
Abstract
Childhood interstitial lung disease (chILD) is a large and heterogeneous group of disorders characterized by diffuse lung parenchymal markings on chest imaging and clinical signs such as dyspnea and hypoxemia from functional impairment. While some children already present in the neonatal period with interstitial lung disease (ILD), others develop ILD during their childhood and adolescence. A timely and accurate diagnosis is essential to gauge treatment and improve prognosis. Supportive care can reduce symptoms and positively influence patients' quality of life; however, there is no cure for many of the chILDs. Current therapeutic options include anti-inflammatory or immunosuppressive drugs. Due to the rarity of the conditions and paucity of research in this field, most treatments are empirical and based on case series, and less than a handful of small, randomized trials have been conducted thus far. A trial on hydroxychloroquine yielded good safety but a much smaller effect size than anticipated. A trial in fibrotic disease with the multitargeted tyrosine kinase inhibitor nintedanib showed similar pharmacokinetics and safety as in adults. The unmet need for the treatment of chILDs remains high. This article summarizes current treatments and explores potential therapeutic options for patients suffering from chILD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
No specific therapy is approved yet for childhood interstitial lung disease (chILD). |
Improved understanding of disease pathogenesis is instrumental to the development of novel therapies. |
The disease course is highly heterogeneous, and when treatment is required, immunosuppressive and anti-inflammatory drugs are often the first-line therapies. |
Multi-stakeholder collaboration is key to develop novel drugs and move the field of chILD forward. |
1 Introduction
The term childhood interstitial lung disease (chILD) was coined to collect a large and heterogeneous group of rare and ultra-rare entities manifesting during childhood. Despite being uncommon, the burden of chILD is high for both caregivers and the health system [1, 2]. Indeed, chILD has a high morbidity and mortality rate, and a global prevalence of 1.6–46 per million [3,4,5,6]. Interstitial lung disease (ILD) in children is approximately ten times rarer and, at the same time, 100 times less studied and published than adult ILD [7]. The diagnosis of chILD should be suspected if at least three of the following four elements are present for more than 4 weeks in the absence of a respiratory tract infection: (1) symptoms like exercise intolerance, tachypnea, (dry) cough; (2) respiratory signs including dyspnea, crackles on lung auscultation, failure to thrive; (3) respiratory insufficiency with hypoxia/low oxygen saturation; and (4) diffuse parenchymal lung abnormalities on chest computed tomography (CT) scan or chest X-ray [8].
When chILD is suspected, a detailed anamnesis and family history (with emphasis on siblings/relatives with a history of ILD or early death from lung disease), clinical examination searching for potential rheumatological, immunological, or dermatological manifestations, and a chest X-ray should be performed, before referring the patient to an expert center. Additional clinical signs may include wall deformity or pectus excavatum (for example, in patients with surfactant disorders), or digital clubbing.
As with adult ILD, the diagnostic algorithm includes non-invasive (i.e., pulmonary function tests, assessment of ventilation and oxygenation, and chest imaging) and invasive procedures (i.e., bronchoscopy, and lung biopsy). Moreover, genetic testing has been implemented in most expert centers, thus improving diagnostic accuracy and reducing the need for invasive diagnostic modalities. Despite considerable progress in recent years, disease pathogenesis remains unknown in many of the chILDs, making the development of efficacious treatments challenging [9, 10]. Moreover, the rarity of each condition and the fact that patients are often living in geographically dispersed areas represent additional important hurdles when planning clinical trials among many other obstacles [11]. In this regard, the importance of collaboration among expert centers with the aim of establishing patient registries and databases cannot be overemphasized [12]. In this review, we explore the landscape of pharmacological treatment of chILD.
2 ChILD Classification
Although the term chILD has the advantage of allowing easy communication and is popular, it has several disadvantages, including lumping together many conditions with completely different presentations, treatments, and outcomes, even including conditions involving the lung interstitium only indirectly or very mildly, like in patients with persistent tachypnea of infancy [13]. Thus, the need for a simple classification system emerged over the years. In 2004, a task force conducted by the European Respiratory Society, which included respiratory physicians and basic scientists, reviewed 185 cases of chILD and classified them with a system similar to that used for adult disease [14]. A few years later, a new classification scheme based on lung histology was proposed for ILD in children < 2 years of age [15, 16]. This classification system was later extended to all pediatric age groups [17]. Recently, an etiological classification system combining pediatric and adult lung ILD in a single system was proposed [18]. The system differentiates four main categories, i.e., lung‐only (native parenchymal) disorders, systemic disease‐related disorders, exposure-related disorders, and vascular disorders. Of particular importance are those conditions closely linked to lung development and thus representing the majority of “typical” chILD, such as those in children not surviving into adulthood, those infrequently diagnosed at adult age, and those that transition into adulthood, as is now being seen more and more [19]. They include “Diffuse developmental disorders (A1),” usually resulting in death within the neonatal period; “Growth abnormalities with deficient alveolarization (A2),” such as lung hypoplasia, chronic lung disease of prematurity (bronchopulmonary dysplasia [BPD]), and others with a somewhat better prognosis; and “Infant conditions of undefined etiology (A3),” comprising the overall most frequent diagnoses in these children, i.e., neuroendocrine cell hyperplasia of infancy (NEHI) (or more correctly labeled as persistent tachypnoea of infancy [PTI]) [13, 20, 21]. Lastly, “ILD related to the alveolar surfactant region (A4)” includes surfactant dysfunction disorders [22] and pulmonary alveolar proteinosis (PAP) [23]. Patients with the latter diagnosis now often reach adulthood or are diagnosed at adult age [24,25,26]. Many other conditions, in particular “ILD related to systemic disease processes (B1),” with examples like sarcoidosis [27] and connective tissue diseases [28], are ILDs with increasing frequency in adulthood.
Recently, the Children's Interstitial and Diffuse Lung Disease Research Network (chILDRN) published data regarding a prospective registry including 683 individuals enrolled from different centers in the United States. NEHI was the most frequent diagnosis (23%), the second being ILD associated with connective tissue or immune‐mediated disorders (16.5%). Notably, 11% of cases of chILD remained “unclassified” (Table 1) [29].
3 Genetic Background
Despite intrinsic peculiarities, chILDs often share similar clinical and radiological manifestations and may be difficult to differentiate from one another. Genetic testing can identify disease-causing mutations, thus reducing the need for invasive diagnostic procedures (Table 2) [30]. The number of genetic etiologies identified as causes of ILD in children continues to grow and includes genes involved in surfactant production (SFTPB, SFTPC, ABCA3, and NKX2-1) [31,32,33,34,35] and catabolism (CSF2RA and CSF2RB) [36, 37], immune regulation (COPA) [38, 39], and lung development (FLNA and TBX4, among others) [40, 41]. Some chILDs are associated with high mortality, whereas others have a favorable outcome. For instance, surfactant protein B (SP-B) deficiency has the worst prognosis, whereas variants within SFTPC may lead to a range of phenotypes and prognoses (Fig. 1) [42]. On the contrary, most but not all children with PTI/NEHI tend to experience a favorable prognosis; therefore, a limited follow-up to 10 or 15 years of age needs to be considered [43].

Seventeen-year-old girl with SP-C deficiency and interstitial lung disease. CT scans show extensive ground glass opacity and traction bronchiectasis throughout both lungs. CT computed tomography, SP-C surfactant protein C
4 Current Therapeutic Options for ChILD
Corticosteroids, hydroxychloroquine, and azithromycin are the most common pharmacological treatments for patients with chILD [44]. Less frequently used medications include azathioprine, cyclophosphamide, and colchicine [45]. Acute exacerbations in chILD were treated with β-lactam antibiotics (54%), systemic glucocorticosteroids (25%), inhaled bronchodilators (24%), or macrolides (19%) [46].
Intravenous pulse methylprednisolone (10–30 mg/kg) is used in critical patients or those needing ventilation [47]. Chronic treatments with oral glucocorticosteroids in children are done rarely, and the frequency and (potential) severity of side effects of corticosteroids must always be considered [48]. Consequently, close clinical monitoring is mandatory, including bone density, periodic complete blood count, and growth measurements [49].
