
Abstract
Introduction
The aim of the study was to demonstrate the bioequivalence, and compare the safety and tolerability of MSB11022, a proposed biosimilar of adalimumab, when delivered by either an autoinjector (AI) or a pre-filled syringe (PFS).
Methods
In this pharmacokinetic (PK), parallel group, open-label study, 216 healthy volunteers were randomised 1:1 to receive a single subcutaneous injection of a 40 mg/0.8 mL dose of MSB11022 administered via AI or PFS. Coprimary PK endpoints were maximum observed concentration (Cmax), area under the concentration–time curve (AUC) from time 0 to the last quantifiable concentration (AUC0–t), and AUC from time 0 extrapolated to infinity (AUC0–inf). PK equivalence between the AI and PFS administration methods was declared if the 90% confidence intervals (CIs) for the ratio of geometric least square means was entirely contained within the 80–125% equivalence margin for all coprimary endpoints. Safety and tolerability were also evaluated.
Results
The 90% CI for the three coprimary PK endpoints (Cmax, AUC0–t and AUC0–inf) were entirely contained within the predefined equivalence margins of 80–125%. Mean serum concentration–time profiles were similar following injection via AI or PFS. Treatment-emergent adverse events (TEAEs) were comparable across both treatment groups. Study device-related TEAEs were reported by 11.3% and 13.1% of subjects in the AI and PFS treatment groups, respectively. Study drug-related TEAEs were reported by 28.3% and 34.6% of subjects in the AI and PFS treatment groups, respectively. Few subjects experienced injection-site reactions, mainly pain and erythema, regardless of the administration method.
Conclusion
Delivery of MSB11022 via an AI is bioequivalent to delivery via a PFS. The safety and tolerability profile of MSB11022 was comparable across administration methods. The development of an AI for MSB11022 provides a choice of self-injection devices available to patients, potentially improving treatment compliance.
Trial Registration
ClinicalTrials.gov trial identifier: NCT04018599.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study ? |
Self-injection can be challenging for patients who require treatment with biologics for chronic immune-mediated inflammatory diseases. |
Offering a choice of devices, including pre-filled syringes (PFS) or autoinjectors (AIs), can address patient needs and potentially improve adherence to therapy. |
When transitioning from a PFS to an AI, some regulatory authorities require a pharmacokinetic bridging study to demonstrate similar delivery of the drug product to the same biospace. |
What was learned from the study ? |
Bioequivalence and comparable safety and tolerability were demonstrated for delivery of MSB11022 with an AI versus delivery with a PFS. |
Administration of the proposed adalimumab biosimilar, MSB11022, via an AI represents an alternative delivery option to a PFS in clinical practice. |
Introduction
Adalimumab (Humira®; AbbVie Inc., North Chicago, IL, USA), an anti-tumour necrosis factor alpha (TNFα) monoclonal antibody, is indicated for the treatment of rheumatoid arthritis (RA), ankylosing spondylitis, psoriasis, Crohn’s disease, ulcerative colitis, psoriatic arthritis, juvenile idiopathic arthritis, uveitis and hidradenitis suppurativa [1, 2]. MSB11022 (Idacio®; Fresenius Kabi, Runcorn, UK), a proposed biosimilar of adalimumab, is currently available in Europe in three different presentations (pre-filled syringes PFS], vial and autoinjector [AI] and administered in a citrate-based formulation [3, 4]. The same formulation has also been approved in other countries, including Australia, New Zealand and Canada; regulatory approvals in many other countries are ongoing [5,6,7]. In a pre-clinical setting, MSB11022 demonstrated physicochemical and functional similarity to reference adalimumab [8]. Subsequently, pharmacokinetic (PK) equivalence was established for MSB11022 and reference adalimumab (both EU and US approved) during a phase 1 trial [9]. Equivalent efficacy, and comparable safety and immunogenicity were observed between MSB11022 and reference adalimumab in AURIEL-PsO, a phase 3, multicentre, randomised, equivalence trial in patients with moderate-to-severe chronic plaque-type psoriasis [10]. An acetate-buffered formulation of MSB11022 was subsequently developed and shown to be bioequivalent to citrate-buffered MSB11022 with a comparable safety and immunogenicity profile in a phase 1 study (unpublished data). In AURIEL-RA (a phase 3, multicentre, randomised, descriptive study), the safety, immunogenicity, and efficacy profiles of acetate-buffered MSB11022 were demonstrated to be comparable with that of reference adalimumab in patients with moderately-to-severely active RA [11]. Subsequently, process changes were made to the acetate-buffered MSB11022 and PK comparability was demonstrated to citrate-buffered MSB11022 and to reference adalimumab. All studies described above were performed using PFS presentation of MSB11022.
