
Abstract
Introduction
Abiraterone acetate (AA) is approved for treatment of metastatic castration-resistant prostate cancer. The originator AA (OAA) formulation has been associated with AUC and C max increases of 10- and 17-fold, respectively, when administered following a high-fat meal relative to the fasted state. AA fine particle (AAFP) is a proprietary formulation (utilizing SoluMatrix Fine Particle Technology™) that has enhanced dissolution properties. It was designed to increase the oral bioavailability of AA, and to potentially reduce variability in drug exposure and food effects compared with OAA. In healthy subjects, AAFP 500 mg was previously shown to be bioequivalent to OAA 1000 mg when taken in fasted conditions.
Methods
Healthy male subjects (N = 25) fasted 10 h overnight before randomization in a crossover design received a single 500-mg dose of AAFP under fasted or fed conditions.
Results
The extent of AAFP drug exposure was significantly (P < 0.001) greater under fed versus fasted conditions. Following a single dose of AAFP, relative bioavailability measured by the geometric mean ratio of AUC and C max of abiraterone indicated that the extent of drug absorption increased under fed conditions by approximately 4.5- and 6.5-fold, respectively, versus under fasted conditions. AAFP was safe and well-tolerated. AAFP under fed conditions has higher bioavailability and is not bioequivalent to AAFP under fasted conditions. The food effect with AAFP 500 mg was approximately 50% less than what has been reported for OAA 1000 mg.
Conclusions
Although diminished, the potential for excessive abiraterone plasma concentrations in patients who are noncompliant with dosing instructions remains.
Funding
Churchill Pharmaceuticals LLC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Food has a complicated effect on the pharmacokinetic and pharmacodynamic profiles of many drugs. The extent of absorption of orally administered drugs with or without food appears to be determined by the drug’s solubility and membrane permeability, among other parameters [1]. Administration of certain drugs with food has the potential to dramatically alter drug absorption, especially after meals with a high fat content [2]. This potential impact of food on drug absorption underlines the importance of conducting food effect studies early in the oncology drug development process under well-defined and standardized fed and fasted conditions, as reviewed by Parsad and Ratain [3].
The oral bioavailability of lipophilic drugs is known to be limited by in vivo dissolution rate, which in turn is related to drug particle size [4, 5]. Taking lipophilic drugs with a high-fat meal is also known to increase their rate of dissolution [6]. Abiraterone acetate (AA), an orally available, highly lipophilic drug [7], is approved for the treatment of metastatic castration-resistant prostate cancer. AA (1000 mg; two 500-mg tablets or four 250-mg tablets) is administered once daily in combination with prednisone 5 mg (orally, twice daily). Dosing instructions indicate that AA must be taken on an empty stomach and no food should be consumed for at least 2 h before, or at least 1 h after, administering the drug. The AA tablets should be swallowed whole with water and should not be crushed or chewed [7]. Several studies have investigated the effect of food on AA absorption and bioavailability [8,9,10,11,12] and established that consumption of a high-fat meal before administration of the originator AA (OAA) formulation markedly increases abiraterone absorption [9, 10, 12].
To evaluate the food effect of AA, Li et al. used a modeling approach to simulate a clinical oncology population pharmacokinetic trial. They found that food effect was a statistically significant covariate on oral clearance for abiraterone [8]. Other studies have found substantial food effect in healthy subjects when AA was given following a meal, with plasma abiraterone exposure increasing by 2.9- to 10-fold [9, 10, 12]. For example, Chi et al. reported that in healthy subjects administered the OAA formulation, the area under the plasma concentration-time curve (AUC) and maximum measured plasma concentration (C max) of abiraterone, the active metabolite of AA, increased by 10- and 17-fold, respectively, when given shortly after a high-fat meal relative to the fasted state [10]. Biorelevant in vitro studies suggest that intestinal fluids, such as bile salt [6, 10] produced during the fed state, may facilitate the absorption of abiraterone, thus producing the food effect observed in pharmacokinetic trials [9]. The large bioavailability changes recognized in the reference product with regard to meal fat content [10] may lead to differences in safety, tolerability, and efficacy.
AA fine particle (AAFP) is a novel proprietary formulation of AA utilizing SoluMatrix Fine Particle Technology™ (which is licensed from iCeutica, Inc. to Churchill Pharmaceuticals for use with AAFP) and is designed to improve oral bioavailability and reduce food effects compared with the OAA formulation. This technology provides improved uniformity of very small drug particles resulting in a high ratio of surface area-to-mass. The higher surface area-to-mass ratio can increase the in vivo dissolution rate of lipophilic, poorly water-soluble, active pharmaceutical ingredients such as AA that often have dissolution rate-limited oral bioavailability. In a previous companion study of healthy male subjects, AAFP 500 mg was shown to be bioequivalent to OAA 1000 mg (within 80–125% of C max and AUC) when taken under fasted conditions [13]. The present study investigates the effect of a high-fat meal on AAFP pharmacokinetic parameters and bioavailability.
