
Abstract
Childhood absence epilepsy (CAE) is a common pediatric epilepsy syndrome with distinct seizure semiology, electroencephalography (EEG) features, and treatment. A diagnosis of CAE can be obtained during an office visit with a careful history, physical exam including prolonged hyperventilation, and a routine EEG. The treatment of choice for CAE with absence seizures only is ethosuximide. Valproic acid and lamotrigine are also effective treatments for many patients, but when compared to ethosuximide, valproic acid has more adverse effects and lamotrigine is less effective. Attention to predictors of response to treatment, including clinical, electrographic, and genetic factors, is increasing. Refractory CAE occurs in fewer than half of patients, and treatment strategies are available, though efficacy data are lacking. Careful assessment and treatment of psychosocial comorbidities is essential in caring for patients with CAE.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Ethosuximide, valproate, and lamotrigine each have a role as treatments for childhood absence epilepsy. |
Care of children with childhood absence epilepsy should go beyond drug treatment and address associated behavioral and psychosocial conditions as well. |
1 Introduction
Childhood absence epilepsy (CAE) is a well-known and common pediatric epilepsy syndrome affecting 10–17% of all children with epilepsy [1]. Seizures usually begin between 4 and 10 years of age, with a peak around 6–7 years, in a previously healthy and typically developing child. CAE occurs more often in girls than in boys [2]. Seizures occur many times daily and consist of brief staring spells, sometimes with rhythmic eye blinking or motor automatisms, lasting seconds, with immediate return to the baseline level of awareness and activity. On electroencephalography (EEG), seizures are characterized by a highly recognizable pattern of generalized (bilateral, symmetric and synchronous) 3 Hz spike and wave discharges (see Fig. 1). This article reviews the current evidence and practical considerations for treating CAE.

A typical absence seizure on electroencephalogram, characterized by 3 Hz generalized spike wave discharges, with abrupt onset and offset, lasting several seconds
2 Diagnosis
CAE can be diagnosed during an outpatient clinic visit with a careful history, physical exam including hyperventilation, and a routine EEG [3]. A history of staring is common in children, and staring spells are not always absence seizures or other seizure types. Absence seizures typically cause sudden cessation of activity that is easily recognizable. Elements of history that suggest absence seizures include sudden loss of facial expression; repetitive movements (such as lip smacking or blinking); events occurring regardless of setting (not only while watching television or in the car for example, where staring is more likely behavioral); and events that occur in the midst of an activity such as chewing or playing. Compared to staring spells that are not seizures, absence seizures are more likely to be noted by a professional such as a teacher rather than a parent, and are not responsive to external stimuli such as touch [4].
Other important elements of history include other seizure types, developmental history, and age of onset. Other seizure types (for example, generalized tonic–clonic, myoclonic, or atonic) may distinguish a diagnosis of CAE from other epilepsy types in which absence seizures might be prominent (such as myoclonic astatic epilepsy in a young child, and juvenile myoclonic epilepsy in an older child) [5]. Development is typically grossly normal in a child in CAE, though comorbid attentional deficits or other subtle behavioral or cognitive impairments might be present at onset [6, 7]. In a child with early onset absence epilepsy (onset under the age of 4 years) or a child with absence seizures and an abnormal neurologic exam or substantial developmental impairments, considering the possibility of an underlying diagnosis of glucose transporter 1 deficiency syndrome (GLUT1 DS) is particularly important because more targeted therapy, specifically the ketogenic diet, can be pursued [8]. A similar but distinct syndrome where absence seizures predominate is juvenile absence epilepsy (JAE), which occurs in children 10–15 years old and is characterized by less frequent absence seizures (sometimes occurring a few times daily or less than daily) as well as the occurrence of generalized tonic–clonic seizures in 80% or more of children with the disorder [9].
An essential component of the physical exam of a child with suspected absence seizures is hyperventilation. Hyperventilation provokes the occurrence of absence seizures; asking a child to blow on a pinwheel or a strip of paper for 2–3 min during a clinic visit is a simple way to elicit an absence seizure. A routine EEG, which includes hyperventilation, will confirm a clinically suspected diagnosis in an untreated patient [10].
3 First-Line Treatment
Three antiepileptic medications have been commonly used as first-line agents for CAE—ethosuximide (ETX), valproic acid (VPA), and lamotrigine (LTG) [11]—but not until recently has robust evidence emerged on the comparative effectiveness of these drugs. The 2010 Childhood Absence Epilepsy study provided class I evidence for the use of ETX as the optimal initial treatment for CAE [12]. This double-blind, randomized, controlled trial enrolled 453 children between the ages of 4 and 10 years with newly diagnosed and untreated CAE, who had absence seizures only and no history of generalized tonic–clonic seizures. Participants were randomly assigned to treatment with ETX (n = 156), VPA (n = 148), or LTG (n = 149). The primary outcome measure was freedom from failure, defined as the absence of clinically apparent seizures (based on parent report) or electrographic seizures at 16–20 weeks of treatment, and the absence of treatment-limiting adverse effects. Seven participants were ineligible after randomization, and the analysis cohort included 446 children. The freedom-from-failure rates for ETX and VPA were similar (53% and 58%, respectively, p = 0.35), and were higher than the freedom-from-failure rate for LTG (29%, p < 0.001 when compared to both ETX and VPA). Discontinuation rates due to adverse events were not significantly different between the three drugs. Post-treatment attention dysfunction, measured using the Connor’s Continuous Performance Test, was more common with VPA than ETX (49% and 33%, respectively, p = 0.03). The study concluded that ETX and VPA are more effective than LTG for initial monotherapy of CAE, and of these two, ETX is the most optimal treatment because it is associated with a lower risk of adverse effects on attention.
