
Abstract
Imaging of biological and molecular processes has provided the platform for evaluating the hallmarks of cancer, such as metabolism, proliferation, tissue invasion, angiogenesis, apoptosis and hypoxia, and in turn for assessing the efficacy of treatments including novel targeted therapies. Cross-sectional imaging methods can measure response to chemotherapy and radiotherapy by measuring changes in tumour volume. Imaging modalities such as positron emission tomography and functional magnetic resonance imaging can non-invasively detect early molecular changes in response to therapy, provide guidance for therapy optimisation, and predict response to treatments and clinical outcome. In an era of escalating drug trial costs, with high attrition rates of early-phase studies, the development of an imaging biomarker can contribute to optimisation of proof of concept and patient stratification. In this review, we examine the current molecular imaging modalities used to assess pharmacodynamics and therapy response and highlight some novel emerging imaging strategies.
Similar content being viewed by others


Introduction
The emergence of tumour-targeted therapies, in contrast to conventional chemotherapy and radiotherapy treatments, has contributed to the discovery and validation of novel oncology biomarkers. A biomarker is defined as a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes or pharmacological responses to a therapeutic intervention [1].
Examples of tumour-targeted strategies include the use of monoclonal antibodies, such as trastuzumab, targeting ERBB2 receptors in breast cancer [2] and bevacizumab antibody to vascular endothelial growth factor (VEGF) in colorectal cancer [3]. These agents have led to improved progression-free and/or overall survival benefit in recent clinical trials. In addition, small-molecule inhibitors of membrane-bound tyrosine kinase receptors, such as sorafenib, a target of the VEGF receptor and raf kinase, have been developed as novel strategies for renal cancer [4]. Other examples include c-kit inhibition with imatinib in gastrointestinal stromal tumours [5] and epidermal growth factor receptor (EGFR) inhibition with erlotinib in lung cancer [6]. Molecularly-targeted drug therapies typically lead to cytostasis rather than cytoreduction which, instead, occurs with conventional treatments. In this setting, conventional cross-sectional imaging techniques such as computed tomography (CT) or magnetic resonance imaging (MRI) have limited power for the assessment of response.
Clinical CT and MRI provide anatomical information regarding the tumour size. Response is defined according to the criteria of the World Health Organisation (WHO) [7] and the Response Evaluation Criteria in Solid Tumours (RECIST) [8, 9]. The WHO criteria require assessment of tumour size in two dimensions; however, variations in WHO criteria-based reporting have resulted in widespread adoption of the RECIST, which allow tumour size to be measured in one dimension. The RECIST response categories include complete response, partial response, stable disease and progressive disease. In clinical trials, these imaging biomarkers define overall response and progression-free survival and are currently used as surrogate end points of treatments, including targeted treatments [10]. However, with novel targeted therapies, changes in tumour size early in treatment are minimal and may not be adequately measured using the WHO criteria and RECIST. Alternatively, novel imaging biomarkers can be used to provide information at the cellular and molecular level by non-invasively detecting changes in tumour biology such as target expression, alterations in cellular metabolism, perturbed cell signalling and other molecular characteristics [11]. These functional/molecular imaging biomarkers fall under the umbrella of molecular imaging and can potentially be used to diagnose disease, guide treatment selection, measure early response to treatment and follow disease progression.
Current imaging modalities adapted for molecular imaging include radionuclide imaging, e.g. positron emission tomography (PET) and single-photon emission computed tomography (SPECT), functional MRI and magnetic resonance spectroscopy (MRS), as well as ultrasound and optical imaging.
PET imaging
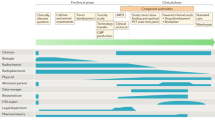
Positron emission tomography is a nuclear medicine imaging modality that provides a molecular or functional image of biological processes such as blood flow, metabolism, cell surface receptor expression, angiogenesis, hypoxia, proliferation and apoptosis. Clinically, PET/CT is used for the diagnosis and evaluation of response to treatment in cancer and is the primary focus of this review, which provides an overview of the tracers currently under development or used in clinical imaging as pharmacodynamic and response markers. Table 1 summarises the clinical targets discussed in this review.
The PET radiotracer most commonly used in clinical oncology is the glucose analogue, 18F-2-fluoro-2-deoxyglucose (18F-FDG). In highly metabolically active cells, such as cancerous cells, glucose uptake and utilisation, termed the ‘Warburg effect’ is increased. FDG is taken up by cells via facilitative transport, primarily via the glucose transporter isoforms GLUT-1 and GLUT-3 [12], phosphorylated by hexokinase to FDG-6-phosphate and trapped within the cell; glucose-6-phosphate, on the other hand, is metabolised in the glycolytic pathway. High levels of glucose transporters as well as increased levels of hexokinase and therefore increased glucose consumption in tumours, are responsible for the high levels of FDG uptake which can be detected by PET [13]. FDG PET is currently used in oncology for staging and re-staging malignancy and for assessing therapy response. In the context of FDG PET response assessments, a significant change in FDG uptake is seen, for example, following a single cycle of chemotherapy in lymphoma [14]. While changes in FDG uptake with cytoreductive therapies may reflect a loss of cell viability/cellularity, it has been hypothesised that changes in FDG uptake with a number of specific targeted therapies reflect redistribution of GLUT receptors from the plasma membrane to the cytosol consequent to the anti-cell signalling action of these drugs and subsequently changes in glucose metabolism due to effects of the targeted treatments on gene or protein expression [15–17]. Despite its wide adoption in the clinical setting, FDG PET is not effective in all tumour types and a better understanding of this heterogeneous uptake will be relevant for a better understanding of tumour biology. For example, well-differentiated prostate adenocarcinoma and renal cell carcinoma are less metabolically active and show low FDG uptake [18]. By contrast, more aggressive, poorly differentiated hepatocellular tumours show high FDG uptake compared with well-differentiated tumours [19]; this situation is complicated by the high expression of phosphatases that can reverse phosphorylation and trapping of FDG within background normal liver, together with the notion that the process of carcinogenesis is multi-stage rather than simply binary (early stage/aggressive). In tumours of the brain, a high background uptake by surrounding normal tissue can mask tumour uptake [20], and high uptake is also noted in the presence of infected or inflamed tissue; all these are factors that can limit the accuracy of FDG PET [21]. Furthermore, after treatment, high uptake by infiltrating immune cells can mask the decreased uptake by the dying tumour cells [22]. There is, therefore, an urgent need to develop novel imaging biomarkers able to measure other biological processes in tumours, such as proliferation, apoptosis, vasculature dynamics and receptor expression, and therefore to provide readouts of on-drug pharmacological effects of mechanism-based anti-cancer drugs. The following section describes such molecular imaging biomarker approaches.
Measurement of tumour cell proliferation
Tumour cells exhibit increased cellular proliferation and corresponding increased nucleoside metabolism that can be exploited for imaging by PET. In this regard, 11C-thymidine was initially developed to detect tumour cell proliferation [23]. The 11C-radioisotope, however, has a short half-life of approximately 20 min and can therefore only be used in the presence of an on-site cyclotron. For this reason, 18F-fluorothymidine (18F-FLT) was developed, exploiting the longer radioactive half-life of fluorine-18. FLT is a pyrimidine analogue that enters cells via ENT1-mediated facilitated transport and is phosphorylated into FLT monophosphate by thymidine kinase 1 (TK1), a key enzyme of the thymidine salvage pathway for DNA synthesis. FLT monophosphate is trapped in the cell, but not incorporated into DNA during the S-phase. TK1 enzymatic activity is increased in rapidly proliferating cells and therefore FLT PET provides an indirect measurement of proliferation. The validity of FLT PET, in comparison with the Ki-67 cellular biomarker, as a readout of proliferation was recently confirmed in a comparative meta-analysis [24]. FLT uptake is therefore a surrogate measure of cellular proliferation and can be visualised in many tumours. For cancer detection and staging, 18F-FLT did not show superiority over 18F-FDG due to its lower uptake in solid tumours [17].
Fluorothymidine positron emission tomography has been found to be beneficial in the assessment of response to therapy in breast cancer. Patients underwent FLT PET scans a week after chemotherapy; a decrease in FLT corresponded to clinical response and a decrease in Ki-67 and SUVmax, a measure of maximal radiotracer accumulation, discriminated between clinical response and stable disease patients (Fig. 1) [25].

18F-FLT PET images in responding and non-responding patients. a Pre-treatment and b post-treatment images of a patient with grade II lobular carcinoma that responded to treatment. c Pre-treatment and d post-treatment images of a patient with grade II invasive ductal carcinoma that did not respond to treatment. Reproduced from Kenny et al. [25] with permission (color figure online)
Fluorothymidine positron emission tomography has limited use in gastrointestinal tumours due to its physiological retention in the liver. However, 18F-FLT has been used in pharmacodynamic assessment of rectal tumours. In a study of ten patients with rectal cancer treated with a long course of preoperative chemoradiotherapy, FLT uptake was observed in the rectal tumours before the start of the chemoradiotherapy and decreased significantly post-treatment in both responding and non-responding tumours, suggesting that FLT PET may not provide a valid surrogate marker of response to therapy [26]. Although this study failed to demonstrate a biomarker of response, a recent study also measured 18F-FLT (and 18F-FDG) uptake at baseline and early time points in 15 patients with rectal cancer undergoing neoadjuvant chemotherapy and concluded that a high percentage change in 18F-FLT uptake during therapy was predictive of improved disease-free survival [27]. These two studies in rectal cancer patients highlight the need to establish the ideal PET imaging biomarker (i.e. proliferation versus metabolism marker) and an optimal time point for imaging (mid-treatment versus end of treatment) linked to survival as the outcome variable.
The use of molecularly targeted treatments that induce cytostatic and not cytotoxic effects, including inhibitors of signal transduction targets such as EGFR, c-kit, platelet-derived growth factor receptor (PDGFR) and vascular endothelial growth factor receptor (VEGFR), leads to decreases in cellular proliferation, which are more appropriately measured using FLT. In non-smokers with advanced or recurrent adenocarcinoma of the lung (a good prognostic group for treatment with EGFR tyrosine kinase inhibitors), FLT PET predicted the response to gefitinib 7 days after initiating treatment and FLT responders showed a longer time to progression [28]. With regard to the comparative predictive values of FDG and FLT PET, in a study of non-small cell lung cancer patients treated with erlotinib, a change in FDG PET 1 week after therapy was predictive of non-progression 6 weeks post-therapy, with metabolic response found to be associated with improved progression-free survival and overall survival. By comparison, FLT PET response predicted improved progression-free survival, but did not correlate with non-progression on treatment or overall survival [29].
