
Abstract
Epilepsy is one of the most common and disabling chronic neurological disorders. Antiseizure medications (ASMs), previously referred to as anticonvulsant or antiepileptic drugs, are the mainstay of symptomatic epilepsy treatment. Epilepsy is a multifaceted complex disease and so is its treatment. Currently, about 30 ASMs are available for epilepsy therapy. Furthermore, several ASMs are approved therapies in nonepileptic conditions, including neuropathic pain, migraine, bipolar disorder, and generalized anxiety disorder. Because of this wide spectrum of therapeutic activity, ASMs are among the most often prescribed centrally active agents. Most ASMs act by modulation of voltage-gated ion channels; by enhancement of gamma aminobutyric acid-mediated inhibition; through interactions with elements of the synaptic release machinery; by blockade of ionotropic glutamate receptors; or by combinations of these mechanisms. Because of differences in their mechanisms of action, most ASMs do not suppress all types of seizures, so appropriate treatment choices are important. The goal of epilepsy therapy is the complete elimination of seizures; however, this is not achievable in about one-third of patients. Both in vivo and in vitro models of seizures and epilepsy are used to discover ASMs that are more effective in patients with continued drug-resistant seizures. Furthermore, therapies that are specific to epilepsy etiology are being developed. Currently, ~ 30 new compounds with diverse antiseizure mechanisms are in the preclinical or clinical drug development pipeline. Moreover, therapies with potential antiepileptogenic or disease-modifying effects are in preclinical and clinical development. Overall, the world of epilepsy therapy development is changing and evolving in many exciting and important ways. However, while new epilepsy therapies are developed, knowledge of the pharmacokinetics, antiseizure efficacy and spectrum, and adverse effect profiles of currently used ASMs is an essential component of treating epilepsy successfully and maintaining a high quality of life for every patient, particularly those receiving polypharmacy for drug-resistant seizures.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Epilepsy is a multifaceted complex disease and so is its treatment. |
We review the pharmacology of the ~ 30 approved antiseizure medications, including their preclinical and clinical efficacy, pharmacokinetics, and mechanisms of action. |
We summarize the available data on the > 30 novel epilepsy therapies that are in the preclinical or clinical drug development pipeline, including new potentially disease-modifying treatments. |
1 Introduction
Epilepsy is one of the most common and disabling chronic neurological disorders, affecting approximately 1% of the general population. Epilepsy affects all age groups and is characterized by an enduring predisposition to generate epileptic seizures and the associated cognitive, psychological, and social consequences [1].
Epilepsy is not a specific disease, or even a single syndrome, but rather a complex group of disorders with widely varying types of epileptic seizures, ranging from nonconvulsive to convulsive and focal to generalized [2].
The causes of epilepsy are only partially understood and include a variety of insults that perturb brain function, including acquired causes (e.g., stroke or traumatic brain injury [TBI]), infectious diseases (such as neurocysticercosis and cerebral malaria), autoimmune diseases, and genetic mutations [1].
There is currently no cure, so symptomatic pharmacological treatment remains the mainstay of therapy for people with epilepsy [3].
By definition, antiseizure medications (ASMs) prevent or suppress the generation, propagation, and severity of epileptic seizures. The term “antiseizure medication” has replaced the old term “anticonvulsant drugs” because epilepsy therapies suppress not only convulsive but also nonconvulsive seizures [4, 5]. Furthermore, the term “antiseizure medication” more and more replaces the term “antiepileptic drug” because such drugs provide symptomatic treatment only and have not been demonstrated to alter the course of epilepsy [1, 6].
Achieving complete seizure control is the most important objective in the treatment of epilepsy. For this goal, ASMs are administered chronically to prevent seizure recurrence in patients with spontaneous recurrent seizures (SRS). In addition, ASMs are being used to treat status epilepticus (SE) and interrupt acute symptomatic seizures in response to a variety of causes, including intoxication. However, despite the availability of numerous ASMs with different mechanisms of action (MOAs), both SRS and SE may be resistant to treatment in about 30% of all patients with epilepsy [7,8,9,10]. Interestingly, seizure freedom outcomes have not changed much since 1939, the year that phenytoin came into use, in spite of the development of numerous novel ASMs in recent decades [9,10,11]. Mechanisms of ASM resistance are incompletely understood [12].
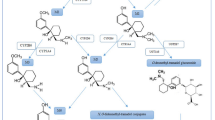
Epilepsy is a multifaceted complex disease and so is its treatment. About 30 different ASMs are available for the treatment of epilepsy (Fig. 1). For the treatment of epilepsy, the initial ASM should be individualized based on the epilepsy syndrome and seizure type, the adverse effects profile, the pharmacokinetic profile, potential interactions with other drugs, comorbidities that the ASM may affect, the age of the patient, reproductive considerations, and cost [1].

Chemical structures of clinically approved antiseizure drugs discussed in this review
We review the pharmacology of ASMs, including their preclinical efficacy, pharmacokinetics, and MOAs, and their clinical efficacy. Rather than discussing each of the ~ 30 ASMs separately, we highlight commonalities and differences as well as general principles in their pharmacology. Furthermore, we review novel epilepsy therapies that are in the preclinical or clinical drug development pipeline.
2 The Development of Antiseizure Medications
Early drugs (such as potassium bromide and phenobarbital), which were discovered by serendipity, had relatively unfavorable efficacy-to-tolerability profiles. This changed with the event of drug screening in animal seizure models in the 1930s, initiated by H. Houston Merritt and Tracy J. Putnam. These scientists, working at the Neurological Unit of the Boston City Hospital, used an electroshock seizure model in cats for drug screening for ASM efficacy, leading to the discovery of phenytoin as the first nonsedating ASM [13].
Phenytoin (5,5-diphenylhydantoin) was first synthesized in 1908 as a barbiturate derivative in Germany by Heinrich Biltz and subsequently resynthesized by an American chemist at Parke-Davis in 1923 in Detroit. Screening of phenytoin did not reveal sedative side effects such as those seen with sedative/hypnotic barbiturates, so Parke-Davis discarded this compound as a useful drug. In 1936, phenytoin's antiseizure properties were identified by Putnam and Merritt, who also evaluated its clinical value in a number of patients in the period 1937–1940 [14].
The history of phenytoin is considered a keystone event for drug discovery and development and the beginning of modern ASM development because it demonstrated that (1) systematic screening of large numbers of compounds may lead to a hit with the desired effect and (2) an antiseizure effect determined in an animal model can be translated to patients.
As illustrated in Fig. 2, the discovery and subsequent success of phenytoin led to the systematic search for chemically related and unrelated compounds with antiseizure efficacy and, subsequently, to the marketing of more than ten novel ASMs, which are commonly referred to as the “first generation” of ASMs because they were derived mainly by modification of the barbiturate structure. They include mephobarbital, primidone, oxazolidinediones such as trimethadione, and succinimides such as ethosuximide.

Modified from Löscher and Schmidt [11]. For further details, see Löscher et al. [30]. ACTH adrenocorticotropic hormone
Introduction of antiseizure drugs (ASMs) to the market from 1853 to 2020. Licensing varied from country to country. Figure shows the year of first licensing or first mention of clinical use in Europe, the USA, or Japan. We have not included all derivatives of listed ASMs nor ASMs used solely for the treatment of status epilepticus. The first generation of ASMs, entering the market from 1857 to 1958, included potassium bromide, phenobarbital, and a variety of drugs mainly derived by modification of the barbiturate structure, including phenytoin, primidone, trimethadione, and ethosuximide. The second-generation ASMs, including carbamazepine, valproate, and benzodiazepines, which were introduced between 1960 and 1975, differed chemically from the barbiturates. The era of the third-generation ASMs started in the 1980s with “rational” (target-based) developments such as progabide, vigabatrin, and tiagabine, i.e., drugs designed to selectively target a mechanism thought to be critical for the occurrence of epileptic seizures. Note that some drugs have been removed from the market.
The second-generation ASMs, including carbamazepine, valproate, and benzodiazepines, which were introduced between 1960 and 1975 (Fig. 2), differed chemically from the cyclic ureides (barbiturates, hydantoins, succinimides, oxazolidinediones; see Fig. 1) and exhibited superior tolerability to cyclic ureide-based structures [15].
The era of the third-generation ASMs started in the 1980s with the ‘‘rational’’ development of drugs such as progabide and vigabatrin, i.e., drugs that were designed to selectively target a mechanism (GABAergic inhibition) thought to be critical for ictogenesis [16]. Several of the new drugs that have been introduced since the 1980s have advantages over the older ASMs in terms of pharmacokinetics and drug–drug interactions, and some drugs have better tolerability and potentially fewer long-term adverse effects and reduced teratogenicity, although this remains to be proven. However, as mentioned, new drugs have not increased the percentage of seizure-free patients [1, 8, 10, 11].
The development of third-generation ASMs was spurred largely by the Anticonvulsant Screening Program, currently known as the Epilepsy Therapy Screening Program (ETSP), set up in 1975 by J. Kiffin Penry at the National Institutes of Neurological Disorders and Stroke of the National Institutes of Health [17]. Throughout its history, the program has tested over 32,000 compounds from more than 600 pharmaceutical firms and other organizations and has played a major role in the development of felbamate, topiramate, lacosamide, retigabine, and cannabidiol and a contributory role in the development of vigabatrin, lamotrigine, oxcarbazepine, and gabapentin [17,18,19].
One of the most recent third-generation ASMs is cenobamate (Fig. 2), which was approved in 2019 for the treatment of patients with focal-onset seizures. In randomized controlled trials, cenobamate produced high seizure-free rates (20/111 subjects [18%] treated with the highest [400 mg/day] dose during a 12-week maintenance period), suggesting that this novel ASM can outperform existing options [20]. This has so far been borne out in long-term open-label extension studies [21]. However, further safety studies and clinical experiences are needed to determine its clinical value.
It is important to note that significant methodological changes in clinical ASM trials were introduced over the eight decades since the discovery of phenytoin [22]. Today, the randomized, double-blind, placebo-controlled adjunctive therapy trial in patients with drug-resistant focal seizures continues to be the primary tool to obtain regulatory approval of novel ASMs. Because of the existence of ~30 ASMs on the market, this creates major hurdles to demonstrating the efficacy of any novel compound, discouraging pharmaceutical companies from investing in ASM development [22, 23]. The ASM market is crowded, and the costs of drug development are steadily increasing. As a result, many of the large pharmaceutical companies previously active in epilepsy, such as GlaxoSmithKline, Novartis, and Pfizer, have withdrawn from the field. This has increased interest, particularly among small- and medium-sized companies, in developing novel molecules for orphan indications (i.e., rare genetic epilepsies) where unmet needs are particularly large [22]. In fact, five of the 11 ASMs introduced after 2005 (vs. none of the ten ASMs licensed between 1989 and 2005) have been licensed exclusively for the treatment of orphan disorders such as Dravet syndrome (stiripentol, cannabidiol, fenfluramine), Lennox–Gastaut syndrome (rufinamide, cannabidiol), and tuberous sclerosis complex (TSC; everolimus, cannabidiol).
As shown in Fig. 3, ASMs have a wide clinical spectrum of indications in both epileptic and nonepileptic disorders. Because of this wide spectrum of therapeutic activity, ASMs are among the most often prescribed centrally active agents [24, 25]. We compare the preclinical and clinical profiles of ASMs in the treatment of epileptic seizures.

The clinical spectrum of antiseizure drugs. For details see text. i.v. intravenous
3 The Preclinical Profile of Antiseizure Medications in the Treatment of Epilepsy
During preclinical development, novel ASMs are typically being tested in a battery of animal models of seizures and epilepsy [15, 19, 26,27,28]. Only compounds that exert antiseizure activity at doses far below those inducing behavioral adverse effects such as sedation or ataxia are developed further.
A typical battery of rodent seizure models is shown in Table 1, including the maximal electroshock seizures (MES) test for identifying efficacy against generalized tonic-clonic seizures, the 6-Hz seizure test and chronic kindling models for identifying activity against focal-onset seizures, and genetic rat models for identifying activity against generalized absence seizures, i.e. the GAERS (Genetic Absence Epilepsy Rat from Strasbourg) model and the WAG/Rij (Wistar Albino Glaxo from Rijswijk) model. The MES and 6-Hz tests are models in which acute seizures are induced by transcorneal electrical stimulation in normal mice or rats, whereas the kindling and genetic absence models use animals that exhibit chronic epilepsy-like brain alterations [29]. Previously, seizures induced by the convulsant pentylenetetrazole (PTZ) have been used as a model for identifying compounds acting against absence seizures, but the PTZ model produced too many false-positive and false-negative results so has been largely abandoned [27].