The therapeutic potential of biologics is not always achieved in clinical practice and this is often related to suboptimal treatment compliance [12, 13]. Patient-reported factors can be associated with treatment non-compliance and these can include the type of medication delivery device used [12]. Self-injection of biologics via PFS and, more recently, AI devices offer increased flexibility and independence for patients, as well as reduced costs compared with infusions [14]. However, self-injection via PFS may be associated with challenges from a patient’s perspective, including needle phobia, concerns related to pain, risk of incorrect treatment administration, as well as the potential difficulties associated with using a self-injection device while suffering with arthritic hand pain [12, 14].
Having a choice of self-injection devices can help patients to select a device that addresses their individual challenges and lifestyles, with some patients preferring a PFS and others electing to use an AI [12, 14]. This may increase patient tolerance of self-injection and potentially improve adherence to treatment [14]. Many patients have indicated a preference for AIs over PFS, as they perceive AIs to offer decreased injection-site pain, simplicity of use, increased convenience, quicker administration, increased portability, easier grip and an overall improved treatment experience [12, 14,15,16,17,18]. Additionally, studies have demonstrated a preference for an AI compared with a PFS for patients in the advanced stages of RA and in other patients who may lack dexterity of the hand (e.g. those with psoriatic arthritis and ankylosing spondylitis) [15,16,17,18,19].
To address some of the possible limitations mentioned previously, and to increase the range of self-injection devices available for patients, an AI administration device for MSB11022 has been developed. This study was undertaken to investigate the bioequivalence and compare the safety and tolerability of a single subcutaneous (s.c.) injection of 40 mg of MSB11022 when delivered by either an AI or a PFS.
Methods
Study Design
This was a phase 1, randomised, open-label, two-arm, parallel-group, single-dose study conducted at two sites in the US (ClinicalTrials.gov trial identifier: NCT04018599). The study was conducted in accordance with the International Conference on Harmonisation Good Clinical Practice Guidelines and the Declaration of Helsinki, and the protocol was approved by an institutional review board. Written informed consent was required prior to the conduct of any study procedures. Participants were screened within 4 weeks prior to their dose of MSB11022 and were resident in the research centre from the day prior to dosing until 8 days afterwards. Participants returned for outpatient visits on days 9 and 11, followed by weekly ambulatory visits up to day 43, a penultimate visit on day 57 and an end-of-study visit on day 71 (Fig. 1).

Study design. AI Autoinjector, EOS end of study, PFS pre-filled syringe, PK pharmacokinetic, R 1:1 randomisation
Participants
The study enrolled healthy volunteers of either non-childbearing potential or using adequate contraception. Participants were eligible if they were aged 18–55 years, weighed 50.0–100.0 kg and had a body mass index of 18.5–30.0 kg/m2. Exclusion criteria included prior exposure to any TNFα inhibitor therapy (including adalimumab or adalimumab biosimilars) and receipt of any medication (excluding paracetamol, and for female participants, oral contraceptives and hormone replacement therapy) within 2 weeks (or longer if the medication had a long half-life) prior to administration of the trial medication.
Randomisation and Treatment
Participants were randomised 1:1 to receive a single s.c. injection of a 40 mg/0.8 mL dose of MSB11022 (in acetate buffer) administered either by BD Physioject™ AI (Fig. 2) or BD Hypak™ PFS. The solution for injection in the two devices is identical in terms of formulation and manufacturing process. The injection depth of the two devices is comparable, therefore the delivered dose is expected to be similar, resulting in comparable PK exposure. Randomisation was performed via an Interactive Web Response System (IWRS) using a permuted block size and was stratified by clinical site and body weight categories (50.0 to ≤ 80.0 kg and > 80.0 to ≤ 100.0 kg) as assessed on day −1. The IWRS assigned a kit number and injection site (lower abdomen or thigh) to the participant, corresponding to the treatment to be administered. Injections via an AI or a PFS were administered after breakfast in the morning to participants at the designated injection site by a trained staff member. Both the single-use, disposable, AI pen and the standard PFS included a 1-mL syringe.