On 12-month follow-up of the original cohort, ETX and VPA remained superior to LTG in freedom-from-failure rates, but the VPA group had higher rates of drug discontinuation and attention dysfunction [13]. VPA-associated weight gain also became apparent at 12 months of follow-up. Across all treatment groups, slightly more than one-third of all participants remained on the initial treatment. Almost two-thirds of the 125 subjects with treatment failure due to persistent seizures were in the LTG group. The largest subgroup (42%) of the 115 subjects who discontinued treatment due to adverse effects was in the VPA group.
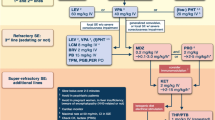
Therefore, based on the CAE trial, ETX is the drug of choice as initial monotherapy for CAE, when absence seizures are the only seizure type [14], but there are specific treatment considerations for each drug, which are discussed below. A proposed algorithm for treatment of CAE is contained in Fig. 2.

Proposed treatment algorithm for childhood absence epilepsy. AE adverse effects, ETX ethosuximide, GTC generalized tonic–clonic, LTG lamotrigine, VPA valproic acid
3.1 Ethosuximide
ETX is 2-ethyl-2-methyl-succinamide, in clinical use since 1958. It has a narrow clinical spectrum—it suppresses absence seizures, but not generalized tonic–clonic seizures or focal onset seizures. The available evidence suggests that the main mechanism of action of ETX is blockade of transient, low-threshold calcium currents produced by T-type calcium channels in thalamic neurons, thereby preventing the synchronized firing of corticothalamic neurons that produce the spike wave discharges of absence seizures [15].
Oral ingestion of ETX in capsule or syrup form leads to rapid absorption from the gastrointestinal tract and near-complete bioavailability, with peak levels occurring 3–5 h after intake [16]. Metabolism is primarily hepatic; the principal cytochrome p450 oxidase involved is CYP3A. The elimination half-life is 30–40 h in children, with linear kinetics and no auto-induction—thus, 7–10 days of daily intake of a stable dose leads to steady-state levels [17]. ETX is susceptible to the effects of enzyme-inducing and enzyme-inhibiting antiepileptic medications [18]. For example, co-administration of the enzyme inhibitor VPA may lead to elevated serum and brain ETX levels, though their additive anticonvulsant effects may be greater than any additive neurotoxic effects [19].
Although gastrointestinal side effects are by far the most commonly reported side effects with ETX, they are rarely the sole reason for drug discontinuation [12]. These benign and fully reversible side effects include abdominal discomfort, vomiting, diarrhea, and hiccups. Measures that may improve gastrointestinal discomfort include taking the medication with food and switching from liquid to the capsule form. The neurologic side effects and adverse reactions of ETX can include headaches, sedation, fatigue, insomnia, ataxia, or extrapyramidal reactions. Rarely, clinically apparent behavioral disturbances may occur, including nervousness, irritability, depression, hallucinations, and even psychosis. Blood dyscrasias occur rarely with ETX; clinical alertness to possible signs and symptoms is a more useful and more cost effective way of recognizing these rare adverse drug reactions than routine monitoring of blood counts. ETX-induced lupus is a rare adverse drug reaction, appearing in the medical literature mostly as single case reports [20]. Symptoms typically resolve with cessation of ETX, but recovery may be prolonged. Other idiosyncratic adverse drug reactions include allergic rashes, which typically resolve with medication withdrawal.
ETX can be taken orally as a syrup (250 mg/mL) or a capsule (250 mg). The initial target dosage of ETX is 20–30 mg/kg/day, usually divided into two doses. The initial dosage is typically around 10 mg/kg/day, with upward titration to the target every week (Table 1). If capsules are preferred but the patient’s weight requires 125-mg increments, the capsules can be frozen and then cut in half. Neither routine monitoring of blood levels nor routine monitoring of blood counts, liver function, or other laboratory tests is recommended. Assessing potential adverse effects with specific questions at a follow-up visit is the most effective way of identifying issues requiring dose adjustment, further laboratory tests, or drug discontinuation [21].
3.2 Valproate
VPA is the drug of choice as initial monotherapy in CAE when a generalized tonic–clonic seizure has occurred, because ETX is not effective against seizure types beyond absence seizures. VPA is also preferred as a second treatment when ETX fails, whether due to efficacy or tolerability reasons, based on a crossover extension phase of the CAE study [22]. In this second trial, 209 participants from the original trial were enrolled and randomized to ETX, VPA, or LTG as a second monotherapy treatment after the first drug had failed. The results were similar to those of the original trial: ETX and VPA were superior to LTG regarding freedom-from-failure rate at a 16- to 20-week time point (63% and 65%, respectively, compared to 45%, p = 0.051) and at a 12-month time point (57% and 49%, respectively, compared to 36%, p = 0.062).