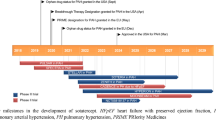
Hydroxychloroquine is an antimalarial drug used in several autoimmune diseases, including arthritis [50]; historically, it has been used also in some chILDs, although its role in this disease remains controversial. Between 1984 and 2013, 85 case reports and small case series reported on children with different forms of ILD who were treated with chloroquine or hydroxychloroquine. In 35 cases, the effect was beneficial, while in the remaining, the results were unsatisfactory or inconclusive [51]. Hydroxychloroquine is generally safe, but its chronic use may be associated with visual loss, making regular visual assessment mandatory [52]. The effect of hydroxychloroquine in patients with chILD has been recently evaluated in a phase 2a, randomized, double-blind, placebo-controlled, multinational study [53, 54]. The primary endpoint of presence or absence of response to treatment as assessed by oxygenation (calculated from a change in transcutaneous O2 saturation of ≥ 5%, respiratory rate ≥ 20%, or level of respiratory support) did not differ between hydroxychloroquine and placebo. Acknowledging major limitations such as the small study population (n = 26), the heterogeneity of included patients, the treatment duration (12 weeks followed by an open observation period of 12 weeks), and the lack of lung function data below the age of 6 years, the authors warned that prescription of hydroxychloroquine in daily practice needs to be reassessed. It is very likely that the beneficial effect of hydroxychloroquine is mutation specific, particularly in ABCA3 deficiency [55].
Azithromycin is an anti-inflammatory and immunomodulatory antibiotic that is used in a range of respiratory diseases; however, the evidence favoring its utility in chILD is limited to less than a handful of case reports [56]. Although widely used, no clinical trial of azithromycin has been conducted in chILD, and its role remains marginal and empirical; the potential risk of microbial resistance to azithromycin should also be considered [57]. Although prospective data are needed, several case reports have reported improvements of adult ILD associated with ABCA3 deficiency following azithromycin treatment, suggesting a possible benefit to be evaluated in selected chILDs [24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,, 58].
Whole lung lavage (WLL) is the standard treatment in PAP, but this procedure is available only in specialized centers. Indeed, special techniques are necessary in infants, due to the small airway dimensions [59,60,61]. In addition, although safe and efficacious, WLL is invasive and time-consuming [62].
Specific therapies with a mechanistically plausible role exist, but they are applicable only in a minority of conditions (biologics [i.e., rituximab], immunomodulatory therapies in connective tissue disorders, or stem cell transplant in alveolar proteinosis) [63, 64].
5 Non-pharmacological Treatments
Supportive care is similarly important for children with ILD. As with adult disease, chILD patients with gas exchange impairment may benefit from supplemental oxygen, whereas children with severe respiratory failure may benefit from invasive or non-invasive ventilation [65]. Patients with chILD may display poor somatic growth, thus needing specific nutritional support. Lessons learned from BPD and cystic fibrosis (CF) suggest that growth should be closely monitored also in patients with chILD.
Similar to adult ILD patients, preventing further damage to the lung is critically important. Pneumonia and other infections impart an important morbidity and mortality burden on children with ILD. Therefore, vaccination (pneumococcal and annual influenza), avoidance of harmful environmental exposures (such as second-hand smoke), and appropriate personal hygiene (both for children and caregivers) are strongly recommended [16].
Gastroesophageal reflux disease (GERD), a common comorbidity in adult ILD patients, has been investigated also in chILD. In a study by Dziekiewicz and colleagues, the prevalence of GERD among children with ILD (n = 18) aged 0.2–11.6 years was 50% [66].
Lung transplantation is rarely indicated but has been successfully performed in cases of SP-B and SP-C deficiency, in patients carrying variants within ABCA3 and NKX2-1, and in children with chronic pneumonitis of infancy. In comparison with adolescents, children are more often transplanted for ILD and precapillary pulmonary hypertension than for respiratory diseases, but with a similar in-hospital mortality [67]. A single-lobe lung transplantation has been successfully performed from a living donor to a patient with ABCA3 disorder [68].
6 Treatment of Specific Conditions
To date, there is no therapy specifically approved for chILD. However, in 2015, the chILD-EU collaboration created standard operating procedures and protocols for a staged investigation of chILD. In addition, participating centers across Europe developed a Delphi consensus process with the aim of harmonizing treatment protocols such as the use of intravenous and oral corticosteroids, and add-on therapies such as hydroxychloroquine and azithromycin [17]. Thus, the development of efficacious and well-tolerated drugs is a particularly urgent need [69]. Because of the rarity of these diseases, many barriers exist to drug development for chILD, including the low economic benefits for the pharma industry and limited funding for researchers. Yet, the burden of chILD on the healthcare system is very high [1]. At present, the management of patients with chILD largely relies on case series investigating the effect of anti-inflammatory and immunomodulatory drugs to prevent the development of lung fibrosis. In this regard, most of the drugs used for chILD derive from the treatment of adult disease [70].
6.1 Fibrosing ILD
Two drugs with pleiotropic antifibrotic effects (pirfenidone and nintedanib) can reduce the rate of functional decline—as assessed by forced vital capacity (FVC)—and disease progression in adult patients with idiopathic pulmonary fibrosis (IPF) [71, 72] and ILD that progress despite appropriate treatment (nintedanib) [73]. The similarities between childhood and adult diseases provided the rationale for assessing the efficacy of antifibrotic drugs approved for adult diseases also in chILD. In a recent phase 2, double-blind, randomized, placebo-controlled trial (NCT04093024—InPedILD trial) [74], 39 patients aged 6–17 years with fibrosing ILD on chest CT and clinically significant disease were randomized 2:1 to receive nintedanib (n = 26) or placebo (n = 13) for 24 weeks. Co-primary endpoints were the area under the plasma concentration–time curve at steady state at weeks 2 and 26 and the proportion of patients with treatment-emergent adverse events at week 24. Two patients (7.7%) discontinued nintedanib because of adverse events. As with adult patients, diarrhea was the most frequent adverse event associated with nintedanib, being reported in 38.5% of cases (compared to 15.4% in the placebo group). Mean change in FVC % predicted at week 24 was 0.3% in the nintedanib group versus −0.9% in the placebo group. A stabilization of peripheral oxygen saturation (SpO2) at rest over 24 weeks in the nintedanib group was also observed; both results were not statistically significant, although the study was not powered to assess efficacy. Overall, in children and adolescent with fibrosing ILD, a weight-based regimen resulted in exposure to nintedanib similar to adult patients, with an acceptable safety and tolerability profile. An open-label extension to assess long-term safety and tolerability of nintedanib in children and adolescents with ILD is currently recruiting (NCT05285982) [75].
6.2 Telomere-Related Diseases
Similar to adult disease, short telomeres and variations in telomere-related genes can manifest as ILD also in children [76, 77]. A phase 1/2 study has shown that danazol, a synthetic sex hormone with androgenic properties, leads to telomere elongation in patients with telomere diseases (NCT01441037) [78]. A multicenter, phase 2, double-blind, placebo-control trial that is currently ongoing will assess the safety and efficacy of danazol (in combination with standard of care) in adults and pediatric patients with pulmonary fibrosis associated with short telomeres. With regard to the pediatric population (age < 16 years), enrollment in the trial is limited to patients with a diagnosis of dyskeratosis congenita who will receive danazol 2 mg/kg/day, while adult patients will receive a dosage of 4 mg/kg/day (TELOSCOPE—NCT04638517) [79]. The primary endpoint is the annual change in absolute telomere length from baseline. The efficacy of danazol is also investigated in the French ANDROTELO study (NCT03710356). The results should be available soon, but without the chILD population.
