
Abstract
Introduction
The efficacy and safety of ixekizumab, an anti-interleukin-17A antibody, in patients with severe symptoms of psoriatic arthritis are largely unexplored. We report the efficacy and safety of ixekizumab in a post hoc analysis of the SPIRIT-P1 trial.
Methods
Patients were treated with placebo, ixekizumab 80 mg every 2 weeks (Q2W) or 4 weeks (Q4W), or adalimumab 40 mg Q2W for 24 weeks. In this subgroup analysis of SPIRIT-P1, the population with severe psoriatic arthritis was defined using the modified composite psoriatic activity index total score > 7 and peripheral arthritis score = 3 (> 4 tender or swollen joint count and ≥ 0.5 Health Assessment Questionnaire-Disability Index). Efficacy was measured by joint and skin endpoints including disease progression.
Results
In the severe population, significantly more patients (p ≤ 0.001) treated with ixekizumab than placebo achieved 20% improvement according to the American College of Rheumatology criteria (ACR 20): 63.3% for ixekizumab Q4W, 60.4% for ixekizumab Q2W, and 24.5% for placebo. Statistically greater responses compared with placebo were observed in the severe population for ACR 50, ACR 70, ACR core set, disease activity index for psoriatic arthritis (DAPSA) low disease activity and DAPSA remission, and 28-joint disease activity score using C-reactive protein, as well as Psoriasis Area and Severity Index (PASI) 75, PASI 90, and PASI 100 (p ≤ 0.001). Efficacy findings and the safety profile of ixekizumab in the severe population were consistent with those of the overall population, with no new safety concerns identified.
Conclusions
In patients with severe psoriatic arthritis, 24 weeks of treatment with ixekizumab resulted in improvements in both joint and skin symptoms. The safety profile in the severe population was consistent with the established safety profile of ixekizumab.
Trial Registration
ClinicalTrials.gov identifier, NCT01695239.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Psoriatic arthritis (PsA) is a chronic inflammatory disease associated with joint inflammation and psoriasis and is commonly treated using disease-modifying antirheumatic drugs such as ixekizumab (an interleukin-17A antagonist). |
Currently, efficacy and safety evidence of ixekizumab is insufficient for the management of patients with severe PsA and efficacy analyses in patients with severe PsA treated with biologics or targeted conventional synthetic disease-modifying antirheumatic drugs are lacking. |
We report the efficacy and safety of ixekizumab in patients with severe PsA symptoms in a post hoc analysis of the SPIRIT-P1 trial. |
What was learned from the study? |
Treatment with ixekizumab improved joint and skin symptoms in patients with severe disease activity with an adequate safety profile, consistent with findings in the overall study population. |
These findings support the use of ixekizumab for the treatment of patients with severe PsA symptoms. |
Introduction
Psoriatic arthritis (PsA) is a chronic, heterogeneous, inflammatory disease associated with symptoms such as peripheral arthritis, enthesitis, dactylitis, spondylitis, and skin disease [1, 2]. PsA can lead to progressive joint destruction and functional disability. Patients with PsA experience physical and psychosocial difficulties and reduced quality of life [3, 4].
Ixekizumab is a specific inhibitor of the interleukin (IL)-17A cytokine and is approved in adults for the treatment of active PsA, active ankylosing spondylitis, and moderate-to-severe plaque psoriasis [5]. In the phase 3 SPIRIT-P1 and SPIRIT-P2 studies, ixekizumab demonstrated efficacy both as monotherapy and with background conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) or methotrexate following 24 and 52 weeks of treatment [6,7,8,9]. Furthermore, ixekizumab showed sustained efficacy in treating patients with PsA for up to 3 years, with a long-term safety profile consistent with previous reports [10].
In patients with PsA, optimal improvements in health-related quality of life are dependent on successful treatment of both joint and skin symptoms [11]. In the 52-week SPIRIT-H2H study, ixekizumab was superior to adalimumab in improvement of joint and skin disease (American College of Rheumatology [ACR] 50 and Psoriasis Area and Severity Index [PASI] 100) in patients with PsA and inadequate response to csDMARDs [12, 13]. According to the European League Against Rheumatism recommendations for the pharmacological management of PsA, an IL-17 inhibitor may be preferred for patients with peripheral arthritis and skin involvement [14]. In a post hoc analysis of the SPIRIT-H2H trial, ixekizumab was shown to be more effective than adalimumab in simultaneously achieving ACR 50 and PASI 100, regardless of baseline severity of psoriasis [15].