With regard to tumours of the central nervous system, since 18F-FDG shows high uptake in normal brain tissue, 18F-FLT has been preferred for the detection of brain tumours, as it demonstrates low uptake in normal brain tissue; a dynamic imaging sequence is required to discriminate blood–brain barrier transit. In the assessment of responses to targeted treatments, 18F-FLT PET and MRI were used to predict outcome following treatment with bevacizumab in a study of 30 patients with recurrent malignant gliomas. Patients underwent FLT PET scans at baseline, and 2 and 6 weeks after treatment. The results showed that early and late changes in 18F-FLT PET were more predictive of overall survival than MRI criteria. 18F-FLT changes were also predictive of progression-free survival. A multivariate analysis of outcome predictors identified the 6-week 18F-FLT PET scan as the best predictor of overall survival [30].
Although FLT is not currently in use for cancer diagnosis (due to its low uptake compared with FDG), it may be more useful in predicting response to new treatments. The limitations of FLT PET include its uptake in rapidly dividing bone marrow cells and its elimination through the liver, both of which limit the assessment of lesions that are near these organs. New methods including use of kinetic spatial filters [31, 32] aim to overcome the high liver background uptake problem. More studies are required to validate this latter application, especially with respect to molecular targeted therapies. The unknown relative flux through de novo and salvage pathways for thymidine monophosphate synthesis could also be regarded as a limitation to the use of FLT PET for measurement of proliferation, as FLT PET only assesses the salvage pathway. The ‘FLT flare’ effect (attributed to enhanced ENT1 activity) that ensues when inhibitors of the de novo pathway, such as thymidylate synthase inhibitors, are used has been exploited as a pharmacodynamic imaging biomarker of these inhibitors [33–35].
Lipid metabolism
Choline
Aberrant de novo lipid synthesis is another hallmark of many cancers, and a number of PET tracers have been developed that target this metabolic pathway; these include 11C-choline and 11C-acetate. Phosphatidylcholine is an important component of phospholipids in the cell membrane, choline being the precursor in the synthesis of phosphatidylcholine by choline kinase. In neoplastic tissues, levels of both phosphocholine and choline kinase, which catalyses the phosphorylation of choline, are elevated [36]. Radiotracers that target choline have clinical applications in tumours that are well differentiated and have low glucose uptake. Like 11C-thymidine, 18F-fluoromethylcholine has also been developed for PET imaging of choline metabolism. Choline and phosphocholine, as well as the product of choline oxidation, contribute to the PET signal acquired, which is thus a signal of both transport and phosphorylation of choline [37]. Choline oxidation competes with the specific choline kinase reactions, complicating PET image interpretation. The difference in the levels of total choline and its metabolites in normal versus tumour samples is exploited using MRS. The association of MRS with conventional MRI greatly enhances diagnostic accuracy. Total choline levels are elevated in tumours and, on this basis, MRS has been used in the diagnosis of cancer and to help in the detection of tumour recurrence. The use of the choline resonance in MRS for assessing the response to anti-cancer therapy is based on decreases in the total choline signal or in choline metabolite ratios [38].
The first study of 11C-choline was conducted in brain tumour patients [39]. In current clinical studies, 11C-choline PET imaging and more recently 18F-fluorocholine are used in prostate cancer for the identification of locally recurrent and metastatic disease [40]. This is in part due to the low sensitivity and specificity of 18F-FDG in this tumour group, as well as the lower levels of radioactivity excreted into the bladder (particularly for 11C-choline). The current choline radiotracers used in PET imaging, including 11C-choline and 18F-fluorocholine, have poor metabolic stability in vivo, being readily oxidised to betaine analogues by choline oxidase; this makes data interpretation difficult due to the presence of metabolites detected soon after injection [41]. To overcome these difficulties, we have developed a novel radiotracer, 18F-fluoromethyl (1,2-2H4)-choline (18F-D4-choline), which features improved metabolic stability, while retaining phosphorylation potential [37, 42]. A comparison of 11C-choline, 11C-(1,2-2H4)-choline (11C-D4 choline) and 18F-D4-choline has been undertaken [43]. Both deuteration and fluorination led to increased metabolic stability of 18F-D4-choline. In addition, 18F-D4 choline uptake was observed in relation to choline kinase α expression in three different tumour types, confirming its utility in different tumour types [43]. 18F-D4-choline has also been assessed as a response biomarker in vitro.
In September 2012, the Food and Drug Administration (FDA) approved the use of 11C-choline as an imaging agent for the detection of recurrent prostate cancer at the Mayo Clinic, USA (http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm319201.htm).
11C-choline PET is currently indicated clinically in patients who have raised prostate specific antigen (PSA) following treatment for prostate cancer and when other conventional imaging tests, e.g. CT, are negative. This FDA approval is based on a systematic review of four published studies [44–46]. In addition to soft tissue imaging in prostate cancer, 18F-fluorocholine (including 18F-fluoromethylcholine and 18F-fluoroethylcholine) may be applied for imaging of both lytic and early sclerotic bone metastases from prostate cancer, particularly when 18F-fluoride is negative [47].
Radiolabelled choline has been investigated as a pharmacodynamic biomarker in vitro in HT29 human colon carcinoma cells treated with geldanamycin. In these cells, geldanamycin inhibited extracellular signal-regulated kinase 1 and 2 phosphorylation. In addition, this was associated with a decrease in both methyl-14C-choline uptake and methyl-14C-phosphocholine production. On the basis of these results it was suggested that choline radiolabelled with a carbon-11 isotope could be utilised as a pharmacodynamic marker for the evaluation of geldanamycin analogues [48]. In addition, human HCT116 colon carcinoma xenografts were treated with PD0325901, a mitogenic extracellular kinase inhibitor for 10 days. A reduction in tumour radiotracer uptake was demonstrated and correlated to choline kinase α expression at cytostatic drug doses [49].
In clinical studies evaluating response with 11C-choline, breast cancer patients were examined to assess reproducibility of 11C-choline and the effect of trastuzumab. All tumour lesions were visualised by 11C-choline PET. In response to trastuzumab, 11C-choline uptake was decreased significantly within three lesions in two patients who also had a clinical response [50], providing information on the initial utility of 11C-choline in breast cancer and the effect of targeted treatments on choline uptake.
The utility of 11C-choline PET for pharmacodynamic/response assessment has also been reported in prostate cancer patients. Six patients with biopsy-proven prostate cancer were imaged at baseline with 11C-choline PET and were subsequently started on bicalutamide treatment (150 mg/day). A second choline scan was performed 4 months later and prior to commencing local radiotherapy. A decrease in SUVmax was noted in all patients treated with bicalutamide therapy, but no significant relationship was found between bicalutamide therapy duration and percentage change in 11C-choline uptake (Fig. 2) [51]. Although the diagnostic utility of 11C-choline PET was not proven, the study demonstrated its usefulness for monitoring response to anti-androgenic therapy [51], suggesting its potential application for testing novel anti-androgens, including abiraterone.

11C-choline in response to anti-androgenic therapy. Fused 11C-choline PET/CT images in two patients (a and b, respectively) studied before and after bicalutamide therapy (150 mg/day). Despite a substantial difference in 11C-choline SUVmax values before therapy (SUVmax = 4.5 and 11.9 for a and b, respectively), a considerable reduction in 11C-choline uptake is evident in both patients after treatment (SUVmax = 1.8 and 4.1 for a and b, respectively). Reproduced from Giovacchini et al. [51] with permission (color figure online)
Acetate
Acetate is a metabolic substrate for cholesterol and lipid synthesis. It is thought to be converted into acetyl-CoA by acetyl-CoA synthetase. In the anabolic pathway, acetyl-CoA can be used to synthesise cholesterol and fatty acids and therefore the cell membrane. In the catabolic pathway, acetate is oxidised by mitochondria in the tricarboxylic acid cycle to CO2 and H2O to produce energy [52]. Tumour cells overexpress fatty acid synthetase (FAS), thus converting most of the acetate to fatty acids, and subsequently incorporating them into intracellular membranes which, in turn, are important for tumour growth, metastasis, as well as being associated with poor prognosis [53]. FAS also protects cells against apoptosis and therefore has a tumour-promoting function [54].
Fatty acid synthetase is overexpressed in prostate cancer tumours and the pharmacological inhibition of the FAS pathway has been studied using 11C-acetate. In an in vitro study examining three different prostate tumour cell lines, cells were treated with C75 (an inhibitor of FAS) and 5-(tetradecycloxy)-2-furoic acid (TOFA, an inhibitor of acetyl-CoA carboxylase). A decrease in cellular accumulation of 11C-acetate was noted with these treatments. A positive correlation between 11C-acetate uptake in solid tumours and FAS expression, determined by immunohistochemistry, was observed in vivo [55].
A further in vivo study examined the capacity of radiolabelled acetate to predict FAS-targeted therapy outcome. 1-11C-acetate uptake corresponded to the level of FAS expression in different prostate cancer cell lines and tumour xenografts derived from these. Furthermore, 1-11C-acetate uptake predicted response to orlistat (a selective inhibitor of FAS) therapy; high FAS-expressing LNCap cells, which demonstrated the highest radiotracer uptake, showed the most pronounced decrease in treatment-induced cell viability [53].
As regards molecularly targeted drugs, only a clinical case report of the use of 11C-acetate PET has been published; in a patient with metastatic renal carcinoma, early functional inhibition was noted by 11C-acetate PET after just 14 days of therapy with sunitinib and found to correlate with partial and complete remission [56].
In prostate cancer patients with bone metastases, 11C-acetate was compared with 18F-FDG in the assessment of response to therapy (anti-androgen therapy or chemotherapy): 11C-acetate was found to detect quantitatively more bone metastases in these patients. Patients underwent pre- and post-treatment acetate scans and demonstrated a response to therapy that matched the decline in tumour:normal SUV as evaluated by 11C-acetate PET. This study highlights the potential utility of 11C-acetate PET for monitoring response in patients with bone metastases [57], but it is necessary to consider the biological heterogeneity of bone metastases and also to compare the effectiveness of this tracer with results obtained using 11C-choline and 18F-fluorocholine imaging in prostate cancer. Furthermore, it should also be compared with novel radiotracers specific for prostate biology, such as androgen receptor (AR) and prostate membrane-specific antigen (PMSA) [58], in the context of pharmacodynamics and response.
The advantages of 11C-acetate are that it can be used to visualise tumours of the urinary tract and can distinguish neoplasm from inflammation. Its limitations include its short half-life of 20 min and, therefore, the fact that not all institutions are able to produce it. In addition, with respect to novel targeted therapy agents, data on response to therapy are lacking and this aspect needs to be further examined.