Methods
This phase 1 clinical study investigating a single-dose of AAFP 500 mg under both fed and fasted conditions in healthy male subjects was conducted in compliance with the International Conference on Harmonisation Good Clinical Practice guidelines and the Declaration of Helsinki. Registration of this study in ClinicalTrials.gov was not necessary based on exclusion criteria for phase 1 drug trials (https://clinicaltrials.gov/ct2/manage-recs/fdaaa#footnote1). All subjects provided written informed consent before any treatment was initiated, and the protocol was reviewed and approved by an institutional review board (Aspire IRB, 11491 Woodside Avenue, Santee, California 92071, USA).
Study Population
Normal healthy male subjects aged 18–50 years with a body mass index between 18 and 30 kg/m2 who met additional inclusion/exclusion criteria were enrolled in the study. Subjects were required to be in good health based on physical examination, recording of vital signs, electrocardiogram, and clinical laboratory testing (hematology, biochemistry, and urinalysis). Subjects who used prescription medications or tested positive for drugs of abuse and alcohol at the screening visit were excluded from the study.
Study Design
This was a single-center, randomized, open-label, two-period crossover study of food effects. In period 1 of the study, enrolled subjects were admitted to the study site (PAREXEL International, Early Phase Clinical Unit, Baltimore, MD, USA) the evening before the morning dose of AAFP. All subjects underwent a 10-h overnight fast. The following morning, subjects were randomized to receive AAFP 500 mg (4 × 125-mg AAFP tablets) under either fed or fasted conditions. Subjects randomized to the fed condition consumed an entire standard high-fat FDA breakfast (two eggs fried in butter, two strips of bacon, two slices of toast with butter, 113.4 g of hash brown potatoes, and 226.8 g of whole milk) [14] 30 min before AAFP 500 mg administration. Subjects randomized to the fasted condition were administered AAFP 500 mg with 240 mL of water. This group remained in the fasted state until 4 h after taking the dose of AAFP.
In period 2 of the study, the subjects returned to the same study site following a 7-day washout period. The same study procedures as in period 1 were performed; however, the subjects followed the order of their randomly assigned treatment sequences and crossed over to the opposite fed or fasted condition from period 1. In this study, each subject served as his own control.
Subjects were confined to the study site for collection of blood samples for pharmacokinetic evaluation of plasma abiraterone concentration until 48 h after AAFP administration for both periods 1 and 2. Blood samples (approximately 6 mL) were collected in each study period at − 0.75 h, and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, and 48 h after dosing. AAFP tablets were manufactured by Mayne Pharma Group Ltd. for Churchill Pharmaceuticals LLC.
Blood Sample Collection and Bioanalytical Assay
Blood samples (approximately 6 mL) for pharmacokinetic analyses were collected into 6-mL tripotassium ethylenediaminetetraacetic acid-containing tubes and immediately placed on ice. Plasma was separated by centrifugation at 4 °C, transferred to appropriately labeled polypropylene specimen containers, frozen at − 20 °C within 1 h of collection, then stored at − 20 °C. Samples were then shipped on dry ice to the bioanalytical laboratory for analysis. Plasma samples were analyzed for abiraterone concentrations using a validated bioanalytical assay (Sannova Analytical, Somerset, NJ, USA).
The samples were extracted using the liquid–liquid extraction method and analyzed using reverse-phase liquid chromatography. The analytes were detected using tandem mass spectrometry. The lower limit of quantitation for abiraterone was 0.651 ng/mL. The upper limit of quantitation for abiraterone was 203.519 ng/mL. Plasma drug concentrations identified as below the limit of quantification were entered as zero. A calibration curve consisting of 2 control blanks, 2 zero standards, and 10 nonzero calibration standards covering a concentration range of 0.651–203.519 ng/mL for abiraterone were analyzed with every sample batch. Quality control standards for abiraterone were also analyzed with every sample batch. Internal standard peak area ratio values were used to set up the calibration curve and to determine quality control and unknown sample concentrations. Linear regression for abiraterone was used to obtain the best fit of the data for the calibration curve.