At present, there is insufficient evidence and an unmet medical need regarding the efficacy and safety of ixekizumab in patients with severe symptoms of PsA including severe peripheral arthritis. Furthermore, there are currently no broadly accepted criteria defining the severity of PsA, nor reports of efficacy analyses in patients with severe PsA treated with biologics or csDMARDs. We report a subgroup analysis of the SPIRIT-P1 trial examining the efficacy and safety of ixekizumab in patients with severe PsA symptoms including severe peripheral arthritis, defined as modified composite psoriatic activity index (mCPDAI) total score > 7 [16] and peripheral arthritis score = 3 (> 4 tender joint count or swollen joint count [17], and ≥ 0.5 Health Assessment Questionnaire-Disability Index [HAQ-DI]).
Methods
Study Design and Patients
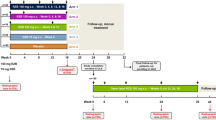
The SPIRIT-P1 study design has been described previously [9]. Briefly, SPIRIT-P1 was a phase 3, multicenter, randomized, double-blind, clinical trial comparing the efficacy and safety of ixekizumab 80 mg every 4 weeks (Q4W) or every 2 weeks (Q2W) and an active reference arm of adalimumab (Humira®; AbbVie) 40 mg Q2W, with placebo in patients not previously treated with biologic agents for plaque psoriasis and PsA. All treatments were administered via subcutaneous injection, with patients randomized to the ixekizumab treatment group receiving a starting dose of 160 mg. Patients with an inadequate response at week 16, regardless of their treatment group, were required to add or modify concurrent treatment, with investigators/study personnel/patients blinded to the inadequate response criteria. Inadequate responders remained on their original dose of ixekizumab, or if receiving adalimumab or placebo, were re-randomized to ixekizumab Q4W or Q2W in a 1:1 ratio.
Enrolled patients were adults (≥ 18 years) who fulfilled the Classification Criteria for Psoriatic Arthritis, had active PsA defined as the presence of ≥ 3 of 68 tender joints and ≥ 3 of 66 swollen joints, had either ≥ 1 PsA-related hand or foot joint erosion on centrally read X-rays or C-reactive protein (CRP) > 6 mg/l, and active psoriatic skin lesions (plaque) or a documented history of plaque psoriasis. Patients with symptoms consistent with axial involvement were not excluded from the SPIRIT-P1 study.
As PsA can affect various domains in addition to peripheral arthritis, we defined PsA severity by considering multiple clinical domains. The total mCPDAI score is a composite measure defining the severity of individual domains including peripheral arthritis, skin, enthesitis, dactylitis, and axial manifestation, but not including Ankylosing Spondylitis Quality of Life (ASQoL). It has previously been reported that the cutoff for high disease activity for CPDAI is ‘8’ [18]. In SPIRIT-P1, mCPDAI (excluding ASQoL) was used as one of the secondary endpoints. Taken together, for this subgroup analysis of SPIRIT-P1, we defined the severe population using both the total and peripheral arthritis scores. Hence, patients with a mCPDAI total score > 7 [16] and peripheral arthritis score = 3 (the highest score) were defined as having severe symptoms.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committees at all sites where these studies were conducted and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study, and all participants consented to the publication of study results. Protocols and consent forms were approved by the institutional review board or ethics committee of each site, including the Western Institutional Review Board (SPIRIT-P1). A listing of individual sites for SPIRIT-P1 is included in the supplement of the primary manuscript [9]. SPIRIT-P1 was registered at ClinicalTrials.gov, NCT01695239.
Study Assessments
Efficacy was measured by the proportion of patients achieving ACR 20, ACR 50, and ACR 70 responses from week 0 to week 24; change from baseline in ACR core set at weeks 1, 12, and 24; the proportion of patients with change from baseline in van der Heijde modified total Sharp score (mTSS) from week 0 to week 24; the proportion of structural damage progression, cutoffs ≤ 0, ≤ 0.5, and ≤ 0.95 (the smallest detectable change [9]), at week 24; the proportion of patients achieving PASI 75, PASI 90, and PASI 100 at week 24; the proportion of patients achieving disease activity index for psoriatic arthritis (DAPSA) low disease activity (LDA) (> 4 and ≤ 14) and DAPSA remission (≤ 4) from week 0 to 24; and change from baseline in 28-joint Disease Activity Score using C-reactive protein (DAS28-CRP) from week 0 to 24.
Safety was assessed by the proportion of patients experiencing treatment-emergent adverse events (TEAEs), serious adverse events, and study discontinuation due to adverse events (AEs). AEs of special interest included cytopenia, infection, serious infections, injection site reactions, hepatic events, allergic reaction/hypersensitivity, depression, and malignancy.