Apoptosis
Apoptosis (organised, energy-dependent cell death) is a process essential for the maintenance of tissue homeostasis, through the elimination of unwanted cells during growth and differentiation. Evasion of apoptosis is classed as a hallmark of cancer [59], and cancer treatments including chemotherapy, radiotherapy as well as molecular targeted treatments induce tumour death through apoptosis [60]. Apoptosis is characterised, in terms of morphological changes, by cell shrinkage, membrane blebbing and compartmentalisation of intracellular contents into small membrane-bound apoptotic bodies. At the molecular level, the apoptosis pathway is activated by the death receptor (extrinsic) pathway or the mitochondria-directed (intrinsic) pathway, through the activation of effector caspases that execute apoptosis [61]. Another biochemical change of apoptosis is the rapid transport of phosphatidylserine residues from the inner to the outer surface of the plasma membrane. These biochemical changes can be exploited to image apoptosis non-invasively following cancer treatments.
The first imaging biomarker that was used to evaluate apoptosis clinically was 99mTc-annexin-V, detected by scintigraphy. This tracer binds with high affinity (sub nM) to phosphatidylserine groups that are exposed on the cell surface during the process of apoptosis. In an initial study of 15 patients with lung cancer, breast cancer and lymphoma treated with just one cycle of chemotherapy, increased radiolabelled annexin-V uptake was observed in seven patients and was associated with a favourable response on conventional imaging [62]. 99mTc-annexin-V scintigraphy was also used to determine response to radiotherapy in 11 patients with follicular lymphoma. A baseline scan before radiotherapy and a post-treatment scan 24 h after radiotherapy were obtained. In ten patients, an increase in 99mTc-annexin-V was noted following treatment, which correlated with the cytological assessment of apoptosis and the clinical outcome [63]. In another study of 33 patients, 99mTc-HYNIC-rh-annexin-V scintigraphy (TAVS) was used to predict response to therapy. Patients with lymphoma, leukaemia or head and neck cancer were scheduled for chemotherapy, radiotherapy or combined chemoradiotherapy depending on tumour type. Baseline and early post-treatment scans were conducted. A post-treatment increase in radiotracer uptake was associated with complete or partial tumour response, while no significant increase was noted in tumours that exhibited stable or progressive disease [64]. These studies document the effect of chemotherapy and radiotherapy on apoptosis and validate the use of annexin-V apoptosis imaging as a surrogate biomarker of therapy and response. A poor specificity for apoptosis over necrosis, slow clearance from non-target tissues and problems with radiolabelling have been reported as limitations of radiolabelled annexin-V imaging [65].
The ApoSense family of compounds is a novel class of small non-peptide molecules which have been developed to discriminate apoptotic from viable cells. In the early stages of the apoptotic process, these compounds selectively bind to membranes and accumulate in the cytoplasm of apoptotic cells [66]. A healthy volunteer study using the ApoSense radiotracer 18F-ML-10 has been successfully completed, with the radiotracer found to show a favourable dosimetry, biodistribution, stability and safety profile, with binding to apoptotic sites in the testes of the men observed [67]. Furthermore, in a trial to assess response of brain metastases to whole-brain radiotherapy, patients underwent 18F-ML-10-PET scans at baseline and after ten fractions of radiotherapy. Conventional MRI was performed 6–8 weeks after radiotherapy. A positive correlation was noted between early changes in 18F-ML-10 and late changes in tumour anatomical dimensions [68].
Imaging of the activated executioner caspase-3 is an attractive alternative strategy. Small-molecule PET radioligands based on the isatin-5-sulfonamides class of caspase-3 inhibitors have been developed to image the committal stage of programmed cell death. A library of compounds with the isatin-5-sulfonamide scaffold screened for activated caspase-3 inhibitory affinity led to the development of ICMT-11, with subnanomolar affinity for activated caspase-3, reduced lipophilicity and easy radiolabelling [69]. In the in vivo study of murine lymphoma xenografts, increased signal intensity of the radiotracer by PET imaging was noted following treatment with cyclophosphamide and was associated with increased apoptosis (Fig. 3) [69].

18F-ICMT-11 PET imaging analysis. 38C13 xenograft-bearing mice were treated with 100 mg/kg cyclophosphamide, CPA (n = 3) or vehicle (n = 3) for 24 h and subsequently subjected to 60 min dynamic 18F-ICMT-11 PET imaging. a 18F-ICMT-11 PET images of two representative 38C13 xenograft-bearing mice treated with CPA or vehicle. White arrowheads indicate the tumour. b Tumour and blood were removed after the scan and analysed for 18F-ICMT-11 tissue uptake. c The tumour time versus radioactivity curve (TAC). d Semi-quantitative imaging variables extracted from the TAC. Data are mean ± SEM. Reproduced from Nguyen et al. [69] with permission (color figure online)
A further study investigated temporal and spatial assessment of apoptosis by 18F-ICMT-11. The pharmacodynamics of a single dose of the alkylating agent cyclophosphamide, or of the mechanism-based small-molecule SMAC mimetic birinapant, was examined. With cyclophosphamide and birinapant treatment, 18F-ICMT-11 uptake peaked at 24 and 6 h, respectively. Voxel-based spatiotemporal analysis of tumour suggested that discrete areas of caspase-3 activation could be detected by 18F-ICMT-11 and uptake correlated with caspase-3 activation measured in vivo [70]. A first human study in healthy volunteers has been completed, showing favourable dosimetry and no adverse effects and the radiotracer is currently progressing to patient trials [71].
Hypoxia
Hypoxia (deficiency in tissue oxygenation) in solid tumours is a characteristic associated with aggressive tumour types; it contributes to radiotherapy and chemotherapy resistance and increased metastatic activity, and is therefore a surrogate of poor patient prognosis [72]. The effects of hypoxia on the behaviour of gene expression lead not only to suppression of apoptosis and promotion of angiogenesis, invasiveness and metastasis, but also to genomic instability [73].
The development of PET tracers for the non-invasive assessment of tumour hypoxia led to the use of hypoxia imaging to predict response to treatment and to direct cytotoxic therapies and radiotherapy adaptations [74]. Initially, radiolabelled 2-nitroimidazole compounds were used for imaging hypoxia by PET [75]. Depending on the level of hypoxia in the cells, these compounds are reduced to reactive intermediate metabolites. The metabolites bind intracellular proteins and therefore accumulate in hypoxic cells. The best-studied radiotracer in this group is 18F-fluoromisonidazole (FMISO) [76, 77]. FMISO is sufficiently lipophilic, readily crossing the cellular membrane. Although the radiotracer has been utilised in several tumour types to image hypoxia, its drawbacks include poor tissue uptake and slow cellular washout. Due to it slow clearance, the contrast between hypoxic and normal tumours is low, limiting response assessment with this tracer.
Other nitroimidazole radiotracers were subsequently developed, including 18F-fluoroetanidazole (18F-FEZA) [78], 18F-fluoroazomycin-arabinofuranoside (18F-FAZA) [79, 80], 3-[18F]fluoro-2-(4-((2-nitro-1H-imidazol-1-yl)methyl)-1H-1,2,3-triazol-1-yl)propan-1-ol (18F-HX4) [81, 82] and 18F-fluoroerythronitroimidazole (18F-FETNIM) [83].
18F-FMISO has been used as a predictive or prognostic indicator of response in a number of clinical studies with mixed outcomes. In a study of 28 patients with head and neck cancer, 18F-FMISO PET was used in the assessment of those undergoing chemoradiotherapy. Patients underwent one 18F-FDG PET and two 18F-FMISO baseline scans and a mid-treatment 18F-FMISO PET scan. Despite detection of hypoxia on 18F-FMISO PET scans in 18 of 20 patients, the patients achieved excellent loco-regional control of the cancer with treatment. Two patients had persistent hypoxia detected on the mid-treatment 18F-FMISO PET scan, but this did not translate into poor outcome. In one patient who experienced loco-regional failure, no evidence of residual hypoxia was detected on the mid-treatment 18F-FMISO scan. This study therefore suggests that absence or presence of hypoxia as determined by mid-therapy 18F-FMISO PET scan cannot be related to clinical outcomes [84].
This conclusion was not supported by another study, in which 18F-FMISO was used as a predictor of radiotherapy outcome in head and neck cancer patients. Thirty lesions from 17 patients were evaluated. SUVmax and tumour-to-muscle ratios were measured as hypoxia indicators. Patients with high uptake of 18F-FMISO exhibited poor local control rates compared with patients showing low uptake of 18F-FMISO. In addition, disease-specific survival was lower in patients with high 18F-FMISO, as defined by SUVmax [85]. This study, however, used baseline hypoxia values only, whereas the previous study [84] also examined mid-treatment hypoxia levels to determine treatment outcomes. Standardisation of image acquisition times, as well as analysis techniques will be required to further validate 18F-FMISO PET detection of hypoxia.
The effect of sunitinib on hypoxia in metastatic renal cancer was assessed with 18F-FMISO. Fifty-three patients underwent 18F-FMISO scans at baseline and 1 month after treatment with sunitinib. Sunitinib was noted to decrease hypoxia in initially hypoxic metastases. In addition, tumour hypoxia noted on the baseline scans was associated with a shorter progression-free survival, but change in tumour hypoxia (i.e. in the related pharmacodynamic readout) was not associated with progression-free or overall survival [86].
18F-FMISO has been utilised in breast cancer to predict primary endocrine (letrozole) resistance. A total of 45 lesions were evaluated in 20 patients. Patients underwent both 18F-FDG and 18F-FMISO PET scans at baseline and after endocrine therapy. The correlation between baseline 18F-FDG and clinical outcome was not significant. However, baseline 18F-FMISO uptake showed a strong positive correlation with clinical outcome and allowed the prediction of 88 % of the cases of progressive disease. A poor correlation was noted between 18F-FMISO and hypoxia-inducible factor 1α [87]. Interestingly, this study and the sunitinib study in renal cancer [86] support the validity of baseline hypoxia assessment as a tool for predicting treatment outcomes.
Another interesting radiotracer in hypoxia imaging is based on a metal complex of radioactive copper with tracers having a bis (thiosemicarbazone) core. Bis (thiosemicarbazone) ligands react with copper and form low molecular weight stable and neutral complexes capable of being taken up by cells. Cu(II)-diacetyl-bis(N4-methylthiosemicarbazone) (Cu-ATSM) is highly lipophilic and therefore membrane permeable. In hypoxic cells, Cu-ATSM undergoes reduction and is converted to [Cu(I)-ATSM]- becoming entrapped in hypoxic cells due to its negative charge [88].