VPA is a broad-spectrum antiepileptic medication that has been used in the United States since 1978, and appears to have multiple mechanisms of action, including raising gamma-aminobutyric acid (GABA) levels in the brain, blocking voltage-sensitive sodium channels, and activating calcium-dependent potassium conductance [23]. However, the specific mechanism by which VPA prevents absence seizures is unknown.
The rate of absorption of VPA from the gastrointestinal tract is variable depending on the formulation. With liquid or uncoated tablets, peak levels occur around 1–2 h. Time to peak levels after sprinkle capsules can be longer—up to 4 h. VPA is highly bound to serum proteins and has a relatively small volume of distribution, meaning VPA has a lower preference for binding to proteins outside of the blood compartment. As the total concentration of VPA increases, the free fraction increases. The half-life of VPA given as monotherapy in children is around 12 h, and is lower when given with other antiepileptic drugs that induce liver metabolism [17].
VPA has a number of potential adverse effects; some are dose related and some are idiosyncratic, but a higher dose may increase the risk. Neurologic side effects in children with CAE are uncommon, but may include a fine high-frequency tremor. Altered mental status can be a VPA side effect, but would be unusual in children with CAE on typical VPA doses. Increased appetite and weight gain may occur and rarely constitute treatment-limiting side effects with long-term VPA treatment. Rare gastrointestinal adverse drug reactions include pancreatitis and the well-known but rare occurrence of hepatic failure [24]. Because the risk factors for hepatic failure (age less than 2 years, polypharmacy, polymerase gamma 1 mutation) are inconsistent with or rare in CAE, the risk of this adverse drug reaction in CAE is extremely low; if VPA is needed, then the potential benefit likely outweighs the risk. Liver failure is not typically identified through routine testing of transaminases; a better method is to counsel parents on signs of illness, including persistent vomiting without diarrhea and jaundice, which should prompt immediate medical attention. Pancreatitis is also a rare, idiosyncratic adverse reaction, and is reversible with cessation of VPA. Thrombocytopenia is a dose-dependent side effect of VPA, and thus may be dose limiting. VPA may also cause platelet dysfunction, without thrombocytopenia, or possibly fibrinogen depletions and coagulation factor deficiencies. Therefore, discontinuing VPA about 1 month prior to elective surgery is recommended (particularly if the surgery has a risk of substantial blood loss). Potential metabolic and endocrine side effects/adverse reactions include carnitine depletion, hyperammonemia, hyperinsulinemia, polycystic ovarian syndrome, vitamin D deficiency and decreased bone mineral density, and hypothyroidism. Hair loss is an idiosyncratic adverse reaction, and can sometimes be improved with the use of a selenium-containing shampoo or oral selenium supplementation. Finally, while it is not typically a consideration in the CAE age range, VPA is known to have a higher teratogenicity risk than other antiepileptic drugs, and a higher risk of neurodevelopmental impairments including autism after fetal exposure [25]. While there are no current blanket recommendations for weaning a child with CAE off of VPA when she reaches puberty due to the risk of teratogenicity, conversations about these risks and avoiding pregnancy while taking VPA are generally recommended.
VPA has multiple oral formulations including syrup, sprinkle capsules, tablets, and extended-release tablets. For children in the CAE age group, the most common formulation administered is likely sprinkle capsules, which can be swallowed directly or twisted open and poured into a spoonful of a puree/soft food for administration (such as apple sauce, yogurt, or ice cream). The initial target dosage of VPA for CAE is typically 20–30 mg/kg/day divided into two doses, but the dosage may be increased as needed up to 60 mg/kg/day. The target serum level is the level at which absence seizures stop occurring, both by parent report and by EEG—that is, there is no need to increase the dose to reach an arbitrarily set serum level. However, for most patients, the therapeutic range is 50–100 µg/dL, and in the absence of side effects, serum levels can be pushed even higher to 120 µg/dL, or perhaps even 150 µg/dL. A key dose-dependent side effect of VPA is thrombocytopenia. In our practice, a platelet count below 100,000 is the threshold for avoiding further increases in VPA in most circumstances. A typical titration schedule would consist of weekly increases from 10 mg/kg/day to 15 mg/kg/day, to 20–25 mg/kg/day.
While many clinicians assess a complete blood count and liver transaminases at regular intervals, typically every 6 months, during VPA use, there is little evidence to show that routine testing will uncover clinically significant laboratory abnormalities in the absence of symptoms [26, 27]. In addition, sometimes routine testing reinforces the misperception of patients and families that VPA use causes cumulative liver damage over time. We recommend assessing a complete blood count at least once after the goal dose of VPA is reached to evaluate for thrombocytopenia, and if serum levels are pushed above 100 µg/dL, though there is very limited evidence for parameters for testing [28]. In addition, symptoms of liver dysfunction including nausea, emesis, lethargy, or altered mental status should prompt evaluation of liver enzymes and a serum ammonia level.