The advantage of using batteries of animal models as shown in Table 1 is their translational value, which is superior to various other areas of neurology [30]. Thus, starting with phenytoin, all ASMs shown in Figs. 1 and 2 were discovered using animal models, such as MES or kindling. The best predictivity of clinical activity is obtained by using amygdala-kindled rats, which correctly predicted the efficacy of numerous ASMs against focal-onset seizures in patients (Table 1). The term “kindling” is used for the progressive development of seizures in response to a previously subconvulsant stimulus administered in a repeated and intermittent fashion [31]. Kindling can be achieved by electrical stimulation of limbic brain regions such as the amygdala, by transcorneal application of electrical stimuli, or by convulsants such as PTZ. The best-characterized and predictive model is amygdala kindling [29]. Importantly, testing of novel compounds in the kindling model was more predictive of clinical efficacy than testing in the MES test, as for instance demonstrated by vigabatrin, levetiracetam, and tiagabine (Table 1). The finding of Löscher and Hönack [32] that levetiracetam is particularly effective in the amygdala-kindling model was essential in the further development of this compound, which is now one of the most widely used ASMs [33].
As shown in Table 1, ASMs differ markedly in their efficacy in animal models. ASMs can be grouped into three categories: (1) ASMs with a narrow spectrum of efficacy such as ethosuximide (only active against absence seizures) or vigabatrin (active in the kindling model but not the other models shown in Table 1); (2) ASMs that mainly act in MES and focal-onset seizure models (the vast majority of compounds shown in Table 1), and (3) ASMs with a broad spectrum of efficacy such as the benzodiazepines, brivaracetam, topiramate, valproate, and alkyl-carbamates such as cenobamate. At least in part, the preclinical spectrum of antiseizure efficacies resembles the clinical spectrum (Table 1). For instance, ethosuximide is only effective in the GAERS model and almost exclusively used for the treatment of absence seizures in humans; phenytoin and carbamazepine act mainly against focal-onset and primarily generalized tonic-clonic seizures in animal models and patients, and benzodiazepines and valproate exhibit a broad spectrum of preclinical and clinical efficacy.
In addition to the preclinical models illustrated in Table 1, specific animal models for pediatric genetic epilepsies, such as Lennox–Gastaut syndrome, infantile spasms (West syndrome), Dravet syndrome, and TSC can be used to discover novel ASMs for the difficult-to-treat seizures in these syndromes [34]. As described, several ASMs, including cannabidiol, rufinamide, stiripentol, everolimus, and fenfluramine, are almost exclusively used in such pediatric epilepsies (Table 1). Furthermore, infantile spasms, which rarely respond to usual ASMs, are treated with high doses of adrenocorticotropic hormone (ACTH) or prednisone for the rapid and complete elimination of these seizures. Efficacy has been demonstrated in prospective controlled studies [35], but it is not fully understood how these drugs work for this condition. Current preclinical models of pediatric epilepsies include mouse, rat, and zebrafish models carrying the mutations that are responsible for the genetic epilepsies as well as in vitro models, such as induced pluripotent stem cells, which are increasingly used for screening novel compounds for the treatment of epileptic encephalopathies [36].
4 The Clinical Profile and Efficacy of Antiseizure Medications in the Treatment of Epilepsy
Although ASMs share a common property of suppressing seizures, they all have different pharmacologic profiles that are relevant when selecting and prescribing these agents in patients with epilepsy and other conditions. This includes a spectrum of antiseizure efficacy against different types of seizures and epilepsies (Table 1), MOA, pharmacokinetic properties, propensity for drug–drug interactions, and side effect profiles and toxicities.
As shown in Fig. 1, ASMs markedly differ in their chemical structures, ranging from barbiturate-like compounds to γ-aminobutyric acid (GABA) derivatives and branched fatty acids. Often, the success of a novel ASM initiates the synthesis and development of additional compounds from the same chemical family (Fig. 1), as exemplified by cyclic ureides (barbiturate-like ASMs such as phenobarbital and primidone, hydantoins such as phenytoin and fosphenytoin, oxazolidinediones such as trimethadione and paramethadione, and succinimides such as ethosuximide and methsuximide), iminostilbenes (carbamazepine, oxcarbazepine, eslicarbazepine acetate), benzodiazepines (clonazepam, clobazam, diazepam, lorazepam, midazolam), piracetam derivatives (levetiracetam, brivaracetam), and alkyl-carbamates (felbamate, retigabine, cenobamate).
The clinical use of ASMs is tailored first by the patient’s type of epilepsy [2]. Only certain ASMs are effective in generalized epilepsies (GE). These include valproate, levetiracetam, lamotrigine, topiramate, zonisamide, felbamate, perampanel, and lacosamide. Seizure types within the broad grouping of GE include primary generalized tonic and tonic-clonic seizures, absence seizures, myoclonic seizures, and atonic seizures [37]. Although all the ASMs mentioned are effective against generalized tonic/tonic-clonic seizures, some, such as lamotrigine may be less effective against absence seizures and not effective against myoclonic seizures. Levetiracetam is effective in generalized tonic-clonic seizures but not against absence, tonic, or atonic seizures (although it is commonly used off-label with those seizures).
Our knowledge remains insufficient to marry an ASM’s known antiseizure MOA in animals to the treatment of specific seizure types in humans, primarily because the mechanisms of ictogenesis in humans are still largely unknown. Thus, ASMs effective in GE include ASMs with diverse known MOAs, including sodium channel blocking (lamotrigine, lacosamide), presynaptic neurotransmitter release modulation (levetiracetam), antiglutamatergic activity (perampanel), and multiple MOAs (valproate, topiramate, zonisamide, felbamate, cannabinoids) [38]. Yet, certain other ASMs with similar MOA may be ineffective in GE (e.g., the sodium channel blockers carbamazepine, oxcarbazepine or phenytoin), which may in fact sometimes exacerbate GE-related seizures [39]. Why one sodium channel blocker is effective in GE and others are not remains unknown. In some instances, the use or non-use of an ASM may be dictated by the regulatory approval process rather than biology. For instance, brivaracetam, closely related to levetiracetam, which is approved for the treatment of GE, is effective in several animal models of GE [40] but is not approved for the treatment of GE because the necessary clinical studies have not been done.
The second, largest group of epilepsies are focal epilepsies with focal seizures, with or without evolution to bilateral tonic-clonic seizures (previously known as secondary generalization). Nearly all medications on the market are effective in focal seizures, again, without a clear coupling of known MOA and putative mechanisms of ictogenesis of focal seizures.
The third group includes special epilepsy syndromes, which may be treated by a limited number of ASMs. These syndromes include rare childhood epilepsies, comprising some genetic epilepsies. For absence seizures associated with childhood or juvenile absence epilepsy, both examples of GE, ethosuximide is the drug of choice, followed by valproate and other ASMs used for GE [41]. Ethosuximide has a unique MOA of T-type calcium channel modulation (see Sect. 10). Infantile spasms, primary generalized seizures of infancy seen with a number of different and often catastrophic causes of epilepsy respond uniquely to the hormone ACTH or to prednisone and to vigabatrin [42, 43]. Lennox–Gastaut syndrome, a syndrome with multiple seizure types, developmental delay, and characteristic slow spike and wave electroencephalogram (EEG) characteristics that can be caused by multiple etiologies, responds to the benzodiazepine clobazam and to cannabidiol, amongst others [44]. TSC, which can also result in multiple seizure types, can be treated specifically and mechanistically by the mechanistic target of rapamycin (mTOR) inhibitor everolimus [45] in addition to multiple other medications [46]. Surprisingly, this mechanistically very targeted form of treatment appears to be no more effective than treatment with other ASMs whose MOA is unrelated to the cause of TSC. A rare genetic form of severe epilepsy, Dravet syndrome, can similarly be treated by clobazam and cannabidiol but with only modest results [47, 48]. In 80% of cases, this condition is caused by de novo mutations in the gene responsible for voltage-gated sodium channel protein SCNA1 or 2, which results in loss of function of small inhibitory neurons, increase in hyperexcitability, and seizures that are very difficult to treat [49]. Treatment with sodium channel blockers exacerbates seizures in Dravet syndrome. Seizures in Dravet syndrome appear to be significantly more responsive to fenfluramine than to all other ASMs [50, 51] (see below), a weight loss medication with serotonergic MOA.
Often, novel ASMs resulting from the structural variation of older ASMs differ in their pharmacology from the older drugs in terms of potency, efficacy, spectrum of activity, and tolerability. However, most novel (third-generation) ASMs are not more effective than older drugs [8, 12]. Thus, analysis of a longitudinal cohort study of adolescents and adults with newly diagnosed epilepsy attending a specialist clinic in Glasgow, Scotland, indicated that levetiracetam, zonisamide, eslicarbazepine acetate, and lacosamide are as efficacious as carbamazepine for focal epilepsy [3]. There has been no gain in efficacy with second-generation or third-generation ASMs over valproate for GEs and unclassified epilepsies [3]. In fact, most second- and third-generation ASMs are less efficacious than valproate in those epilepsies. Similar results on the comparative efficacies of ASMs were obtained by network meta-analyses of monotherapy studies [52, 53]. Indeed, the widespread use and the unsurpassed clinical efficacy of carbamazepine and valproate made them benchmarks for comparison with third-generation ASMs [11].
It has been argued that one of the major reasons for the apparent failure to discover drugs with higher efficacy is that, with few exceptions, all ASMs have been discovered using the same conventional animal models, particularly the MES test in rodents, which served as a critical gatekeeper [11].
Evaluation of most new ASMs for treatment of epilepsy has followed broadly similar randomized, double-blind, placebo-controlled study designs in which the new ASM or placebo is added to baseline medications in patients with refractory epilepsy; patients are then treated for ~3 months, and seizure frequency is compared between active treatment and pretreatment baseline periods between the ASM- and placebo-treated groups [10, 54, 55]. Standard primary efficacy outcomes are median percent seizure frequency reduction and proportion of patients who achieve ≥50% seizure frequency reduction, the 50% responder rate. Secondary efficacy outcomes sometimes include 75% responder rate and seizure freedom. Results of pivotal studies of different new ASMs cannot be directly compared, but it is striking that, until recently, the outcome figures were very similar for most of the new ASMs. Most ASMs achieve 20–30% median seizure frequency reduction over and above placebo effect and a 30–50% responder rate [10, 55,56,57,58]. In the more recent studies, 75% responder rate has been achieved in about 20% of patients. Typically, seizure freedom rate is low, ranging from 2 to 5% [59, 60].
Recently, a possible breakthrough may have been achieved for two new medications. In adults with refractory focal epilepsy, treatment with a new ASM, cenobamate, resulted in seizure freedom of 21% of patients treated with the highest approved dose, 400 mg/day, during the 12-week maintenance period (20/111; 18% of all patients when those who discontinued the study during the titration period were included) [61]. The seizure freedom was sustained in an open-label extension study with treatment lasting up to 4 years [21]. Cenobamate has two known MOAs: a block of the “persistent current” of the voltage-gated sodium channels and a weak positive allosteric modulation of GABA-A receptors [62]. In children with Dravet syndrome, treatment with the serendipitously discovered weight-loss medication fenfluramine similarly resulted in an 8% seizure freedom during the entire 14-week treatment period, which was also sustained long term [50]. Fenfluramine acts primarily as a serotonin releasing agent but also positively modulates different subtypes of serotonin receptors and the sigma 1 receptor [38]. In both cenobamate and fenfluramine, it is unknown whether the known MOAs are responsible for the notably higher efficacy rates of these medications compared with all other new ASMs.
