
Abstract
Background and Objective
Venlafaxine hydrochloride extended-release (ER) capsules are commonly used to treat depression and anxiety disorders. Evaluation of the bioequivalence of generic formulations with reference products is essential to ensure therapeutic equivalence. The objective of this study was to evaluate the bioequivalence, safety, and tolerability of Chinese-manufactured venlafaxine hydrochloride extended-release capsules compared with USA-manufactured EFFEXOR® XR in healthy Chinese volunteers under fed conditions.
Methods
A randomized, open-label, single-dose, crossover study was conducted. Subjects were randomly assigned to receive the test formulation (one 150-mg ER capsule manufactured in China) or the reference formulation (one 150-mg ER capsule manufactured in the USA). The bioequivalence of the two drugs was assessed using the area under the plasma concentration–time curve from time zero to the last sampling time (AUC0–t) and the maximum observed concentration (Cmax).
Results
A total of 28 subjects were enrolled and randomly assigned to receive a single dose of either the test or reference capsule. All the subjects completed the study and were included in the pharmacokinetic (PK) and safety analyses. The mean AUC0–t and Cmax of venlafaxine and its active metabolite O-desmethylvenlafaxine were comparable between the test and reference products with both parameters close to 100% and the corresponding 90% confidence intervals within the specified 80–125% bioequivalence boundary. Safety was also assessed between the two products and all adverse events (AEs) in this study were mild in severity.
Conclusions
Both the test and reference venlafaxine hydrochloride ER capsules were bioequivalent and showed a similar safety and tolerability profile in the population studied.
Clinical Trials Registration
This study was registered at the Drug Clinical Trial Registration and Information Publicity Platform (http://www.chinadrugtrials.org.cn/index.html) with registration number CTR20211243, date: June 1, 2021.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The bioequivalence of Chinese- and USA-manufactured venlafaxine (150 mg) extended-release capsules was demonstrated under fed conditions in healthy Chinese volunteers. |
1 Introduction
Major depressive disorder (MDD), a common psychiatric condition characterized by depressed mood, affects about 4.4% of the world’s population [1]. Antidepressants are one of the mainstays of depression treatment with large market demand. Venlafaxine is a third-generation selective serotonin and norepinephrine reuptake inhibitor (SNRI) antidepressant that exerts a dual antidepressant effect by blocking the reuptake of norepinephrine and serotonin (5-HT) [2].
Venlafaxine has been reported to be well absorbed after oral administration, at least 92% of a single dose of venlafaxine is absorbed after oral administration and the absolute bioavailability of venlafaxine is 45% [3]. Taking it with food may reduce its absorption rate, but it does not affect the degree of absorption [3,4,5]. Venlafaxine undergoes significant first-pass metabolism by CYP2D6 in the intestine and liver to produce O-desmethylvenlafaxine (also known as desvenlafaxine, ODV), which is further converted to N-desmethylvenlafaxine (NDV) by CYP2C19 [6]. Approximately 5% of an administered dose is excreted in the urine as unchanged drug, with <30% excreted as the active metabolite, ODV [7].
Both venlafaxine and its active metabolite, ODV, can partially antagonize the reuptake of 5-HT and norepinephrine, and has a certain effect on the reuptake of dopamine, with antidepressant effect [3, 8]. Thus, the US Food and Drug Administration (FDA) guidelines suggest to measure the concentrations of the original drug venlafaxine and its active metabolite ODV in plasma in the equivalence study of venlafaxine hydrochloride extended-release capsules [9]. The mean t½ of venlafaxine is 2~13 h and that of ODV is 10~19 h [10,11,12,13], necessitating administration two or three times daily to maintain adequate plasma drug concentrations [14]. A microencapsulated, extended-release formulation of venlafaxine was developed to allow once-daily administration [15].
Venlafaxine had an initially increasing dose–efficacy relationship up to around 75~150 mg, followed by a more modest increase [16]. Thus, the 150-mg dosage of venlafaxine sustained-release capsule is widely used in the clinical treatment of various types of depression with the advantages of fast onset [17], less adverse reactions [18], a larger tolerated dose range [16, 19], and a higher clinical cure rate for patients with major depressive disorder [20].
It was first developed by Wyeth and first approved by the US FDA in October 1997, with the trade name ‘EFFEXOR® XR’. However, no generic form of EFFEXOR® XR has been approved in China. The relatively high cost resulted in the medication being prescribed less. A more affordable form of this drug may benefit the depressive disorder population. Based on previous literature, venlafaxine extended-release capsule bioequivalence trials were conducted under fasting and postprandial conditions [21]. However, according to the FDA’s draft guidance on venlafaxine hydrochloride, fasting bioequivalence studies are not recommended due to safety concerns [9]. Therefore, we compared the bioequivalence and safety of venlafaxine extended-release capsule 150 mg produced by Hubei Duorui Pharmaceutical Company with EFFEXOR® XR in healthy Chinese subjects under fed conditions.