6.3 Disorders of Surfactant Dysfunction
Ivacaftor and genistein, two drugs approved for CF, have been evaluated as potential treatments in patients carrying ABCA3 variants, the rationale being that the ABCA3 gene has some degree of homology with CFTR, with the cystic fibrosis transmembrane conductance regulator (CFTR) also being an ABC transporter (Fig. 2). In CF, both ivacaftor and genistein increase CFTR channel opening, which translates to several beneficial effects, including lung function, surfactant function, weight gain, and fertility. Disorders related to ABCA3 dysfunction lead to respiratory distress syndrome, early death, and chronic ILD in children and adults [24]. Kinting and colleagues have shown that disease-causing misfolding ABCA3 variants can be rescued in vitro by the bithiazole correctors C13 and C17 as well as by the chemical chaperone trimethylamine N-oxide and low temperature [80]. Moreover, in A549 cells expressing ABCA3 variants, the same authors demonstrated that ivacaftor and genistein can rescue variants N568D, F629L, and G667R [81]. These observations make CFTR potentiators a potential therapeutic option for patients suffering from surfactant deficiency caused by ABCA3 variants. Other genes involved in surfactant production include SFTPA, SFTPB, and SFTPC, with variants within these genes leading to abnormal surfactant production and clearance [82]. Cyclosporine A (CsA), a calcineurin inhibitor, has recently been suggested as a new potential candidate for ABCA3-specific molecular correction using high-content screening [83] and was used in association with pirfenidone in a child suffering from systemic lupus erythematosus ILD. An improvement in symptoms, pulmonary function, and chest CT images was observed after 2 years of treatment [84]. Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a cytokine with a key role in surfactant physiology, and GM-CSF−/− mice display defective clearance of surfactant by alveolar macrophages, leading to PAP [85]. The safety and efficacy of inhaled sargramostim, a recombinant human GM-CSF, were assessed in children with hereditary PAP caused by bi-allelic variants in CSF2RA or CSF2RB (NCT01511068). However, the study was prematurely discontinued because of slow recruitment. The effect of inhaled sargramostim in patients with PAP was also assessed in a phase 2, multicenter, randomized, double-blind, placebo-controlled trial conducted in Japan (NCT02835742). The study enrolled 64 patients, including patients aged 16–18 years [86]. The frequency and severity of the adverse events did not differ significantly between the sargramostim and placebo groups. The mean change in the alveolar–arterial oxygen gradient between baseline and week 25, the primary endpoint, was significantly better in the GM-CSF group than in the placebo group (− 4.50 ± 9.03 mmHg vs. 0.17 ± 10.50 mmHg; p = 0.02). However, inhaled GM-CSF provided no clinical benefits. On the other hand, in a more recent double-blind, placebo-controlled trial in adult patients with autoimmune PAP, molgramostim (an Escherichia coli-produced recombinant GM-CSF formulated as a nebulizer solution) resulted in greater improvements in pulmonary gas transfer and functional health status than placebo, with similar rates of adverse events [87]. Inhaled GM-CSF has also been used in pediatric patients with autoimmune PAP, among other treatments [88].

Seven-year-old boy ABCA3-related interstitial lung disease. CT scans show bilateral ground glass opacity. CT computed tomography
Following anecdotal case reports [89, 90], methionine has recently been evaluated in patients with PAP carrying pathogenic variants within MARS (NCT03887169) [91]. MARS encodes the methionyl–transfer RNA synthetase (MetRS), and the addition of methionine to the culture medium restores MetRS function in mutated yeast [92]. The study enrolled four children who were evaluated for respiratory, hepatic, and inflammation-related outcomes. For all patients, methionine supplementation was associated with respiratory improvement, reduced liver dysfunction, and reduced need for WLL. While encouraging, these data need to be validated in prospectively enrolled, larger populations of patients.
6.4 SAVI and COPA Disorders
STING-associated vasculopathy with onset in infancy (SAVI) is a genetic autoinflammatory disease secondary to perpetual STING (STimulator of INterferon Genes) activation. The disease, which is characterized by small vessel inflammation, generally has a neonatal or infantile-onset [93]. Recently, a phase 2/3 multicenter, open-label study (NCT04517253) [94] investigated the role of baricitinib, a Janus kinase (JAK)-1/2 inhibitor, in adult and pediatric Japanese patients with SAVI and other diseases, such as Nakajo–Nishimura syndrome (NNS) and Aicardi-Goutières syndrome (AGS). Nine patients were enrolled, including three with SAVI. At the end of the maintenance period (52 weeks), all patients experienced an improvement in their symptoms, and one patient reported a serious drug-related adverse event. COPA syndrome is a rare, genetic autoimmune disorder that is caused by dysfunctional coatomer associate protein subunit alpha (COPα), a protein that functions in the retrograde transport from the Golgi to the endoplasmic reticulum [39]. COPA syndrome can affect multiple organs, especially the lungs, joints, and kidneys [39]. Recent data have linked COPA mutations to STING-dependent interferon signaling. The JAK inhibitors baricitinib and ruxolitinib have recently been suggested as promising therapeutic options for patients with COPA syndrome, but additional data are needed to corroborate these preliminary findings [95,96,97].
7 Emerging Therapies
7.1 Gene Transfer Therapies
Gene transfer may allow the application of recent advances in molecular biology to clinical practice [98]. Synthetic gene transfer vectors have been tested in experimental models both in vitro and in vivo in patients with chILDs, including those related to dysfunctional SFTPC, SFTPB, and ABCA3. Viral vectors (i.e., retroviral vectors, lentiviral vectors, adeno-associated viral [AAV] vectors, or adenoviral [Ad]-based vectors) have shown great potential in gene modulation, particularly for SP-B deficiency [99,100,101]. Nuclease-encoding, chemically modified mRNA is able to deliver site-specific nucleases in a mouse model of SP-B deficiency and improve survival of the animals. Kang and co-workers have recently shown that an AAV vector can restore surfactant activity and improve survival in SP-B knockout mice [102]. However, the development of a delivery vector requires knowledge of the precise disease target(s), transgene expression, and vector design. For instance, Ad-based vectors provide robust expression and a relatively large carrying capacity (~ 10 kb). Conversely, lentiviral vectors have a packaging capacity of at least 7.5 kb. Moreover, Ad-based vectors transduce both dividing and non-dividing cells with good tissue tropism and flexibility. Viral vectors have been evaluated in a range of diseases, including hereditary PAP with CSF2RA mutations. Hetzel and colleagues have shown that a lentiviral vector was able to induce Csf2ra complementary DNA (cDNA) expression in Csf2ra−/− macrophages, leading to restoration of GM-CSF signaling in hereditary PAP macrophages. The lentiviral vector had no adverse effects in the intended target cells, supporting testing lentivirus-mediated gene transfer therapy in hereditary PAP in humans [103].
7.2 Mesenchymal Stromal Cells
Cell therapy has fueled significant interest as a treatment for a range of respiratory diseases. Due to their low immunogenicity, easy isolation, and fleet differentiation in multiple lineages, mesenchymal stem cells (MSCs) are attractive therapeutic strategies also for chILD [104]. MSCs can be easily obtained from various fonts, including amniotic fluid, bone marrow, skeletal muscle, spleen, and lung. Previous studies in vitro and in vivo have explored the ability of MSCs to differentiate into alveolar epithelial cells, with the aim of assessing their potential utility in human lung disease [105]. Ahn and colleagues [106] conducted a phase 2, double-blind, placebo-controlled trial to assess the efficacy of intratracheal transplantation of human umbilical cord blood-derived MSCs (hUC-MSCs) (NCT01828957) in patients with BPD, a chronic lung disease limited to infants, typically caused by prolonged ventilation [107]. The study enrolled 66 premature infants aged between 23 and 28 gestational weeks. After 1 week, hUCB-MSC therapy significantly reduced the levels of several pro-inflammatory cytokines (i.e., interleukin [IL]-1, IL-6, IL-8, tumor necrosis factor [TNF]α, and matrix metallopeptidase [MMP]-9) in the tracheal aspirate fluid compared to placebo. However, the primary outcomes of survival and disease progression were not significantly improved by MSC transplantation. Based on a subgroup analysis suggesting that the secondary outcome of severe BPD was significantly improved in the 23–24 gestational week group, a larger phase 2 study is underway, focusing on infants in this age range (NCT03392467 – PNEUMOSTEM). Several clinical trials are currently evaluating the safety and efficacy of MSCs in BPD (ClinicalTrials.gov). MSCs are administered either intratracheally or intravenously. Induced pluripotent stem cells have been used in a mouse model of PAP induced by CSF2RB deficiency [108]. In addition, Wu and colleagues have shown that hUC-MSCs combined with low-dose pirfenidone reduce bleomycin-induced pulmonary fibrosis in mice more than the two therapies individually [109]. As with other therapeutic approaches, the safety and efficacy of MSCs in chILD need to be validated in larger studies.