5 The Selection of Antiseizure Medications for the Treatment of Epilepsy in Children and Adults
The number of available ASMs has increased rapidly in the past 30 years, giving more choice when initiating therapy but also making drug selection a much more complex process. Major evidence-based guidelines have been developed during this time, assisting clinicians and patients in making appropriate treatment choices in newly diagnosed epilepsy [63, 64]. These include guidelines issued by the International League Against Epilepsy [65, 66], the American Academy of Neurology/American Epilepsy Society [67, 68], and others. These guidelines are based on the best available evidence. However, they are limited by the lack of controlled head-to-head comparative efficacy studies for most ASMs. They may not be a substitute for knowledge, skill, and experience in managing individual patients [63]. Figure 4 shows an extract of these guidelines and common treatment options, including more recent ASMs.

Choice of antiseizure medications (ASMs) in adults and children. Common first monotherapy refers to the first treatment choice in a patient without any specific factors precluding the use of this. Monotherapy alternatives refer to ASMs chosen when certain patient- or ASM-related factors preclude the use of the first-choice ASM. Data from various sources [63, 64, 67, 68] and guidelines discussed in these papers. Note that several additional childhood epilepsy syndromes are not illustrated in this figure. ACTH adrenocorticotropic hormone
The availability of so many ASMs allows for some tailoring of treatment to each patient’s specific situation, even if the relevance of the ASM’s MOA to the patient’s seizures is unknown and the efficacy may be similar to that of many other ASMs. The patient specificity of ASM choices may relate to the ASM’s side effect profile; its potential beneficial or adverse effect on the patient’s comorbid conditions; the potential for drug–drug interactions or lack thereof; ease of use, such as initiation titration and once-daily administration; and specific patient populations such as the elderly, those planning pregnancy, and patients with renal or liver disease [1, 69]. While most ASMs have the potential to cause central nervous system (CNS) side effects, such as somnolence, fatigue, and dizziness, many have ASM-specific side effect potential, which should be avoided in potentially vulnerable patients. For instance, valproate may cause weight gain, hyperandrogenemia, metabolic syndrome, exacerbation of diabetes, polycystic ovarian syndrome, hepatitis, and pancreatitis and should therefore be avoided in patients with these conditions or predisposition for them [70]. Other medications that may cause weight gain include gabapentin, pregabalin, vigabatrin, and benzodiazepines. One common mechanism of these drugs that could explain weight gain is the potentiation of GABAergic inhibition by presynaptic or postsynaptic effects (see Sect. 10).
Phenytoin, phenobarbital, carbamazepine, oxcarbazepine, and lamotrigine have the potential for serious allergic reaction and should be eschewed in patients who have had previous serious or multiple allergic drug reactions [71]. Phenytoin, phenobarbital, carbamazepine, valproate, and zonisamide can cause liver disease. Valproate should be avoided in liver-compromised patients, and caution should be exercised when using the other medications in these patients [72]. Topiramate and zonisamide can both cause renal stones and are therefore not a good choice in patients with a history of renal stones. Levetiracetam can cause or exacerbate depression and anxiety, and both it and perampanel can cause irritability, hostility, and anger and should probably be avoided or used with caution in patients with significant psychiatric disease [8]. Carbamazepine and its derivatives, oxcarbazepine and eslicarbazepine, can cause hyponatremia, which is most common in the elderly treated with antihypertensives such as diuretics or angiotensin-converting enzyme inhibitors [73]. The old hepatic enzyme-inducing medications—phenytoin, phenobarbital, and carbamazepine—and also long-term treatment with valproate can contribute to osteoporosis, particularly in postmenopausal women or immobile patients with epilepsy and severe encephalopathy, and should be avoided in these patients [74]. Phenytoin, phenobarbital, and carbamazepine also have the potential to cause hypoandrogenism and hyposexuality (in both males and females) [75]. Valproate and lamotrigine can cause or exacerbate tremor and are therefore not the drugs of choice for patients with essential tremor.
The potential for secondary effects can also be used to advantage where these secondary effects may be beneficial. Valproate and topiramate are effective antimigraine treatments and are used for dual purpose in patients with epilepsy and migraine [76]. Valproate and lamotrigine are both effective in mood stabilization and treatment of bipolar affective disorder and depression, both common morbidities in epilepsy; carbamazepine and oxcarbazepine are also sometimes used off-label for mood stabilization [77, 78]. Pregabalin and clonazepam have anxiolytic effects and may be used for comorbid anxiety [77]. Topiramate, zonisamide, and felbamate may cause weight loss; topiramate and zonisamide can be used beneficially in patients with epilepsy and obesity. Notably, topiramate, one of a number of serendipitously discovered ASMs, was initially developed for the treatment of type 2 diabetes mellitus [79]. Phenobarbital, gabapentin, pregabalin, and perampanel all have sedating effects, which can help with insomnia, another common comorbidity of epilepsy [80]. Gabapentin, pregabalin, carbamazepine, and oxcarbazepine may be effective in painful neuropathy [81, 82]. Carbamazepine and oxcarbazepine are used for the treatment of trigeminal neuralgia, pregabalin is indicated for the treatment of fibromyalgia, and carbamazepine, gabapentin, and pregabalin are used for the treatment of restless leg syndrome [83,84,85]. Primidone and topiramate are treatments for essential tremor [86].
Valproate, phenobarbital, and topiramate increase the risk of major congenital malformations in babies born to people with epilepsy and should therefore be avoided in those planning to conceive or who are pregnant [87]. Valproate in addition negatively impacts fetal neurocognitive development, reducing the child’s intelligence quotient and increasing the risk for autism [87, 88]. Conversely, lamotrigine and levetiracetam have been shown to have no increase in the risk of major congenital malformation and are ASMs of choice for people planning pregnancy. The elderly are often more sensitive to the adverse events of ASMs and are also often on multiple other medications [89]. ASMs with a good side effect profile and little or no interaction with other drugs are of advantage in this population. These include levetiracetam, gabapentin, pregabalin, lamotrigine, and lacosamide.
For patients with renal disease, drugs that are renally excreted should be used with caution or avoided. These include, amongst others, levetiracetam, lacosamide, gabapentin, and pregabalin. In patients with liver disease, liver-metabolized medications such as phenytoin, phenobarbital, carbamazepine, valproate clobazam, and cannabidiol are best avoided [88].
Drug–drug interactions and pharmacokinetics are also important in the choice of ASM. This can be complicated, but a number of the new ASMs have little or no drug–drug interactions and straightforward pharmacokinetics [90]. These include levetiracetam, brivaracetam, lacosamide, gabapentin, and pregabalin. Medications that are easy to use, with quick straightforward titration or no titration (e.g., levetiracetam, brivaracetam, oxcarbamazepine, eslicarbazepine, lacosamide, and zonisamide as well as the older ASMs phenytoin, phenobarbital, and carbamazepine) may be easier for a patient to use and adhere to than medications with more complicated slower initiation, which may be necessary to mitigate the side effect potential, for instance with lamotrigine, topiramate, perampanel, or cenobamate.
6 Resistance to Antiseizure Medications in Patients with Epilepsy
An unresolved problem is the drug resistance of many types of epilepsy, including temporal lobe epilepsy (TLE), the most common type of epilepsy in adults [9]. More than 50% of patients with TLE do not become seizure free with the current ASMs, despite the diverse MOAs by which these compounds work (see below). Thus, preclinical models reflecting such ASM resistance were developed and now are used after the drug identification phase shown in Table 1 for further differentiation of novel compounds [19]. Examples are the lamotrigine-resistant amygdala-kindled rat model [91] and amygdala-kindled rats selected for resistance to phenytoin and other ASMs [92]. Such models are, for instance, used in the differentiation phase of the ETSP [19]. It remains to be established whether the implementation of models of ASM resistance will lead to more effective drugs. In this respect, drugs that combine several MOAs may be particularly interesting, as exemplified by the novel ASM cenobamate.
Patients with drug-resistant epilepsy are often treated with more than one ASM. Robust evidence to guide clinicians on when and how to combine ASMs is lacking, and current practice recommendations are largely empirical [93,94,95]. A popular strategy for combination therapy is a pharmacomechanistic approach based on the (perceived) modes of action of ASMs (see Sect. 10). For instance, Deckers et al. [96] reviewed the available animal and human data and concluded that combinations involving a sodium channel modulator and a drug with GABAergic properties appeared to be particularly beneficial. Indeed, one of the few clinically proven synergistic ASM combinations is a combination of lamotrigine and valproate [97, 98]. In general, mainly based on data in animal models, combining ASMs with different MOAs seems to provide greater effectiveness and a lower risk of adverse events than combining ASMs with similar mechanisms [95, 99]. However, one drug specifically developed on this principle, padsevonil, which has a dual action of synaptic vesicle protein (SV)-2A, B, and C modulation and GABA-A receptor potentiation, failed a recently completed phase IIb study, leading to discontinuation of its development.
7 Aggravation of Seizures by Antiseizure Medications
ASMs may also aggravate seizures, including an increase in the frequency or severity of existing seizures, the emergence of new types of seizures, or the occurrence of SE [100,101,102]. Seizure aggravation by ASMs is an infrequent phenomenon, occurring mostly in primary GE treated with drugs that are more efficacious against partial seizures [103]. Thus, a major reason for seizure aggravation is an inappropriate choice of ASMs, which is best documented for the use of carbamazepine in idiopathic generalized and myoclonic epilepsies [101]. Most other ASMs have been reported only occasionally to cause seizure aggravation. In addition to inappropriate choice of ASMs, risk factors for worsening of seizures are polytherapy, excessive ASM doses with some ASMs, high frequency of seizures, epileptic encephalopathy, and cognitive impairment [100,101,102].
8 Use of Antiseizure Medications for Acute Interruption of Seizures, Seizure Clusters, or Status Epilepticus
In addition to using ASMs for long-term oral treatment of patients with SRS, several ASMs are used for acute interruption or prevention of acute symptomatic seizures, seizure clusters, and SE (Fig. 3). Acute symptomatic seizures by definition occur in close proximity to an event and are considered to be situational [104, 105]. Acute symptomatic (or provoked) seizures must be distinguished from unprovoked seizures and may occur as a result of temporary metabolic, toxic, and other systemic illness, e.g., due to illicit drugs, drug withdrawal, toxins, or drug adverse effects or overdose. Furthermore, acute symptomatic seizures (or early seizures) may occur in the first week after a brain lesion or an injury such as stroke, TBI, or infectious encephalitis. While intravenous benzodiazepines are used as rescue treatment for acute symptomatic seizures associated with metabolic, toxic, and other systemic illness, acute symptomatic seizures associated with brain insults such as may occur during the first week after TBI are typically prevented by treatment with ASMs such as levetiracetam, phenytoin, valproate, carbamazepine, or lacosamide after the insult [105].
Seizure clusters, i.e., acute repetitive seizures, are broadly defined as intermittent stereotypic episodes of frequent seizure activity with periods of recovery, thus distinguishing seizure clusters from SE [106,107,108]. While there are different definitions of cluster seizures, the most inclusive one is two or more seizures within 24 h. Cluster seizures are not uncommon, with their frequency estimated in different studies as between ~15 and 70% of patients with epilepsy. Seizure clusters occur despite optimal/maximal oral therapy with ASMs and are distinguishable from a patient’s “normal” seizure pattern. Cluster seizures are a medical emergency unique to patients with epilepsy, whereas SE can occur in any individual, thereby further differentiating these two clinical conditions. Until recently, rectal diazepam gel was the only US FDA-approved rescue medication for seizure clusters. In 2019 and 2020, the FDA approved two nasal sprays, one with diazepam and the other with midazolam, as rescue treatments for seizure clusters in people with epilepsy. In addition, buccal midazolam is approved in European countries for the treatment of prolonged seizures and is under review by the FDA for use in the USA. However, various non-rectal non-intravenous benzodiazepines are safe and effective in treating acute seizures and clusters [107, 108].
SE, the condition of ongoing seizures or repetitive seizure activity without recovery of consciousness between seizures, is a life-threatening emergency that necessitates immediate treatment [109]. The most common treatment protocols for SE specify an intravenous benzodiazepine (either midazolam, lorazepam, or diazepam) as initial ASM therapy, followed—if seizures continue—by fosphenytoin (or phenytoin), valproate, levetiracetam, or, if none of the aforementioned options are available, phenobarbital [110,111,112]. If seizures continue, either second-line therapy is repeated, other medications such as lacosamide or topiramate may be used, or third-line therapy is instituted using intravenous sedation (“therapeutic coma”). Propofol and midazolam are the most commonly used agents, partly because of their short half-life. Barbiturates (pentobarbital or phenobarbital) were common agents in the past but have largely been replaced because of their long half-life, which makes neurological evaluation difficult when the agent is stopped. About 20–40% of patients with SE exhibit treatment resistance despite aggressive treatment [113]. The short-term fatality rates for resistant SE (RSE) have been estimated as between 16 and 39%; mortality after RSE is about three times higher than for nonrefractory SE [113].