In a small pilot study of 17 patients with rectal cancer undergoing chemoradiotherapy, 60Cu-ATSM PET was used to image primary tumour prior to therapy and activity was measured semi-quantitatively by tumour-to-muscle activity ratio. A median tumour-to-muscle activity ratio of 2.6 differentiated between good and bad prognosis groups. Progression and overall survival were worse with hypoxic tumours (tumour-to-muscle activity ratio >2.6) than with non-hypoxic tumours. In addition, tumour-to-muscle activity ratio for tumours that were downstaged was 2.2 vs. 3.3 for non-downstaged tumours. This small study therefore suggests that baseline 60Cu-ATSM PET can predict response to therapy and survival in this tumour type [89]. The drawbacks of 64Cu-ATSM imaging, however, include high lipophilicity and high liver uptake; various chemical modifications are being studied to reduce this lipophilicity.
Angiogenesis
Angiogenesis is a process of new blood vessel formation from existing vasculature and is a hallmark of cancer survival. It promotes tumour growth and survival by allowing an adequate blood supply of oxygen and nutrients and it also aids metastasis [90]. The regulators of angiogenesis include growth factors such as vascular endothelial growth factor (VEGF) and its receptors (VEGFR), integrins and matrix metalloproteinases. In the clinical setting, the VEGF pathway is inhibited by monoclonal antibodies to VEGF, such as bevacizumab, or by small-molecule VEGF tyrosine kinase inhibitors such as pazopanib.
Targets for angiogenesis radiotracers have included VEGF, VEGFR, antibody drugs and αvβ3 integrin. The most widely studied target is αvβ3 integrin, which is expressed on neoangiogenic vessels. The integrin αvβ3 binds to arginine–glycine–aspartic (RGD)-containing components of the extracellular matrix. Therefore, RGD-based labelled peptides have been developed; these show increased affinity and selectivity for integrin αvβ3 and have been utilised for PET imaging. Clinical studies of tracers that bind to αvβ3 integrin have included 18F-galacto-RGD and fluciclatide (18F-AH111585), while only fluciclatide has been used in in vivo pharmacodynamic assessments. In Calu-6 tumours treated with a VEGF tyrosine kinase inhibitor, ZD4190, a decrease of 32 % was noted in fluciclatide uptake [91]. Furthermore, U87-MG human glioblastoma xenografts were assessed with 18F-fluciclatide following treatment with antiangiogenic multi-kinase inhibitor sunitinib. Over a 2-week dosing regimen, a decrease of 18F-fluciclatide uptake was noted as early as 2 days after the start of the regimen in treated tumours compared with controls. This occurred prior to any tumour volume changes [92].
Dynamic 15O-labelled H2O PET is considered the gold standard for imaging of tumour blood flow in patients. In a phase I pharmacodynamic study of a vascular combretastatin A4 phosphate (CA4P) in patients with solid tumours, tissue perfusion was measured using 15O–H2O. Reduction in tumour perfusion by approximately 50 % was noted within 30 min of CA4P infusion. Therefore, rapid changes in tumour vasculature could be assessed by PET perfusion imaging [93]. Two clinical studies have evaluated the efficacy of anti-angiogenic therapy with 15O–H2O. In a PET study of 25 patients, endostatin decreased 15O–H2O-based tumour blood flow by 20 % [94]. In a phase II study of six patients treated with razoxane, no change in tumour perfusion was noted with 15O–H2O PET (true negative) [95].
Receptor imaging
In recent years, there has been a significant development of novel therapies and combination therapies against growth factor receptors. Treatments include tamoxifen and letrozole for oestrogen receptor-expressing breast tumours, and trastuzumab for HER2-expressing tumours. Erlotinib, gefitinib and cetuximab have been approved for treatment of lung and colorectal cancers respectively and octreotide is used in somatostatin receptor-positive cancers. The relatively low response rates for these treatments on conventional imaging have led to interest in the use of imaging biomarker strategies in patient management. In keeping with this concept, PET tracers have been developed to measure target expression of growth factor receptors as well as to evaluate response to therapy.
Oestrogen receptor
The oestrogen receptor (ER) is expressed in over two-thirds of breast cancers and is important in determining therapy as well as prognosis. ER status in tumours is currently evaluated by immunohistochemistry and ER PET imaging provides a non-invasive means of assessing tumours and potentially inaccessible metastatic lesions. The ER PET tracer 18F-fluoro-oestradiol (18F-FES) showed good correlation with expression of ER in tumours [96]. In response to endocrine therapy (aromatase inhibitors for 6 months) in previously heavily treated metastatic breast cancer patients, 18F-FES PET positivity correlated with treatment response [97]. Patients with no uptake of 18F-FES did not exhibit response to treatment. In a quantitative assessment, none of the 15 patients with an initial SUV <1.5 responded to therapy, while 11 of 32 patients with an SUV of 1.5 or more were seen to show a response to hormonal therapy (Fig. 4) [97].

18F-fluoro-oestradiol in response assessment to endocrine therapy. Pre-treatment 18F-fluoro-oestradiol (FES; left) and 18F-fluorodeoxyglucose (FDG; middle) scans and follow-up FDG post-therapy (right). Dashed arrows show normal liver FES uptake. (a; top) Bone metastasis with robust FES and FDG uptake, response at 3 months. (b; bottom) Bone metastasis (solid arrow) with FDG, but no FES uptake; progressive disease at 6 months. Rx, treatment. Reproduced from Linden et al. [97] with permission
In a further study conducted to assess the pharmacodynamic effect of various types of endocrine therapy on 18F-FES uptake, 30 patients with metastatic breast cancer underwent 18F-FES PET prior to and following endocrine therapy with tamoxifen, fulvestrant or aromatase inhibitors. The decrease in 18F-FES uptake (54 %) was more marked with ER inhibitors, tamoxifen and fulvestrant than with aromatase inhibitors (a 15 % decline). In addition, the rate of SUV decrease was greater with tamoxifen than with fulvestrant treatment and found to reflect the response seen clinically (lower efficacy of fulvestrant compared with tamoxifen). However, this study was limited by the confounding factor of prior treatment and by low cohort numbers [98].
Although the imaging of ER and response to treatment has been demonstrated in these small trials, larger trials are needed to validate 18F-FES as a pharmacodynamic marker of endocrine therapy.
FDHT
The AR in prostate cancer can be likened to the ER in breast cancer. Androgen deprivation therapy in prostate cancer has response rates of approximately 90 % [99]. Therefore the AR is a useful target for PET biomarker imaging. In a first-ever feasibility study of 18F-fluoro-5α-dihyrotestosterone (18F-FDHT) in metastatic prostate cancer patients, 97 % of metastatic lesions showed uptake of 18F-FDG, while 78 % showed uptake of 18F-FDHT. In addition, treatment with testosterone in two patients was associated with diminished uptake of 18F-FDHT, suggesting that this radiotracer could be used to determine the pharmacodynamics of drug treatment [100]. In a further study, assessing this radiotracer in metastatic prostate cancer patients, 18F-FDHT was positive in 12 of 19 patients [101]. Patients who had a positive scan and were treated with flutamide showed a decrease in uptake as shown by the SUV (Fig. 5). In addition, patients with positive scans had higher PSA levels than patients with negative scans [101].

18F-FDHT in response assessment to anti-androgen therapy. Anterior (left upper) and posterior (right upper) FDHT PET images at baseline show increased FDHT uptake in several lymph nodes in the supraclavicular, prevascular, paratracheal, precarinal and several retroperitoneal regions (arrows). Anterior (left lower) and posterior (right lower) FDHT PET images performed after flutamide show almost complete blockade of FDHT uptake in the previously seen lymph nodes. Reproduced from Dehdashti et al. [101] with permission
In a phase I/II pharmacodynamic study of MDV3100 (a novel AR antagonist), 18F-FDHT PET was used to assess AR binding. A scan performed 4 weeks post-treatment revealed a decrease versus baseline, in 18F-FDHT uptake following all doses of treatment. Although FDHT response occurred in all patients, correlation with FDG PET revealed a change in SUVmax of 25 % or more in only about 45 % of patients [102]. An important lesson here is that changes in receptor occupancy may precede downstream changes in metabolism.
Current data are not sufficient to determine the true sensitivity of 18F-FDHT in comparison with conventional imaging. In addition, it would be useful to correlate 18F-FDHT uptake with AR expression in tumour tissue. Other radiotracers for imaging the AR pathway are under development. These include 89Zr-J591 [103], designed to quantify changes in PSA expression, 89Zr-5A10, to target free prostatic antigen [104], and 89Zr-transferrin, to measure aberrant MYC signalling and quantify changes in the expression of transferrin receptor [105].
HER2
The ERBB2 (HER2) receptor is a member of the c-erB family of growth receptors and its amplification or overexpression occurs in several tumours and in about 25–30 % of breast cancers [106]. Expression of HER2 is currently determined in tumour samples by immunohistochemical assays and fluorescence in situ hybridisation techniques. Trastuzumab is a humanised monoclonal antibody against HER2 and shows an improved response when given in combination with chemotherapy, as well as improved overall survival rates in metastatic breast cancer patients [2]. Trastuzumab-based imaging probes have been developed to image HER2. In a study of HER2 imaging, used to assess response to an Hsp90 inhibitor, HER2 imaging was achieved by labelling an F(ab′)2 fragment of the HER2 antibody trastuzumab with 68Ga. Hsp90 inhibitors degrade HER2. In BT474 human breast cancer xenografts, HER2 expression determined by imaging, following treatment with 17-allylaminogeldanamycin (17-AAG), showed a decrease of 80 % after 24 h of treatment, before rising to 50 % of its initial expression and thereafter remaining stable for the next 5 days; the control groups instead showed an increase of 20 % in HER2 expression over the same time period [107]. In addition, HER2 expression was correlated with immunoblotting. This study demonstrates the ability of quantitative imaging to assess the pharmacodynamic effect of 17-AAG [107]. Similar data have been replicated with affibodies targeting the HER2 receptor [108].
In a subsequent in vivo study, HER2-overexpressing SKBR3 ovarian tumour cell line xenografts were treated with the Hsp90 inhibitor NVP-AUY922 and the pharmacodynamics determined with 89Zr-trastuzumab. A reduction of 41 % was observed in tumour uptake determined by PET quantification at 144 h post-tracer injection after treatment with NVP-AUY922. In addition, PET results were confirmed by HER2 immunohistochemical staining and ex vivo biodistribution of 89Zr-trastuzumab [109].