Pharmacokinetic Analysis
All subjects who received AAFP 500 mg under both fed and fasted conditions who had sufficient plasma concentrations for calculating C max and AUC from time 0 to the time (t) of the last quantifiable concentration (C t; AUC0–t ) were included in the pharmacokinetic population. The following plasma pharmacokinetic parameters were determined: AUC0–t as calculated by the linear trapezoidal method; AUC from time 0 to infinity (AUC0–∞) approximated by linear trapezoidal summation and extrapolated to infinity by addition of C t/K e; C max; time to maximum measured plasma concentration (T max); apparent elimination rate constant (K e) as determined by linear regression of the terminal points of the log-linear concentration–time curve; and apparent terminal elimination half-life (T 1/2) calculated as loge(2)/K e or 0.693/K e.
Safety Assessments
All subjects who received AAFP were included in the safety population. Assessments included physical examination, recording of vital signs, electrocardiogram, clinical laboratory testing (hematology, biochemistry, and urinalysis), and recording of adverse events (AEs). AE assessments and concomitant medications were assessed throughout the clinical study.
Statistical Analyses
Based on a previous companion study, a sample size of 22 subjects was deemed sufficient to provide statistical power to detect a 20% difference in the reference mean between fed and fasted conditions [13]. Assuming a screen failure/dropout rate of 15%, enrollment of 26 subjects was planned. A parametric general linear model was applied to the pharmacokinetic parameters (AUC, C max, T max, and T 1/2). The model included sequence, subject-within-sequence, period, and condition. The sequence effect was tested using the subject-within-sequence effect, and all other effects were tested using the residual error of the model. Analysis of variance (ANOVA) for a crossover design was used to examine the differences between the two dose conditions (fed vs. fasted). A null hypothesis of zero difference in a parameter between the two conditions was assessed at the 0.05 level, with the alternative hypothesis of nonzero differences. T max was compared for the fed versus fasted conditions using the nonparametric Wilcoxon signed rank test.
To evaluate relative bioavailability, AUC and C max parameters were analyzed on a log scale using the parametric general linear model to assess bioequivalence between AAFP 500 mg under fed versus fasted conditions. The two one-sided t test hypotheses were tested at the 0.05 level by constructing a 90% confidence interval (CI) for the geometric mean ratio. Bioequivalence was concluded if the 90% CI of the ratio was within 0.80 and 1.25 for AUC and C max parameters; otherwise, relative bioavailability for AUCs and C max were expressed based on the least square means ratio values.
Results
This study was conducted between February 18, 2015, and March 30, 2015.
Subjects
Twenty-five subjects were enrolled, randomized, and dosed with AAFP 500 mg without replacement in period 1, and 22 subjects completed the study. Two subjects were discontinued; one for violation of house rules and one for pre-existing increased blood pressure. One subject was lost to follow-up. Subject baseline characteristics and demographics are shown in Table 1.
Pharmacokinetic Parameters of AAFP in Fed and Fasted Conditions
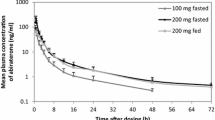
Figure 1 shows the mean plasma concentrations (ng/mL) versus time curves of AAFP 500 mg under fed and fasted conditions. Abiraterone absorption increased following a single dose of AAFP 500 mg under fed conditions compared with the fasted conditions. Peak levels were observed at approximately 2 h post-dose for both fed and fasted conditions.

Arithmetic mean plasma abiraterone concentration–time plots under fed and fasted conditions (linear scale). Error bars indicate standard error. Some error bars are not visible because they are smaller than the size of the symbols
The extent of abiraterone absorption was significantly higher under fed conditions versus fasted conditions measured by abiraterone concentrations in plasma following a single dose of AAFP 500 mg (Table 2, and shown graphically in Fig. 2). Following a single dose of AAFP 500 mg, the median T max was approximately 2 h for fed and fasted conditions. There was no statistically significant (P = 0.99) difference in median elimination T 1/2 under fed conditions (14.7 h) compared with fasted conditions (13.1 h; Table 2).

AUC and C max mean parameters under fed and fasted conditions. Error bars indicate standard error
With reference to fasted conditions, the geometric mean ratios for AUC0–∞, AUC0–t , and C max, respectively, under fed conditions were 442.4, 458.6, and 649.5% (Table 3). The 90% CIs corresponding to the geometric mean ratios of test/reference were well above the upper limit of the 80–125% bioequivalence criteria, indicating that AAFP 500 mg administered under fasted conditions is not bioequivalent to AAFP 500 mg administered under fed conditions.
Safety
Only one treatment-emergent AE related to AAFP 500 mg was reported (testicular pain). It was mild in intensity (grade 1) and resolved without medical intervention by the end of the study. There were no AEs leading to discontinuation, serious AEs, or deaths during the study. No clinically significant biochemical, hematological, or urinalysis changes were observed.