Additional indications of ASMs in the pediatric population include the treatment of neonatal seizures and febrile seizures (Fig. 3). Neonatal seizures are the most frequent neurological event in newborn babies, most commonly due to hypoxic–ischemic encephalopathy as a result of birth asphyxia [114]. Despite suboptimal efficacy, intravenous phenobarbital remains the first-line ASM of choice for interruption of neonatal seizures [115]. In a recent multicenter, randomized, blinded, controlled, phase IIb trial, intravenous phenobarbital was more effective than intravenous levetiracetam for the treatment of neonatal seizures, but higher rates of adverse effects were seen with phenobarbital treatment [116]. There is an urgent need for more effective treatments for neonatal seizures to be developed, and a variety of animal models is used in this respect [117].
Febrile seizures are the most common neurologic disorder of infants and young children, occurring in 2–4% of children aged < 5 years [118]. Febrile seizures are caused by a spike in body temperature, often from an infection. Most febrile seizures are self-limited (“simple febrile seizures”); however, when seizures last longer than 5 minutes (“complex febrile seizures” or “febrile SE”), a benzodiazepine should be administered to break the seizure [118]. A 2018 Cochrane review concluded that intravenous lorazepam and diazepam have similar rates of seizure cessation and respiratory depression [119]. When intravenous access is unavailable, buccal midazolam or rectal diazepam is acceptable.
9 Use of Antiseizure Medications for Nonepileptic Conditions
ASMs are used not only for the treatment of seizures and SE but also for nonepileptic conditions (Fig. 3), including migraine headache, chronic neuropathic pain, mood disorders (such as bipolar disorder), generalized anxiety disorder, schizophrenia, and various neuromuscular syndromes [24, 25, 120, 121]. In many of these conditions, as in epilepsy, the drugs act by modifying the excitability of nerve (or muscle) through effects on voltage-gated sodium and calcium channels or by promoting inhibition mediated by GABAA receptors (see below). Examples of ASMs approved for the treatment of nonepileptic conditions are gabapentin and pregabalin for neuropathic pain, carbamazepine for trigeminal neuralgia, valproate and lamotrigine for bipolar disorder, benzodiazepines for generalized anxiety disorder, and valproate and topiramate for migraine (see also Sect. 5). In addition, combined findings of randomized controlled trials and meta-analyses indicate that pregabalin is efficacious in both acute treatment and relapse prevention in generalized anxiety disorder [121, 122]. Pregabalin was approved for generalized anxiety disorder in the European Union in 2006 [121]. Furthermore, based on randomized controlled trials, zonisamide is considered a safe and efficacious add-on treatment in Parkinson‘s disease [123], whereas no robust efficacy was reported for topiramate or levetiracetam [124]. Preliminary clinical data indicated that cannabidiol improved quality of life but not motor symptoms in patients with Parkinson’s disease [125], and larger randomized controlled trials are ongoing [124].
10 Mechanisms of Action of Antiseizure Medications
In recent years, there have been dramatic advances in our understanding of how ASMs prevent seizures. As shown in Fig. 5 and Table 2, current ASMs act by diverse molecular mechanisms. Based on their molecular targets, ASMs can be categorized into drugs that act quite selectively via a single target (e.g., several of the sodium channel modulators) or act more broadly via several targets (e.g., valproate, topiramate, felbamate, and cenobamate). ASMs that act via several targets are typically also wide-spectrum ASMs in the clinic (Table 1).

Mechanism of action of clinically approved antiseizure medications (ASMs) [162]. Updated and modified from Löscher and Schmidt [167] and Löscher et al. [33]. Asterisks indicate that these compounds act by multiple mechanisms (not all mechanisms shown here). Some ASMs, e.g., fenfluramine, are not shown here, but their mechanism(s) of action are described in Table 2. AMPA α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, GABA γ-aminobutyric acid, GABA-T GABA aminotransferase, GAT-1 GABA transporter 1, KCNQ Kv7 potassium channel family, NMDA N-methyl-D-aspartate, SV2A synaptic vesicle protein 2A
The actions of most ASMs on molecular targets can be categorized into four broad groups [38, 126]: (1) modulation of voltage-gated ion channels, including sodium, calcium, and potassium channels; (2) enhancement of GABA-mediated inhibition through effects on GABAA receptors, the GABA transporter (GAT)-1, GABA transaminase, or the GABA synthesizing enzyme glutamate decarboxylase; (3) inhibition of synaptic excitation mediated by ionotropic glutamate receptors, including N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazole-propionate (AMPA) receptors; and (4) direct modulation of synaptic release through effects on components of the release machinery, including SV2A and the α2δ subunit of voltage-gated calcium channels (Table 2). The result of the interactions at these diverse targets is the modification of the intrinsic excitability properties of neurons or alteration of fast inhibitory or excitatory neurotransmission. By these actions, ASMs reduce the probability of seizure occurrence by modifying the bursting properties of neurons (reducing the capacity of neurons to fire action potentials at a high rate) and reducing synchronization in localized neuronal ensembles. In addition, ASMs inhibit the spread of abnormal firing to adjacent and distant brain sites [126].
Also, inhibition of carbonic anhydrases is involved in the MOA of several ASMs (Table 2). Drugs whose antiseizure action includes carbonic anhydrase inhibition include acetazolamide, topiramate, and zonisamide. Inhibition of carbonic anhydrases reduces the buffering properties of the HCO3−/CO2 buffer system, leading to acidosis at the whole-organism level, including in the brain. The fall in brain pH suppresses neuronal excitability [126]. The protective action of carbonic anhydrase inhibitors in generalized seizures has been attributed to the high pH sensitivity of hyperpolarization-activated and cyclic nucleotide-gated (HCN) channels in thalamocortical neurons. Dysregulation of HCN channels has been strongly implicated in various experimental models of epilepsy, as well as in human epilepsy, including TLE. In addition to carboanhydrase inhibitors, several other ASMs, including lamotrigine and gabapentin, have been reported to modulate the hyperpolarization-activated (Ih) current conducted by HCN channels [127].
It should be considered that the mechanisms of ASMs illustrated in Table 2 and Fig. 5 focus on the primary MOAs of ASMs, where these are known. Many drugs used currently in the treatment of epilepsy have additional, less well-characterized pharmacological effects that manifest at therapeutic concentrations and might contribute to the drug's overall clinical profile [38].
More recently, novel epilepsy therapies have been developed that act by disease-specific mechanisms, including everolimus (inhibition of mTOR signaling in TSC) and cerliponase alfa (for lysosomal enzyme replacement in neuronal ceroid lipofuscinosis type 2) [38]. The latter treatments are examples of “precision medicine,” a relatively new area of disease-specific therapies that may revolutionize the therapy of genetic epilepsies [128]. Indeed, there is now cause for optimism that we are entering a new paradigm where it will be possible to engineer specific treatments for some genetically defined epilepsies using disease-mechanism-targeted small molecules, antisense, gene therapy with viral vectors, and other biological approaches [38]. Such novel therapies may lead to a cure for certain epilepsies [129]. In this respect, it is also important to note that numerous scientists are working on developing novel antiepileptogenic therapies to prevent epilepsy after head injury in patients at risk [130], and antiepileptogenic or disease-modifying therapies are an area of intensive research in childhood epilepsies [131]. However, the role of the pharmaceutical industry in developing antiepileptogenic or disease-modifying therapies for patients at risk is currently low.
11 Are Some Antiseizure Medications also Antiepileptogenic?
It has been suggested that everolimus not only suppresses seizures in patients with TSC but also may have the potential to be a disease-modifying therapy in this disease [132, 133]. TSC is a rare genetic neurocutaneous disorder with epileptic seizures as a common and early presenting symptom. TSC is caused by loss-of-function mutations in the TSC1 or TSC2 genes, which lead to constitutive mTOR activation, resulting in abnormal cerebral cortical development with multiple focal structural malformations [132]. Treatment with the mTOR inhibitor everolimus is thus directly aimed at the underlying dysfunction of the affected cells, which led to the suggestion that it may modify the disease [132]. However, everolimus has not yet fully lived up to its promise as a disease-modifying drug. At least half of patients with TSC with intractable epilepsy have not shown a clinically relevant seizure frequency reduction. Furthermore, there is no evidence yet of a positive effect on the cognitive and neuropsychiatric deficits in patients with TSC [134]. On the other hand, everolimus has demonstrated significant reductions in tumor volume in subependymal giant cell astrocytomas associated with TSC, which led to the approval of the drug for this indication [135].
Concerning disease modification in TSC, recent clinical data with the GABA-T inhibitor vigabatrin are of interest, as they suggest that vigabatrin may have antiepileptogenic effects in TSC [131]. Vigabatrin also partly inhibits mTOR. It is the treatment of choice for infantile spasms, a common early, severe seizure manifestation in TSC. Serial EEGs started shortly after birth have shown that epileptiform activity predictably precedes the onset of seizures. Treatment with vigabatrin starting at the time of appearance of epileptiform activity instead of at the time of onset of seizures reduces the risk of seizures and drug-resistant epilepsy [136].
Given the precedent of preventive clinical trials with vigabatrin for epilepsy in TSC, similar preventive trials with mTOR inhibitors are in the planning stages but have not yet been conducted [131]. One barrier to progress has been the concern for potential adverse effects of mTOR inhibitors in young infants, given the role of the mTOR pathway in normal growth and development.
12 Pharmacokinetics of Antiseizure Medications
Therapy of epilepsy by ASMs necessitates continuous (24/7) maintenance of effective drug levels in the brain over many years. Thus, current ASMs need to meet several pharmacokinetic criteria, including (1) bioavailability after oral administration, (2) sufficiently long half-lives to minimize the frequency of daily drug administrations, and (3) brain target engagement, i.e., sufficient penetration into the brain. To fulfill the third criterion, ASMs are typically small, lipophilic, and uncharged to enable penetration through the blood–brain barrier by passive diffusion [137]. There are some exceptions to this criterion, namely everolimus, which (similar to the prototype mTOR inhibitor rapamycin) only poorly penetrates into the brain, necessitating high plasma levels that may be associated with severe adverse effects. Other examples for relatively poor brain penetration are vigabatrin and valproate, whereas the majority of ASMs are brain permeant [137]. Concerning elimination, all ASMs have sufficiently long half-lives to enable maintenance of active drug levels with one to two administrations per day (Table 3). Several ASMs mainly act by active metabolites Examples are primidone (a prodrug of phenobarbital), fosphenytoin (a prodrug of phenytoin), and eslicarbazepine acetate, which acts as a prodrug of (S)-licarbazepine (i.e., eslicarbazepine), which is also the main active metabolite of oxcarbazepine (Table 3). Other medications act as both parent compounds and active metabolites (e.g., carbamazepine, clobazam, diazepam, cannabidiol).
Table 3 also illustrates the striking interspecies differences in ASM elimination, which must be considered when using such drugs for preclinical rodent studies, in terms of both dosing intervals and interspecies allometric scaling of doses [138]. Such interspecies differences are often ignored or not known when conducting preclinical studies, which may lead to false-negative data. Extrapolation of doses between species is also of crucial importance when estimating the starting dose of novel compounds for clinical trials, necessitating allometric scaling [139]. As indicated in Table 3, vigabatrin differs from other ASMs in that, although its half-life is shorter in rodents than in humans, its pharmacodynamic effects last for days in both rodents and humans through irreversible inhibition of GABA-T [126].