These in vivo studies illustrate the potential of radiolabelled antibodies directed against targeted receptors; clinical trials to validate the pharmacodynamics with imaging are awaited. However, clinical trials of HER2 imaging using macromolecules such as monoclonal antibodies labelled with long-lived radioisotopes including 111In, 124I and 89Zr (to match their slow systemic clearance) have resulted in high radiation exposure for patients. A recent clinical study of a shorter half-life radioisotope, 64Cu-DOTA-trastuzumab, in primary and metastatic HER2 +ve breast cancer patients revealed radiation exposure equivalent to that occurring with conventional 18F-FDG PET. Forty-eight hours was identified as an optimal imaging time point. In addition, both primary and metastatic lesions demonstrated uptake of the radiotracer, although non-specific uptake was noted in the liver, heart and blood. Correlation with immunohistochemistry or fluorescence in situ hybridisation (FISH) would be required to further validate this tracer. This study demonstrates the challenges in identifying the optimal radiotracer for imaging a biological target with a monoclonal antibody, which also involves achieving optimal dosimetry, ensuring sensitive identification of the molecular target in primary and metastatic lesions, and ascertaining the right contrast balance between optimal target visualisation and reduction of non-specific uptake [110].
EGFR
The role of EGFR in colorectal and lung cancer has been highlighted in recent years. EGFR is important in the regulation of cell metabolism, and aberrant EGFR expression or activation contributes to tumour development and progression. Monoclonal antibodies (cetuximab, panitumumab) and small-molecule inhibitors of the tyrosine kinase domain (gefitinib, erlotinib) have been developed to target the EGFR receptor.
A number of imaging strategies have been evaluated to target the external binding domain of the receptor. Anti-EGFR antibodies have been radiolabelled with 64Cu, 111In, 88Y and 125I [111]. Cetuximab conjugates such as DTPA–cetuximab [112] and DTPA–PEG cetuximab have been further developed to image high EGFR-expressing tumours [113].
Erlotinib has been labelled with 11C and evaluated in vivo. In the high EGFR-expressing HC827 cell line, which shows increased sensitivity to erlotinib, high 11C-erlotinib uptake was also observed [114]. In a clinical study examining 11C-labelled 4-N-(3-bromoanilino)-6,7dimethoxyquinazoline (11C-PD153035), an imaging marker for EGFR, patients were imaged at baseline and various time points after the start of erlotinib treatment. Baseline SUV correlated strongly with progression-free and overall survival; patients with a higher SUVmax survived more than twice as long than patients with a lower SUVmax. This small study was useful for identifying patients who were likely to respond to EGFR therapy, but was not able to predict treatment response [115].
Furthermore, small-molecule affibody probes that bind to EGFR have been developed. These have been shown to exhibit rapid uptake, high tumour uptake and high kidney uptake [116]. PET imaging of EGFR currently employs labelled monoclonal antibodies and small-molecule agents. However, these have not demonstrated efficient uptake; in addition, more studies are needed to assess the pharmacodynamics of these targeted agents.
Imaging agents for EGFR and HER2 have included radiolabelled monoclonal antibodies, small molecules, affibodies and nanobodies [117]. In comparison with monoclonal antibodies, the advantages of nanobodies and affibodies include better in vivo clearance as well as their greater amenability to radiolabelling with short-acting radionuclides such as 18F and 99mTc. In addition, nanobody and affibody imaging, due to the small size of the constructs, provide a better readout of tumour protein expression levels than antibody imaging agents [117]. These new technologies in the field of biologically targeted imaging agents certainly need further research.
Conclusion
Conventional imaging of anatomical changes in response to treatment does not provide sufficient information about the tumour characteristics of biology and molecular function. In the era of targeted therapy, it is important to quantify the pharmacodynamics and therapy response in treatments that induce molecular biochemical change, but show limited early lesion size change on conventional imaging.
The use of PET markers of proliferation, hypoxia, angiogenesis and metabolism in the assessment and prediction of response to therapy has advanced significantly. However, 18F-FDG is still the primary PET imaging agent in clinical use for the diagnosis and assessment of response to therapy in most tumours. Radiolabelled choline has only recently been approved for clinical use in a limited number of centres. New radiotracers currently under development, including 18F-D4-choline and 18F-ICMT-11, could potentially show clinical utility and improve both diagnostic and response assessment.
Molecular imaging in combination with anatomical imaging in early clinical trials could provide biomarkers with the potential to inform about biological processes and early responses as well as identify patients who are more likely to benefit from treatments. However, the ideal time for pharmacodynamic assessment varies according to the biological processes imaged. In addition, with increasing use of targeted therapies, response to therapy occurs before cytoreduction (end point for conventional imaging) and, it is important to note, could be detected as early as 24–48 h post-therapy, e.g. in apoptosis imaging. The advantages of imaging biomarkers are that they are “non-invasive” and allow longitudinal measurements over a period of time. However, current imaging biomarkers are not considered surrogate end points. In spite of significant research using radiolabelled imaging biomarkers, most of these probes are not routinely used in clinical practice. The continued development of imaging biomarkers will depend on the achievement of technical and biological validation. According to the European Society of Radiology “White paper on imaging biomarkers” (2010), the development of imaging biomarkers requires standardisation of acquisition and post-processing parameters, so as to ensure reproducibility across different centres. In addition, the validation process must include assessment of sensitivity, specificity and reproducibility, as well as correlation with biological effect and clinical end points. These technical and biological validation steps will allow these methods to be used in the clinic in the confidence that they report specific biological processes, especially in the case of non-response. Finally, academia–industry collaborations will be important in promoting further applications in the development and assessment of novel drugs and in the pursuit of the requirements necessary for imaging biomarkers to be successfully launched for clinical use.
References
Biomarkers Definitions Working Group (2001) Biomarkers and surrogate endpoints: preferred definitions and conceptual framework (2001). Clin Pharmacol Ther 69:89–95. doi:10.1067/mcp.2001.113989
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344:783–792. doi:10.1056/NEJM200103153441101
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350:2335–2342. doi:10.1056/NEJMoa032691
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Freeman S, Schwartz B, Shan M, Simantov R, Bukowski RM (2007) Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 356:125–134. doi:10.1056/NEJMoa060655
van Oosterom AT, Judson IR, Verweij J, Stroobants S, Dumez H, di Donato Paola E, Sciot R, Van Glabbeke M, Dimitrijevic S, Nielsen OS (2002) Update of phase I study of imatinib (STI571) in advanced soft tissue sarcomas and gastrointestinal stromal tumors: a report of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer 38(Suppl 5):S83–S87
Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M, Marrano P, da Cunha Santos G, Lagarde A, Richardson F, Seymour L, Whitehead M, Ding K, Pater J, Shepherd FA (2005) Erlotinib in lung cancer, molecular and clinical predictors of outcome. N Engl J Med 353:133–144. doi:10.1056/NEJMoa050736
WHO Handbook for Reporting Results of Cancer-Treatment, WHO (1979) Public Health Rev 8:411–412
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247. doi:10.1016/j.ejca.2008.10.026
Adminsitration UFaD (2007) Guidance for industry: clinical trial endpoints for the approval of cancer drugs and biologics. www.fda.gov/downloads/Drugs/.../Guidances/ucm071590.pdf. Accessed July 12 2013
Workman P, Aboagye EO, Chung YL, Griffiths JR, Hart R, Leach MO, Maxwell RJ, McSheehy PM, Price PM, Zweit J (2006) Minimally invasive pharmacokinetic and pharmacodynamic technologies in hypothesis-testing clinical trials of innovative therapies. J Natl Cancer Inst 98:580–598. doi:10.1093/jnci/djj162
Merrall NW, Plevin R, Gould GW (1993) Growth factors, mitogens, oncogenes and the regulation of glucose transport. Cell Signal 5:667–675
Juweid ME, Cheson BD (2006) Positron-emission tomography and assessment of cancer therapy. The N Engl J Med 354:496–507. doi:10.1056/NEJMra050276