3.3 Lamotrigine
Prior to the CAE trial, an expert opinion survey identified LTG as a first-line treatment for CAE [11] along with VPA and ETX, though it was less commonly used than the other two. Multiple open-label prospective studies evaluating LTG as initial monotherapy for CAE reported seizure freedom rates in the 50–80% range [29, 30]. However, as discussed in Sect. 3, in the double-blinded randomized design of the CAE trial, LTG fared less well, with approximately one-third of patients achieving seizure freedom without treatment-limiting side effects.
While our practice is to use ETX as the first monotherapy, LTG can be considered as second monotherapy if ETX fails and if VPA is a less appealing choice for a specific patient. LTG, in clinical use since the mid-1990s, is a voltage- and use-dependent sodium channel blocker, but unlike other sodium channel-blocking agents, it must have one or more other mechanisms of action that explain its efficacy in epilepsies with generalized mechanisms of seizure onset which have not yet been clearly characterized. LTG, taken orally, is well absorbed and has nearly 100% bioavailability. LTG undergoes hepatic metabolism and renal excretion in the form of glucuronide conjugates [31].
The key downside to LTG is that initial titration must proceed very slowly to lower the risk of Stevens-Johnson syndrome (SJS), a severe and sometimes life-threatening rash affecting skin and mucus membranes. Because the liver enzyme-inhibiting effects of VPA slows LTG metabolism, titration of LTG in a patient also on VPA begins at an even lower dose, proceeds at a slower rate, and reaches a lower target than in a patient on no other medication.
Initiating LTG in a child as first- or second-line treatment in CAE requires careful counseling on the titration schedule and risk of rash. Although the frequency of allergic skin reactions is actually no higher on LTG compared to carbamazepine or phenytoin, Food and Drug Administration (FDA) labeling of LTG includes a black-box warning about SJS [32]. If a child does develop a rash on LTG that is not clearly attributable to some other cause (such as a contact dermatitis after poison ivy exposure), then LTG should be discontinued and another therapy chosen. Overall LTG is typically well tolerated and appears to have a lower risk of attention and other cognitive side effects compared to ETX and VPA [33]. LTG does have dose-dependent side effects similar to other sodium channel-blocking agents, including blurred vision, diplopia, dizziness, and ataxia [34].
For a child on no other medication, LTG is typically initiated at a dosage of 0.6 mg/kg/day for 2 weeks, then 1.2 mg/kg/day for 2 weeks, and then increased by 0.6 mg/kg/day every 1–2 weeks until a goal dosage of 5–12 mg/kg/day is reached. The dosage forms of LTG include chewable 2-mg, 5-mg, 25-mg tablets, which typically allow for appropriate titration and maintenance dosing even in very young children. If a child will not chew a chewable tablet, the tablets are dissolvable in a small amount of liquid for oral intake.
Serum levels of LTG considered to be in the therapeutic range are between 5 and 15 µg/mL, though dose-dependent side effects may emerge when levels exceed 10–12 µg/mL. Weight-normalized clearance of LTG is higher in children than in adults, so higher mg/kg doses may be needed in children compared to adults to achieve similar serum levels [35, 36].
4 Treatment Options in Medication-Resistant CAE
To discuss approaches to treatment when CAE is resistant to initial medication choices, it may be useful to walk through some scenarios. Evidence for treatment beyond the first and second monotherapy choices is not robust and relies on common practice and experience.
Consider the case of a typically developing 6-year-old girl referred for evaluation after 2 months of episodes, seen by her parents, friends, and teachers, of a behavioral pause, staring, and repetitive mouth movements, lasting only seconds, and with immediate return to her baseline level of awareness and activity. As expected, an EEG records one typical spontaneous seizure and two seizures provoked by hyperventilation, each characterized by 3 Hz generalized spike and wave discharges on a normal background. She initiates ETX with dose titration to 250 mg twice daily, which is about 30 mg/kg/day. At a follow-up visit about a month after she has been on the full dose, she continues to have absence seizures on EEG. Her dose is titrated further, and further again after her parents continue to report seizures, until she reaches a dose of 50 mg/kg/day, and she is having intolerable abdominal discomfort after every dose, leading to decreased appetite and slight weight loss.
At this point, the treating clinician would consider switching from ETX to VPA or LTG. Based on the higher likelihood of efficacy, the clinician chooses VPA, and gradually discontinues ETX. What is the best course of action if VPA does not lead to seizure freedom? We recommend trying a combination of VPA and LTG. In general, combinations of medications can be considered when drug failure is due to lack of efficacy; lack of tolerability requires lowering the dose or stopping the medication altogether. Although there are important pharmacokinetic interactions to consider, VPA and LTG in combination may have a synergistic pharmacodynamic interaction—that is, they may be substantially more effective used together than either one apart, particularly in CAE [37, 38]. Because VPA inhibits LTG metabolism, initiation of LTG when a patient is already on VPA begins at a lower dose, increases in smaller increments, and reaches a lower target dose than dosing in the absence of VPA.