13 Pharmacokinetic Drug–Drug Interactions
Several first-generation ASMs, including carbamazepine, phenytoin, phenobarbital, and primidone, are inducers of isoforms of cytochrome P450 (CYP) enzymes involved in drug metabolism. Primarily, this is clinically relevant with carbamazepine, leading to autoinduction of ASM metabolism during continued treatment and, thus, the development of pharmacokinetic tolerance [140]. Furthermore, the induction of these enzymes can lower the plasma concentration and hence the efficacy of many psychotropic, immunosuppressant, antineoplastic, antimicrobial, and cardiovascular drugs [141]. Importantly, carbamazepine, phenytoin, phenobarbital, oxcarbazepine, eslicarbazepine acetate, felbamate, perampanel (at 12 mg/day), and topiramate (at > 200 mg/day) all increase the metabolic clearance of contraceptive steroids, potentially reducing their efficacy and increasing the risk of unwanted pregnancies [141]; cenobamate may have the same potential. Several of the newer ASMs do not affect hepatic drug-metabolizing enzymes and are renally excreted, resulting in a lower potential for drug interactions [11, 142].
However, pharmacokinetic drug–drug interactions may also occur with third-generation ASMs. A recent example is the interaction between cannabidiol and clobazam; cannabidiol causes a three- to fivefold increase in plasma concentration of clobazam’s active metabolite norclobazam by inhibiting the metabolism of norclobazam during combined treatment [143, 144]. Thus, in four pivotal randomized placebo-controlled trials of adjunctive therapy with cannabidiol in patients with Dravet syndrome and Lennox–Gastaut syndrome, at least part of cannabidiol’s antiseizure effects was due to the inhibited metabolism of norclobazam [143]. In turn, clobazam inhibits the metabolism of cannabidiol, thereby increasing its plasma levels. Similar to cannabidiol, stiripentol, by inhibition of CYP enzymes, can elevate the plasma concentration of norclobazam and other ASMs [145]. Another recent example of complex drug–drug interactions is cenobamate, which decreases plasma concentrations of lamotrigine and carbamazepine and increases levels of phenytoin and phenobarbital and of clobazam’s active metabolite norclobazam [145].
14 Therapeutic Drug Monitoring
Measuring ASM plasma concentrations (therapeutic drug monitoring [TDM]) can have a valuable role in guiding patient management [142, 146]. TDM is useful (1) to establish an individual therapeutic concentration that can subsequently be used to assess potential causes for a change in drug response; (2) as an aid in the diagnosis of clinical toxicity; (3) to assess compliance, particularly in patients with uncontrolled seizures or breakthrough seizures; (4) to guide dosage adjustment in situations associated with increased pharmacokinetic variability (e.g., children, the elderly, patients with associated diseases, drug formulation changes); (5) when a potentially important pharmacokinetic change is anticipated (e.g., in pregnancy, or when an interacting drug is added or removed); and (6) to guide dose adjustments for ASMs with dose-dependent pharmacokinetics, particularly phenytoin [144]. In addition, some ASMs are heavily protein bound in blood, commonly to albumin. These include phenytoin, diazepam, and valproate. For these ASMs, the clinically important blood level is the free (i.e., protein non-bound) level. This may fluctuate according to albumin levels. Thus, in conditions where albumin levels may change, such as during pregnancy, in liver disease, and in the elderly, both total and free levels of these medications should be checked if possible.
Analysis of ASM plasma levels is also useful when translating preclinical to clinical ASM efficacies [138]. In fact, effective plasma ASM levels are remarkably similar in humans and laboratory rodents (rats, mice). However, because of the marked differences in the elimination kinetics of ASMs between humans and rats (Table 3), rodents require much higher doses than humans to achieve and maintain similarly effective ASM levels [138]. Thus, as discussed earlier, interspecies allometric scaling of doses is necessary when extrapolating ASM doses from rodents to humans or vice versa [139].
15 Tolerability and Safety of Antiseizure Medications
Patient tolerability of adverse drug effects is integral to successful treatment [147]. Most modern ASMs are well-tolerated by many patients, which has led to the abandonment of old treatments such as potassium bromide or phenobarbital, which are less tolerable than more modern epilepsy therapies [148]. However, phenobarbital still has an important role in the global management of epilepsy, particularly in resource-poor countries [149]. The most frequently observed adverse effects of ASMs are dose dependent and reversible and include sedation, fatigue, dizziness, coordination disturbances (ataxia, dysarthria, diplopia), tremor, cognitive deficits, mood alterations, and behavioral changes [141, 148]. However, the adverse effect profiles of individual ASMs may differ greatly and are often a determining factor in drug selection because of the similar efficacy rates shown by most ASMs. Arguably the most concerning adverse effects associated with ASM usage are idiosyncratic reactions, such as skin rashes, which can be of sudden onset and sometimes life threatening [148]. Adverse events of ASMs are described in detail in Sect. 5.
Furthermore, possible teratogenic effects of ASMs are of great concern and the risks imposed by the drugs must be weighed against the risks associated with the disorder being treated [150]. For instance, the use of valproate monotherapy in pregnancy is associated with increased risks for spina bifida and other major malformations, and valproate exposure in utero can also result in subsequent impaired cognitive development in the infant and increased risk of autism. These risks are dose (and blood-level) dependent. There is also evidence of dose-dependent teratogenicity with several other ASMs, including phenobarbital and topiramate [148, 150]. Detailed knowledge of the adverse effect profiles of all ASMs is an essential component of treating epilepsy successfully and maintaining a high quality of life for every patient, particularly those receiving polypharmacy for drug-resistant seizures [148].
An important aspect that is often ignored during the preclinical development of novel ASMs is that the chronic brain alterations associated with epilepsy may change the adverse effect profile of drugs [16]. An early example illustrating this problem was that of the competitive antagonists of the NMDA subtype of glutamate receptors, which were well-tolerated in healthy volunteers but induced serious CNS adverse effects in patients with focal epilepsy [16]. This enhanced potential for NMDA receptor antagonists to induce severe adverse effects in epilepsy was correctly predicted in amygdala-kindled rats, i.e., a chronic model of focal epileptogenesis, but not in nonepileptic rodents [16, 151]. Thus, kindled or epileptic animals should be included in preclinical adverse effect testing of novel ASMs [29, 30, 152, 153].
16 Polytherapy vs. Monotherapy
Throughout most of history, treatment of epilepsy has usually involved the use of many agents in combination, that is, polytherapy [154]. Indeed, ASMs were frequently used as polytherapy until evidence from a series of studies in the late 1970s and early 1980s suggested that patients derived as much benefit from monotherapy as from polytherapy [155]. However, the global introduction of numerous new ASMs over the past 30 years as adjunctive treatment in refractory epilepsy has triggered increased interest in optimizing combination therapy [3, 8, 94]. As a general rule, treatment of epilepsy should be started with a single, appropriately chosen ASM, and combination therapy should be reserved for patients refractory to two or more sequential (or alternative) monotherapies [156]. However, most patients with refractory epilepsy take two, three, or even four ASMs [94]. As discussed in Sect. 6, although polytherapy for those who do not benefit from single-drug treatment is the recommended standard, little information is available as to which drugs might work best in combination, so current practice recommendations are largely empirical [93,94,95]. In comparison with monotherapy, polytherapy gives rise to increased adverse effects, drug–drug interactions, poorer compliance, higher cost, and, sometimes, decreased seizure control compared with adequately chosen and dosed monotherapy [156, 157]. In many instances, polytherapy could be avoided by more careful monitoring and supervision of therapy. Polytherapy is clinically useful in a minority of subjects [8] but has been poorly studied despite being a standard treatment strategy for over 100 years [158]. In fact, no evidence-based data show a significant difference in seizure outcome between monotherapy and polytherapy [158]. Because of this, the need for maintaining polypharmacy should be reassessed at regular intervals, and monotherapy should be re-instituted whenever appropriate [156].
17 New Antiseizure Medications in the Preclinical or Clinical Pipeline
As shown in Table 4, > 30 novel ASMs are in the preclinical or clinical drug development pipeline. These compounds act by various mechanisms, including some MOAs that are not shared by approved ASMs. Also, the renaissance of “GABAergic” compounds is interesting to note, including compounds that act as positive allosteric modulators (PAMs), inhibitors of GABA degradation with higher selectivity and tolerability than vigabatrin, and inhibitors of the GABA transporter GAT-1. PAMs that only act as partial or subtype-selective agonists at GABAA receptors are thought to resolve the main disadvantages of previous GABAA receptor agonists, i.e., tolerance and dependence liability. This approach is not new but has been used by several pharmaceutical companies in the 1980/90s in the search for non-sedative anxioselective compounds [159]. Furthermore, one such compound, abecarnil, has been evaluated in patients with photosensitive epilepsy [160]. Whether this approach leads to more effective antiseizure drugs is currently not known. However, one low-affinity partial GABAA receptor agonist, imepitoin, was approved in 2013 for epilepsy treatment in dogs (Fig. 2) and was shown to be as effective as phenobarbital [161]. Novel GABAergic compounds may be particularly interesting for genetic epilepsies with GABA receptor mutations and other alterations in the GABAergic system.
Indeed, in addition to compounds that are developed for the treatment of adult drug-resistant focal epilepsies, an increasing number of new medications are developed for childhood epilepsies, including Dravet and Lennox–Gastaut syndromes. It remains to be proven whether any of these new ASMs is more efficacious than existing ASMs.
As described in Sect. 11, in addition to new ASMs, the development of novel therapeutic strategies to prevent or modify epilepsy is an intensive area of research. This includes evaluation of ASMs such as vigabatrin, perampanel, or eslicarbazepine acetate for antiepileptogenic or disease-modifying potential in patients at risk of developing genetic or acquired epilepsies. Also, as described in Sect. 10, mTOR inhibitors such as everolimus may exert disease-modifying effects in patients with TSC. A novel strategy for epilepsy prevention is to form rationally chosen combinations of repurposed drugs that target several of the processes involved in epileptogenesis [30, 162, 163]. Another interesting approach of disease modification is increasing the brain concentration of the endogenous neuromodulator adenosine by inhibiting its degradation, which can be achieved by inhibitors of the astroglial enzyme adenosine kinase [164].
A new category of novel potentially disease-modifying medications is antisense oligonucleotide therapy, which modulates splicing of pre-messenger RNA transcript to bypass exon nonsense mutations [165]. For instance, nonsense mutations in sodium channel (SCN1A) and GABRG2 account for a proportion of Dravet syndrome. Antisense oligonucleotide therapies under preclinical or clinical development in epilepsy include ataluren, STK-001, and CUR-196 [165]. Furthermore, preclinical findings support gene therapy studies in Dravet syndrome [165].
18 Conclusions and Outlook
The ideal ASM protects against different types of epileptic seizures without adversely affecting the function of the CNS and inducing adverse effects that impair the patient’s quality of life. Because seizure activity represents a subtle functional perturbation of the normal physiologic activity of the nervous system, this goal is difficult to attain. Consequently, CNS adverse effects of ASMs are common. They can have a considerable impact on the quality of life and they contribute to treatment failure. This is probably because all current ASMs have been developed to counteract the hyperexcitability of neurons by targeting mechanisms that also interfere with normal neurotransmission; this is why they all—to a large extent—have similar issues associated with CNS tolerability [30]. Nevertheless, the long-held goal of epilepsy treatment, of “no seizures and no side effects,” can be achieved in a substantial proportion (~50%) of patients with epilepsy.
Epilepsy is a diverse disease, with multiple seizure types and epilepsy syndromes, and is associated with substantial comorbidity, including depression, anxiety, and increased mortality [1]. ASMs are often unable to treat these comorbidities or to reduce the burden of disease in a holistic sense [30]. Furthermore, current ASMs are unable to prevent or reverse the development of drug-resistant epilepsy. A particularly disquieting aspect of current epilepsy treatments is that we have not made substantial progress in seizure freedom despite the development of numerous “modern” (third-generation) ASMs. However, there is some evidence that third-generation ASMs may in some cases be associated with better tolerability, including fewer or no dermatological hypersensitivity reactions [148] and lack of drug–drug interactions, and may possibly be associated with lessening of seizure severity and frequency. Furthermore, very recently, two ASMs were introduced that may achieve seizure freedom in a significant proportion of patients with drug-resistant epilepsy: cenobamate in focal epilepsy and fenfluramine in Dravet syndrome [166].