Autoinjector presentation
Study Assessments
The primary objective of the study was to demonstrate PK equivalence of a single s.c. injection of MSB11022 administered using either an AI or a PFS. The secondary objectives were to compare secondary PK parameters, and overall safety and local tolerability of a single s.c. injection of MSB11022 administered using either an AI or a PFS. The three coprimary PK endpoints were maximum observed concentration (Cmax), area under the concentration–time curve (AUC) from time zero to last quantifiable concentration (AUC0–t), and AUC from time zero extrapolated to infinity (AUC0–inf). Secondary PK endpoints included time to Cmax (tmax), terminal rate constant, terminal half-life and apparent total clearance. Blood samples for PK evaluation were obtained at 0 (pre-dose), 1, 4, 8, 12 and 24 h post-dose. Further samples were obtained every 24 h thereafter until day 9, then at all subsequent outpatient visits (days 11, 15, 22, 29, 36, 43, 57 and at the end-of-study visit on day 71).
Serum concentrations of MSB11022 were determined using a validated enzyme-linked immunosorbent assay that employed a TNFα coated plate, horseradish peroxidase-conjugated anti-human immunoglobulin antibody to detect bound analyte and 3,3′,5,5′-tetramethylbenzidine for colorimetric readout. Colorimetric intensity was determined using a plate reader at wavelengths of 450 nm (detection) and 630 nm (reference) wavelengths. The method was selective, sensitive, precise and accurate for the determination of MSB11022 in serum, with a quantification range of 300–7000 ng/mL. Inter-assay precision and accuracy were evaluated during the study in the 227-sample analysis runs, by including human serum-quality control pools (QCs) at three concentrations spanning the calibration range (low-, mid- and high-QCs) in each plate. Bias at all levels was within ± 3.47%, and the coefficient of variation (CV) at all levels was ≤ 8.66%. Incurred sample re-analysis (ISR) demonstrated reproducible quantitation of the drug in study samples. Overall, 95.4% of the re-analysed samples met the ISR acceptance criteria.
Safety assessments were conducted prior to dosing and regularly until the participant’s final visit. Assessments included adverse events (AEs), predefined AEs of special interest (AESI), clinical laboratory parameters, vital signs, 12-lead electrocardiogram and physical examinations. In this study, AESIs were defined as hypersensitivity reactions (National Cancer Institute-Common Terminology Criteria for Adverse Events [CTCAE] grade ≥ 3 or reported as serious events). Local tolerability assessments at the injection site were conducted at screening (for scars and tattoos potentially obscuring future assessment of injection site) and at 5 min after dosing, then at 4, 8 and 12 h post-dose on day 1, then on days 2, 3, 5, 9, 11, and 15, followed by weekly assessments up to day 43, and final assessments on days 57 and 71. Injection-site reactions (e.g. erythema, rash, tenderness, swelling, itching, bruising or other abnormalities) were assessed by the investigator and graded according to the CTCAE.
Statistical Analyses
Sample size calculation was based on achieving at least 80% power for the 90% confidence interval (CI) for the ratio of geometric means (GM; AI/PFS) to be within the range of 80–125%. The following assumptions were used: a maximum difference of 5% between MSB11022 delivered via an AI compared with a PFS; a geometric CV no larger than 50%; and a drop-out rate of approximately 10%. PK analyses were conducted using the PK analysis set, which consisted of all participants who received MSB11022, had sufficient concentration–time data to calculate at least one valid primary PK parameter and had no major protocol deviation or other events affecting PK assessment. Safety analyses were conducted on the safety analysis set, which consisted of all participants who received at least one dose of MSB11022. Logarithmically transformed PK parameters (AUC0–inf, AUC0–t and Cmax) were compared between treatment groups using an analysis of covariance model with treatment as a fixed effect, and baseline body weight category and injection site as cofactors. The 90% CI for the adjusted treatment GM ratio was derived for each primary PK parameter by exponentiating the 90% CI obtained for the difference between the treatments least squared means, resulting from the analysis of the logarithmically transformed PK parameters. PK equivalence between the AI and PFS administration methods was declared if the 90% CIs for the adjusted GM ratios for Cmax, AUC0–inf and AUC0–t were entirely contained within the 80–125% equivalence margin. Secondary PK parameters, and safety and local tolerability endpoints were summarised using descriptive statistics. In addition, PK parameters were summarised by baseline body weight categories (50 to ≤ 80 kg and > 80 to ≤ 100 kg), study centres and injection sites (lower abdomen and thigh). Phoenix WinNonlin version 8.1 was used for PK parameter derivation. SAS version 9.4 was used for other statistical analyses.