What if VPA fails for lack of tolerability? We recommend a trial of LTG monotherapy; although it is less effective than VPA or ETX, one-third of patients initially randomized to LTG in the CAE trial did experience freedom from failure, and one-third of those randomized to LTG after first drug failure also had freedom from failure at 12 months [22, 33]. There is no established way to convert from VPA to LTG. Our practice for absence epilepsy consists of a 3- to 4-week taper of VPA, simultaneous initiation of LTG on the dosing schedule for concurrent VPA until VPA has been discontinued for a week or longer, and then continued upward titration of LTG by around 0.6 mg/kg per week to a dosage in the 6–8 mg/kg/day range. Temporary use of a benzodiazepine to bridge the conversion is an option, but not necessary in all cases.
If LTG is not efficacious or only partly efficacious at this point, the next treatment to consider is clobazam. Clobazam has US FDA regulatory approval for the treatment of seizures in Lennox-Gastaut syndrome, but is widely used for other indications as well, and has been for many years in countries where it has long been available [39, 40]. Clobazam allosterically activates GABAA receptors, and it binds less avidly to subunits that mediate sedative effects than other benzodiazepines [41]. While there is little to no literature on the use of clobazam in treatment-resistant CAE specifically, it can be considered when first- and second-line treatments fail.
Although the evidence for efficacy in CAE is scant and variably supportive, topiramate and zonisamide may be considered when other treatments fail [42,43,44]. The ketogenic diet has also been used successfully in children with treatment-resistant CAE [45, 46]. Evidence for the usefulness of levetiracetam in childhood absence epilepsy has been mixed, with a few reports of its modest efficacy and one report of absence seizure aggravation by levetiracetam [47,48,49].
Gabapentin is not effective for absence seizures [50]. Oxcarbazepine, carbamazepine, phenytoin, phenobarbital, tiagabine, and vigabatrin may worsen absence seizures or cause absence status epilepticus and should be avoided [51].
5 Prognosis: Treatment Effectiveness
Reported rates of response to medication in CAE vary, ranging from 60% to 95%, depending on the definition of the CAE population studied, duration of the observation period, and how outcomes were measured [52, 53]. In the CAE trial, about half of patients treated with ETX or VPA initially achieved seizure freedom and remained on the medication after 1 year. When the first drug was not successful, about half were successfully treated with ETX or VPA as the second medication [22]. Thus, overall, approximately two-thirds to three-quarters of children newly presenting with CAE responded to the first or second treatment. Similarly, in a population-based study of 86 patients with absence epilepsy, of whom 75 had CAE (and the other 11 JAE), 65% of the CAE patients were successfully treated with the first medication. Of those treated with VPA first, 77% responded compared to 55% of those treated with ETX [52].
Several factors have been identified in the CAE trial and prior observational studies which may predict response to treatment. Older studies of absence epilepsies sometimes studied both typically developing children with what is now considered classic CAE and children with other seizure types or underlying neurologic disorders as well; abnormal development or neurologic status at the onset of seizures is a poor prognostic sign, and these children represent different epilepsy syndromes with different mechanisms. Of those with a more tightly defined CAE (age of onset between 4 and 10 years, normal neurologic status at onset, 3 Hz spike and wave on initial EEG), two key factors predicting less favorable outcome include failure of the first medication and the occurrence of a generalized tonic–clonic seizure before or during initial treatment [52, 53].
EEG features at baseline which are atypical for CAE, such as background slowing, other background abnormalities, or spike wave discharges slower than 3 Hz, are associated with poorer response to treatment and less favorable long-term outcomes [53]. Photosensitivity has also been associated with a higher likelihood of generalized tonic–clonic seizures at onset or later in the course [53, 54]. In children entering the CAE trial, whose baseline EEG had to meet strict entry criteria, longer seizure duration on the initial EEG was associated with better drug response [55]. Specifically, patients whose shortest seizure lasted longer than 7.5 s were more likely to respond to initial treatment than those whose shortest seizure lasted less than 7.5 s. However, patients who had any seizures lasting longer than 20 s were more likely to score poorly on measures of attention.
Baseline seizure semiology characteristics may also help predict response to initial treatment. Absences with marked myoclonic components are uncharacteristic of CAE and may protend a poorer response to treatment [53, 56]. A video EEG analysis of seizures at baseline in the CAE trial noted that no individual semiologic features were associated with differences in treatment outcome, but seizures clustered into four types based on semiology. Patients with the type characterized by a pause/stare (a nearly universal feature), eye involvement (including opening, blinking, and myoclonia), but no motor automatisms were associated with a higher likelihood of failure regardless of treatment arm [57].
The impact of individual genetic characteristics on treatment response is not yet well-understood. The pathophysiology of absence epilepsy involves alterations in the function of T-type calcium channels which mediate the thalamocortical rhythms that are disrupted in absence seizures. A study evaluating the link between genetic variants in genes encoding T-type calcium channel subunits and treatment response in the CAE trial revealed that the presence of the CACNA1H missense variant (rs61734410/P640L), though not in itself a CAE disease-causing variant, was more often associated with lack of response to ETX, a finding supported by accompanying in vitro neurophysiologic studies [58]. In addition, variants in the genes encoding P-glycoprotein, a drug efflux transporter located in the intestine and membranes of brain capillary endothelial cells, may also lead to differential drug response. The presence of the minor allele (T) in the ABCB1 missense variant (rs2032582, T2677G, S893A) was more often associated with lack of response to LTG, a drug which is known to be a substrate of P-glycoprotein.