Our understanding of the mechanisms mediating the development of epilepsy and the causes of drug resistance has grown substantially over the past two decades, providing opportunities for the discovery and development of more efficacious ASMs. For this goal, it is mandatory to revisit ASM discovery and development. The focus should be on new treatments that address key unmet medical needs: that is, drug-resistant epilepsy, comorbidities, refractory SE, and epilepsy prevention. Furthermore, treatments that modify the natural history of epilepsy, rendering the disease less progressive and easier to treat, would be highly welcome given that new-onset epilepsy is progressive in as many as one in three patients [30]. Identifying interventions that will prevent the development of epilepsy in patients at risk, as well as cure the disorder once established, will require a multifaceted approach from basic scientists and clinicians as well as industry [129]. A major incentive for the industry to adopt this approach and to execute it successfully will be the availability of valid and druggable targets, interpretable and target-population-relevant preclinical proof-of-concept studies, disease and target-related biomarkers, diagnostic methodology for the identification of the specific patient populations, and innovative clinical trial designs [30].
Change history
17 August 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40263-021-00853-6
References
Devinsky O, Vezzani A, O’Brien TJ, Jette N, Scheffer IE, De Curtis M, Perucca P. Epilepsy. Nat Rev Dis Prim. 2018;4:18024.
Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshé SL, Nordli DR, Perucca E, Tomson T, Wiebe S, Zhang YH, Zuberi SM. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:512–21.
Chen Z, Brodie MJ, Kwan P. What has been the impact of new drug treatments on epilepsy? Curr Opin Neurol. 2020;33:185–90.
Browne TR. Drug therapy reviews: clinical pharmacology of antiepileptic drugs. Am J Hosp Pharm. 1978;35:1048–56.
Shorvon S, Perucca E, Engel JJ (2020) The treatment of epilepsy. Third edition. In: Shorvon S, Perucca E, Engel JJ (eds) (Oxford: Wiley-Blackwell).
Porter RJ, Dhir A, Macdonald RL, Rogawski MA (2012). Mechanisms of action of antiseizure drugs. In: Stefan H, Theodore WH, editors. Handbook of clinical neurology, vol. 108. Amsterdam: Elsevier. pp. 663–681.
Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–9.
Chen Z, Brodie MJ, Liew D, Kwan P. Treatment outcomes in patients with newly diagnosed epilepsy treated with established and new antiepileptic drugs: a 30-year longitudinal cohort study. JAMA Neurol. 2018;75:279–86.
Janmohamed M, Brodie MJ, Kwan P. Pharmacoresistance—epidemiology, mechanisms, and impact on epilepsy treatment. Neuropharmacology. 2020;168:107790.
Perucca E, Brodie MJ, Kwan P, Tomson T. 30 years of second-generation antiseizure medications: impact and future perspectives. Lancet Neurol. 2020;19:544–56.
Löscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia. 2011;52:657–78.
Löscher W, Potschka H, Sisodiya SM, Vezzani A. Drug resistance in epilepsy: clinical impact, potential mechanisms, and new innovative treatment options. Pharmacol Rev. 2020;72:606–38.
Putnam TJ, Merritt HH. Experimental determination of the anticonvulsant properties of some phenyl derivatives. Science. 1937;85:525–6.
Keppel Hesselink JM, Kopsky DJ. Phenytoin: 80 years young, from epilepsy to breast cancer, a remarkable molecule with multiple modes of action. J Neurol. 2017;264:1617–21.
Löscher W. Animal models of seizures and epilepsy: past, present, and future role for the discovery of antiseizure drugs. Neurochem Res. 2017;42:1873–88.
Löscher W, Schmidt D. Strategies in antiepileptic drug development: is rational drug design superior to random screening and structural variation? Epilepsy Res. 1994;17:95–134.
Porter RJ, Kupferberg HJ. The anticonvulsant screening program of the national institute of neurological disorders and stroke, NIH: history and contributions to clinical care in the twentieth century and beyond. Neurochem Res. 2017;42:1889–93.
Kehne JH. National Institute of Neurological Disorders and Stroke (NINDS) Epilepsy Therapy Screening Program (ETSP). Neurochem Res. 2017;42:1894–903.
Wilcox KS, West PJ, Metcalf CS. The current approach of the epilepsy therapy screening program contract site for identifying improved therapies for the treatment of pharmacoresistant seizures in epilepsy. Neuropharmacology. 2020;166:107811.
French JA. Cenobamate for focal seizures - a game changer? Nat Rev Neurol. 2020;16:133–4.
Klein P, Krauss GL, Aboumatar S, Kamin M. Long-term efficacy and safety of adjunctive cenobamate in patients with uncontrolled focal seizures: open-label extension of a randomized clinical study. Neurology. 2020;94(Suppl. 15):1008.
Perucca E. Antiepileptic drugs: evolution of our knowledge and changes in drug trials. Epileptic Disord. 2019;21:319–29.
Boada CM, French JA, Dumanis SB. Proceedings of the 15th antiepileptic drug and device trials meeting: state of the science. Epilepsy Behav. 2020;111:107189.
Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs for the treatment of nonepileptic conditions. Nat Med. 2004;10:685–92.
Bialer M. Why are antiepileptic drugs used for nonepileptic conditions? Epilepsia. 2012;53(Suppl 7):26–33.
Swinyard EA, Kupferberg HJ. Antiepileptic drugs: detection, quantification, and evaluation. Fed Proc. 1985;44:2629–33.
Löscher W, Schmidt D. Which animal models should be used in the search for new antiepileptic drugs? A proposal based on experimental and clinical considerations. Epilepsy Res. 1988;2:145–81.
Bialer M, White HS. Key factors in the discovery and development of new antiepileptic drugs. Nat Rev Drug Discov. 2010;9:68–82.
Löscher W. Fit for purpose application of currently existing animal models in the discovery of novel epilepsy therapies. Epilepsy Res. 2016;126:157–84.
Löscher W, Klitgaard H, Twyman RE, Schmidt D. New avenues for antiepileptic drug discovery and development. Nat Rev Drug Discov. 2013;12:757–76.
Sato M, Racine RJ, McIntyre DC. Kindling: basic mechanisms and clinical validity. Electroenceph Clin Neurophysiol. 1990;76:459–72.
Löscher W, Hönack D. Profile of ucb L059, a novel anticonvulsant drug, in models of partial and generalized epilepsy in mice and rats. Eur J Pharmacol. 1993;232:147–58.
Löscher W, Gillard M, Sands ZA, Kaminski RM, Klitgaard H. synaptic vesicle glycoprotein 2A ligands in the treatment of epilepsy and beyond. CNS Drugs. 2016;30:1055–77.
Demarest ST, Brooks-Kayal A. From molecules to medicines: the dawn of targeted therapies for genetic epilepsies. Nat Rev Neurol. 2018;14:735–45.
Brunson KL, Avishai-Eliner S, Baram TZ. ACTH treatment of infantile spasms: mechanisms of its effects in modulation of neuronal excitability. Int Rev Neurobiol. 2002;49:185–97.
Niu W, Parent JM. Modeling genetic epilepsies in a dish. Dev Dyn. 2020;249:56–75.
Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, Lagae L, Moshé SL, Peltola J, Roulet PE, Scheffer IE, Zuberi SM. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:522–30.
Sills GJ, Rogawski MA. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology. 2020;168:107966.
Berkovic SF. Aggravation of generalized epilepsies. Epilepsia. 1998;39(Suppl 3):S11–4.
Klitgaard H, Matagne A, Nicolas JM, Gillard M, Lamberty Y, De Ryck M, Kaminski RM, Leclercq K, Niespodziany I, Wolff C, Wood M, Hannestad J, Kervyn S, Kenda B. Brivaracetam: rationale for discovery and preclinical profile of a selective SV2A ligand for epilepsy treatment. Epilepsia. 2016.
Glauser TA, Cnaan A, Shinnar S, Hirtz DG, Dlugos D, Masur D, Clark PO, Capparelli EV, Adamson PC. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. N Engl J Med. 2010;362:790–9.
Song JM, Hahn J, Kim SH, Chang MJ. Efficacy of treatments for infantile spasms: a systematic review. Clin Neuropharmacol. 2017;40:63–84.
Riikonen R. Infantile spasms: outcome in clinical studies. Pediatr Neurol. 2020;108:54–64.
Verrotti A, Striano P, Iapadre G, Zagaroli L, Bonanni P, Coppola G, Elia M, Mecarelli O, Franzoni E, Liso P, Vigevano F, Curatolo P. The pharmacological management of Lennox–Gastaut syndrome and critical literature review. Seizure. 2018;63:17–25.
French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, Curatolo P, de Vries PJ, Dlugos DJ, Berkowitz N, Voi M, Peyrard S, Pelov D, Franz DN. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet. 2016;388:2153–63.
Canevini MP, Kotulska-Jozwiak K, Curatolo P, La Briola F, Peron A, Slowinska M, Strzelecka J, Vignoli A, Jozwiak S. Current concepts on epilepsy management in tuberous sclerosis complex. Am J Med Genet C Semin Med Genet. 2018;178:299–308.
Wheless JW, Fulton SP, Mudigoudar BD. Dravet syndrome: a review of current management. Pediatr Neurol. 2020;107:28–40.
Cross JH, Caraballo RH, Nabbout R, Vigevano F, Guerrini R, Lagae L. Dravet syndrome: treatment options and management of prolonged seizures. Epilepsia. 2019;60(Suppl 3):S39–48.
Mei D, Cetica V, Marini C, Guerrini R. Dravet syndrome as part of the clinical and genetic spectrum of sodium channel epilepsies and encephalopathies. Epilepsia. 2019;60(Suppl 3):S2–7.
Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, Devinsky O, Cross JH, Guerrini R, Talwar D, Miller I, Farfel G, Galer BS, Gammaitoni A, Mistry A, Morrison G, Lock M, Agarwal A, Lai WW, Ceulemans B. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. Lancet. 2019;394:2243–54.
Nabbout R, Mistry A, Zuberi S, Villeneuve N, Gil-Nagel A, Sanchez-Carpintero R, Stephani U, Laux L, Wirrell E, Knupp K, Chiron C, Farfel G, Galer BS, Morrison G, Lock M, Agarwal A, Auvin S. Fenfluramine for treatment-resistant seizures in patients with dravet syndrome receiving stiripentol-inclusive regimens: a randomized clinical trial. JAMA Neurol. 2020;77:300–8.
Campos MSA, Ayres LR, Morelo MRS, Carizio FAM, Pereira LRL. Comparative efficacy of antiepileptic drugs for patients with generalized epileptic seizures: systematic review and network meta-analyses. Int J Clin Pharm. 2018;40:589–98.
Lattanzi S, Zaccara G, Giovannelli F, Grillo E, Nardone R, Silvestrini M, Trinka E, Brigo F. Antiepileptic monotherapy in newly diagnosed focal epilepsy. A network meta-analysis. Acta Neurol Scand. 2019;139:33–41.
Perucca E. From clinical trials of antiepileptic drugs to treatment. Epilepsia Open. 2018;3:220–30.
Golyala A, Kwan P. Drug development for refractory epilepsy: the past 25 years and beyond. Seizure. 2017;44:147–56.
Costa J, Fareleira F, Ascencao R, Borges M, Sampaio C, Vaz-Carneiro A. Clinical comparability of the new antiepileptic drugs in refractory partial epilepsy: a systematic review and meta-analysis. Epilepsia. 2011;52:1280–91.
Hu Q, Zhang F, Teng W, Hao F, Zhang J, Yin M, Wang N. Efficacy and safety of antiepileptic drugs for refractory partial-onset epilepsy: a network meta-analysis. J Neurol. 2018;265:1–11.
Gao L, Xia L, Zhao FL, Li SC. Clinical efficacy and safety of the newer antiepileptic drugs as adjunctive treatment in adults with refractory partial-onset epilepsy: a meta-analysis of randomized placebo-controlled trials. Epilepsy Res. 2013;103:31–44.
Gazzola DM, Balcer LJ, French JA. Seizure-free outcome in randomized add-on trials of the new antiepileptic drugs. Epilepsia. 2007;48:1303–7.
Halford JJ, Edwards JC. Seizure freedom as an outcome in epilepsy treatment clinical trials. Acta Neurol Scand. 2020;142:91–107.
Krauss GL, Klein P, Brandt C, Lee SK, Milanov I, Milovanovic M, Steinhoff BJ, Kamin M. Safety and efficacy of adjunctive cenobamate (YKP3089) in patients with uncontrolled focal seizures: a multicentre, double-blind, randomised, placebo-controlled, dose-response trial. Lancet Neurol. 2020;19:38–48.