Statistical Analyses
Statistical analyses have been previously described for the overall population [9]. Efficacy assessments were conducted on the overall intent-to-treat population (all randomized patients), the severe intent-to-treat population (defined as mCPDAI total score > 7 and peripheral arthritis score = 3), and the non-severe intent-to-treat population. All efficacy endpoints were assessed at a significance level of p < 0.05 with no adjustment for multiplicity.
Post hoc subgroup analyses of categorical endpoints for the severe and non-severe populations were based on a logistic regression analysis with treatment, subgroup (severe/non-severe), and treatment-by-subgroup interaction (tested at the 10% significance level). Patients were considered non-responders (non-responder imputation, NRI) if they did not meet the clinical response criteria for categorical responses or were missing categorical response data at a timepoint of interest. Patients who were eligible for rescue therapy at week 16 were analyzed as non-responders after week 16. Post hoc subgroup analyses of continuous variables for the severe and non-severe populations were based on an analysis of covariance model including treatment, subgroup, treatment-by-subgroup (tested at the 10% significance level), and baseline value of the response variable.
With the exception of analyses for mTSS, a last-observation-carried-forward model was used for all continuous efficacy endpoints. For patients who discontinued study treatment for any reason, the last non-missing observation before discontinuation was carried forward to the corresponding endpoint for evaluation. However, for patients eligible for rescue therapy at week 16, the last non-missing observation up to week 16 was carried forward to the corresponding endpoint for evaluation. Randomized patients without at least one post-baseline observation were not evaluated.
The linear extrapolation method was used for the analyses of mTSS. For patients who discontinued the study or the study treatment, or missed a radiograph for any reason, baseline data and the most recent radiographic data were used for linear extrapolation to impute missing data at subsequent scheduled timepoints. For patients who commenced rescue therapy at week 16, or at any timepoint thereafter, baseline data and the most recent post-baseline radiographic data up to week 16, adjusted for time, were used for linear extrapolation.
Safety analyses were conducted on the overall safety population (all patients who took at least one dose of study medication), the severe safety population (defined as mCPDAI total score > 7 and peripheral arthritis score = 3), and the non-severe safety population using descriptive statistics.
The adalimumab 40 mg Q2W treatment arm served as active reference for comparison with placebo. The study was not powered to test equivalence or non-inferiority of ixekizumab vs. adalimumab. Statistical analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
Patient Characteristics
Of 417 randomized participants in the SPIRIT-P1 study, 347 patients had available CPDAI scores at baseline, 204 had severe disease, and 143 had non-severe disease. Baseline demographics and clinical characteristics in the overall and severe PsA populations are outlined in Table 1. For the severe vs. overall population, the mean age was 49.9 and 49.5 years, respectively, with 44.6 and 45.8% being male. The use of csDMARD and methotrexate therapy (severe vs. overall) was 64.2 vs. 64.0%, and 56.9 vs. 54.2%, respectively. Compared with the overall population, patients with severe PsA reported numerically higher rates of specific disease characteristics at baseline including dactylitis (48.0 vs. 37.6%), enthesitis (86.3 vs. 58.0%), and axial manifestation demonstrated via the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) > 4 (91.9 vs. 76.5%). In terms of skin symptoms, the mean baseline PASI total score was 7.5 in the severe population compared with 6.1 in the overall population. For patients with severe disease vs. the overall population, mean scores were numerically higher for HAQ-DI (1.4 vs. 1.2), patient-assessed joint pain (64.2 vs. 58.9 mm), patient-assessed global disease activity (65.0 vs. 61.4 mm), and physician-assessed global disease activity (60.6 vs. 56.9 mm). Patients with severe PsA also reported numerically higher rates of specific disease characteristics compared with the non-severe population, including dactylitis, enthesitis, tender and swollen joint counts, mTSS, PASI total score, axial manifestation, patient- and physician-assessed global disease activity, and HAQ-DI (Supplementary Material Table S1).
Efficacy
A time course of ACR response rates from week 0 to week 24 in the severe and overall PsA populations is shown in Fig. 1 (Supplementary Material Fig. S1 for the non-severe population). In the severe population, significantly more patients (p ≤ 0.001 or p ≤ 0.05) treated with ixekizumab than placebo achieved ACR 20 at all timepoints through to week 24, consistent with findings in the overall population; ixekizumab Q4W (63.3 and 57.9%), ixekizumab Q2W (60.4 and 62.1%), and placebo (24.5 and 30.2%), for the severe and overall populations, respectively (Fig. 1a). The treatment response to ixekizumab was significantly greater than placebo as early as week 1 (p ≤ 0.01). In terms of ACR 50 and ACR 70, findings in the severe population were consistent with the overall population, with statistically greater responses compared with placebo following treatment with ixekizumab Q4W and ixekizumab Q2W (p ≤ 0.001) (Fig. 1b, c). The active reference treatment arm (adalimumab) also had significantly greater ACR 20, ACR 50, and ACR 70 responses at week 24 compared with placebo (p ≤ 0.001 or p ≤ 0.01) (Fig. 1a–c). Treatment with ixekizumab Q4W and ixekizumab Q2W resulted in significant reductions versus placebo in each component of the ACR core set (p < 0.05) (Fig. 2a–g). In the severe population, these reductions were evident at week 1 and maintained through to week 24, consistent with findings in the overall population.