Romer W, Hanauske AR, Ziegler S, Thodtmann R, Weber W, Fuchs C, Enne W, Herz M, Nerl C, Garbrecht M, Schwaiger M (1998) Positron emission tomography in non-Hodgkin’s lymphoma: assessment of chemotherapy with fluorodeoxyglucose. Blood 91:4464–4471
Witney TH, Kettunen MI, Day SE, Hu DE, Neves AA, Gallagher FA, Fulton SM, Brindle KM (2009) A comparison between radiolabeled fluorodeoxyglucose uptake and hyperpolarized (13)C-labeled pyruvate utilization as methods for detecting tumor response to treatment. Neoplasia 11:574–582
Contractor KB, Aboagye EO (2009) Monitoring predominantly cytostatic treatment response with 18F-FDG PET. J Nucl Med 50(Suppl 1):97S–105S. doi:10.2967/jnumed.108.05727350/Suppl_1/97S
Upadhyay M, Samal J, Kandpal M, Singh OV, Vivekanandan P (2013) The Warburg effect: insights from the past decade. Pharmacol Ther 137:318–330. doi:10.1016/j.pharmthera.2012.11.003
Takahashi N, Inoue T, Lee J, Yamaguchi T, Shizukuishi K (2007) The roles of PET and PET/CT in the diagnosis and management of prostate cancer. Oncology 72:226–233. doi:10.1159/000112946000112946
Khan MA, Combs CS, Brunt EM, Lowe VJ, Wolverson MK, Solomon H, Collins BT, Di Bisceglie AM (2000) Positron emission tomography scanning in the evaluation of hepatocellular carcinoma. J Hepatol 32:792–797. doi:S0168827800802482
Phelps ME, Mazziotta JC (1985) Positron emission tomography: human brain function and biochemistry. Science 228:799–809
Zhuang H, Pourdehnad M, Lambright ES, Yamamoto AJ, Lanuti M, Li P, Mozley PD, Rossman MD, Albelda SM, Alavi A (2001) Dual time point 18F-FDG PET imaging for differentiating malignant from inflammatory processes. J Nucl Med 42:1412–1417
Strauss LG (1996) Fluorine-18 deoxyglucose and false-positive results: a major problem in the diagnostics of oncological patients. Eur J Nucl Med 23:1409–1415
Wells P, Gunn RN, Steel C, Ranicar AS, Brady F, Osman S, Jones T, Price P (2005) 2-[11C]thymidine positron emission tomography reproducibility in humans. Clin Cancer Res 11:4341–4347. doi:10.1158/1078-0432.CCR-04-1818
Chalkidou A, Landau DB, Odell EW, Cornelius VR, O’Doherty MJ, Marsden PK (2012) Correlation between Ki-67 immunohistochemistry and 18F-fluorothymidine uptake in patients with cancer: a systematic review and meta-analysis. Eur J Cancer 48:3499–3513. doi:10.1016/j.ejca.2012.05.001
Kenny L, Coombes RC, Vigushin DM, Al-Nahhas A, Shousha S, Aboagye EO (2007) Imaging early changes in proliferation at 1 week post chemotherapy: a pilot study in breast cancer patients with 3′-deoxy-3′-[18F]fluorothymidine positron emission tomography. Eur J Nucl Med Mol Imaging 34:1339–1347. doi:10.1007/s00259-007-0379-4
Wieder HA, Geinitz H, Rosenberg R, Lordick F, Becker K, Stahl A, Rummeny E, Siewert JR, Schwaiger M, Stollfuss J (2007) PET imaging with [18F]3′-deoxy-3′-fluorothymidine for prediction of response to neoadjuvant treatment in patients with rectal cancer. Eur J Nucl Med Mol Imaging 34:878–883. doi:10.1007/s00259-006-0292-2
Dehdashti F, Grigsby PW, Myerson RJ, Nalbantoglu I, Ma C, Siegel BA (2013) Positron emission tomography with [(18)F]-3′-deoxy-3′fluorothymidine (FLT) as a predictor of outcome in patients with locally advanced resectable rectal cancer: a pilot study. Mol Imaging Biol 15:106–113. doi:10.1007/s11307-012-0566-y
Sohn HJ, Yang YJ, Ryu JS, Oh SJ, Im KC, Moon DH, Lee DH, Suh C, Lee JS, Kim SW (2008) [18F]Fluorothymidine positron emission tomography before and 7 days after gefitinib treatment predicts response in patients with advanced adenocarcinoma of the lung. Clin Cancer Res 14:7423–7429. doi:10.1158/1078-0432.CCR-08-031214/22/7423
Zander T, Scheffler M, Nogova L, Kobe C, Engel-Riedel W, Hellmich M, Papachristou I, Toepelt K, Draube A, Heukamp L, Buettner R, Ko YD, Ullrich RT, Smit E, Boellaard R, Lammertsma AA, Hallek M, Jacobs AH, Schlesinger A, Schulte K, Querings S, Stoelben E, Neumaier B, Thomas RK, Dietlein M, Wolf J (2011) Early prediction of nonprogression in advanced non-small-cell lung cancer treated with erlotinib by using [(18)F]fluorodeoxyglucose and [(18)F]fluorothymidine positron emission tomography. J Clin Oncol 29:1701–1708. doi:10.1200/JCO.2010.32.4939
Schwarzenberg J, Czernin J, Cloughesy TF, Ellingson BM, Pope WB, Geist C, Dahlbom M, Silverman DH, Satyamurthy N, Phelps ME, Chen W (2012) 3′-deoxy-3′-18F-fluorothymidine PET and MRI for early survival predictions in patients with recurrent malignant glioma treated with bevacizumab. J Nucl Med 53:29–36. doi:10.2967/jnumed.111.092387
Gray KR, Contractor KB, Kenny LM, Al-Nahhas A, Shousha S, Stebbing J, Wasan HS, Coombes RC, Aboagye EO, Turkheimer FE, Rosso L (2010) Kinetic filtering of [(18)F]Fluorothymidine in positron emission tomography studies. Phys Med Biol 55:695–709. doi:10.1088/0031-9155/55/3/010
Contractor K, Challapalli A, Tomasi G, Rosso L, Wasan H, Stebbing J, Kenny L, Mangar S, Riddle P, Palmieri C, Al-Nahhas A, Sharma R, Turkheimer F, Coombes RC, Aboagye E (2012) Imaging of cellular proliferation in liver metastasis by [18F]fluorothymidine positron emission tomography: effect of therapy. Phys Med Biol 57:3419–3433. doi:10.1088/0031-9155/57/11/3419
Perumal M, Pillai RG, Barthel H, Leyton J, Latigo JR, Forster M, Mitchell F, Jackman AL, Aboagye EO (2006) Redistribution of nucleoside transporters to the cell membrane provides a novel approach for imaging thymidylate synthase inhibition by positron emission tomography. Cancer Res 66:8558–8564. doi:10.1158/0008-5472.CAN-06-0898
Pillai RG, Forster M, Perumal M, Mitchell F, Leyton J, Aibgirhio FI, Golovko O, Jackman AL, Aboagye EO (2008) Imaging pharmacodynamics of the alpha-folate receptor-targeted thymidylate synthase inhibitor BGC 945. Cancer Res 68:3827–3834. doi:10.1158/0008-5472.CAN-08-013568/10/3827
Kenny LM, Contractor KB, Stebbing J, Al-Nahhas A, Palmieri C, Shousha S, Coombes RC (1078) Aboagye EO (2009) Altered tissue 3′-deoxy-3′-[18F]fluorothymidine pharmacokinetics in human breast cancer following capecitabine treatment detected by positron emission tomography. Clin Cancer Res 15:6649–6657. doi:10.1158/1078-0432.CCR-09-1213
Ramírez de Molina A, Rodríguez-González A, Gutiérrez R, Martínez-Piñeiro L, Sánchez J, Bonilla F, Rosell R, Lacal J (2002) Overexpression of choline kinase is a frequent feature in human tumor-derived cell lines and in lung, prostate, and colorectal human cancers. Biochem Biphys Res Commun 296:580–583
Leyton J, Smith G, Zhao Y, Perumal M, Nguyen QD, Robins E, Arstad E, Aboagye EO (2009) [18F]fluoromethyl-[1,2-2H4]-choline: a novel radiotracer for imaging choline metabolism in tumors by positron emission tomography. Cancer Res 69:7721–7728. doi:10.1158/0008-5472.CAN-09-14190008-5472.CAN-09-1419
Glunde K, Bhujwalla ZM (2011) Metabolic tumor imaging using magnetic resonance spectroscopy. Semin Oncol 38:26–41. doi:10.1053/j.seminoncol.2010.11.001
Hara T, Kosaka N, Shinoura N, Kondo T (1997) PET imaging of brain tumor with [methyl-11C]choline. J Nucl Med 38:842–847
Jadvar H (2011) Prostate cancer: PET with 18F-FDG, 18F- or 11C-acetate, and 18F- or 11C-choline. J Nucl Med 52:81–89. doi:10.2967/jnumed.110.077941
Roivainen A, Forsback S, Grönroos T, Lehikoinen P, Kähkönen M, Sutinen E, Minn H (2000) Blood metabolism of [methyl-11C]choline; implications for in vivo imaging with positron emission tomography. Eur J Nucl Med 27:25–32
Smith G, Zhao Y, Leyton J, Shan B, Nguyen QD, Perumal M, Turton D, Arstad E, Luthra SK, Robins EG, Aboagye EO (2011) Radiosynthesis and pre-clinical evaluation of [(18)F]fluoro-[1,2-(2)H(4)]choline. Nucl Med Biol 38:39–51. doi:10.1016/j.nucmedbio.2010.06.012
Witney TH, Alam IS, Turton DR, Smith G, Carroll L, Brickute D, Twyman FJ, Nguyen QD, Tomasi G, Awais RO (1078) Aboagye EO (2012) Evaluation of deuterated 18F- and 11C-labeled choline analogs for cancer detection by positron emission tomography. Clin Cancer Res 18:1063–1072. doi:10.1158/1078-0432.CCR-11-2462-0432.CCR-11-2462
Scattoni V, Picchio M, Suardi N, Messa C, Freschi M, Roscigno M, Da Pozzo L, Bocciardi A, Rigatti P, Fazio F (2007) Detection of lymph-node metastases with integrated [11C]choline PET/CT in patients with PSA failure after radical retropubic prostatectomy: results confirmed by open pelvic-retroperitoneal lymphadenectomy. Eur Urol 52:423–429. doi:10.1016/j.eururo.2007.03.032
Rinnab L, Mottaghy FM, Simon J, Volkmer BG, de Petriconi R, Hautmann RE, Wittbrodt M, Egghart G, Moeller P, Blumstein N, Reske S, Kuefer R (2008) [11C]Choline PET/CT for targeted salvage lymph node dissection in patients with biochemical recurrence after primary curative therapy for prostate cancer. Preliminary results of a prospective study. Urol Int 81:191–197. doi:10.1159/000144059
Mitchell CR, Lowe VJ, Rangel LJ, Hung JC, Kwon ED, Karnes RJ (2013) Operational characteristics of (11)c-choline positron emission tomography/computerized tomography for prostate cancer with biochemical recurrence after initial treatment. J Urol 189:1308–1313. doi:10.1016/j.juro.2012.10.069
Beheshti M, Vali R, Langsteger W (2007) [18F]fluorocholine PET/CT in the assessment of bone metastases in prostate cancer. Eur J Nucl Med Mol Imaging 34:1316–1317. doi:10.1007/s00259-007-0401-x