Results
Participant Demographics and Baseline Characteristics
A total of 216 healthy volunteers were randomised and followed up between 15 July 2019 and 17 March 2020. The safety analysis set included 106 participants in the AI group and 107 in the PFS group; three participants (AI: n = 1; PFS: n = 2) were randomised but did not receive the study drug. The PK analysis set included 104 participants in each group (Fig. 3). Discontinuations post-dosing were similar between the two groups (AI: n = 4; PFS: n = 3), and none were due to AEs. The baseline demographic characteristics of the study population were similar between the two treatment groups (Table 1).

Participant disposition. AI autoinjector, Cmax maximum observed concentration, PFS pre-filled syringe, PK pharmacokinetic. aThree randomised participants did not receive MSB11022 for the following reasons: infection (AI group: n = 1); nausea and vomiting (AI group: n = 1); and positive drug screen (PFS group: n = 1). bFive treated participants were excluded from the PK analysis set for the following reasons: insufficient concentration–time data to derive at least one PK parameter (PFS group: n = 2; AI group: n = 1); pre-dose serum concentration > 5% of Cmax (PFS group: n = 1); and incomplete dosing (AI group: n = 1). Cmax Maximum observed concentration
Pharmacokinetic Comparison
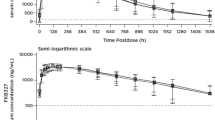
The adjusted GM ratio estimates and 90% CI for the three coprimary PK endpoints (Cmax, AUC0–t and AUC0–inf) were all within the predefined bioequivalence margins of 80–125% (Table 2). Other PK parameters were similar between the two treatment groups (Table 3). Linear and semi-logarithmic plots of the arithmetic mean (standard deviation) serum concentration over time following a single dose of MSB11022, administered via an AI or via a PFS, are shown in Fig. 4. A slight decrease in exposure to MSB11022 was noted for the higher body-weight category, but this was consistent for both devices (Electronic Supplementary Material [ESM] Table 1). PK results were consistent between the two injection sites (ESM Table 2). In the statistical model analysis results, the effect of baseline body-weight category was statistically significant for the three coprimary PK parameters, whereas injection site was not associated with any statistically significant effect.

Mean (SD) serum concentration of MSB11022 on a linear scale (a) and a semi-logarithmic scale (b) following a single 40 mg subcutaneous injection via an autoinjector or a pre-filled syringe (PK analysis set). Data are arithmetic mean. LLOQ = 300 ng/mL; samples below the LLOQ before the last quantifiable data point were set to 0. Concentrations below the LLOQ after the last quantifiable data point were considered as missing. LLOQ Lower limit of quantification, PK pharmacokinetic, SD standard deviation
Safety
No serious AEs or AESIs were recorded during the study in either treatment group. One severe AE was recorded (left-hand fracture), which was not considered by the study investigators to be related to study treatment.
The most commonly reported TEAEs were upper respiratory tract infection (20.7%) and headache (16.4%) (Table 4). These events occurred at a similar frequency between the two study groups. TEAEs related to the study device occurred in a similar proportion of subjects in both study groups (AI: 11.3%; PFS: 13.1%) and were exclusively injection-site reactions. Similarly, the incidence of study drug-related TEAEs was comparable between the study groups (AI: 28.3%; PFS: 34.6%). Overall, injection-site reactions occurred in 13.2% and 17.8% of the AI and PFS treatment groups, respectively. Most of these were either injection-site erythema, affecting 6.6% and 6.5% of the AI and PFS treatment groups, respectively, or injection-site pain, affecting 5.7% and 7.5% of the AI and PFS treatment groups.
Discussion
The most important finding of our study is that it met the predefined criteria for bioequivalence between the AI and PFS delivery methods of MSB11022. A slight inverse relationship between body weight and exposure was observed; however, this was similar for both devices and was as expected based on PK data with the originator, Humira®, and other adalimumab biosimilars [1, 20, 21]. Injection location (lower abdomen vs. thigh) did not appear to have a significant impact on exposure to MSB11022. The safety profile of the AI presentation was comparable to that of the PFS presentation and as expected for a cohort of healthy volunteers. Notably, few participants experienced injection-site reactions regardless of the mode of delivery.
A limitation of the present study is that the mode of delivery could not be blinded, and this may have theoretically affected participants’ perceptions and reporting of AEs, such as injection-site pain. However, the primary objective of the study was to compare PK parameters, and these should not be impacted by the open-label design. Although a double-dummy design could have been utilised, this would have increased the burden on the participants and was not considered appropriate for a study where determining bioequivalence was the primary objective. In this PK study, the treatments were given by trained staff to healthy volunteers and therefore usability of the device by patients with reduced hand function was not intended to be assessed. However, the BD Physioject™ AI has previously been studied in a randomised trial in a population of patients with RA, where a high level of patient acceptance was reported and patients with severe hand disability were able to successfully use the device [22].