Time course of ACR response rates in the overall and severe PsA populations. The percentage of patients achieving a ACR 20, b ACR 50, and c ACR 70 from week 0 to week 24. Patients with inadequate responses to treatment at week 16 or missing data were analyzed as non-responders up to week 24. *p ≤ 0.05 vs. placebo. **p ≤ 0.01. ***p ≤ 0.001 vs. placebo. aAdalimumab was an active reference arm for comparison with placebo. This study was not powered to test for superiority of ixekizumab vs. adalimumab. bSince logistic regression analysis was used for the overall population, the p value could not be calculated at timepoints where no patients in the placebo arm achieved ACR 70 improvement. ACR 20/50/70 American College of Rheumatology response, ADA adalimumab, IXE ixekizumab, PBO placebo, PsA psoriatic arthritis, Q2W every 2 weeks, Q4W every 4 weeks


ACR core set at weeks 1, 12, and 24 for the overall and severe PsA populations. The least-square mean changes in a TJC, b SJC, c Pain VAS, d PtGA VAS, e PhGA VAS, f HAQ-DI, and g CRP scores are shown in the overall and severe populations. *p ≤ 0.05 vs. placebo. **p ≤ 0.01 vs. placebo. ***p ≤ 0.001 vs. placebo. aAdalimumab was an active reference arm for comparison with placebo. This study was not powered to test for superiority of ixekizumab vs. adalimumab. ACR American College of Rheumatology response, ADA adalimumab, CRP C-reactive protein, HAQ-DI Health Assessment Questionnaire-Disability Index, IXE ixekizumab, PBO placebo, PhGA physician’s global assessment of disease activity, PsA psoriatic arthritis, PtGA patient’s global assessment of disease activity, Q2W every 2 weeks, Q4W every 4 weeks, SJC swollen joint count, TJC tender joint count, VAS visual analog scale
In the severe population, the percentage of patients achieving DAPSA LDA (> 4 and ≤ 14) and DAPSA remission (≤ 4) at week 24 was significantly higher in the ixekizumab Q4W and ixekizumab Q2W treatment arms compared with placebo (p ≤ 0.05, p ≤ 0.01, or p ≤ 0.001), and numerically higher in the adalimumab group compared with placebo. Statistical differences were observed as early as week 4 for DAPSA LDA and week 8 for DAPSA remission. These findings are consistent with those for the overall population (Fig. 3). The time course of DAPSA LDA and remission in the non-severe PsA population is outlined in Supplementary Material Fig. S2.

Time course of DAPSA LDA and DAPSA remission in the overall and severe PsA populations. Change from baseline in DAPSA LDA (score > 4 and ≤ 14) and DAPSA remission (score ≤ 4) from week 0 to week 24 in the overall population (a and c) and severe populations (b and d). *p ≤ 0.05 vs. placebo. **p ≤ 0.01 vs. placebo. ***p ≤ 0.001 vs. placebo. Adalimumab was an active reference arm for comparison with placebo. This study was not powered to test for superiority of ixekizumab vs. adalimumab. ADA adalimumab, DAPSA disease activity index for psoriatic arthritis, LDA low disease activity, PBO placebo, Q2W every 2 weeks, Q4W every 4 weeks
At week 24, mean reduction in the level of disease activity observed in DAS28-CRP in the severe population was statistically greater in the ixekizumab treatment arms and adalimumab arm compared with placebo (all p ≤ 0.001; Table 2, Supplementary Material Fig. S3). Among patients with psoriasis at baseline affecting ≥ 3% of the body surface area, a significantly greater percentage of patients achieved PASI 75, PASI 90, and PASI 100 at week 24 for the ixekizumab Q4W and ixekizumab Q2W treatment groups, and PASI 75 for adalimumab, compared with the placebo group in the severe and overall populations (p ≤ 0.001 or p ≤ 0.05; Table 2).