Liu D, Hutchinson OC, Osman S, Price P, Workman P, Aboagye EO (2002) Use of radiolabelled choline as a pharmacodynamic marker for the signal transduction inhibitor geldanamycin. Br J Cancer 87:783–789. doi:10.1038/sj.bjc.6600558
Leyton J, Smith G, Lees M, Perumal M, Nguyen QD, Aigbirhio FI, Golovko O, He Q, Workman P, Aboagye EO (2008) Noninvasive imaging of cell proliferation following mitogenic extracellular kinase inhibition by PD0325901. Mol Cancer Ther 7:3112–3121. doi:10.1158/1535-7163.MCT-08-02647/9/3112
Kenny LM, Contractor KB, Hinz R, Stebbing J, Palmieri C, Jiang J, Shousha S, Al-Nahhas A, Coombes RC (1078) Aboagye EO (2010) Reproducibility of [11C]choline-positron emission tomography and effect of trastuzumab. Clin Cancer Res 16:4236–4245. doi:10.1158/1078-0432.CCR-10-0468-0432.CCR-10-0468
Giovacchini G, Picchio M, Coradeschi E, Scattoni V, Bettinardi V, Cozzarini C, Freschi M, Fazio F, Messa C (2008) [(11)C]choline uptake with PET/CT for the initial diagnosis of prostate cancer: relation to PSA levels, tumour stage and anti-androgenic therapy. Eur J Nucl Med Mol Imaging 35:1065–1073. doi:10.1007/s00259-008-0716-2
Swinnen JV, Van Veldhoven PP, Timmermans L, De Schrijver E, Brusselmans K, Vanderhoydonc F, Van de Sande T, Heemers H, Heyns W, Verhoeven G (2003) Fatty acid synthase drives the synthesis of phospholipids partitioning into detergent-resistant membrane microdomains. Biochem Biophys Res Commun 302:898–903. doi:S0006291X03002651
Yoshii Y, Furukawa T, Oyama N, Hasegawa Y, Kiyono Y, Nishii R, Waki A, Tsuji AB, Sogawa C, Wakizaka H, Fukumura T, Yoshii H, Fujibayashi Y, Lewis JS, Saga T (2013) Fatty acid synthase is a key target in multiple essential tumor functions of prostate cancer: uptake of radiolabeled acetate as a predictor of the targeted therapy outcome. PLoS ONE 8:e64570. doi:10.1371/journal.pone.0064570PONE-D-13-05378
Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, Inazuka F, Grisanzio C, Palescandolo E, Shin E, Fiore C, Xie W, Kung AL, Febbo PG, Subramanian A, Mucci L, Ma J, Signoretti S, Stampfer M, Hahn WC, Finn S, Loda M (2009) Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Inst 101:519–532. doi:10.1093/jnci/djp030djp030
Vavere AL, Kridel SJ, Wheeler FB, Lewis JS (2008) 1-11C-acetate as a PET radiopharmaceutical for imaging fatty acid synthase expression in prostate cancer. J Nucl Med 49:327–334. doi:10.2967/jnumed.107.046672
Maleddu A, Pantaleo MA, Castellucci P, Astorino M, Nanni C, Nannini M, Busato F, Di Battista M, Farsad M, Lodi F, Boschi S, Fanti S, Biasco G (2009) 11C-acetate PET for early prediction of sunitinib response in metastatic renal cell carcinoma. Tumori 95:382–384
Yu EY, Muzi M, Hackenbracht JA, Rezvani BB, Link JM, Montgomery RB, Higano CS, Eary JF, Mankoff DA (2011) C11-acetate and F-18 FDG PET for men with prostate cancer bone metastases: relative findings and response to therapy. Clin Nucl Med 36:192–198. doi:10.1097/RLU.0b013e318208f14000003072-201103000-00005
Evans MJ (2012) Measuring oncogenic signaling pathways in cancer with PET: an emerging paradigm from studies in castration-resistant prostate cancer. Cancer Discov 2:985–994. doi:10.1158/2159-8290.CD-12-01782159-8290.CD-12-0178
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Storey S (2008) Targeting apoptosis: selected anticancer strategies. Nat Rev Drug Discov 7:971–972. doi:10.1038/nrd2662nrd2662
Hengartner MO (2000) The biochemistry of apoptosis. Nature 407:770–776. doi:10.1038/35037710
Belhocine T, Steinmetz N, Hustinx R, Bartsch P, Jerusalem G, Seidel L, Rigo P, Green A (2002) Increased uptake of the apoptosis-imaging agent (99m)Tc recombinant human Annexin V in human tumors after one course of chemotherapy as a predictor of tumor response and patient prognosis. Clin Cancer Res 8:2766–2774
Haas RL, de Jong D, Valdés Olmos RA, Hoefnagel CA, van den Heuvel I, Zerp SF, Bartelink H, Verheij M (2004) In vivo imaging of radiation-induced apoptosis in follicular lymphoma patients. Int J Radiat Oncol Biol Phys 59:782–787. doi:10.1016/j.ijrobp.2003.11.017S0360301603023125
Kartachova M, Haas RL, Olmos RA, Hoebers FJ, van Zandwijk N, Verheij M (2004) In vivo imaging of apoptosis by 99m Tc-Annexin V scintigraphy: visual analysis in relation to treatment response. Radiother Oncol 72:333–339. doi:10.1016/j.radonc.2004.07.008S0167-8140(04)00296-8
Reshef A, Shirvan A, Akselrod-Ballin A, Wall A, Ziv I (2010) Small-molecule biomarkers for clinical PET imaging of apoptosis. J Nucl Med 51:837–840. doi:10.2967/jnumed.109.063917
Damianovich M, Ziv I, Heyman SN, Rosen S, Shina A, Kidron D, Aloya T, Grimberg H, Levin G, Reshef A, Bentolila A, Cohen A, Shirvan A (2006) ApoSense: a novel technology for functional molecular imaging of cell death in models of acute renal tubular necrosis. Eur J Nucl Med Mol Imaging 33:281–291. doi:10.1007/s00259-005-1905-x
Hoglund J, Shirvan A, Antoni G, Gustavsson SA, Langstrom B, Ringheim A, Sorensen J, Ben-Ami M, Ziv I (2011) 18F-ML-10, a PET tracer for apoptosis: first human study. J Nucl Med 52:720–725. doi:10.2967/jnumed.110.081786
Allen AM, Ben-Ami M, Reshef A, Steinmetz A, Kundel Y, Inbar E, Djaldetti R, Davidson T, Fenig E, Ziv I (2012) Assessment of response of brain metastases to radiotherapy by PET imaging of apoptosis with 18F-ML-10. Eur J Nucl Med Mol Imaging 39:1400–1408. doi:10.1007/s00259-012-2150-8
Nguyen QD, Smith G, Glaser M, Perumal M, Arstad E, Aboagye EO (2009) Positron emission tomography imaging of drug-induced tumor apoptosis with a caspase-3/7 specific [18F]-labeled isatin sulfonamide. Proc Natl Acad Sci USA 106:16375–16380. doi:10.1073/pnas.0901310106
Nguyen QD, Lavdas I, Gubbins J, Smith G, Fortt R, Carroll LS, Graham MA (1078) Aboagye EO (2013) Temporal and spatial evolution of therapy-induced tumor apoptosis detected by caspase-3-selective molecular imaging. Clin Cancer Res 19:3914–3924. doi:10.1158/1078-0432.CCR-12-3814-0432.CCR-12-3814
Challapalli A, Kenny LM, Hallett WA, Kozlowski K, Tomasi G, Gudi M, Al-Nahhas A, Coombes RC, Aboagye EO (2013) 18F-ICMT-11, a caspase-3-specific PET tracer for apoptosis: biodistribution and radiation dosimetry. J Nucl Med 54:1551–1556. doi:10.2967/jnumed.112.118760
Vaupel P, Mayer A (2007) Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev 26:225–239. doi:10.1007/s10555-007-9055-1
Wilson WR, Hay MP (2011) Targeting hypoxia in cancer therapy. Nat Rev Cancer 11:393–410. doi:10.1038/nrc3064nrc3064
Padhani AR, Krohn KA, Lewis JS, Alber M (2007) Imaging oxygenation of human tumours. Eur Radiol 17:861–872. doi:10.1007/s00330-006-0431-y
Grierson JR, Link JM, Mathis CA, Rasey JS, Krohn KA (1989) A radiosynthesis of fluorine-18 fluoromisonidazole. J Nucl Med 30:343–350
Hwang DR, Dence CS, Bonasera TA, Welch MJ (1989) No-carrier-added synthesis of 3-[18F]fluoro-1-(2-nitro-1-imidazolyl)-2-propanol. A potential PET agent for detecting hypoxic but viable tissues. Int J Rad Appl Instrum A 40:117–126
Rasey JS, Koh WJ, Evans ML, Peterson LM, Lewellen TK, Graham MM, Krohn KA (1996) Quantifying regional hypoxia in human tumors with positron emission tomography of [18F]fluoromisonidazole: a pretherapy study of 37 patients. Int J Radiat Oncol Biol Phys 36:417–428. doi:S0360301696003252
Barthel H, Wilson H, Collingridge DR, Brown G, Osman S, Luthra SK, Brady F, Workman P, Price PM, Aboagye EO (2004) In vivo evaluation of [18F]fluoroetanidazole as a new marker for imaging tumour hypoxia with positron emission tomography. Br J Cancer 90:2232–2242. doi:10.1038/sj.bjc.66018626601862
Valtorta S, Belloli S, Sanvito F, Masiello V, Di Grigoli G, Monterisi C, Fazio F, Picchio M, Moresco RM (2013) Comparison of 18F-fluoroazomycin-arabinofuranoside and 64Cu-diacetyl-bis(N4-methylthiosemicarbazone) in preclinical models of cancer. J Nucl Med 54:1106–1112. doi:10.2967/jnumed.112.111120
Tran LB, Bol A, Labar D, Jordan B, Magat J, Mignion L, Gregoire V, Gallez B (2012) Hypoxia imaging with the nitroimidazole 18F-FAZA PET tracer: a comparison with OxyLite, EPR oximetry and 19F-MRI relaxometry. Radiother Oncol 105:29–35. doi:10.1016/j.radonc.2012.04.011
Dubois LJ, Lieuwes NG, Janssen MH, Peeters WJ, Windhorst AD, Walsh JC, Kolb HC, Ollers MC, Bussink J, van Dongen GA, van der Kogel A, Lambin P (2011) Preclinical evaluation and validation of [18F]HX4, a promising hypoxia marker for PET imaging. Proc Natl Acad Sci USA 108:14620–14625. doi:10.1073/pnas.11025261081102526108
van Loon J, Janssen MH, Ollers M, Aerts HJ, Dubois L, Hochstenbag M, Dingemans AM, Lalisang R, Brans B, Windhorst B, van Dongen GA, Kolb H, Zhang J, De Ruysscher D, Lambin P (2010) PET imaging of hypoxia using [18F]HX4: a phase I trial. Eur J Nucl Med Mol Imaging 37:1663–1668. doi:10.1007/s00259-010-1437-x
Lehtio K, Oikonen V, Nyman S, Gronroos T, Roivainen A, Eskola O, Minn H (2003) Quantifying tumour hypoxia with fluorine-18 fluoroerythronitroimidazole ([18F]FETNIM) and PET using the tumour to plasma ratio. Eur J Nucl Med Mol Imaging 30:101–108. doi:10.1007/s00259-002-1016-x