7.3 Future Perspectives
In the past few years, genetic testing and whole genome sequencing (WGS) have increased both our ability to diagnose chILD and our understanding of disease pathobiology [110]. New insights have also emerged regarding disease-associated biomarkers. For example, unique protein signatures are shown in the bronchoalveolar lavage fluid (BALF) of NEHI patients and in other surfactant disorders [111]. Moreover, a number of blood biomarkers deeply investigated in adult ILD, including mucin-5B (MUC-5B) and Krebs von den Lungen-6 (KL-6), could also be useful in chILD to predict the risk of disease development and progression [112, 113]. However, research focused on gene-to-protein translation is also needed. Because of the limited availability of human lung tissue (especially in children) and with animal models of lung fibrosis recapitulating only partially the complexity of human disease, lung “organoids” of varying cellular components have recently emerged as novel strategies to model, among other organs, the lung and airway [114]. Organoids are promising tools for studying complex cellular interactions, thus mimicking disease environments; indeed, they arise from colonies generated by single cells and maintain the genomic profile of the parent tissue. However, current organoid models cannot reproduce in toto the physiological repertoire of their respective organs [114].
8 Conclusion
The interest in childhood ILD has increased substantially in recent years, fueled mainly by genetic discoveries and the development of large national and international registries collecting rare and ultra-rare cases (Europe: chILD-EU; France: Respirare; USA: children; Australia: chILDRN). These consortia have also disseminated new knowledge on the management of these diseases and have improved the care for these children. Controlled studies of accepted but unproven treatments and novel treatments are urgently needed. To this end, participation of children in adult drug evaluation programs, the obligate implementation of pediatric investigational plans for all drugs introduced in adults, and the set-up of trials for conditions mainly prevalent in pediatrics must be realized. Continuing the build-up of international collaborations between expert centers and large databases of phenotypically well-defined patients is instrumental to any progress in these rare conditions.
References
Seidl E, Schwerk N, Carlens J, Wetzke M, Cunningham S, Emiralioğlu N, Kiper N, Lange J, Krenke K, Ullmann N, Krikovszky D, chILD-EU collaborators, Maqhuzu P, Griese CA, Schwarzkopf L, Griese M. Healthcare resource utilisation and medical costs for children with interstitial lung diseases (chILD) in Europe. Thorax. 2022;77(8):781–9. https://doi.org/10.1136/thoraxjnl-2021-217751.
Niemitz M, Schwerk N, Goldbeck L, Griese M. Development and validation of a health-related quality of life questionnaire for pediatric patients with interstitial lung disease. Pediatr Pulmonol. 2018;53(7):954–63. https://doi.org/10.1002/ppul.24018.
Young L, et al. A national registry for childhood interstitial and diffuse lung diseases in the United States. (2018).
Griese M, Seidl E, Hengst M, Reu S, Rock H, Anthony G, Kiper N, Emiralioğlu N, Snijders D, Goldbeck L, Leidl R, Ley-Zaporozhan J, Krüger-Stollfuss I, Kammer B, Wesselak T, Eismann C, Schams A, Neuner D, MacLean M, Nicholson AG, Lauren M, Clement A, Epaud R, de Blic J, Ashworth M, Aurora P, Calder A, Wetzke M, Kappler M, Cunningham S, Schwerk N, Bush A, the other chILD-EU collaborators. International management platform for children’s interstitial lung disease (chILD-EU). Thorax. 2018;73(3):231–9. https://doi.org/10.1136/thoraxjnl-2017-210519.
Casamento K, et al. Assessing the feasibility of a web-based registry for multiple orphan lung diseases: the Australasian Registry Network for Orphan Lung Disease (ARNOLD) experience. Orphanet J Rare Dis. 2016;11:1–6.
Torrent-Vernetta A, Gaboli M, Castillo-Corullón S, Mondéjar-López P, Sanz Santiago V, Costa-Colomer J, Osona B, Torres-Borrego J, de la Serna-Blázquez O, Bellón Alonso S, Caro Aguilera P, Gimeno-DíazdeAtauri Á, Valenzuela Soria A, Ayats R, Martinde Vicente C, Velasco González V, Moure González JD, Canino Calderín EM, Pastor-Vivero MD, Villar Álvarez MÁ, Rovira-Amigo S, Iglesias Serrano I, Díez Izquierdo A, de MirMessa I, Gartner S, Navarro A, Baz-Redón N, Carmona R, Camats-Tarruella N, Fernández-Cancio M, Rapp C, Dopazo J, Griese M, Moreno-Galdó A, ChILD-Spain Group. Incidence and prevalence of children’s diffuse lung disease in Spain. Arch Bronconeumol. 2022;58(1):22–9. https://doi.org/10.1016/j.arbres.2021.06.001. (English, Spanish).
Griese M. Chronic interstitial lung disease in children. Eur Respir Rev. 2018;27(147): 170100. https://doi.org/10.1183/16000617.0100-2017.
Nathan N, Griese M, Michel K, Carlens J, Gilbert C, Emiralioglu N, Torrent-Vernetta A, Marczak H, Willemse B, Delestrain C, Epaud R, ERS CRC chILD-EU group. Diagnostic workup of childhood interstitial lung disease. Eur Respir Rev. 2023;32(167):220188. https://doi.org/10.1183/16000617.0188-2022.
Cunningham S, Jaffe A, Young LR. Children’s interstitial and diffuse lung disease. Lancet Child Adolesc Health. 2019;3(8):568–77. https://doi.org/10.1016/S2352-4642(19)30117-8. (Epub 2019 Jun 18).
Clement A, Nathan N, Epaud R, et al. Interstitial lung diseases in children. Orphanet J Rare Dis. 2010;5:22. https://doi.org/10.1186/1750-1172-5-22.
Del Álamo M, Bührer C, Fisher D, Griese M, Lingor P, Palladini G, Sireau N, Hivert V, Sangiorgi L, Guillot F, Halftermeyer J, Soucková L, Nosková K, Demlová R. Identifying obstacles hindering the conduct of academic-sponsored trials for drug repurposing on rare-diseases: an analysis of six use cases. Trials. 2022;23(1):783. https://doi.org/10.1186/s13063-022-06713-y.
Ring AM, Schwerk N, Kiper N, Aslan AT, Aurora P, Ayats R, Azevedo I, Bandeira T, Carlens J, Castillo-Corullon S, Cobanoglu N, Elnazir B, Emiralioğlu N, Eyuboglu TS, Fayon M, Gursoy TR, Hogg C, Kötz K, Karadag B, Látalová V, Krenke K, Lange J, Manali ED, Osona B, Papiris S, Proesmann M, Reix P, Roditis L, Rubak S, Rumman N, Snijders D, Stehling F, Weiss L, Yalcın E, Zirek F, Bush A, Clement A, Griese M, Buchvald FF, Nathan N, Nielsen KG. Diffuse alveolar haemorrhage in children: an international multicentre study. ERJ Open Res. 2023;9(2):00733–2022. https://doi.org/10.1183/23120541.00733-2022. (Erratum in: ERJ Open Res. 2023 May 30;9(3)).
Rauch D, Wetzke M, Reu S, Wesselak W, Schams A, Hengst M, Kammer B, Ley-Zaporozhan J, Kappler M, Proesmans M, Lange J, Escribano A, Kerem E, Ahrens F, Brasch F, Schwerk N, Griese M, PTI (Persistent Tachypnea of Infancy) Study Group of the Kids Lung Register. Persistent tachypnea of infancy. Usual and aberrant. Am J Respir Crit Care Med. 2016;193(4):438–47. https://doi.org/10.1164/rccm.201508-1655OC.
Clement A, ERS Task Force. Task force on chronic interstitial lung disease in immunocompetent children. Eur Respir J. 2004;24(4):686–97. https://doi.org/10.1183/09031936.04.00089803.
Deutsch GH, Young LR, Deterding RR, Fan LL, Dell SD, Bean JA, Brody AS, Nogee LM, Trapnell BC, Langston C, Pathology Cooperative Group, ChILD Research Co-operative, et al. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med. 2007;176:1120–8.