Guignet M, Campbell A, White HS. Cenobamate (XCOPRI®): can preclinical and clinical evidence provide insight into its mechanism of action? Epilepsia. 2020;61:2329–39.
Stephen LJ, Brodie MJ. Selection of antiepileptic drugs in adults. Neurol Clin. 2009;27:967–92.
Boon P, Ferrao SS, Jansen AC, Lagae L, Legros B, Weckhuysen S. Recommendations for the treatment of epilepsy in adult and pediatric patients in Belgium: 2020 update. Acta Neurol Belg. 2021;121:241–257.
Glauser T, Ben Menachem E, Bourgeois B, Cnaan A, Chadwick D, Guerreiro C, Kalviainen R, Mattson R, Perucca E, Tomson T. ILAE treatment guidelines: evidence-based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2006;47:1094–120.
Glauser T, Ben Menachem E, Bourgeois B, Cnaan A, Guerreiro C, KÃlviÃinen R, Mattson R, French JA, Perucca E, Tomson T. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2013;54:551–63.
Kanner AM, Ashman E, Gloss D, Harden C, Bourgeois B, Bautista JF, Abou-Khalil B, Burakgazi-Dalkilic E, Llanas PE, Stern J, Hirtz D, Nespeca M, Gidal B, Faught E, French J. Practice guideline update summary: Efficacy and tolerability of the new antiepileptic drugs I: treatment of new-onset epilepsy: report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2018;91:74–81.
Kanner AM, Ashman E, Gloss D, Harden C, Bourgeois B, Bautista JF, Abou-Khalil B, Burakgazi-Dalkilic E, Llanas PE, Stern J, Hirtz D, Nespeca M, Gidal B, Faught E, French J. Practice guideline update summary: Efficacy and tolerability of the new antiepileptic drugs II: treatment-resistant epilepsy: report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2018;91:82–90.
Burakgazi E, French JA. Treatment of epilepsy in adults. Epileptic Disord. 2016;18:228–39.
Tomson T, Battino D, Perucca E. Valproic acid after five decades of use in epilepsy: time to reconsider the indications of a time-honoured drug. Lancet Neurol. 2016;15:210–8.
Wyllie E. Wyllie's treatment of epilepsy. In: Wyllie E, editor. Philadelphia: Wolters Kluwer; 2021.
Vidaurre J, Gedela S, Yarosz S. Antiepileptic drugs and liver disease. Pediatr Neurol. 2017;77:23–36.
Lu X, Wang X. Hyponatremia induced by antiepileptic drugs in patients with epilepsy. Expert Opin Drug Saf. 2017;16:77–87.
Diemar SS, Sejling AS, Eiken P, Andersen NB, Jörgensen NR. An explorative literature review of the multifactorial causes of osteoporosis in epilepsy. Epilepsy Behav. 2019;100:106511.
Klein P, Herzog AG. Endocrine aspects of partial epilepsy. in the comprehensive management and treatment of epilepsy. In: Schachter SC, Schomer DL, editors. San Diego: Academic Press; 1997. pp. 207–232.
Parikh SK, Silberstein SD. Current status of antiepileptic drugs as preventive migraine therapy. Curr Treat Opt Neurol. 2019;21:16.
Goh KK, Chang SC, Chen CH, Lu ML. Therapeutic strategies for treatment-resistant depression: state of the art and future perspectives. Curr Pharm Des. 2020;26:244–52.
Kanner AM. Management of psychiatric and neurological comorbidities in epilepsy. Nat Rev Neurol. 2016;12:106–16.
Faught E. Topiramate in the treatment of partial and generalized epilepsy. Neuropsychiatr Dis Treat. 2007;3:811–21.
Shvarts V, Chung S. Epilepsy, antiseizure therapy, and sleep cycle parameters. Epilepsy Res Treat. 2013;2013:670682.
Vinik A. Clinical review: use of antiepileptic drugs in the treatment of chronic painful diabetic neuropathy. J Clin Endocrinol Metab. 2005;90:4936–45.
Goodman CW, Brett AS. A clinical overview of off-label use of gabapentinoid drugs. JAMA Intern Med. 2019;179:695–701.
Cruccu G, Di Stefano G, Truini A. Trigeminal neuralgia. N Engl J Med. 2020;383:754–62.
Tzadok R, Ablin JN. Current and emerging pharmacotherapy for fibromyalgia. Pain Res Manag. 2020;2020:6541798.
Anguelova GV, Vlak MHM, Kurvers AGY, Rijsman RM. Pharmacologic and nonpharmacologic treatment of restless legs syndrome. Sleep Med Clin. 2020;15:277–88.
Ondo WG. Current and emerging treatments of essential tremor. Neurol Clin. 2020;38:309–23.
Tomson T, Battino D, Bromley R, Kochen S, Meador K, Pennell P, Thomas SV. Executive summary: management of epilepsy in pregnancy: a report from the international league against epilepsy task force on women and pregnancy. Epilepsia. 2019;60:2343–5.
Kellogg M, Meador KJ. Neurodevelopmental effects of antiepileptic drugs. Neurochem Res. 2017;42:2065–70.
Sen A, Jette N, Husain M, Sander JW. Epilepsy in older people. Lancet. 2020;395:735–48.
Anderson G. Pharmacokinetics and drug interactions. In: Wyllie's treatment of epilepsy. In: Wyllie E, editor. Philadelphia: Wolters and Kluwer; 2021. pp. 520–531.
Srivastava AK, Alex AB, Wilcox KS, White HS. Rapid loss of efficacy to the antiseizure drugs lamotrigine and carbamazepine: a novel experimental model of pharmacoresistant epilepsy. Epilepsia. 2013;54:1186–94.
Löscher W, Rundfeldt C, Hönack D. Pharmacological characterization of phenytoin-resistant amygdala-kindled rats, a new model of drug-resistant partial epilepsy. Epilepsy Res. 1993;15:207–19.
Kwan P, Brodie MJ. Combination therapy in epilepsy: when and what to use. Drugs. 2006;66:1817–29.
Brodie MJ, Sills GJ. Combining antiepileptic drugs—rational polytherapy? Seizure. 2011;20:369–75.
Verrotti A, Lattanzi S, Brigo F, Zaccara G. Pharmacodynamic interactions of antiepileptic drugs: from bench to clinical practice. Epilepsy Behav. 2020;104:106939.
Deckers CL, Czuczwar SJ, Hekster YA, Keyser A, Kubova H, Meinardi H, Patsalos PN, Renier WO, van Rijn CM. Selection of antiepileptic drug polytherapy based on mechanisms of action: the evidence reviewed. Epilepsia. 2000;2000(41):1364–74.
Brodie MJ, Yuen AWC. Lamotrigine substitution study: evidence for synergism with sodium valproate? Epilepsy Res. 1997;26:423–32.
Pisani F, Oteri G, Russo MF, Di Perri R, Perucca E, Richens A. The efficacy of valproate-lamotrigine comedication in refractory complex partial seizures: evidence for a pharmacodynamic interaction. Epilepsia. 1999;40:1141–6.
Blaszczyk B, Miziak B, Czuczwar P, Wierzchowska-Cioch E, Pluta R, Czuczwar SJ. A viewpoint on rational and irrational fixed-drug combinations. Expert Rev Clin Pharmacol. 2018;11:761–71.
Perucca E, Gram L, Avanzini G, Dulac O. Antiepileptic drugs as a cause of worsening seizures. Epilepsia. 1998;39:5–17.
Gayatri NA, Livingston JH. Aggravation of epilepsy by anti-epileptic drugs. Dev Med Child Neurol. 2006;48:394–8.
Somerville ER. Some treatments cause seizure aggravation in idiopathic epilepsies (especially absence epilepsy). Epilepsia. 2009;50(Suppl 8):31–6.
Sazgar M, Bourgeois BF. Aggravation of epilepsy by antiepileptic drugs. Pediatr Neurol. 2005;33:227–34.
Beghi E, Carpio A, Forsgren L, Hesdorffer DC, Malmgren K, Sander JW, Tomson T, Hauser WA. Recommendation for a definition of acute symptomatic seizure. Epilepsia. 2010;51:671–5.
Gunawardane N, Fields M. Acute symptomatic seizures and provoked seizures: to treat or not to treat? Curr Treat Opt Neurol. 2018;20:41.
Haut SR. Seizure clusters: characteristics and treatment. Curr Opin Neurol. 2015;28:143–50.
Jafarpour S, Hirsch LJ, GaÃnza-Lein M, Kellinghaus C, Detyniecki K. Seizure cluster: definition, prevalence, consequences, and management. Seizure. 2019;68:9–15.
Gidal B, Klein P, Hirsch LJ. Seizure clusters, rescue treatments, seizure action plans: Unmet needs and emerging formulations. Epilepsy Behav. 2020;112:107391.
Lowenstein DH. The management of refractory status epilepticus: an update. Epilepsia. 2006;47(Suppl 1):35–40.
Glauser T, Shinnar S, Gloss D, Alldredge B, Arya R, Bainbridge J, Bare M, Bleck T, Dodson WE, Garrity L, Jagoda A, Lowenstein D, Pellock J, Riviello J, Sloan E, Treiman DM. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr. 2016;16:48–61.
Kapur J, Elm J, Chamberlain JM, Barsan W, Cloyd J, Lowenstein D, Shinnar S, Conwit R, Meinzer C, Cock H, Fountain N, Connor JT, Silbergleit R. Randomized trial of three anticonvulsant medications for status epilepticus. N Engl J Med. 2019;381:2103–13.
Chamberlain JM, Kapur J, Shinnar S, Elm J, Holsti M, Babcock L, Rogers A, Barsan W, Cloyd J, Lowenstein D, Bleck TP, Conwit R, Meinzer C, Cock H, Fountain NB, Underwood E, Connor JT, Silbergleit R. Efficacy of levetiracetam, fosphenytoin, and valproate for established status epilepticus by age group (ESETT): a double-blind, responsive-adaptive, randomised controlled trial. Lancet. 2020;395:1217–24.
Rossetti AO, Lowenstein DH. Management of refractory status epilepticus in adults: still more questions than answers. Lancet Neurol. 2011;10:922–30.
Silverstein FS, Jensen FE. Neonatal seizures. Ann Neurol. 2007;62:112–20.
Donovan MD, Griffin BT, Kharoshankaya L, Cryan JF, Boylan GB. Pharmacotherapy for neonatal seizures: current knowledge and future perspectives. Drugs. 2016;76:647–61.
Sharpe C, Reiner GE, Davis SL, Nespeca M, Gold JJ, Rasmussen M, Kuperman R, Harbert MJ, Michelson D, Joe P, Wang S, Rismanchi N, Le NM, Mower A, Kim J, Battin MR, Lane B, Honold J, Knodel E, Arnell K, Bridge R, Lee L, Ernstrom K, Raman R, Haas RH. Levetiracetam versus phenobarbital for neonatal seizures: a randomized controlled trial. Pediatrics. 2020;145.
Millar LJ, Shi L, Hoerder-Suabedissen A, Molnár Z. Neonatal hypoxia ischaemia: mechanisms, models, and therapeutic challenges. Front Cell Neurosci. 2017;11:78.
Smith DK, Sadler KP, Benedum M. Febrile seizures: risks, evaluation, and prognosis. Am Fam Physician. 2019;99:445–50.
McTague A, Martland T, Appleton R. Drug management for acute tonic-clonic convulsions including convulsive status epilepticus in children. Cochrane Database Syst Rev. 2018;1:CD001905.
Alles SRA, Smith PA. Etiology and pharmacology of neuropathic pain. Pharmacol Rev. 2018;70:315–47.
Garakani A, Murrough JW, Freire RC, Thom RP, Larkin K, Buono FD, Iosifescu DV. Pharmacotherapy of anxiety disorders: current and emerging treatment options. Front Psychiatry. 2020;11:595584.
Baldwin DS, Ajel K, Masdrakis VG, Nowak M, Rafiq R. Pregabalin for the treatment of generalized anxiety disorder: an update. Neuropsychiatr Dis Treat. 2013;9:883–92.