Structural disease progression, as measured by mTSS, is outlined in Table 3. Compared with placebo, a numerically greater percentage of patients in the ixekizumab treatment arms and adalimumab arm at week 24 experienced no structural progression as defined by thresholds of ≤ 0.5 or ≤ 0.95 in the severe population; these findings reached statistical significance in the overall population (Table 3). Change from baseline in mTSS through to week 24 was similar in both the severe and overall populations for all treatment groups, as shown by cumulative probability plots (Fig. 4, Supplementary Material Fig. S4 for the non-severe population).

Change from baseline in structural progression evaluated using cumulative probability plots for mTSS in the overall and severe PsA populations. Change from baseline in mTSS from week 0 to week 24 as analyzed by cumulative probability plots in the a overall population and b severe population. Analyses based on CPDAI severity at baseline. aSmallest detectable change [9]. ADA adalimumab, IXE ixekizumab, mTSS modified total sharp score, PBO placebo, PsA psoriatic arthritis, Q2W every 2 weeks, Q4W every 4 weeks
Safety Profile
A greater percentage of patients receiving ixekizumab or adalimumab reported at least one TEAE compared with patients receiving placebo (Table 4). Similar percentages of TEAEs were observed for the severe and overall populations; 66.7 and 66.4% in the ixekizumab Q4W arm, 63.8 and 65.7% in the ixekizumab Q2W arm, 67.4 and 64.4% in the adalimumab arm, and 45.3 and 47.2% in the placebo arm, respectively. The majority of TEAEs were mild or moderate in intensity. The most common TEAEs in the severe and overall populations are outlined in Supplementary Material Table S2. There were no cases of serious injection site reactions. The percentage of patients in the severe population who experienced serious infections was low (2.8%) and similar to the overall populations (2.0%). No deaths occurred.
The rate of study treatment discontinuation due to AEs was low and similar between all treatment groups in both the severe and overall populations. Four patients in the severe population discontinued study treatment due to an AE; one in the ixekizumab Q4W arm due to a positive interferon gamma release assay, one in the ixekizumab Q2W arm due to depression, and two in the placebo arm (one due to asthenia and one due to injection site pain).
Discussion
As PsA can present with a number of characteristic clinical manifestations, we considered it vital to define the severity of disease in terms of several clinical domains in addition to peripheral arthritis. In patients with severe disease from the SPIRIT-P1 study (defined as mCPDAI total score > 7 and peripheral arthritis score = 3), treatment with 80 mg of ixekizumab (Q4W or Q2W) significantly improved the signs and symptoms of PsA, with no new safety concerns identified. Overall, findings in the severe population were broadly consistent with those observed in the overall population [9]. Compared with the non-severe population, the rate of dactylitis and axial symptoms were numerically greater in the severe population. In a previous report, dactylitis and axial symptoms are related to disease severity of PsA [19, 20], supporting our definition of severe PsA for the purpose of this subgroup analysis.
In the severe population at baseline, patient- and physician-reported disease activities (including pain visual analog scale [VAS], global disease activity VAS, and HAQ-DI), were numerically greater compared with the overall population. This finding is similar to previous reports confirming CPDAI as a validated and disease-specific tool for the assessment of disease activity in PsA [16].
Consistent with the overall population, ixekizumab demonstrated statistically significant improvements compared with placebo in terms of ACR 20, ACR 50, and ACR 70 in patients with severe disease. Significant improvements with ixekizumab were also observed in the ACR core set, including tender and swollen joint count, HAQ-DI, physicians’ and patients’ global assessment of disease, and CRP. For most endpoints, treatment responses to ixekizumab were observed as early as weeks 1 and 2 and maintained through to week 24. Of note, CRP decreased maximally at week 1 with the response sustained until week 24. This finding supports the rapid treatment response of ixekizumab in patients with PsA, regardless of disease severity.
In addition to improvements in disease activity, our findings confirm that ixekizumab inhibits radiographic progression in patients with severe PsA, as measured by DAPSA LDA and remission, DAS28-CRP, and mTSS. These clinically meaningful findings are consistent with observations in the overall population. An international task force for axial and peripheral spondyloarthritis (including PsA) recommended DAPSA and minimal disease activity as endpoints to assess disease activity [21]. Furthermore, a recent post hoc analysis in tofacitinib-treated patients with PsA showed a correlation between achieving DAPSA LDA/DAPSA remission and improved HAQ-DI and Short Form (36 Items) Health Survey Physical Component Score after 6 months, and slightly reduced radiographic progression at 12 months [22]. This study supports DAPSA and very low disease activity as being useful tools for evaluating disease activity and treatment response in PsA.