Kurland G, Deterding RR, Hagood JS, Young LR, Brody AS, Castile RG, Dell S, Fan LL, Hamvas A, Hilman BC, Langston C, Nogee LM, Redding GJ, American Thoracic Society Committee on Childhood Interstitial Lung Disease (chILD) and the chILD Research Network. An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med. 2013;188(3):376–94. https://doi.org/10.1164/rccm.201305-0923ST.
Bush A, Cunningham S, de Blic J, et al. European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax. 2015;70:1078–84.
Griese M. Etiologic classification of diffuse parenchymal (interstitial) lung diseases. J Clin Med. 2022;11:1747. https://doi.org/10.3390/jcm11061747.
Koucký V, Pohunek P, Vašáková M, Bush A. Transition of patients with interstitial lung disease from paediatric to adult care. ERJ Open Res. 2021;7(2):00964–2020. https://doi.org/10.1183/23120541.00964-2020.
Dervaux M, Thumerelle C, Fabre C, Abou-Taam R, Bihouee T, Brouard J, Clement A, Delacourt C, Delestrain C, Epaud R, Ghdifan S, Hadchouel A, Houdouin V, Labouret G, Perisson C, Reix P, Renoux MC, Troussier F, Weiss L, Mazenq J, Nathan N, Dubus JC. Long-term evolution of neuroendocrine cell hyperplasia of infancy: the FRENCHI findings. Eur J Pediatr. 2023;182(2):949–56.
Seidl E, Carlens J, Schwerk N, Wetzke M, Marczak H, Lange J, Krenke K, Mayell SJ, Escribano A, Seidenberg J, Ahrens F, Hebestreit H, Nährlich L, Sismanlar T, Aslan AT, Snijders D, Ullmann N, Kappler M, Griese M. Persistent tachypnea of infancy: follow up at school age. Pediatr Pulmonol. 2020;55(11):3119–25. https://doi.org/10.1002/ppul.25004. (Epub 2020 Aug 13).
Singh J, Jaffe A, Schultz A, Selvadurai H. Surfactant protein disorders in childhood interstitial lung disease. Eur J Pediatr. 2021;180(9):2711–21.
Trapnell BC, Nakata K, Bonella F, Campo I, Griese M, Hamilton J, Wang T, Morgan C, Cottin V, McCarthy C. Pulmonary alveolar proteinosis. Nat Rev Dis Primers. 2019;5(1):16.
Li Y, Seidl E, Knoflach K, Gothe F, Forstner ME, Michel K, Pawlita I, Gesenhues F, Sattler F, Yang X, Kroener C, Reu-Hofer S, Ley-Zaporozhan J, Kammer B, Krüger-Stollfuß I, Dinkel J, Carlens J, Wetzke M, Moreno-Galdó A, Torrent-Vernetta A, Lange J, Krenke K, Rumman N, Mayell S, Sismanlar T, Aslan A, Regamey N, Proesmans M, Stehling F, Naehrlich L, Ayse K, Becker S, Koerner-Rettberg C, Plattner E, Manali ED, Papiris SA, Campo I, Kappler M, Schwerk N, Griese M. ABCA3-related interstitial lung disease beyond infancy. Thorax. 2023;78(6):587–95. https://doi.org/10.1136/thorax-2022-219434.
Manali ED, Legendre M, Nathan N, Kannengiesser C, Coulomb-L’Hermine A, Tsiligiannis T, Tomos P, Griese M, Borie R, Clement A, Amselem S, Crestani B, Papiris SA. Bi-allelic missense ABCA3 mutations in a patient with childhood ILD who reached adulthood. ERJ Open Res. 2019;5(3):00066–2019. https://doi.org/10.1183/23120541.00066-2019.
van Moorsel CHM, van der Vis JJ, Grutters JC. Genetic disorders of the surfactant system: focus on adult disease. Eur Respir Rev. 2021;30(159): 200085. https://doi.org/10.1183/16000617.0085-2020.
Nathan N, Sileo C, Calender A, Pacheco Y, Rosental PA, Cavalin C, Macchi O, Valeyre D, Clement A, French Sarcoidosis Group (GSF), Silicosis Research Group. Paediatric sarcoidosis. Paediatr Respir Rev. 2019;29:53–9.
Tarvin SE, O’Neil KM. Systemic lupus erythematosus, Sjögren syndrome, and mixed connective tissue disease in children and adolescents. Pediatr Clin North Am. 2018;65(4):711–37.
Nevel RJ, Deutsch GH, Craven D, Deterding R, Fishman MP, Wambach JA, Casey A, Krone K, Liptzin DR, O'Connor MG, Kurland G, Taylor JB, Gower WA, Hagood JS, Conrad C, Tam-Williams JB, Fiorino EK, Goldfarb S, Sadreameli SC, Nogee LM, Montgomery G, Hamvas A, Laguna TA, Bansal M, Lew C, Santiago M, Popova A, De A, Chan M, Powers MR, Josephson MB, Camburn D, Voss L, Li Y, Young LR; chILD Registry Collaborative. The US national registry for childhood interstitial and diffuse lung disease: report of study design and initial enrollment cohort. Pediatr Pulmonol. 2023.
Nogee LM. Genetic basis of children’s interstitial lung disease. Pediatr Allergy Immunol Pulmonol. 2010;23:15–24.
Hamvas A. Inherited surfactant protein-B deficiency and surfactant protein-C associated disease: clinical features and evaluation. Semin Perinatol. 2006;30:316–26.
Doan M, Guillerman R, Dishop M, Nogee L, Langston C, Mallory G, Sockrider M, Fan L. Clinical, radiological and pathological features of ABCA3 mutations in children. Thorax. 2008;63:366–73.
Patel NJ, Jankovic J. NKX2-1-related disorders. 2014 [updated 2016 Jul 29]. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993–2023.
Kröner C, Reu S, Teusch V, Schams A, Grimmelt AC, Barker M, Brand J, Gappa M, Kitz R, Kramer BW, Lange L, Lau S, Pfannenstiel C, Proesmans M, Seidenberg J, Sismanlar T, Aslan AT, Werner C, Zielen S, Zarbock R, Brasch F, Lohse P, Griese M. Genotype alone does not predict the clinical course of SFTPC deficiency in paediatric patients. Eur Respir J. 2015;46(1):197–206. https://doi.org/10.1183/09031936.00129414.
Kröner C, Wittmann T, Reu S, Teusch V, Klemme M, Rauch D, Hengst M, Kappler M, Cobanoglu N, Sismanlar T, Aslan AT, Campo I, Proesmans M, Schaible T, Terheggen-Lagro S, Regamey N, Eber E, Seidenberg J, Schwerk N, Aslanidis C, Lohse P, Brasch F, Zarbock R, Griese M. Lung disease caused by ABCA3 mutations. Thorax. 2017;72(3):213–20. https://doi.org/10.1136/thoraxjnl-2016-208649.
Hildebrandt J, Yalcin E, Bresser HG, Cinel G, Gappa M, Haghighi A, Kiper N, Khalilzadeh S, Reiter K, Sayer J, Schwerk N, Sibbersen A, Van Daele S, Nübling G, Lohse P, Griese M. Characterization of CSF2RA mutation related juvenile pulmonary alveolar proteinosis. Orphanet J Rare Dis. 2014;26(9):171.
Suzuki T, Maranda B, Sakagami T, Catellier P, Couture CY, Carey BC, Chalk C, Trapnell BC. Hereditary pulmonary alveolar proteinosis caused by recessive CSF2RB mutations. Eur Respir J. 2011;37(1):201–4. https://doi.org/10.1183/09031936.00090610.
Gao X, Michel K, Griese M. Interstitial lung disease in immunocompromised children. Diagnostics (Basel). 2022;13(1):64. https://doi.org/10.3390/diagnostics13010064.
Kumrah R, Mathew B, Vignesh P, Singh S, Rawat A. Genetics of COPA syndrome. Appl Clin Genet. 2019;8(12):11–8.
Galambos C, Mullen MP, Shieh JT, Schwerk N, Kielt MJ, Ullmann N, Boldrini R, Stucin-Gantar I, Haass C, Bansal M, Agrawal PB, Johnson J, Peca D, Surace C, Cutrera R, Pauciulo MW, Nichols WC, Griese M, Ivy D, Abman SH, Austin ED, Danhaive O. Phenotype characterisation of TBX4 mutation and deletion carriers with neonatal and paediatric pulmonary hypertension. Eur Respir J. 2019;54(2):1801965. https://doi.org/10.1183/13993003.01965-2018.