Discussion
The extent of drug exposure for AAFP 500 mg was significantly greater under fed conditions compared with fasted conditions. The relative bioavailability measured by the geometric mean ratio of AUC and C max parameters of abiraterone indicated that relative to fasted conditions, drug exposure of a single dose of AAFP 500 mg was increased under fed conditions by approximately 4.5-fold for AUC and 6.5-fold for C max. In a previous study, the 500-mg dose of AAFP was shown to be bioequivalent to the 1000-mg dose of OAA under fasted conditions [13]. In a food effect study of OAA by Chi et al., the AUC0–∞ and C max of abiraterone in healthy subjects given the 1000-mg dose of the OAA formulation increased by 10- and 17-fold, respectively, when given shortly after a high-fat meal relative to the fasted state [10]. Comparing these results with those from the present study, the food effect with AAFP 500 mg shows an approximate 50% decrease versus OAA 1000 mg. The application of the proprietary technology used in the manufacture of AAFP provides a smaller drug particle size and is intended to increase dissolution in vivo [15], which may explain the improved bioavailability of AAFP under fasted conditions compared with OAA [13]. The improved bioavailability may be important for highly lipophilic drugs such as AA, which needs to be administered under fasted conditions.
Food restriction requirements with medications may lead to inappropriate use or nonadherence. In a survey of cancer patients, 43% of patients who received agents with known significant food effects reported that they did not consider the timing of meals before dosing, and 23% were unaware of the food-drug effects of their medication [16, 17]. Reducing the extent of food effects could ameliorate differences in drug exposure and drug level variability. Consequently, the reduced food effect with AAFP could potentially result in less exposure-related drug toxicity. Although no exposure-safety relationship for abiraterone has been demonstrated, a formulation of AA that could be given at a lower dose to achieve similar drug exposure with less food effect could provide safety benefits for patients who may not strictly adhere to the time intervals needed around meals.
The systemic exposure of abiraterone is subject to a large inter-individual variability. Carton et al. looked at the PK/PD relationship between abiraterone trough concentrations and prostate-specific antigen (PSA) levels in patients with metastatic castration-resistant prostate cancer after 3 months of treatment [18]. Their results showed that the average plasma abiraterone trough concentration was significantly higher in PSA responders versus nonresponders when OAA was administered under fasted conditions. A phase 2 study (ClinicalTrials.gov identifier: NCT02737332) in castration-resistant prostate cancer patients comparing the safety and biomarkers of efficacy (testosterone and PSA levels) of AAFP 500 mg with OAA 1000 mg under modified fasting conditions is ongoing. A secondary objective of the study will include an analysis of the steady-state trough concentration of abiraterone at various time points with the two AA formulations.
Limitations of this study include the lack of direct comparison between OAA and AAFP under fed and fasted conditions. This was a single-dose study and therefore does not show whether reduced food effect leads to reduced toxicity at steady state. Additionally, food effects were assessed in healthy volunteers, pharmacokinetic parameters in a patient population may differ due to concomitant drug use or disease state. As mentioned, an ongoing phase 2 trial in patients with castration-resistant prostate cancer is underway that will provide direct comparative data between AAFP and OAA in the intended patient population of metastatic castrate-resistant prostate cancer, and will assess key pharmacodynamic and safety indicators.
Conclusions
AAFP 500 mg under fed conditions has higher bioavailability than under fasted conditions. At doses under fasted conditions (AAFP 500 mg and OAA 1000 mg), the food effect for AAFP 500 mg is approximately 50% less than what has been reported for OAA 1000 mg in healthy volunteers. Although there was a large food effect with AAFP 500 mg, it was less than that with OAA and may reduce the potential for excessive abiraterone plasma concentrations in patients who are noncompliant with dosing instructions. The clinical impact of this requires further investigation.
References
Kawai Y, Fujii Y, Tabata F, et al. Profiling and trend analysis of food effects on oral drug absorption considering micelle interaction and solubilization by bile micelles. Drug Metab Pharmacokinet. 2011;26(2):180–91.
Welling PG. Effects of food on drug absorption. Annu Rev Nutr. 1996;16:383–415.
Parsad S, Ratain MJ. Food effect studies for oncology drug products. Clin Pharmacol Ther. 2017;101:606–12.
Fincher JH. Particle size of drugs and its relationship to absorption and activity. J Pharm Sci. 1968;57:1825–35.
Zhi J, Rakhit A, Patel IH. Effects of dietary fat on drug absorption. Clin Pharmacol Ther. 1995;58:487–91.
Singh BN. Effects of food on clinical pharmacokinetics. Clin Pharmacokinet. 1999;37(3):213–55.