In terms of cutaneous symptoms, similar observations were seen in the severe and overall populations. For patients with baseline psoriatic lesions involving ≥ 3% body surface area, the majority of patients in both the severe and overall populations receiving ixekizumab achieved PASI 75 and PASI 90, with up to half of patients also achieving PASI 100 (statistically significantly higher than placebo). In addition, responses to ixekizumab were observed as early as week 2.
Taken together, our efficacy findings are consistent with previous studies of ixekizumab in patients with moderate-to-severe psoriasis, including real-world data from clinical settings [23, 24]. Findings are also consistent with those observed in the SPIRIT program. In SPIRIT-P2, conducted in patients with PsA and with previously inadequate responses to tumor necrosis factor inhibitors, both ixekizumab arms showed significantly higher proportions of participants with ACR 20 (53% in the Q4W and 48% in the Q2W) than the placebo group (19%) [6, 25].
In terms of the adalimumab treatment arm, numerical differences compared with the ixekizumab treatment arms were observed for several endpoints including ACR responses, DAS28-CRP, DAPSA LDA, and DAPSA remission in the severe population. In addition, while a greater proportion of adalimumab-treated patients in the severe population achieved PASI 75 at week 24 compared with placebo, there were no significant differences between the adalimumab and placebo treatment arms at week 24 in terms of PASI 90 and PASI 100. A subgroup analysis of the SPIRIT-H2H trial showed significant ACR 50 and PASI 100 improvements with ixekizumab compared with adalimumab without the use of concomitant methotrexate [13]. Hence, it appears that ixekizumab delivers consistent efficacy in several clinical domains of PsA regardless of concomitant methotrexate use whilst the efficacy of adalimumab is increased by the concomitant use of methotrexate [13]. In the current study, the frequency of concomitant use of methotrexate was 56.9% in the severe population and 54.2% in the overall population, which may have affected the efficacy outcomes of adalimumab. In this study, even in the severe population, response to treatment was observed rapidly, and efficacy was maintained through to week 24. Conversely, in the cumulative probability plots of mTSS, the proportion of patients without progression was numerically higher in the adalimumab treatment arm in both the severe and overall populations. In previous clinical trials of adalimumab in PsA patients, some patients showed a decrease in mTSS [26]. As IL-17A plays a role in inducing osteoclast precursors leading to increased sensitivity to RANKL signaling [27], but tumor necrosis factor-α directly affects osteocyte RANKL expression and increases osteoclastogenesis [28], it can be speculated that these differential effects on osteoclast differentiation may affect radiographic results.
The safety profile of ixekizumab in patients with severe disease was consistent with findings in the overall population. The most common TEAEs were injection site reaction, injection site erythema, and nasopharyngitis, with most TEAEs being mild to moderate in intensity (note, this study was conducted with the original formulation of ixekizumab, not the citrate-free formulation shown to reduce injection site pain [29]). The safety findings are consistent with long-term safety data from 21 clinical trials of patients with psoriasis, PsA, and axial spondyloarthritis treated with ixekizumab, where both injection site reaction and nasopharyngitis were the most common TEAEs (along with upper respiratory tract infection) [30]. Interestingly, long-term safety data from the three SPIRIT studies demonstrated that the rate of injection site reactions decreased with longer ixekizumab exposure [31].
As the SPIRIT-P1 study was restricted to patients who were naive to biologic therapy, the current findings cannot be generalized to patients with a history of failed therapy, loss of efficacy to therapy, or intolerance to anti-tumor necrosis factor agents. An additional factor for consideration is the relatively limited number of patients in each subgroup of the severe population. Overall, the findings from this study provide healthcare providers and patients with additional information on the efficacy and safety profile associated with the use of ixekizumab in cases of severe PsA.
Conclusions
In patients with severe PsA, 24 weeks of treatment with ixekizumab improved key clinical outcomes of PsA. No new safety findings were identified in patients with severe PsA, and the safety profile was consistent with the current understanding of ixekizumab. Overall, the present findings support the use of ixekizumab for the treatment of patients with severe PsA, a population not previously characterized in terms of response to IL-17A inhibition.
Data Availability
Eli Lilly and Company provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, and blank or annotated case report forms, will be provided in a secure data-sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
Change history
16 May 2024
A Correction to this paper has been published: https://doi.org/10.1007/s40744-024-00673-2
References
Gladman DD. Psoriatic arthritis. Dermatol Ther. 2009;22:40–55.
de Vlam K, Gottlieb AB, Mease PJ. Current concepts in psoriatic arthritis: pathogenesis and management. Acta Derm Venereol. 2014;94:627–34.