Meliota G, Vairo U, Ficarella R, Milella L, Faienza MF, D’Amato G. Cardiovascular, brain, and lung involvement in a newborn with a novel FLNA mutation: a case report and literature review. Adv Neonatal Care. 2022;22(2):125–31.
Yurdakök M. Inherited disorders of neonatal lung diseases. Turk J Pediatr. 2004;46(2):105–14.
Liptzin DR, Pickett K, Brinton JT, Agarwal A, Fishman MP, Casey A, Towe CT, Taylor JB, Kurland G, Hagood JS, Wambach J, Srivastava R, Al-Saleh H, Dell SD, Young LR, Deterding RR. Neuroendocrine cell hyperplasia of infancy. Clinical score and comorbidities. Ann Am Thorac Soc. 2020;17(6):724–8.
Dinwiddie R, Sharief N, Crawford O. Idiopathic interstitial pneumonitis in children: a national survey in the United Kingdom and Ireland. Pediatr Pulmonol. 2002;34(1):23–9. https://doi.org/10.1002/ppul.10125.
Hines EJ, Walsh M, Armes JE, Douglas T, Chawla J. Interstitial lung disease in infancy: a general approach. J Paediatr Child Health. 2016;52:370–6.
Seidl E, Schwerk N, Carlens J, Wetzke M, Emiralioğlu N, Kiper N, Lange J, Krenke K, Szepfalusi Z, Stehling F, Baden W, Hämmerling S, Jerkic SP, Proesmans M, Ullmann N, Buchvald F, Knoflach K, Kappler M, chILD EU collaborators, Griese M. Acute exacerbations in children’s interstitial lung disease. Thorax. 2022;77(8):799–804. https://doi.org/10.1136/thoraxjnl-2021-217941.
Desmarquest P, Tamalet A, Fauroux B, Boule M, Boccon-Gibod L, Tournier G, Clement A. Chronic interstitial lung disease in children: response to high-dose intravenous methylprednisolone pulses. Pediatr Pulmonol. 1998;26(5):332–8. https://doi.org/10.1002/(sici)1099-0496(199811)26:5%3c332::aid-ppul5%3e3.0.co;2-q.
de Benedictis FM, Bush A. Corticosteroids in respiratory diseases in children. Am J Respir Crit Care Med. 2012;185(1):12–23.
Breuer O, Schultz A. Side effects of medications used to treat childhood interstitial lung disease. Paediatr Respir Rev. 2018;28:68–79.
Rainsford KD, Parke AL, Clifford-Rashotte M, et al. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology. 2015;23:231–69. https://doi.org/10.1007/s10787-015-0239-y.
Braun S, Ferner M, Kronfeld K, Griese M. Hydroxychloroquine in children with interstitial (diffuse parenchymal) lung diseases. Pediatr Pulmonol. 2015;50(4):410–9.
Modjtahedi BS, Movassagh N, Gandhi N, Morse LS, Maibach HI. Screening for hydroxychloroquine toxicity in children. Cutan Ocul Toxicol. 2013;32(4):344.
Griese M, Köhler M, Witt S, Sebah D, Kappler M, Wetzke M, Schwerk N, Emiralioglu N, Kiper N, Kronfeld K, et al. Prospective evaluation of hydroxychloroquine in pediatric interstitial lung diseases: Study protocol for an investigator-initiated, randomized controlled, parallel-group clinical trial. Trials. 2020;21:307.
Griese M, Kappler M, Stehling F, Schulze J, Baden W, Koerner-Rettberg C, Carlens J, Prenzel F, Nährlich L, Thalmeier A, Sebah D, Kronfeld K, Rock H, Ruckes C, HCQ-study group, Wetzke M, Seidl E, Schwerk N. Randomized controlled phase 2 trial of hydroxychloroquine in childhood interstitial lung disease. Orphanet J Rare Dis. 2022;17(1):289.
Yang X, Forstner M, Rapp CK, Rothenaigner I, Li Y, Hadian K, Griese M. ABCA3 deficiency-variant-specific response to hydroxychloroquine. Int J Mol Sci. 2023;24(9):8179. https://doi.org/10.3390/ijms24098179.
Thouvenin G, Nathan N, Epaud R, Clement A. Diffuse parenchymal lung disease caused by surfactant deficiency: dramatic improvement by azithromycin. BMJ Case Rep. 2013;2013:bcr2013009988.
Bush A. Azithromycin is the answer in paediatric respiratory medicine, but what was the question? Paediatr Respir Rev. 2020;34:67–74.
Klay D, Grutters JC, van der Vis JJ, Platenburg MGJP, Kelder JC, Tromp E, van Moorsel CHM. Progressive disease with low survival in adult patients with pulmonary fibrosis carrying surfactant-related gene mutations: an observational study. Chest. 2023;163(4):870–80. https://doi.org/10.1016/j.chest.2022.11.002.
Reiter K, Schoen C, Griese M, Nicolai T. Whole-lung lavage in infants and children with pulmonary alveolar proteinosis. Paediatr Anaesth. 2010;20(12):1118–23. https://doi.org/10.1111/j.1460-9592.2010.03442.x.
Paschen C, Reiter K, Stanzel F, Teschler H, Griese M. Therapeutic lung lavages in children and adults. Respir Res. 2005;6(1):138. https://doi.org/10.1186/1465-9921-6-138.
Bush A, Pabary R. Pulmonary alveolarproteinosis in children. Breathe (Sheff). 2020;16(2): 200001. https://doi.org/10.1183/20734735.0001-2020.
Wilson CA, Wilmshurst SL, Black AE. Anesthetic techniques to facilitate lung lavage for pulmonary alveolar proteinosis in children-new airway techniques and a review of the literature. Paediatr Anaesth. 2015;25(6):546–53.
Seidl E, Schramm D, Schön C, Reiter K, Pawlita I, Kappler M, Reu-Hofer S, Hauck F, Albert M, Griese M. Pulmonary alveolar proteinosis due to heterozygous mutation in OAS1: whole lung lavages for long-term bridging to hematopoietic stem cell transplantation. Pediatr Pulmonol. 2022;57(1):273–7. https://doi.org/10.1002/ppul.25728.
Griese M, Zarbock R, Costabel U, Hildebrandt J, Theegarten D, Albert M, Thiel A, Schams A, Lange J, Krenke K, Wesselak T, Schön C, Kappler M, Blum H, Krebs S, Jung A, Kröner C, Klein C, Campo I, Luisetti M, Bonella F. GATA2 deficiency in children and adults with severe pulmonary alveolar proteinosis and hematologic disorders. BMC Pulm Med. 2015;12(15):87. https://doi.org/10.1186/s12890-015-0083-2.
Khirani S, Nathan N, Ramirez A, Aloui S, Delacourt C, Clément A, Fauroux B. Work of breathing in children with diffuse parenchymal lung disease. Respir Physiol Neurobiol. 2015;15(206):45–52.
Dziekiewicz MA, Karolewska-Bochenek K, Dembiński Ł, Gawronska A, Krenke K, Lange J, Banasiuk M, Kuchar E, Kulus M, Albrecht P, Banaszkiewicz A. Gastroesophageal reflux disease in children with interstitial lung disease. Adv Exp Med Biol. 2016;912:57–64.
Iablonskii P, Carlens J, Mueller C, Aburahma K, Niehaus A, Boethig D, Franz M, Floethmann K, Sommer W, Optenhoefel J, Tudorache I, Greer M, Koeditz H, Jack T, Hansmann G, Kuehn C, Horke A, Hansen G, Haverich A, Warnecke G, Avsar M, Salman J, Bobylev D, Ius F, Schwerk N. Indications and outcome after lung transplantation in children under 12 years of age: a 16-year single center experience. J Heart Lung Transplant. 2022;41(2):226–36.