2 Materials and Methods
2.1 Study Design
We employed an open, single-center, randomized, two-period crossover, single-dose trial design for this study. A total of 87 volunteers were initially recruited to this trial, of which 28 subjects met the inclusion criteria and were randomly assigned to either the Test formulation (T)-Reference formulation (R) group (T-R group) or the R-T group, in a 1:1 ratio. The sequence of study events is visually depicted in Fig. S1 (see electronic supplementary material [ESM]). Each subject received a single dose of either the T formulation, containing venlafaxine 150 mg (manufactured by Hubei Duorui Pharmaceutical, Wuhan, PR China) or the R formulation, containing venlafaxine 150 mg (manufactured by Pfizer Pharmaceutical Co., Ltd, United States) 30 minutes after consuming a high-calorie breakfast. The high-fat breakfast comprised 1000 kcal total calories, of which fat accounted for 50%. Subsequently, lunch was provided to the subjects 4 h after dosing. Followed by another meal 10 h after drug administration. Throughout the study, strict control over the subjects’ food intake was maintained. A washout period of 8 days was implemented between the two treatment periods.
This study adhered to the guidelines outlined by the International Conference on Harmonization (ICH) for Good Clinical Practice and the Helsinki Declaration of 1964, along with its subsequent revisions. Approval from the ethics committee at Wuhan Pulmonary Hospital was obtained prior to the commencement of the study (approved No. of ethic committee. 2021004). Informed consent was obtained from all eligible participants before any research procedures were conducted.
2.2 Subjects
This investigation involved a cohort of Chinese individuals, both male and female, aged 18–65 years with a body mass index of 19.0–27.0 kg/m2, and a weight of ≥50.0 kg for males and ≥45.0 kg for females. Prior to enrollment, all potential participants underwent a thorough evaluation of their medical history, physical and mental condition, and clinical laboratory results, such as blood and urine biochemistry and hematology test, to confirm their overall well-being. Subjects demonstrated an understanding of the purpose, protocol, and possible adverse events (AEs) of the study drugs and provided informed consent prior to participating in the clinical trial.
Individuals who had any significant medical disorder or condition that could potentially affect the absorption of the drug, had engaged in smoking or alcohol abuse within the past 3 months, or had a positive nicotine test were not included in the study. Additionally, subjects who had donated blood or experienced extensive blood loss exceeding 200 mL within the 3 months prior to the trial, unable to tolerate venous puncture for blood collection or high-fat meals or had a history of milk diarrhea (lactose intolerance) were also excluded. Moreover, pregnant or lactating women were not eligible for participation, and individuals who were known to be hypersensitive to venlafaxine or other specified substances were also excluded.
2.3 Study Endpoints
AUC0–t, the AUC from time zero to infinity (AUC0–∞), and the Cmax were the primary endpoints of the study, while the secondary endpoints were time to peak drug concentration (Tmax), apparent terminal half-life (t½), AUC from time to the last quantifiable concentration extrapolated to infinity expressed as a percentage of AUC0–∞ (AUCextra%), terminal elimination rate constant (λz), apparent total body clearance of the drug after extravascular administration (CL/f), and apparent volume of distribution in the terminal phase after extravascular administration (Vz/f). Safety analysis included assessment of AEs, vital signs (blood pressure, pulse rate, body temperature, and respiration), clinical laboratory tests (biochemistry, hematology, and urinalysis), electrocardiogram (ECG), and physical examination.
AEs were classified by system organ class (SOC) and preferred term (PT) using the Medical Dictionary for Regulatory Activities (MedDRA) version 26.1 classification system and graded according to severity and potential relationship to the study drug using the Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 guidelines. In accordance with the World Health Organisation (WHO) guidelines and with reference to the methodology proposed by KARCH and LASAGNA in 1977 [22], which is now widely used, we classified the evaluation of the causal relevance of adverse drug reactions (ADRs) into six levels: positive, probable, possible, probably irrelevant, to be evaluated, and unable to be evaluated.