Conclusions
The present study has demonstrated the bioequivalence and comparable safety profile of MSB11022 when administered by an AI or a PFS. This confirms that administration of the proposed adalimumab biosimilar, MSB11022, via an AI represents an alternative delivery option to a PFS in clinical practice, increasing the range of self-injection devices available to patients.
References
EMC. Humira 40 mg/0.4 ml pre-filled syringe and pre-filled pen. Summary of product characteristics (SmPC). 2021. https://www.medicines.org.uk/emc/medicine/31860#gref. Accessed 8 Nov 2021.
US FDA. HUMIRA® (adalimumab) injection, for subcutaneous use. 2021. https://www.rxabbvie.com/pdf/humira.pdf. Accessed 8 Nov 202l.
EMC. Idacio 40 mg solution for injection in pre-filled pen. Summary of product characteristics (SmPC). 2021. https://www.medicines.org.uk/emc/product/11307/smpc#gref. Accessed 11 Feb 2021.
EMA. Idacio – EPAR all authorised presentations. https://www.ema.europa.eu/en/documents/all-authorised-presentations/idacio-epar-all-authorised-presentations_en.pdf. Accessed 11 Feb 2021.
Australian Government: Department of Health. Therapeutic Goods Administration: Idacio. 2020. https://www.tga.gov.au/apm-summary/idacio. Accessed 11 Feb 2021.
New Zealand Government. New Zealand data sheet: Idacio 40 mg adalimumab (rch) solution for injection. 2020. https://www.medsafe.govt.nz/Profs/Datasheet/I/Idacioinj.pdf. Accessed 11 Feb 2021.
Fresenius Kabi. Fresenius Kabi Canada launches IDACIO® (adalimumab injection) a biosimilar to HUMIRA® (adalimumab) for the treatment of multiple chronic inflammatory conditions. 2021. https://www.newswire.ca/news-releases/fresenius-kabi-canada-launches-idacio-r-adalimumab-injection-a-biosimilar-to-humira-r-adalimumab-for-the-treatment-of-multiple-chronic-inflammatory-conditions-822638421.html. Accessed 24 Feb 2022.
Magnenat L, Palmese A, Fremaux C, et al. Demonstration of physicochemical and functional similarity between the proposed biosimilar adalimumab MSB11022 and Humira®. MAbs. 2017;9(1):127–39.
Hyland E, Mant T, Vlachos P, et al. Comparison of the pharmacokinetics, safety, and immunogenicity of MSB11022, a biosimilar of adalimumab, with Humira® in healthy subjects. Br J Clin Pharmacol. 2016;82(4):983–93.
Hercogová J, Papp KA, Chyrok V, Ullmann M, Vlachos P, Edwards CJ. AURIEL-PsO: a randomized, double-blind phase III equivalence trial to demonstrate the clinical similarity of the proposed biosimilar MSB11022 to reference adalimumab in patients with moderate-to-severe chronic plaque-type psoriasis. Br J Dermatol. 2020;182(2):316–26.
Edwards CJ, Monnet J, Ullmann M, Vlachos P, Chyrok V, Ghori V. Safety of adalimumab biosimilar MSB11022 (acetate-buffered formulation) in patients with moderately-to-severely active rheumatoid arthritis. Clin Rheumatol. 2019;38(12):3381–90.
Maniadakis N, Toth E, Schiff M, et al. A targeted literature review examining biologic therapy compliance and persistence in chronic inflammatory diseases to identify the associated unmet needs, driving factors, and consequences. Adv Ther. 2018;35(9):1333–55.
Rubin DT, Mittal M, Davis M, Johnson S, Chao J, Skup M. Impact of a patient support program on patient adherence to adalimumab and direct medical costs in Crohn’s disease, ulcerative colitis, rheumatoid arthritis, psoriasis, psoriatic arthritis, and ankylosing spondylitis. J Manag Care Spec Pharm. 2017;23(8):859–67.
van den Bemt BJF, Gettings L, Domańska B, Bruggraber R, Mountian I, Kristensen LE. A portfolio of biologic self-injection devices in rheumatology: how patient involvement in device design can improve treatment experience. Drug Deliv. 2019;26(1):384–92.