ZYTIGA® (abiraterone acetate) tablets [prescribing information]. Horsham, PA; Janssen Biotech, Inc; 2017. https://www.zytigahcp.com/shared/product/zytiga/zytiga-prescribing-information.pdf. Accessed 1 May 2017.
Li CH, Sherer EA, Lewis LD, Bies RR. Clinical trial simulation to evaluate population pharmacokinetics and food effect: capturing abiraterone and nilotinib exposures. J Clin Pharmacol. 2015;55(5):556–62.
Geboers S, Stappaerts J, Mols R, et al. The effect of food on the intraluminal behavior of abiraterone acetate in man. J Pharm Sci. 2016;105(9):2974–81.
Chi KN, Spratlin J, Kollmannsberger C, et al. Food effects on abiraterone pharmacokinetics in healthy subjects and patients with metastatic castration-resistant prostate cancer. J Clin Pharmacol. 2015;55(12):1406–14.
Bernard A, Vaccaro N, Acharya M, et al. Impact on abiraterone pharmacokinetics and safety: open-label drug–drug interaction studies with ketoconazole and rifampicin. Clin Pharmacol Drug Dev. 2015;4(1):63–73.
Inoue K, Shishido A, Vaccaro N, et al. Pharmacokinetics of abiraterone in healthy Japanese men: dose-proportionality and effect of food timing. Cancer Chemother Pharmacol. 2015;75(1):49–58.
Goldwater R, Hussaini A, Zhang Y, Bosch B, Nemeth P. Comparison of a novel formulation of abiraterone acetate vs the originator formulation in healthy male subjects: two randomized, open-label, crossover studies. Clin Pharmacokinet. 2017;56(7):803–13.
Food and Drug Administration. Guidance for Industry. Food-effect bioavailability and fed bioequivalence studies. http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm070241.pdf. Published December 2002. Accessed 16 Sep 2016.
Desjardins PJ, Olugemo K, Solorio D, Young CL. Pharmacokinetic properties and tolerability of low-dose SoluMatrix diclofenac. Clin Ther. 2015;37(2):448–61.
Lin H, Suri A, Webb I, Aggarwal S. Relationships between food effects, patient adherence to treatment, and pharmacokinetics of oral anticancer drugs. J Clin Oncol. 2014;32(suppl):e17614.
Muluneh B, Alexander M, Deal A, et al. Prospective evaluation of perceived barriers to medication adherence by patients on oral antineoplastics. J Clin Oncol. 2012;30(Suppl 15):6042.
Carton E, Noe G, Huillard O, et al. Relation between plasma trough concentration of abiraterone and prostate-specific antigen response in metastatic castration-resistant prostate cancer patients. Eur J Cancer. 2017;72:54–61.
Acknowledgements
This study was funded and the journal’s article processing charges were paid by Churchill Pharmaceuticals LLC. Editorial support (assembling tables and figures, collating author comments, copyediting, fact checking, and referencing) and graphic services were provided by Gautam Bijur, PhD, and Nancy Price, PhD, of AOI Communications, LP and were funded by Churchill Pharmaceuticals LLC.
All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published. The datasets during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Yuxin Zhang, Xtiers Consulting, Inc. conducted the statistical analyses. Sharon Reinhard, Churchill Pharmaceuticals LLC assisted in the study design. PAREXEL conducted the clinical study. Sannova Analytical Inc. performed the bioanalytical analyses. Tata Solutions, Inc. conducted the pharmacokinetic analyses. The authors would like to thank the study subjects and the site staff who made this study possible.
Disclosures
Alexander Papangelou declares that he has no conflict of interest. Anthony J. Olszanski declares he is a paid consultant of Churchill Pharmaceuticals LLC. Cy Aaron Stein declares he is a paid consultant of Churchill Pharmaceuticals LLC. Anthony J. Olszanski has received support for travel to meetings for the study and manuscript preparation. Bill Bosch is an employee of Churchill Pharmaceuticals LLC. Paul Nemeth is an employee of Churchill Pharmaceuticals LLC. Bill Bosch has declared he has stock options and patents.
Compliance with Ethics Guidelines
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content To view enhanced content for this article go to http://www.medengine.com/Redeem/B9CCF0602F1ABC93.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Papangelou, A., Olszanski, A.J., Stein, C.A. et al. The Effect of Food on the Absorption of Abiraterone Acetate from a Fine Particle Dosage Form: A Randomized Crossover Trial in Healthy Volunteers. Oncol Ther 5, 161–170 (2017). https://doi.org/10.1007/s40487-017-0054-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40487-017-0054-2