Ritchlin CT, Colbert RA, Gladman DD. Psoriatic arthritis. N Engl J Med. 2017;376:957–70.
Taylor WJ. Impact of psoriatic arthritis on the patient: through the lens of the WHO International Classification of Functioning, Health, and Disability. Curr Rheumatol Rep. 2012;14:369–74.
Food and Drug Administration. TALTZ (ixekizumab) injection, for subcutaneous use. 2016. Updated March 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125521s020lbl.pdf. Accessed Apr 18, 2023.
Nash P, Kirkham B, Okada M, et al. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet. 2017;389:2317–27.
van der Heijde D, Gladman DD, Kishimoto M, et al. Efficacy and safety of ixekizumab in patients with active psoriatic arthritis: 52-week results from a phase III study (SPIRIT-P1). J Rheumatol. 2018;45:367–77.
Genovese MC, Combe B, Kremer JM, et al. Safety and efficacy of ixekizumab in patients with PsA and previous inadequate response to TNF inhibitors: week 52 results from SPIRIT-P2. Rheumatology (Oxford). 2018;57:2001–11.
Mease PJ, van der Heijde D, Ritchlin CT, et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis. 2017;76:79–87.
Coates LC, Mease P, Kronbergs A, et al. Efficacy and safety of ixekizumab in patients with active psoriatic arthritis with and without concomitant conventional disease-modifying antirheumatic drugs: SPIRIT-P1 and SPIRIT-P2 3-year results. Clin Rheumatol. 2022;41:3035–47.
Kavanaugh A, Gottlieb A, Morita A, et al. The contribution of joint and skin improvements to the health-related quality of life of patients with psoriatic arthritis: a post hoc analysis of two randomised controlled studies. Ann Rheum Dis. 2019;78:1215–9.
Mease PJ, Smolen JS, Behrens F, et al. A head-to-head comparison of the efficacy and safety of ixekizumab and adalimumab in biological-naïve patients with active psoriatic arthritis: 24-week results of a randomised, open-label, blinded-assessor trial. Ann Rheum Dis. 2020;79:123–31.
Smolen JS, Sebba A, Ruderman EM, et al. Efficacy and safety of ixekizumab with or without methotrexate in biologic-naïve patients with psoriatic arthritis: 52-week results from SPIRIT-H2H study. Rheumatol Ther. 2020;7:1021–35.
Gossec L, Baraliakos X, Kerschbaumer A, et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2019 update. Ann Rheum Dis. 2020;79:700–12.
Kristensen LE, Okada M, Tillett W, et al. Ixekizumab demonstrates consistent efficacy versus adalimumab in biologic disease-modifying anti-rheumatic drug-naïve psoriatic arthritis patients regardless of psoriasis severity: 52-week post hoc results from SPIRIT-H2H. Rheumatol Ther. 2022;9:109–25.
Acosta Felquer ML, Szentpetery A, Elmamoun M, Gallagher P, FitzGerald O, Soriano ER. Composite psoriatic disease activity index (CPDAI), defining remission and disease activity states using data from daily clinical practice [abstract]. Arthritis Rheumatol. 2016; 68(suppl 10).
Mumtaz A, Gallagher P, Kirby B, et al. Development of a preliminary composite disease activity index in psoriatic arthritis. Ann Rheum Dis. 2011;70:272–7.
Helliwell PS, FitzGerald O, Fransen J. Composite disease activity and responder indices for psoriatic arthritis: a report from the GRAPPA 2013 meeting on development of cutoffs for both disease activity states and response. J Rheumatol. 2014;41:1212–7.
Dubash S, Alabas OA, Michelena X, et al. Dactylitis is an indicator of a more severe phenotype independently associated with greater SJC, CRP, ultrasound synovitis and erosive damage in DMARD-naive early psoriatic arthritis. Ann Rheum Dis. 2022;81:490–5.
Ogdie A, Blachley T, Lakin PR, et al. Evaluation of clinical diagnosis of axial psoriatic arthritis (PsA) or elevated patient-reported spine pain in CorEvitas’ PsA/Spondyloarthritis Registry. J Rheumatol. 2022;49:281–90.
Smolen JS, Schöls M, Braun J, et al. Treating axial spondyloarthritis and peripheral spondyloarthritis, especially psoriatic arthritis, to target: 2017 update of recommendations by an international task force. Ann Rheum Dis. 2018;77:3–17.
Schneeberger EE, Citera G, Nash P, et al. Comparison of disease activity index for psoriatic arthritis (DAPSA) and minimal disease activity (MDA) targets for patients with psoriatic arthritis: a post hoc analysis of data from phase 3 tofacitinib studies. Semin Arthritis Rheum. 2023;58: 152134.