Kivitz A, Cohen S, Dowd JE, et al. Clinical assessment of pain, tolerability, and preference of an autoinjection pen versus a prefilled syringe for patient self-administration of the fully human, monoclonal antibody adalimumab: the TOUCH trial. Clin Ther. 2006;28(10):1619–29.
Demary W, Schwenke H, Rockwitz K, et al. Subcutaneously administered methotrexate for rheumatoid arthritis, by prefilled syringes versus prefilled pens: patient preference and comparison of the self-injection experience. Patient Prefer Adherence. 2014;8:1061–71.
Hsiao B, Fraenkel L. Patient preferences for rheumatoid arthritis treatment. Curr Opin Rheumatol. 2019;31(3):256–63.
Borrs-Blasco J, Gracia-Prez A, Rosique-Robles JD, Caster MDE, Abad FJ. Acceptability of switching adalimumab from a prefilled syringe to an autoinjection pen. Expert Opin Biol Ther. 2010;10(3):301–7.
Ghil J, Zielińska A, Lee Y. Usability and safety of SB5 (an adalimumab biosimilar) prefilled syringe and autoinjector in patients with rheumatoid arthritis. Curr Med Res Opin. 2019;35(3):497–502.
Puri A, Niewiarowski A, Arai Y, et al. Pharmacokinetics, safety, tolerability and immunogenicity of FKB327, a new biosimilar medicine of adalimumab/Humira, in healthy subjects. Br J Clin Pharmacol. 2017;83(7):1405–15.
Ramael S, Van Hoorick B, Tiessen R, et al. Similar pharmacokinetics of the adalimumab (Humira®) biosimilar BI 695501 whether administered via subcutaneous autoinjector or prefilled syringe (VOLTAIRE®-AI and VOLTAIRE®-TAI): Phase 1, randomized, open-label, parallel-group trials. Rheumatol Ther. 2018;5(2):403–21.
Schwarzenbach F, Trong MD, Grange L, et al. Results of a human factors experiment of the usability and patient acceptance of a new autoinjector in patients with rheumatoid arthritis. Patient Prefer Adherence. 2014;8:199–209.
Acknowledgements
The authors would like to thank the participants who took part in the study.
Funding
Sponsorship for this study and Rapid Service Fee were funded by Fresenius Kabi SwissBioSim.
Medical Writing and/or Editorial Assistance
Medical writing support and editorial assistance was provided by Claire Stoker, PhD, of Arc, a division of Spirit Medical Communications Group Limited, and Stephanie Carter, CMPP, PhD, a contract writer working on behalf of Arc, and were funded by Fresenius Kabi, in accordance with Good Publication Practice 3 guidelines.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole and have given their approval for this version to be published.
Author Contributions
Ahad Sabet, Daniel S. Dickerson, Eugenia E. Kunina, Anna Lucia Buccarello and Joëlle Monnet made substantial contributions to the conception or design of the work or the acquisition, analysis of interpretation of data. All authors were responsible for critically revising the work for important intellectual content and for making all content and editorial decisions. All authors had final approval of the manuscript version to be published and are accountable for all aspects of the work in ensuring the accuracy and integrity of this manuscript.
Disclosures
Ahad Sabet and Daniel S. Dickerson are employees of PRA Health Sciences. Joëlle Monnet and Anna Lucia Buccarello are employees of, and report personal fees from, Fresenius Kabi SwissBioSim during the conduct of the study. Eugenia E. Kunina is now an employee of Arena Pharmaceuticals Development GmbH, Zug, Switzerland, but was an employee of, and reports personal fees from, Fresenius Kabi SwissBioSim during the conduct of the study.
Compliance with Ethics Guidelines
The study was conducted in accordance with the International Conference on Harmonisation Good Clinical Practice Guidelines and the Declaration of Helsinki, and the protocol was approved by the Midlands Independent Review Board (ESM Table 3). Written informed consent was required prior to the conduct of any study procedures.
Data Availability
The datasets generated during and/or analysed during the current study are not publicly available due to ongoing regulatory filing activities. Once the regulatory application is complete, datasets will be available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the Electronic Supplementary Material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Sabet, A., Dickerson, D.S., Kunina, E.E. et al. A Randomised Controlled Trial Comparing the Pharmacokinetics and Tolerability of the Proposed Adalimumab Biosimilar MSB11022 Delivered via Autoinjector and Pre-filled Syringe in Healthy Subjects. Rheumatol Ther 9, 693–704 (2022). https://doi.org/10.1007/s40744-022-00432-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-022-00432-1