6 Prognosis: Long-Term Outcomes
A well-recognized feature of CAE is the high rate of long-term seizure remission compared to other childhood onset epilepsies. However, the occurrence of long-term remission is still unclear because studies have used varying definitions of CAE and have varied in length of follow-up. In studies reporting on more narrowly defined CAE, remission rates have ranged from 56 to 95% after multiple years of observation time [1, 59,60,61]. Remission typically occurs between 10 and 14 years of age, but may occur at younger ages as well.
Factors associated with increased risk of persistent seizures or later occurrence of seizures (such as generalized tonic–clonic seizures or myoclonic seizures) include generalized tonic–clonic seizures or other seizure types at presentation or during the first months of treatment of CAE, absence status epilepticus, EEG abnormalities such as background slowing, and family history of generalized tonic–clonic seizures [53, 62]. Approximately 15% of children with CAE will develop juvenile myoclonic epilepsy during adolescence, with generalized tonic seizures, myoclonic seizures, and sometimes persistent absence seizures [59]. Although antiepileptic medications are generally not considered disease modifying (that is, the medications treat seizures, but have no effect on long-term prognosis for remission), there is some evidence that suggests initial treatment with ETX is more often associated with remission than initial treatment with VPA [63].
CAE is often thought of as a benign, self-limited epilepsy, but there are significant cognitive, behavioral and psychiatric comorbidities that must be detected early and addressed separately. Baseline rates of inattention are 30–40% and do not improve with successful treatment of seizures [6]. One quarter may have subtle cognitive or language impairments, and more than half are found to have psychiatric diagnoses when formally assessed, particularly attention deficit hyperactivity disorder (ADHD) and anxiety [64]. Children with absence epilepsy compared to children with a non-neurologic chronic disease are less likely to graduate high school, and are more likely to be under-employed. These children are more likely to require special classes and repeat a grade, more likely to have an unplanned pregnancy, and more likely to report psychiatric and emotional problems [65]. We recommend screening for behavioral, psychologic, and educational issues at the time of diagnosis, with referral for further behavioral health services as needed. Follow-up visits, even when seizures are well controlled on medication, should include discussion of potential comorbid conditions.
7 Weaning off Medication
The current widespread practice in CAE is to maintain patients on antiepileptic medication for 2 years after achieving seizure freedom, which is the common practice for childhood onset epilepsy in general [66]. At the end of these 2 years, an EEG can help determine risk of relapse with medication withdrawal. It is important that this EEG includes hyperventilation and captures sleep, both of which can bring out EEG abnormalities not seen during quiet wakefulness. While a brief abnormal discharge during sleep may not preclude drug withdrawal, presence of generalized spike-wave discharges during hyperventilation indicates a high risk of recurrence off of medication, and might prompt continued therapy for another year or longer. The utility of another EEG after medication discontinuation for determining the risk of short-term seizure recurrence or long-term prognosis is an unanswered question in CAE, though there is some evidence for this practice in childhood epilepsy more generally [67]. In our experience, some clinicians obtain an EEG after medication discontinuation in patients whose absence seizures were difficult for parents or teachers to recognize in the past, or in patients who are nearing driving age.
8 Conclusion
CAE is a common pediatric epilepsy syndrome with recognizable features and natural history. The syndrome is the subject of one of the few comparative effectiveness, randomized, controlled treatment trials carried out in pediatric epilepsy, which provided information on not only optimal treatment, but also on factors affecting response to treatment. In a patient without generalized tonic-clonic seizures at onset, ETX remains the initial treatment of choice. Many but not all patients with CAE will become seizure free on the first or second medication. Many but not all patients with CAE will also reach terminal remission (seizure freedom after medication has been discontinued), but remain at risk over a lifetime for psychosocial comorbidities.
References
Loiseau P, Duché B, Pédespan JM. Absence epilepsies. Epilepsia. 1995;36:1182–6.
Waaler PE, Blom BH, Skeidsvoll H, Mykletun A. Prevalence, classification, and severity of epilepsy in children in Western Norway. Epilepsia. 2000;41:802–10.
L C et al. Differential diagnosis of staring spells in children: a video-EEG study. Pediatr Neurol. 14:199–202 (1996).
Rosenow F, et al. Staring spells in children: descriptive features distinguishing epileptic and nonepileptic events. J Pediatr. 1998;133:660–3.
Holmes GL, McKeever M, Adamson M. Absence seizures in children: clinical and electroencephalographic features. Ann Neurol. 1987;21:268–73.
Masur D, et al. Pretreatment cognitive deficits and treatment effects on attention in childhood absence epilepsy. Neurology. 2013;81:1572–80.
Shinnar RC, et al. Pretreatment behavior and subsequent medication effects in childhood absence epilepsy. Neurology. 2017;89:1698–706.
Larsen J, et al. The role of SLC2A1 mutations in myoclonic astatic epilepsy and absence epilepsy, and the estimated frequency of GLUT1 deficiency syndrome. Epilepsia. 2015;56:e203–8.