2.4 Blood Sampling and Bioanalytical Methods
Blood samples were collected at various time points throughout the study, including before a high-calorie breakfast (baseline), and at multiple time intervals after dosing (1, 2, 3, 4, 4.5, 5, 5.5, 6, 6.5, 7, 7.5, 8, 9, 10, 11, 12, 13, 14, 16, 18, 24, 36, 48, and 72 h on days 1–4 of each period) for PK measurement. The collected blood samples were subjected to centrifugation at a temperature of 25 ℃ for 10 min at a speed of 1700 g. Subsequently, the plasma samples were separated within 100 min and stored at a temperature below −60 ℃ until further analysis.
The ACQUITY UPLC/Xevo TQ MS/MS system (Waters Corp., USA) was utilized to determine the plasma concentration of venlafaxine and its metabolite ODV [23]. The lower limit of quantification (LLOQ) for venlafaxine and ODV was determined to be 1 ng/mL. The inter-assay variation for all samples was <20%. The noncompartmental method was employed to calculate the PK parameters of venlafaxine and its main metabolite ODV.
2.5 Statistical Methods
Phoenix WinNonlin software (version 7.0) was used to calculate PK parameters using a non-atrioventricular model. According to the drug grouping, the analysis results of the main PK parameters were summarized by sample size, arithmetic mean, standard deviation, coefficient of variation, minimum and maximum, and geometric mean. The key PK parameters including Cmax, Tmax, t½, λz, AUC0–t, and AUC0–∞ were statistically analyzed, and the difference is considered statistically significant when p < 0.05. AUC and Cmax require logarithmic conversion followed by analysis of variance for random-effects models. ANOVA for random-effects models breaks down total variation into sequential variation, periodic variation, treatment variation (variation caused by the test and reference formulations), between-individual variation, and within-individual variation (residuals).
The mixed linear model confidence interval method was used for bioequivalence evaluation. The 90% confidence intervals of the PK parameters Cmax, AUC0–t, and AUC0–∞ of the test formulation are within the range of 80.00~125.00% of the corresponding parameters of the reference formulation, and the bioequivalence of the two formations may be convicted. SAS (version 9.4) software was used for statistical analysis of safety variables.
3 Results
3.1 Subject Characteristics
A screening process was conducted on a total of 87 volunteers, out of which 28 individuals (comprising 21 males and 7 females) successfully enrolled and completed the entire clinical trial. The remaining 59 individuals were excluded from the study due to screening failures, as illustrated in the Table S1. The most common reason for screening failures was ‘eligibility criteria not met’. All 28 randomized subjects received the study drug, completed the study, and were included in the PK and safety analysis sets. Detailed characteristics of the participants can be found in the provided Table 1.
3.2 Pharmacokinetics of Venlafaxine and O-Desmethylvenlafaxine
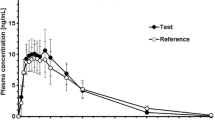
The mean plasma concentration-time profiles of the test and reference formulations after a single dose are shown in Fig. 1. After a single oral dose administration of venlafaxine extended-release capsule under fed conditions, mean venlafaxine plasma concentrations increased and peaked at approximately 4.5 h post-dose for both products. However, the mean ODV plasma concentrations peaked at approximately 9.5 h for the test formulation and 10 h for the reference formulation, respectively. Both the venlafaxine and ODV plasma concentrations declined in a multi-exponential manner over 72 h, and the mean profiles were almost superimposable between the two products.

The drug–time curve of venlafaxine (A) and O-desmethylvenlafaxine (B) under fed conditions. The plasma drug concentrations are presented as mean ± SD. R reference formulation, SD standard deviation, T test formulation
The PK parameters for venlafaxine and ODV for the test and reference drugs under fed condition are summarized in Table 2. The mean Cmax, AUC0–t, AUC0–∞, and Tmax were comparable between the test and reference products. The Cmax, Tmax and AUC0–t boxplots for venlafaxine and ODV are shown in Fig. S2 (see ESM). No significant differences in the aforementioned PK parameters were observed between the test and reference products in the fed condition. The significance analysis of pharmacokinetic parameters Cmax, AUC0–t, and AUC0–∞ effects of different formulations, dosing cycles, and dosing sequences of venlafaxine hydrochloride extended-release capsule was performed using a random effects ANOVA model. The results showed that there were no significant differences (p > 0.05) for dosing sequence, formulation effect, and cycle effect of venlafaxine. For desvenlafaxine, there were significant differences (p < 0.05) for effect of dosing sequence and cycle, significant differences (p < 0.05) for formulation effect Cmax, and no significant differences (p > 0.05) for AUC0–t and AUC0–∞. (As shown in Table 3.)