Kumata S, Matsuda Y, Oishi H, Sado T, Niikawa H, Watanabe T, Noda M, Hoshikawa Y, Sakurada A, Saito-Koyama R, Niizuma H, Kitazawa H, Akiba M, Sasahara Y, Okada Y. Living donor lobar lung transplant for a patient with lung disease caused by ABCA3 gene mutations: a case report. Transplant Proc. 2022;54(10):2803–6. https://doi.org/10.1016/j.transproceed.2022.07.020.
Hime NJ, Zurynski Y, Fitzgerald D, Selvadurai H, Phu A, Deverell M, Elliott EJ, Jaffe A. Childhood interstitial lung disease: a systematic review. Pediatr Pulmonol. 2015;50(12):1383–92. https://doi.org/10.1002/ppul.23183. (Epub 2015 Apr 30).
Deterding RR, DeBoer EM, Cidon MJ, Robinson TE, Warburton D, Deutsch GH, Young LR. Approaching clinical trials in childhood interstitial lung disease and pediatric pulmonary fibrosis. Am J Respir Crit Care Med. 2019;200(10):1219–27. https://doi.org/10.1164/rccm.201903-0544CI.
King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, Lederer DJ, Nathan SD, Pereira CA, Sahn SA, Sussman R, Swigris JJ, Noble PW, ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–92. https://doi.org/10.1056/NEJMoa1402582. (Epub 2014 May 18. Erratum in: N Engl J Med. 2014 Sep 18;371(12):1172).
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B, Collard HR, INPULSIS Trial Investigators. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–82. https://doi.org/10.1056/NEJMoa1402584. (Epub 2014 May 18. Erratum in: N Engl J Med. 2015 Aug 20;373(8):782).
Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, Richeldi L, Kolb M, Tetzlaff K, Stowasser S, Coeck C, Clerisme-Beaty E, Rosenstock B, Quaresma M, Haeufel T, Goeldner RG, Schlenker-Herceg R, Brown KK, INBUILD Trial Investigators. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718–27. https://doi.org/10.1056/NEJMoa1908681.
Deterding R, Young LR, DeBoer EM, Warburton D, Cunningham S, Schwerk N, Flaherty KR, Brown KK, Dumistracel M, Erhardt E, Bertulis J, Gahlemann M, Stowasser S, Griese M, InPedILD trial investigators. Nintedanib in children and adolescents with fibrosing interstitial lung diseases. Eur Respir J. 2023;61(2):2201512. https://doi.org/10.1183/13993003.01512-2022.
Gozal D, Kolb M. Nintedanib in chILD: a small step, yes… but at least a step forward in a marathon! Eur Respir J. 2023;61(2):2201797. https://doi.org/10.1183/13993003.01797-2022.
Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13(10):693–704. https://doi.org/10.1038/nrg3246. (Epub 2012 Sep 11. Erratum in: Nat Rev Genet. 2013 Mar;14(3):235).
Savage SA, Alter BP. Dyskeratosis congenita. Hematol Oncol Clin North Am. 2009;23:215–31.
Townsley DM, Dumitriu B, Liu D, Biancotto A, Weinstein B, Chen C, Hardy N, Mihalek AD, Lingala S, Kim YJ, Yao J, Jones E, Gochuico BR, Heller T, Wu CO, Calado RT, Scheinberg P, Young NS. Danazol treatment for telomere diseases. N Engl J Med. 2016;374(20):1922–31. https://doi.org/10.1056/NEJMoa1515319.
Mackintosh JA, Pietsch M, Lutzky V, Enever D, Bancroft S, Apte SH, Tan M, Yerkovich ST, Dickinson JL, Pickett HA, Selvadurai H, Grainge C, Goh NS, Hopkins P, Glaspole I, Reynolds PN, Wrobel J, Jaffe A, Corte TJ, Chambers DC. TELO-SCOPE study: a randomised, double-blind, placebo-controlled, phase 2 trial of danazol for short telomere related pulmonary fibrosis. BMJ Open Respir Res. 2021;8(1): e001127. https://doi.org/10.1136/bmjresp-2021-001127.
Kinting S, Höppner S, Schindlbeck U, Forstner ME, Harfst J, Wittmann T, Griese M. Functional rescue of misfolding ABCA3 mutations by small molecular correctors. Hum Mol Genet. 2018;27(6):943–53.
Kinting S, Li Y, Forstner M, Delhommel F, Sattler M, Griese M. Potentiation of ABCA3 lipid transport function by ivacaftor and genistein. J Cell Mol Med. 2019;23(8):5225–34. https://doi.org/10.1111/jcmm.14397.
Whitsett JA, Wert SE, Weaver TE. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu Rev Med. 2010;61:105–19.
Forstner M, Lin S, Yang X, Kinting S, Rothenaigner I, Schorpp K, Li Y, Hadian K, Griese M. High-content screening identifies cyclosporin A as a novel ABCA3-specific molecular corrector. Am J Respir Cell Mol Biol. 2022;66(4):382–90. https://doi.org/10.1165/rcmb.2021-0223OC.
Deng L, Chen Y, Hu X, Zhou J, Zhang Y. Case report: successful treatment of refractory interstitial lung disease with cyclosporine A and pirfenidone in a child with SLE. Front Immunol. 2021;4(12): 708463. https://doi.org/10.3389/fimmu.2021.708463.
Bonella F, Borie R. Targeted therapy for pulmonary alveolar proteinosis: the time is now. Eur Respir J. 2022;59(4):2102971. https://doi.org/10.1183/13993003.02971-2021.
Tazawa R, Ueda T, Abe M, Tatsumi K, Eda R, Kondoh S, Morimoto K, Tanaka T, Yamaguchi E, Takahashi A, Oda M, Ishii H, Izumi S, Sugiyama H, Nakagawa A, Tomii K, Suzuki M, Konno S, Ohkouchi S, Tode N, Handa T, Hirai T, Inoue Y, Arai T, Asakawa K, Sakagami T, Hashimoto A, Tanaka T, Takada T, Mikami A, Kitamura N, Nakata K. Inhaled GM-CSF for pulmonary alveolar proteinosis. N Engl J Med. 2019;381(10):923–32. https://doi.org/10.1056/NEJMoa1816216.
Trapnell BC, Inoue Y, Bonella F, Morgan C, Jouneau S, Bendstrup E, Campo I, Papiris SA, Yamaguchi E, Cetinkaya E, Ilkovich MM, Kramer MR, Veltkamp M, Kreuter M, Baba T, Ganslandt C, Tarnow I, Waterer G, Jouhikainen T, IMPALA Trial Investigators. Inhaled molgramostim therapy in autoimmune pulmonary alveolar proteinosis. N Engl J Med. 2020;383(17):1635–44. https://doi.org/10.1056/NEJMoa1913590.
Griese M, Panagiotou P, Manali ED, Stahl M, Schwerk N, Costa V, Douros K, Kallieri M, Urbantat RM, von Bernuth H, Kolilekas L, Morais L, Ramos A, Landwehr K, Knoflach K, Gothe F, Reiter K, Papaevangelou V, Kaditis AG, Kanaka-Gantenbein C, Papiris SA. Autoimmune pulmonary alveolar proteinosis in children. ERJ Open Res. 2022;8(1):00701–2021. https://doi.org/10.1183/23120541.00701-2021. (eCollection 2022 Jan).
Lenz D, Stahl M, Seidl E, Schöndorf D, Brennenstuhl H, Gesenhues F, Heinzmann T, Longerich T, Mendes MI, Prokisch H, Salomons GS, Schön C, Smith DEC, Sommerburg O, Wagner M, Westhoff JH, Reiter K, Staufner C, Griese M. Rescue of respiratory failure in pulmonary alveolar proteinosis due to pathogenic MARS1 variants. Pediatr Pulmonol. 2020;55(11):3057–66. https://doi.org/10.1002/ppul.25031. (Epub 2020 Sep 7).
Rips J, Meyer-Schuman R, Breuer O, et al. MARS variant associated with both recessive interstitial lung and liver disease and dominant Charcot–Marie–Tooth disease. Eur J Med Genet. 2018;61:616–20. https://doi.org/10.1016/j.ejmg.2018.04.005.