Reutens DC, Berkovic SF. Idiopathic generalized epilepsy of adolescence: are the syndromes clinically distinct? Neurology. 1995;45:1469–76.
Sadleir LG, Farrell K, Smith S, Connolly MB, Scheffer IE. Electroclinical features of absence seizures in childhood absence epilepsy. Neurology. 2006;67:413–8.
Wheless JW, Clarke DF, Carpenter D Treatment of pediatric epilepsy: expert opinion, 2005. J Child Neurol. 20:59–60 (2005).
Glauser TA, et al. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. N Engl J Med. 2010;362:790–9.
Glauser TA, et al. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy: initial monotherapy outcomes at 12 months. Epilepsia. 2013;54:141–55.
Kanner AM, et al. Practice guideline update summary: efficacy and tolerability of the new antiepileptic drugs I: treatment of new-onset epilepsy: report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2018;91:74–81.
Coulter DA, Huguenard JR, Prince DA. Characterization of ethosuximide reduction of low-threshold calcium current in thalamic neurons. Ann Neurol. 1989;25:582–93.
Buchanan RA, Fernandez L, Kinkel AW Absorption and elimination of ethosuximide in children. J Clin Pharmacol J. 9:393–8.
Battino D, Estienne M, Avanzini G Clinical pharmacokinetics of antiepileptic drugs in paediatric patients. Part I: phenobarbital, primidone, valproic acid, ethosuximide and mesuximide. Clin. Pharmacokinet. 29, 257–286 (1995).
Smith GA, McKauge L, Dubetz D, Tyrer JH, Eadie MJ. Factors influencing plasma concentrations of ethosuximide. Clin Pharmacokinet. 1979;4:38–52.
Bourgeois BF. Combination of valproate and ethosuximide: antiepileptic and neurotoxic interaction. J Pharmacol Exp Ther. 1988;247:1128–32.
Crespel A, Velizarova R, Agullo M, Gélisse P. Ethosuximide-induced de novo systemic lupus erythematosus with anti-double-strand DNA antibodies: a case report with definite evidence. Epilepsia. 2009;50:2003.
Wyllie E, Wyllie R. Routine laboratory monitoring for serious adverse effects of antiepileptic medications: the controversy. Epilepsia. 1990;32:S74–9.
Cnaan A, et al. Second monotherapy in childhood absence epilepsy. Neurology. 2017;88:182–90.
Johnston D. Valproic acid: update on its mechanisms of action. Epilepsia. 1984;25:S1–4.
Zaccara G, Franciotta D, Perucca E. Idiosyncratic adverse reactions to antiepileptic drugs. Epilepsia. 2007;48:1223–44.
Tomson T, et al. Valproate in the treatment of epilepsy in girls and women of childbearing potential. Epilepsia. 2015;56:1006–19.
Camfield C, Camfield P, Smith E, Tibbles JAR. Asymptomatic children with epilepsy: little benefit from screening for anticonvulsant-induced liver, blood, or renal damage. Neurology. 1986;36:838–41.
AZ C, BD M, EC W Specific safety and tolerability considerations in the use of anticonvulsant medications in children. Drug Healthc Patient Saf 4:39–54 (2012).
Buoli M, Serati M, Botturi A, Altamura AC The risk of thrombocytopenia during valproic acid therapy: a critical summary of available clinical data. Drugs 18, (2018).
Frank LM, et al. Lamictal (lamotrigine) monotherapy for typical absence seizures in children. Epilepsia. 1999;40:973–9.
Holmes GL, et al. Lamotrigine monotherapy for newly diagnosed typical absence seizures in children. Epilepsy Res. 2008;82:124–32.
Rambeck B, Wolf P. Lamotrigine clinical pharmacokinetics. Clin Pharmacokinet. 1993;25:433–43.
Arif H, et al. Comparison and predictors of rash associated with 15 antiepileptic drugs. Neurology. 2007;68:1701–9.
Glauser TA, et al. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. N Engl J Med. 2010;362:790–9.
ES et al Lamotrigine in treatment of 120 children with epilepsy. Epilepsia 35, 359–367 (1994).
Chen C, Casale EJ, Duncan B, Culverhouse EH, Gilman J. Pharmacokinetics of lamotrigine in children in the absence of other antiepileptic drugs. Pharmacotherapy. 1999;19:437–41.
van Dijkman SC, de Jager NCB, Rauwé WM, Danhof M. Effect of age-related factors on the pharmacokinetics of lamotrigine and potential implications for dose optimisation in epilepsy patients. Clin. Pharmacokinet. 2018;1–15:56. https://doi.org/10.3303/cet0917181.
Brodie MJ, Yuen AWC. Lamotrigine substitution study: evidence for synergism with sodium valproate? Epilepsy Res. 1997;26:423–32.
Hirsch E, Panayiotopoulos C. Childhood absence epilepsy and related syndromes. In: Bureau, R., Dravet, C., Genton, P., Tassinari C, Wolf P (eds) Epileptic syndromes in infancy, childhood, and adolescence. John Libbey Eurotext LTD, 2005, pp 315–335
Perry MS, et al. Clobazam for the treatment of intractable epilepsy in children. J Child Neurol. 2013;28:34–9.