Griffiths CEM, Reich K, Lebwohl M, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386:541–51.
Gönülal M, Altunay İK, Doğan S, Türkmen M, Balcı DD, Öztürkcan S. Ixekizumab for the treatment of the patients with moderate to severe plaque psoriasis: clinical data from a real-world experience. Dermatol Ther. 2022;35: e15955.
Nash P, Behrens F, Orbai AM, et al. Ixekizumab is efficacious when used alone or when added to conventional synthetic disease-modifying antirheumatic drugs (cDMARDs) in patients with active psoriatic arthritis and previous inadequate response or intolerance to tumour necrosis factor inhibitors. RMD Open. 2018;4: e000692.
Gladman DD, Mease PJ, Ritchlin CT, et al. Adalimumab for long-term treatment of psoriatic arthritis: forty-eight week data from the adalimumab effectiveness in psoriatic arthritis trial. Arthritis Rheum. 2007;56:476–88.
Adamopoulos IE, Chao CC, Geissler R, et al. Interleukin-17A upregulates receptor activator of NF-κB on osteoclast precursors. Arthritis Res Ther. 2010;12:R29.
Marahleh A, Kitaura H, Ohori F, et al. TNF-α directly enhances osteocyte RANKL expression and promotes osteoclast formation. Front Immunol. 2019;10:2925.
Eli Lilly and Company. Lilly's Taltz® (ixekizumab) now available in new, citrate-free formulation to reduce injection site pain for improved patient experience. News Release. Published August 8, 2022. https://investor.lilly.com/news-releases/news-release-details/lillys-taltzr-ixekizumab-now-available-new-citrate-free. Accessed Apr 18, 2023.
Genovese MC, Mysler E, Tomita T, et al. Safety of ixekizumab in adult patients with plaque psoriasis, psoriatic arthritis and axial spondyloarthritis: data from 21 clinical trials. Rheumatology (Oxford). 2020;59:3834–44.
Combe B, Rahman P, Kameda H, et al. Safety results of ixekizumab with 1822.2 patient-years of exposure: an integrated analysis of 3 clinical trials in adult patients with psoriatic arthritis. Arthritis Res Ther. 2020;22:14.
Acknowledgements
The authors thank the clinical trial participants and their caregivers, investigators, and site staff.
Medical Writing, Editorial, and Other Assistance
Project management was provided by Naotsugu Akashi of Eli Lilly Japan K.K. Medical writing and editorial assistance were provided by Lisa Cossens and Antonia Baldo of Syneos Health and funded by Eli Lilly Japan K.K.
Funding
This study was funded by Eli Lilly and Company, including the journal’s Rapid Service fee.
Author information
Authors and Affiliations
Contributions
Kohei Hagimori and Ayako Konomi were involved in conceptualization. Hideto Kameda, Kohei Hagimori and Yoji Morisaki were involved in the study design. Thorsten Holzkämper was involved in data acquisition. Yoji Morisaki was involved in data analysis. All authors were involved in data interpretation and contributed to writing and critical revision of the manuscript. All authors participated sufficiently in the work to agree to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Hideto Kameda declares consulting fees, speaker fees, honoraria, and/or research grants from AbbVie, Asahi Kasei, BMS, Chugai, Eisai, Eli Lilly and Company, Janssen, Mitsubishi-Tanabe, Novartis, Pfizer, Sanofi, Taisho, and UCB. Kohei Hagimori, Yoji Morisaki, and Ayako Konomi are full-time employees of Eli Lilly Japan K.K and stockholders of Eli Lilly and Company. Thorsten Holzkaemper is a full-time employee of Lilly Deutschland GmbH. Hiroaki Dobashi is a speaker for BMS, Chugai, Eli Lilly and Company, GSK, MSD, Novartis, Pfizer, and UCB Pharma.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committees at all sites where these studies were conducted and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study, and all participants consented to the publication of study results. Protocols and consent forms were approved by the institutional review board or ethics committee of each site, including the Western Institutional Review Board (SPIRIT-P1). A listing of individual sites for SPIRIT-P1 are included in the supplement of the primary manuscript [9]. SPIRIT-P1 was registered at ClinicalTrials.gov, NCT01695239.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kameda, H., Hagimori, K., Morisaki, Y. et al. Ixekizumab Efficacy in Patients with Severe Peripheral Psoriatic Arthritis: A Post Hoc Analysis of a Phase 3, Randomized, Double-Blind, Placebo-Controlled Study (SPIRIT-P1). Rheumatol Ther 10, 1683–1703 (2023). https://doi.org/10.1007/s40744-023-00605-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-023-00605-6