Lee N, Nehmeh S, Schoder H, Fury M, Chan K, Ling CC, Humm J (2009) Prospective trial incorporating pre-/mid-treatment [18F]-misonidazole positron emission tomography for head-and-neck cancer patients undergoing concurrent chemoradiotherapy. Int J Radiat Oncol Biol Phys 75:101–108. doi:10.1016/j.ijrobp.2008.10.049
Kikuchi M, Yamane T, Shinohara S, Fujiwara K, Hori SY, Tona Y, Yamazaki H, Naito Y, Senda M (2011) 18F-fluoromisonidazole positron emission tomography before treatment is a predictor of radiotherapy outcome and survival prognosis in patients with head and neck squamous cell carcinoma. Ann Nucl Med 25:625–633. doi:10.1007/s12149-011-0508-9
Hugonnet F, Fournier L, Medioni J, Smadja C, Hindie E, Huchet V, Itti E, Cuenod CA, Chatellier G, Oudard S, Faraggi M (2011) Metastatic renal cell carcinoma: relationship between initial metastasis hypoxia, change after 1 month’s sunitinib, and therapeutic response: an 18F-fluoromisonidazole PET/CT study. J Nucl Med 52:1048–1055. doi:10.2967/jnumed.110.084517
Cheng J, Lei L, Xu J, Sun Y, Zhang Y, Wang X, Pan L, Shao Z, Liu G (2013) 18F-fluoromisonidazole PET/CT: a potential tool for predicting primary endocrine therapy resistance in breast cancer. J Nucl Med 54:333–340. doi:10.2967/jnumed.112.111963
Fujibayashi Y, Taniuchi H, Yonekura Y, Ohtani H, Konishi J, Yokoyama A (1997) Copper-62-ATSM: a new hypoxia imaging agent with high membrane permeability and low redox potential. J Nucl Med 38:1155–1160
Dietz DW, Dehdashti F, Grigsby PW, Malyapa RS, Myerson RJ, Picus J, Ritter J, Lewis JS, Welch MJ, Siegel BA (2008) Tumor hypoxia detected by positron emission tomography with 60Cu-ATSM as a predictor of response and survival in patients undergoing Neoadjuvant chemoradiotherapy for rectal carcinoma: a pilot study. Dis Colon Rectum 51:1641–1648. doi:10.1007/s10350-008-9420-3
Carmeliet P (2000) Mechanisms of angiogenesis and arteriogenesis. Nat Med 6:389–395. doi:10.1038/74651
Morrison MS, Ricketts SA, Barnett J, Cuthbertson A, Tessier J, Wedge SR (2009) Use of a novel Arg-Gly-Asp radioligand, 18F-AH111585, to determine changes in tumor vascularity after antitumor therapy. J Nucl Med 50:116–122. doi:10.2967/jnumed.108.056077
Battle MR, Goggi JL, Allen L, Barnett J, Morrison MS (2011) Monitoring tumor response to antiangiogenic sunitinib therapy with 18F-fluciclatide, an 18F-labeled alphaVbeta3-integrin and alphaV beta5-integrin imaging agent. J Nucl Med 52:424–430. doi:10.2967/jnumed.110.077479
Anderson HL, Yap JT, Miller MP, Robbins A, Jones T, Price PM (2003) Assessment of pharmacodynamic vascular response in a phase I trial of combretastatin A4 phosphate. J Clin Oncol 21:2823–2830. doi:10.1200/JCO.2003.05.186
Herbst RS, Mullani NA, Davis DW, Hess KR, McConkey DJ, Charnsangavej C, O’Reilly MS, Kim HW, Baker C, Roach J, Ellis LM, Rashid A, Pluda J, Bucana C, Madden TL, Tran HT, Abbruzzese JL (2002) Development of biologic markers of response and assessment of antiangiogenic activity in a clinical trial of human recombinant endostatin. J Clin Oncol 20:3804–3814
Anderson H, Yap JT, Wells P, Miller MP, Propper D, Price P, Harris AL (2003) Measurement of renal tumour and normal tissue perfusion using positron emission tomography in a phase II clinical trial of razoxane. Br J Cancer 89:262–267. doi:10.1038/sj.bjc.66011056601105
Mintun MA, Welch MJ, Siegel BA, Mathias CJ, Brodack JW, McGuire AH, Katzenellenbogen JA (1988) Breast cancer: PET imaging of estrogen receptors. Radiology 169:45–48
Linden HM, Stekhova SA, Link JM, Gralow JR, Livingston RB, Ellis GK, Petra PH, Peterson LM, Schubert EK, Dunnwald LK, Krohn KA, Mankoff DA (2006) Quantitative fluoroestradiol positron emission tomography imaging predicts response to endocrine treatment in breast cancer. J Clin Oncol 24:2793–2799. doi:10.1200/JCO.2005.04.3810
Linden HM, Kurland BF, Peterson LM, Schubert EK, Gralow JR, Specht JM, Ellis GK, Lawton TJ, Livingston RB, Petra PH, Link JM, Krohn KA (1078) Mankoff DA (2011) Fluoroestradiol positron emission tomography reveals differences in pharmacodynamics of aromatase inhibitors, tamoxifen, and fulvestrant in patients with metastatic breast cancer. Clin Cancer Res 17:4799–4805. doi:10.1158/1078-0432.CCR-10-3321-0432.CCR-10-3321
Sharifi N, Gulley JL, Dahut WL (2005) Androgen deprivation therapy for prostate cancer. JAMA 294:238–244. doi:10.1001/jama.294.2.238
Larson SM, Morris M, Gunther I, Beattie B, Humm JL, Akhurst TA, Finn RD, Erdi Y, Pentlow K, Dyke J, Squire O, Bornmann W, McCarthy T, Welch M, Scher H (2004) Tumor localization of 16beta-18F-fluoro-5alpha-dihydrotestosterone versus 18F-FDG in patients with progressive, metastatic prostate cancer. J Nucl Med 45:366–373
Dehdashti F, Picus J, Michalski JM, Dence CS, Siegel BA, Katzenellenbogen JA, Welch MJ (2005) Positron tomographic assessment of androgen receptors in prostatic carcinoma. Eur J Nucl Med Mol Imaging 32:344–350. doi:10.1007/s00259-005-1764-5
Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, Rathkopf D, Shelkey J, Yu EY, Alumkal J, Hung D, Hirmand M, Seely L, Morris MJ, Danila DC, Humm J, Larson S, Fleisher M, Sawyers CL (2010) Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet 375:1437–1446. doi:10.1016/S0140-6736(10)60172-9S0140-6736(10)
Holland JP, Divilov V, Bander NH, Smith-Jones PM, Larson SM, Lewis JS (2010) 89Zr-DFO-J591 for immunoPET of prostate-specific membrane antigen expression in vivo. J Nucl Med 51:1293–1300. doi:10.2967/jnumed.110.076174
Ulmert D, Evans MJ, Holland JP, Rice SL, Wongvipat J, Pettersson K, Abrahamsson PA, Scardino PT, Larson SM, Lilja H, Lewis JS, Sawyers CL (2012) Imaging androgen receptor signaling with a radiotracer targeting free prostate-specific antigen. Cancer Discov 2:320–327. doi:10.1158/2159-8290.CD-11-03162159-8290.CD-11-0316
Holland JP, Evans MJ, Rice SL, Wongvipat J, Sawyers CL, Lewis JS (2012) Annotating MYC status with 89Zr-transferrin imaging. Nat Med 18:1586–1591. doi:10.1038/nm.2935
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL (1987) Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235:177–182
Smith-Jones PM, Solit DB, Akhurst T, Afroze F, Rosen N, Larson SM (2004) Imaging the pharmacodynamics of HER2 degradation in response to Hsp90 inhibitors. Nat Biotechnol 22:701–706. doi:10.1038/nbt968nbt968
Kramer-Marek G, Kiesewetter DO, Capala J (2009) Changes in HER2 expression in breast cancer xenografts after therapy can be quantified using PET and (18)F-labeled affibody molecules. J Nucl Med 50:1131–1139. doi:10.2967/jnumed.108.057695
Nagengast WB, de Vries EG, Hospers GA, Mulder NH, de Jong JR, Hollema H, Brouwers AH, van Dongen GA, Perk LR, de Lubde Hooge MN (2007) In vivo VEGF imaging with radiolabeled bevacizumab in a human ovarian tumor xenograft. J Nucl Med 48:1313–1319. doi:10.2967/jnumed.107.041301
Tamura K, Kurihara H, Yonemori K, Tsuda H, Suzuki J, Kono Y, Honda N, Kodaira M, Yamamoto H, Yunokawa M, Shimizu C, Hasegawa K, Kanayama Y, Nozaki S, Kinoshita T, Wada Y, Tazawa S, Takahashi K, Watanabe Y, Fujiwara Y (2013) 64Cu-DOTA-trastuzumab PET imaging in patients with HER2-positive breast cancer. J Nucl Med 54:1869–1875. doi:10.2967/jnumed.112.118612
Mishani E, Abourbeh G, Eiblmaier M, Anderson CJ (2008) Imaging of EGFR and EGFR tyrosine kinase overexpression in tumors by nuclear medicine modalities. Curr Pharm Des 14:2983–2998
Goldenberg A, Masui H, Divgi C, Kamrath H, Pentlow K, Mendelsohn J (1989) Imaging of human tumor xenografts with an indium-111-labeled anti-epidermal growth factor receptor monoclonal antibody. J Natl Cancer Inst 81:1616–1625
Wen X, Wu QP, Ke S, Ellis L, Charnsangavej C, Delpassand AS, Wallace S, Li C (2001) Conjugation with (111)In-DTPA-poly(ethylene glycol) improves imaging of anti-EGF receptor antibody C225. J Nucl Med 42:1530–1537
Memon AA, Jakobsen S, Dagnaes-Hansen F, Sorensen BS, Keiding S, Nexo E (2009) Positron emission tomography (PET) imaging with [11C]-labeled erlotinib: a micro-PET study on mice with lung tumor xenografts. Cancer Res 69:873–878. doi:10.1158/0008-5472.CAN-08-31180008-5472.CAN-08-3118
Meng X, Loo BW Jr, Ma L, Murphy JD, Sun X, Yu J (2011) Molecular imaging with 11C-PD153035 PET/CT predicts survival in non-small cell lung cancer treated with EGFR-TKI: a pilot study. J Nucl Med 52:1573–1579. doi:10.2967/jnumed.111.092874
Nordberg E, Friedman M, Gostring L, Adams GP, Brismar H, Nilsson FY, Stahl S, Glimelius B, Carlsson J (2007) Cellular studies of binding, internalization and retention of a radiolabeled EGFR-binding affibody molecule. Nucl Med Biol 34:609–618. doi:10.1016/j.nucmedbio.2007.05.010
Corcoran EB, Hanson RN (2013) Imaging EGFR and HER2 by PET and SPECT: a Review. Med Res Rev. doi:10.1002/med.21299
Conflict of interest
Shairoz Merchant, Timothy H. Witney, Eric O. Aboagye declare no conflict of interest.
Human and animal studies
This article does not contain any new studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Color figures online at http://link.springer.com/article/10.1007/s40336-014-0049-z
Rights and permissions
About this article
Cite this article
Merchant, S., Witney, T.H. & Aboagye, E.O. Imaging as a pharmacodynamic and response biomarker in cancer. Clin Transl Imaging 2, 13–31 (2014). https://doi.org/10.1007/s40336-014-0049-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40336-014-0049-z