Klehm J, et al. Clobazam: effect on frequency of seizures and safety profile in different subgroups of children with epilepsy. Pediatr Neurol. 2014;51:60–6.
Sankar R. GABA(A) receptor physiology and its relationship to the mechanism of action of the 1,5-benzodiazepine clobazam. CNS Drugs. 2012;26:229–44.
Cross JH. Topiramate monotherapy for childhood absence seizures: an open label pilot study. Seizure. 2002;11:406–10.
Piña-Garza JE, Schwarzman L, Wiegand F, Hulihan J. A pilot study of topiramate in childhood absence epilepsy. Acta Neurol Scand. 2011;123:54–9.
Wilfong A, Schultz R. Zonisamide for absence seizures. Epilepsy Res. 2005;64:31–4.
Groomes LB, et al. Do patients with absence epilepsy respond to ketogenic diets? J Child Neurol. 2011;26:160–5.
Thammongkol S, et al. Efficacy of the ketogenic diet: which epilepsies respond? Epilepsia. 2012;53:e55–9.
Fattore C, et al. A multicenter, randomized, placebo-controlled trial of levetiracetam in children and adolescents with newly diagnosed absence epilepsy. Epilepsia. 2011;52:802–9.
Verrotti A, et al. Levetiracetam in absence epilepsy. Dev Med Child Neurol. 2008;50:850–3.
Auvin S, et al. Aggravation of absence seizure related to levetiracetam. Eur J Paediatr Neurol. 2011;15:508–11.
Trudeau V, et al. Gabapentin in naive childhood absence epilepsy: results from two double-blind, placebo-controlled, multicenter studies. J Child Neurol. 1996;11:470–5.
Somerville ER. Some treatments cause seizure aggravation in idiopathic epilepsies (especially absence epilepsy). Epilepsia. 2009;50:31–6.
Wirrell E, Camfield C, Camfield P, Dooley J. Prognostic significance of failure of the initial antiepileptic drug in children with absence epilepsy. Epilepsia. 2001;42:760–3.
Grosso S, et al. Childhood absence epilepsy: evolution and prognostic factors. Epilepsia. 2005;46:1796–801.
Loiseau P, et al. Long-term prognosis in two forms of childhood epilepsy: typical absence seizures and epilepsy with rolandic (centrotemporal) EEG foci. Ann Neurol. 1983;13:642–8.
Dlugos D, et al. Pretreatment EEG in childhood absence epilepsy. Neurology. 2013;81:150–6.
Panayiotopoulos CP, Obeid T, Waheed G. Differentiation of typical absence seizures in epileptic syndromes. A video EEG study of 224 seizures in 20 patients. Brain. 1989;112:1039–56.
Kessler SK, et al. Pretreatment seizure semiology in childhood absence epilepsy. Neurology. 2017;89:56.
Glauser TA, et al. Pharmacogenetics of antiepileptic drug efficacy in childhood absence epilepsy. Ann Neurol. 2017;81:444–53.
Wirrell EC, Camfield CS, Camfield PR, Gordon KE, Dooley JM. Long-term prognosis of typical childhood absence epilepsy: remission or progression to juvenile myoclonic epilepsy. Neurology. 1996;47:912–8.
Callenbach PMC, et al. Long-term outcome of childhood absence epilepsy: Dutch Study of Epilepsy in Childhood. Epilepsy Res. 2009;83:249–56.
Sato S, Dreifuss FE, Penry JK, Kirby DD, Palesch Y. Long-term follow-up of absence seizures. Neurology. 1983;33:1590–5.
Sato S, Dreifuss FE, Penry JK. Prognostic factors in absence seizures. Neurology. 1976;26:788–96.
Berg AT, Levy SR, Testa FM, Blumenfeld H. Long-term seizure remission in childhood absence epilepsy: might initial treatment matter? Epilepsia. 2014;55:551–7.
Caplan R, et al. Childhood absence epilepsy: behavioral, cognitive, and linguistic comorbidities. Epilepsia. 2008;49:1838–46.
Wirrell EC, et al. Long-term psychosocial outcome in typical absence epilepsy. Sometimes a wolf in sheeps’ clothing. Arch Pediatr Adolesc Med. 1997;151:152–8.
Shinnar S, et al. Discontinuing antiepileptic drugs in children with epilepsy: a prospective study. Ann Neurol. 1994;35:534–45.
Verrotti A, et al. Antiepileptic drug withdrawal in childhood epilepsy: what are the risk factors associated with seizure relapse? Eur J Paediatr Neurol. 2012;16:599–604.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the publication of this review. Dr. Kessler has received funding from the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health (NIH) for work related to Childhood Absence Epilepsy.
Conflict of interest
Dr. Kessler and Dr McGinnis have no conflicts of interest to disclose.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kessler, S.K., McGinnis, E. A Practical Guide to Treatment of Childhood Absence Epilepsy. Pediatr Drugs 21, 15–24 (2019). https://doi.org/10.1007/s40272-019-00325-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40272-019-00325-x