K. Chemiluminescence determination of catecholamines in human blood plasma using 1,2-bis(3-chlorophenyl)ethylenediamine as pre-column derivatizing reagent for liquid chromatography. Anal Chim Acta. 2000;403(1–2):155–60.
Nalewajko E, Wiszowata A, Kojło A. Determination of catecholamines by flow-injection analysis and high-performance liquid chromatography with chemiluminescence detection. J Pharm Biomed Anal. 2007;43(5):1673–81.
Huang F, Li J, Shi HL, Wang TT, Muhtar W, Du M, Zhang BB, Wu H, Yang L, Hu ZB, Wu XJ. Simultaneous quantification of seven hippocampal neurotransmitters in depression mice by LC-MS/MS. J Neurosci Methods. 2014;229:8–14.
Zhang MY, Hughes ZA, Kerns EH, Lin Q, Beyer CE. Development of a liquid chromatography/tandem mass spectrometry method for the quantitation of acetylcholine and related neurotransmitters in brain microdialysis samples. J Pharm Biomed Anal. 2007;44(2):586–93.
Zhao XE, Suo YR. Simultaneous determination of monoamine and amino acid neurotransmitters in rat endbrain tissues by pre-column derivatization with high-performance liquid chromatographic fluorescence detection and mass spectrometric identification. Talanta. 2008;76(3):690–7.
European Medicines Agency. ICH Guideline M10 on Bioanalytical Method Validation and study sample analysis. Science Medicines Health; 2022. p. 1–45.
Gad SC, Spainhour CB, Shoemake C, Pallman DRS, Stricker-Krongrad A, Downing PA, Seals RE, Eagle LA, Polhamus K, Daly J. Tolerable levels of nonclinical vehicles and formulations used in studies by multiple routes in multiple species with notes on methods to improve utility. Int J Toxicol. 2016;35(2):95–178.
Citti C, Braghiroli D, Vandelli MA, Cannazza G. Pharmaceutical and biomedical analysis of cannabinoids: a critical review. J Pharm Biomed Anal. 2018;147:565–79.
Renard CE, Dailly E, David DJP, Hascoet M, Bourin M. Monoamine metabolism changes following the mouse forced swimming test but not the tail suspension test. Fundam Clin Pharmacol. 2003;17(4):449–55.
Marx W, McGuinness AJ, Rocks T, Ruusunen A, Cleminson J, Walker AJ, Gomes-da-Costa S, Lane M, Sanches M, Diaz AP, Tseng PT, Lin PY, Berk M, Clarke G, O’Neil A, Jacka F, Stubbs B, Carvalho AF, Quevedo J, Soares JC, Fernandes BS. The kynurenine pathway in major depressive disorder, bipolar disorder, and schizophrenia: a meta-analysis of 101 studies. Mol Psychiatry Springer, US. 2021;26(8):4158–78.
Pawlowski T, Pawlak D, Inglot M, Zalewska M, Marciniak D, Bugajska J, Janocha-Litwin J, Malyszczak K. The role of anthranilic acid in the increase of depressive symptoms and major depressive disorder during treatment for hepatitis c with pegylated interferon-α2a and oral ribavirin. J Psychiatry Neurosci. 2021;46(1):E166–75.
Bartoli F, Misiak B, Callovini T, Cavaleri D, Cioni RM, Crocamo C, Savitz JB, Carrà G. The kynurenine pathway in bipolar disorder: a meta-analysis on the peripheral blood levels of tryptophan and related metabolites. Mol Psychiatry. 2021;26(7):3419–29.
Jenny M, Schröcksnadel S, Überall F, Fuchs D. The potential role of cannabinoids in modulating serotonergic signaling by their influence on tryptophan metabolism. Pharmaceuticals. 2010;3(8):2647–60.
Acknowledgements
This work was supported by funds from Fundação para a Ciência e a Tecnologia (FCT), Portugal, European Union, MCTES, and Fundo Social Europeu (FSE) in the scope of the Centro 2020 Regional Operational Programme (Carla’s PhD research grant 2021.06135.BD and Mariana’s PhD research grant 2020.6582.BD). This work was also supported by FCT Exploratory Project 2022.01066.PTDC, COMPETE and FEDER funds, FCT, Portugal. Figures 1 and 2 were created with BioRender.com.
Funding
Open access funding provided by FCT|FCCN (b-on).
Author information
Authors and Affiliations
Contributions
Carla Fonseca: Data curation; Validation, Investigation, Writing—original draft, Visualization. Susana Cunha: Investigation. Ricardo Ferreira: Investigation. Mariana Lapo Pais: Investigation. Joana Gonçalves: Resources. Antoni Camins: Writing—review & editing, Supervision. Miren Ettcheto: Writing—review & editing, Supervision. Rui Barbosa: Supervision. Ana Fortuna: Writing—review & editing, Supervision, Project administration, Funding acquisition, Resources, Validation.
Corresponding author
Ethics declarations
Conlict of interest
The authors declare that they have no competing interests.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Carla, F., Susana, C., Ricardo, F. et al. Cannabinoid Modulation of Monoamine Levels in Mouse Brain: Unveiling Neurochemical Dynamics through an Innovative High-performance Liquid Chromatography-Fluorescence Detection Bioanalysis. J. Anal. Test. (2024). https://doi.org/10.1007/s41664-024-00307-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41664-024-00307-0