Hadchouel A, Drummond D, Pontoizeau C, Aoust L, Hurtado Nedelec MM, El Benna J, Gachelin E, Perisson C, Vigier C, Schiff M, Lacaille F, Molina TJ, Berteloot L, Renolleau S, Ottolenghi C, Tréluyer JM, de Blic J, Delacourt C. Methionine supplementation for multi-organ dysfunction in MetRS-related pulmonary alveolar proteinosis. Eur Respir J. 2022;59(4):2101554. https://doi.org/10.1183/13993003.01554-2021.
Hadchouel A, Wieland T, Griese M, Baruffini E, Lorenz-Depiereux B, Enaud L, Graf E, Dubus JC, Halioui-Louhaichi S, Coulomb A, et al. Biallelicmutations of methionyl-tRNA synthetase cause aspecific type of pulmonary alveolar proteinosisprevalent on Reunion island. Am J Hum Genet. 2015;96:826–31.
Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, Lee CR, DiMattia MA, Cowen EW, Gonzalez B, Palmer I, DiGiovanna JJ, Biancotto A, Kim H, Tsai WL, Trier AM, Huang Y, Stone DL, Hill S, Kim HJ, St Hilaire C, Gurprasad S, Plass N, Chapelle D, Horkayne-Szakaly I, Foell D, Barysenka A, Candotti F, Holland SM, Hughes JD, Mehmet H, Issekutz AC, Raffeld M, McElwee J, Fontana JR, Minniti CP, Moir S, Kastner DL, Gadina M, Steven AC, Wingfield PT, Brooks SR, Rosenzweig SD, Fleisher TA, Deng Z, Boehm M, Paller AS, Goldbach-Mansky R. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371(6):507–18.
Kanazawa N, Ishii T, Takita Y, Nishikawa A, Nishikomori R. Efficacy and safety of baricitinib in Japanese patients with autoinflammatory type I interferonopathies (NNS/CANDLE, SAVI, And AGS). Pediatr Rheumatol Online J. 2023;21(1):38.
Krutzke S, Rietschel C, Horneff G. Baricitinib in therapy of COPA syndrome in a 15-year-old girl. Eur J Rheumatol. 2020;7(Suppl1):S78–81.
Frémond ML, Nathan N. COPA syndrome, 5 years after: where are we? Jt Bone Spine. 2021;88(2): 105070.
Frémond ML, Legendre M, Fayon M, Clement A, Filhol-Blin E, Richard N, Berdah L, Roullaud S, Rice GI, Bondet V, Duffy D, Sileo C, Ducou le Pointe H, Begueret H, Coulomb A, Neven B, Amselem S, Crow Y, Nathan N. Use of ruxolitinib in COPA syndrome manifesting as life-threatening alveolar haemorrhage. Thorax. 2020;75(1):92–5.
Pelizzo G, et al. Mesenchymal stromal cells for the treatment of interstitial lung disease in children: a look from pediatric and pediatric surgeon viewpoints. Cells. 2021;10(12):3270.
Mahiny AJ, Dewerth A, Mays LE, Alkhaled M, Mothes B, Malaeksefat E, Loretz B, Rottenberger J, Brosch DM, Reautschnig P, Surapolchai P, Zeyer F, Schams A, Carevic M, Bakele M, Griese M, Schwab M, Nürnberg B, Beer-Hammer S, Handgretinger R, Hartl D, Lehr CM, Kormann MS. In vivo genome editing using nuclease-encoding mRNA corrects SP-B deficiency. Nat Biotechnol. 2015;33(6):584–6. https://doi.org/10.1038/nbt.3241. (Epub 2015 May 18).
Cooney AL, Wambach JA, Sinn PL, McCray PB Jr. Gene therapy potential for genetic disorders of surfactant dysfunction. Front Genome Ed. 2022;14(3): 785829. https://doi.org/10.3389/fgeed.2021.785829.
Presti S, Parisi GF, Papale M, Gitto E, Manti S, Leonardi S. Interstitial lung disease in children: “Specific Conditions of Undefined Etiology” becoming clearer. Children (Basel). 2022;9(11):1744. https://doi.org/10.3390/children9111744.
Kang MH, van Lieshout LP, Xu L, Domm JM, Vadivel A, Renesme L, et al. A lung tropic AAV vector improves survival in a mouse model of surfactant B deficiency. Nat Commun. 2020;11:3929. https://doi.org/10.1038/s41467-020-17577-8.
Hetzel M, et al. Function and safety of lentivirus-mediated gene transfer for CSF2RA-deficiency. Hum Gene Therapy Methods. 2017;28(6):318–29.
Brennan LC, O’Sullivan A, MacLoughlin R. Cellular therapy for the treatment of paediatric respiratory disease. Int J Mol Sci. 2021;22(16):8906.
Liu J, Peng D, You J, Zhou O, Qiu H, Hao C, Chen H, Fu Z, Zou L. Type 2 alveolar epithelial cells differentiated from human umbilical cord mesenchymal stem cells alleviate mouse pulmonary fibrosis through β-catenin-regulated cell apoptosis. Stem Cells Dev. 2021;30(13):660–70. https://doi.org/10.1089/scd.2020.0208.
Ahn SY, Chang YS, Lee MH, Sung SI, Lee BS, Kim KS, Kim AR, Park WS. Stem cells for bronchopulmonary dysplasia in preterm infants: a randomized controlled phase II trial. Stem Cells Transl Med. 2021;10(8):1129–37. https://doi.org/10.1002/sctm.20-0330.
Ehrenkranz RA, Walsh MC, Vohr BR, Jobe AH, Wright LL, Fanaroff AA, Wrage LA, Poole K, National Institutes of Child Health and Human Development Neonatal Research, Network. Validation of the National Institutes of Health consensus definition of bronchopulmonary dysplasia. Pediatrics. 2005;116(6):1353–60. https://doi.org/10.1542/peds.2005-0249.
Mucci A, Lopez-Rodriguez E, Hetzel M, Liu S, Suzuki T, Happle C, Ackermann M, Kempf H, Hillje R, Kunkiel J, Janosz E, Brennig S, Glage S, Bankstahl JP, Dettmer S, Rodt T, Gohring G, Trapnell B, Hansen G, Trapnell C, Knudsen L, Lachmann N, Moritz T. iPSC-derived macrophages effectively treat pulmonary alveolar proteinosis in Csf2rb-deficient mice. Stem Cell Reports. 2018;11(3):696–710. https://doi.org/10.1016/j.stemcr.2018.07.006.
Wu X, Gou H, Zhou O, Qiu H, Liu H, Fu Z, Chen L. Human umbilical cord mesenchymal stem cells combined with pirfenidone upregulates the expression of RGS2 in the pulmonary fibrosis in mice. Respir Res. 2022;23(1):270. https://doi.org/10.1186/s12931-022-02192-6.
Vece TJ, Wambach JA, Hagood JS. Childhood rare lung disease in the 21st century: “-omics” technology advances accelerating discovery. Pediatr Pulmonol. 2020;55(7):1828–37.
Deterding RR, Wagner BD, Harris JK, DeBoer EM. Pulmonary aptamer signatures in children’s interstitial and diffuse lung diseases. Am J Respir Crit Care Med. 2019;200(12):1496–504.
Nathan N, Corvol H, Amselem S, Clement A. Biomarkers in interstitial lung diseases. Paediatr Respir Rev. 2015;16:219–24.
Kilinc AA, Arslan A, Yildiz M, Kucur M, Adrovic A, Barut K, Sahin S, Cokugras H, Kasapcopur O. Serum KL-6 level as a biomarker of interstitial lung disease in childhood connective tissue diseases: a pilot study. Rheumatol Int. 2020;40(10):1701–6.
Zhao Z, Chen X, Dowbaj AM, et al. Organoids. Nat Rev Methods Primers. 2022;2:94. https://doi.org/10.1038/s43586-022-00174-y.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Author contributions
The authors contributed equally to the work. All authors have critically revised and approved the final version of the manuscript.
Availability of data
Not applicable.
Funding
Open access funding provided by Università degli Studi di Padova within the CRUI-CARE Agreement. No sources of financial assistance were used to conduct the study/analysis described in the article and/or used to assist with the preparation of the manuscript.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bernardinello, N., Griese, M., Borie, R. et al. Emerging Treatments for Childhood Interstitial Lung Disease. Pediatr Drugs 26, 19–30 (2024). https://doi.org/10.1007/s40272-023-00603-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-023-00603-9