Li C, Xue L, Liu Y, Yang Z, Chi S, Xie A. Zonisamide for the treatment of Parkinson disease: a current update. Front Neurosci. 2020;14:574652.
Gonzalez-Latapi P, Bhowmick SS, Saranza G, Fox SH. Non-dopaminergic treatments for motor control in Parkinson’s disease: an update. CNS Drugs. 2020;34:1025–44.
Chagas MH, Eckeli AL, Zuardi AW, Pena-Pereira MA, Sobreira-Neto MA, Sobreira ET, et al. Cannabidiol can improve complex sleep-related behaviours associated with rapid eye movement sleep behaviour disorder in Parkinson’s disease patients: a case series. J Clin Pharm Ther. 2014;39:564–6.
Rogawski MA, Löscher W, Rho JM. Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harb Perspect Med. 2016.
Brennan GP, Baram TZ, Poolos NP. Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels in epilepsy. Cold Spring Harb Perspect Med. 2016;6:a022384.
Sisodiya SM. Precision medicine and therapies of the future. Epilepsia. 2021;62(Suppl.2):S90–S105.
Carvill GL, Dulla CG, Lowenstein DH, Brooks-Kayal AR. The path from scientific discovery to cures for epilepsy. Neuropharmacology. 2020;167:107702.
Löscher W. The holy grail of epilepsy prevention: preclinical approaches to antiepileptogenic treatments. Neuropharmacology. 2020;167:107605.
Jozwiak S, Kotulska-Jozwiak K, Bebin M, Wong M. Modifying genetic epilepsies—results from studies on tuberous sclerosis complex and their potential mpact. Neuropharmacology. 2020;166:107908.
Jeong A, Wong M. mTOR inhibitors in children: current indications and future directions in neurology. Curr Neurol Neurosci Rep. 2016;16:102.
van der Poest CE, Jansen FE, Braun KPJ, Peters JM. Update on Drug Management of Refractory Epilepsy in Tuberous Sclerosis Complex. Paediatr Drugs. 2020;22:73–84.
Overwater IE, Rietman AB, van Eeghen AM, de Wit MCY. Everolimus for the treatment of refractory seizures associated with tuberous sclerosis complex (TSC): current perspectives. Ther Clin Risk Manag. 2019;15:951–5.
Franz DN, Weiss BD. Molecular therapies for tuberous sclerosis and neurofibromatosis. Curr Neurol Neurosci Rep. 2012;12:294–301.
Kotulska K, Kwiatkowski DJ, Curatolo P, Weschke B, Riney K, Jansen F, Feucht M, Krsek P, Nabbout R, Jansen AC, Wojdan K, Sijko K, Glowacka-Walas J, Borkowska J, Sadowski K, Domanska-Pakiela D, Moavero R, Hertzberg C, Hulshof H, Scholl T, Benova B, Aronica E, de Ridder J, Lagae L, Joszwiak S. Prevention of epilepsy in infants with tuberous sclerosis complex in the EPISTOP trial. Ann Neurol. 2021;89:304–14.
Löscher W, Friedman A. Structural, molecular and functional alterations of the blood-brain barrier during epileptogenesis and epilepsy: a cause, consequence or both? Int J Mol Sci. 2020;21:591.
Löscher W. The pharmacokinetics of antiepileptic drugs in rats: consequences for maintaining effective drug levels during prolonged drug administration in rat models of epilepsy. Epilepsia. 2007;48:1245–58.
Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27–31.
Löscher W, Schmidt D. Experimental and clinical evidence for loss of effect (tolerance) during prolonged treatment with antiepileptic drugs. Epilepsia. 2006;47:1253–84.
Perucca E, Meador KJ. Adverse effects of antiepileptic drugs. Acta Neurol Scand Suppl. 2005;181:30–5.
Patsalos PN, Spencer EP, Berry DJ. Therapeutic drug monitoring of antiepileptic drugs in epilepsy: a 2018 update. Ther Drug Monit. 2018;40:526–48.
Bialer M, Perucca E. Does cannabidiol have antiseizure activity independent of its interactions with clobazam? An appraisal of the evidence from randomized controlled trials. Epilepsia. 2020;61:1082–9.
Klein P, Tolbert D, Gidal BE. Drug–drug interactions and pharmacodynamics of concomitant clobazam and cannabidiol or stiripentol in refractory seizures. Epilepsy Behav. 2019;99:106459.
Sommerfeld-Klatta K, Zielinska-Psuja B, Karazniewcz-Lada, Glowka FK. New methods used in pharmacokinetics and therapeutic monitoring of the first and newer generations of antiepileptic drugs (AEDs). Molecules. 25;2020.
Patsalos PN, Berry DJ, Bourgeois BF, Cloyd JC, Glauser TA, Johannessen SI, Leppik IE, Tomson T, Perucca E. Antiepileptic drugs–best practice guidelines for therapeutic drug monitoring: a position paper by the subcommission on therapeutic drug monitoring, ILAE Commission on Therapeutic Strategies. Epilepsia. 2008;49:1239–76.
Cramer JA. Tolerability of antiepileptic drugs: can we determine differences? Epilepsy Behav. 2012;23:187–92.
Brodie MJ. Tolerability and safety of commonly used antiepileptic drugs in adolescents and adults: a clinician’s overview. CNS Drugs. 2017;31:135–47.
Brodie MJ, Kwan P. Current position of phenobarbital in epilepsy and its future. Epilepsia. 2012;53(Suppl 8):40–6.
Tomson T, Battino D. Teratogenic effects of antiepileptic drugs. Lancet Neurol. 2012;11:803–13.
Löscher W, Hönack D. The novel competitive N-methyl-D-aspartate (NMDA) antagonist CGP 37849 preferentially induces phencyclidine-like behavioral effects in kindled rats: attenuation by manipulation of dopamine, alpha-1 and serotonin1A receptors. J Pharmacol Exp Ther. 1991;257:1146–53.
Klitgaard H, Matagne A, Lamberty Y. Use of epileptic animals for adverse effect testing. Epilepsy Res. 2002;50:55–65.
Meldrum B. Do preclinical seizure models preselect certain adverse effects of antiepileptic drugs. Epilepsy Res. 2002;50:33–40.
Leppik IE. Rational polypharmacy. Amsterdam: Elsevier; 1996.
Reynolds EH, Shorvon SD. Monotherapy or polytherapy for epilepsy? Epilepsia. 1981;22:1–10.
Perucca E. General principles of medical management. In: Shorvon S, Perucca E, Engel JJ, editors. The treatment of epilepsy. 3rd ed. Oxford: Wiley-Blackwell; 2009. p. 121–39.
Guberman A. Monotherapy or polytherapy for epilepsy? Can J Neurol Sci. 1998;25:S3–8.
Schmidt D. Drug treatment strategies for epilepsy revisited: starting early or late? One drug or several drugs? Epileptic Disord. 2016;18:356–66.
Skolnick P. Anxioselective anxiolytics: on a quest for the Holy Grail. Trends Pharmacol Sci. 2012;33:611–20.
Kasteleijn-Nolst Trenité DG, Groenwold RH, Schmidt B, Löscher W. Single dose efficacy evaluation of two partial benzodiazepine receptor agonists in photosensitive epilepsy patients: a placebo-controlled pilot study. Epilepsy Res. 2016;122:30–6.
Rundfeldt C, Löscher W. The pharmacology of imepitoin: the first partial benzodiazepine receptor agonist developed for the treatment of epilepsy. CNS Drugs. 2014;28:29–43.
Schidlitzki A, Bascunana P, Srivastava PK, Welzel L, Twele F, Töllner K, Käufer C, Gericke B, Feleke R, Meier M, Polyak A, Ross TL, Gerhauser I, Bankstahl JP, Johnson MR, Bankstahl M, Löscher W. Proof-of-concept that network pharmacology is effective to modify development of acquired temporal lobe epilepsy. Neurobiol Dis. 2020;134:104664.
Welzel L, Bergin D, Schidlitzki A, Twele F, Johne A, Klein P, Löscher W. Systematic evaluation of rationally chosen multitargeted drug combinations: a combination of low doses of levetiracetam, atorvastatin and ceftriaxone exerts antiepileptogenic effects in a mouse model of acquired epilepsy. Neurobiol Dis. 2021;149:105227.
Boison D, Jarvis MF. Adenosine kinase: a key regulator of purinergic physiology. Biochem Pharmacol. 2020;114321.
Steriade C, French J, Devinsky O. Epilepsy: key experimental therapeutics in early clinical development. Expert Opin Investig Drugs. 2020;29:373–83.
Klein P, Tyrlikova I. No prevention or cure of epilepsy as yet. Neuropharmacology. 2020;168:107762.
Löscher W, Schmidt D. Epilepsy: perampanel-new promise for refractory epilepsy? Nat Rev Neurol. 2012;8:661–2.
Pitkänen A, Buckmaster PS, Galanopoulou AS, Moshé SL. Models of seizures and epilepsy. 2nd ed. London: Academic Press; 2017.
Wirrell EC, Nabbout R. Recent advances in the drug treatment of Dravet syndrome. CNS Drugs. 2019;33:867–81.
Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5:553–64.
Löscher W, Sills GJ, White HS. The ups and downs of alkyl-carbamates in epilepsy therapy: How does cenobamate differ? Epilepsia. 2021 (in press).
Baraban SC, Löscher W. What new modeling approaches will help us identify promising drug treatments? Adv Exp Med Biol. 2014;813:283–94.
Kaur H, Kumar B, Medhi B. Antiepileptic drugs in development pipeline: a recent update. eNeurologicalSci. 2016;4:42–51.
Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Tomson T, White HS. Progress report on new antiepileptic drugs: a summary of the fourteenth eilat conference on new antiepileptic drugs and devices (EILAT XIV). I. Drugs in preclinical and early clinical development. Epilepsia. 59;2018:1811–41.
Auvin S. Lennox–Gastaut syndrome: new treatments and treatments under investigation. Rev Neurol (Paris). 2020;176:444–7.
Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Perucca P, Tomson T, White HS. Progress report on new antiepileptic drugs: a summary of the fifteenth eilat conference on new antiepileptic drugs and devices (EILAT XV). I. Drugs in preclinical and early clinical development. Epilepsia. 2020;61:2340–64.
Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Perucca P, Tomson T, White HS. Progress report on new antiepileptic drugs: a summary of the fifteenth eilat conference on new antiepileptic drugs and devices (EILAT XV). II. Drugs in more advanced clinical development. Epilepsia. 2020;61:2365–85.
Acknowledgements
This review is dedicated to the late Professor Hans-Hasso Frey, who acted as a mentor for W. Löscher throughout his scientific career.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access Publishing enabled and organized by Projekt DEAL. The open access publication was supported by the Deutsche Forschungsgemeinschaft and University of Veterinary Medicine Hannover, Foundation, within the funding program. No sources of funding were used to conduct this study or prepare this manuscript.
Conflict of interest
WL and PK are co-founders as well as CFO and CSO, respectively, of PrevEp, Inc. (Bethesda, MD, USA). PrevEp did not fund this review and played no role in the writing of the review. WL was involved in the development of levetiracetam (UCB Pharma) and imepitoin (Elbion/Boehringer Ingelheim); has received consultancy fees from Lundbeck, AC Immune, Clexio Biosciences, UCB Pharma, Pragma Therapeutics, Boehringer Ingelheim, Pfizer, and Johnson & Johnson; and has served on the advisory boards of Grünenthal, UCB Pharma, and Angelini Pharma. PK receives grant support from CURE/US Department of Defense; has received consulting or speaker fees from or been on the advisory boards of Abbot, Aquestive, Arvelle, Eisai, Greenwich Pharmaceuticals, Neurelis, SK Life Science, Sunovion, and UCB Pharma; and is on the medical advisory board of Alliance-Stratus and the scientific advisory board of OB Pharma.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
WL and PK performed the literature search and wrote the manuscript. Both authors read and approved the final manuscript.
Additional information
The original online version of this article was revised: In the original publication, Figure 1 was incorrectly published and this has been corrected now.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Löscher, W., Klein, P. The Pharmacology and Clinical Efficacy of Antiseizure Medications: From Bromide Salts to Cenobamate and Beyond. CNS Drugs 35, 935–963 (2021). https://doi.org/10.1007/s40263-021-00827-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-021-00827-8