3.3 Bioequivalence
The bioequivalence assessment and analysis of the main PK parameters of 28 subjects were analyzed. Geometric mean ratio (GMR) was used for the bioequivalence evaluation as reported in Table 4. The statistical assessment of the mean primary PK parameters (Cmax, AUC0–t, and AUC0–∞) between the test and reference formulations for venlafaxine and ODV were comparable under fed conditions. The point estimates for all three parameters were close to 100% and the corresponding 90% CIs were well within the established 80–125% bioequivalence limit.
3.4 Safety
The data in this safety evaluation are based on data from 28 subjects in the safety analysis set. In this study, a total of 15 subjects had AEs; the AE rate was 53.6% (15/28) and a total of 27 AEs occurred, 12 cases in the T-formulation group and 15 cases in the R-formulation group. Nineteen cases were classified as probably related to the drug, seven cases were classified as possibly related to the drug, and one case was classified as unlikely related to the drug. The results of the adverse events were as follows: 91.7% (11/12 cases) of the T group recovered, while 8.3% (1/12 cases) showed improvement. In the R group, 86.7% (13/15 cases) recovered and 13.3% (2/15 cases) improved. It should be noted that all adverse events observed in this study were of mild severity. The statistical results of adverse events in each system are classified by drug category (Table 5).
In this study, the number of subjects with adverse drug reactions (ADRs) was 20, with a prevalence of 71.4% (20/28); the number of cases from the T-formulation group was 10, the incidence rate was 35.7% (10/28), and the number of cases from the R-formulation group was 10, with an incidence rate of 35.7% (10/28) (different types of adverse reactions and adverse reactions with different drugs may occur in the same subject). A total of 26 adverse drug reactions occurred including abnormal liver function, increased systolic blood pressure, dizziness, headache, tremors, laryngeal pain, yawning, euphoria, nausea, canker ulcers, and abdominal pain. Supplemental information contains a comprehensive list of adverse events experienced by each participant. Please refer to the ESM (Table S2) for further details.
4 Discussion
Major depressive disorder (MDD) is a common type of depressive disorder characterized by a persistent low mood, a lack of positive affect, and a loss of interest in usually pleasurable activities (anhedonia) that is different from the patient’s usual self and causes significant distress or impairment for ≥2 weeks [24, 25]. Treatment usually includes pharmacological therapy with antidepressants, such as selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs). Venlafaxine is the first of the SNRIs that provides dose-dependent norepinephrine reuptake inhibition [16], with a dosage of 150 mg/day or higher sufficient to produce noradrenergic activity. It has an acute onset of down-regulation of β-adrenergic receptors and low affinity for the postsynaptic receptors [26, 27], which might be a mechanism underlying the early onset of action [28].
In this study, the PK parameters for venlafaxine showed that the 90% confidence intervals of the PK parameters Cmax, AUC0–t, and AUC0–∞ of the T and R formulations were 88.93~99.47%, 94.51~102.34%, and 94.88~102.16%, respectively. The PK parameters for ODV analysis showed that the 90% confidence intervals of the PK parameters Cmax, AUC0–t, and AUC0–∞ of the T and R formulations were 91.02~96.50%, 97.17~101.44%, and 98.25~102.72%, respectively. The bioequivalence range of venlafaxine and ODV are all within 80.00~125.00%, which met the acceptance criteria of bioequivalence, that is, the T and R formulation had bioequivalence under the conditions of postprandial administration. However, the Tmax for venlafaxine in both the test and reference formulations was found to be 4.5 h. The peak time for ODV was 9.5 h in the test formulation and 10 h in the reference formulation. In addition, the t½ of venlafaxine and ODV in the test formulation were slightly longer in the reference formulation, with values of 10.78 ± 3.46 h and 12.97 ± 2.85 h, respectively, compared with 9.15 ± 2.13 h and 11.63 ± 2.2 h, respectively, in the reference formulation.
In terms of safety, the most common AE in this study was nausea, with an incidence of 14.3% (4/28) in the T formulation and 17.9% (5/28) in the R formulation, which is consistent with previous literature [29]. This was followed by dizziness (7.1% [2/28] in the T formulation vs 0% in the R formulation). All adverse reactions were mild and recovered spontaneously without any intervention, indicating that venlafaxine demonstrates reasonable safety, acceptability, and tolerability [30]. Therefore, it is important to pay particular attention to the occurrence of nausea as an adverse reaction in the clinical use of venlafaxine. This finding has important implications for patients, providing more options and reducing the cost of treatment. This means that patients can choose to use domestic or imported venlafaxine according to the actual situation, without worrying about the treatment effect and safety.
We also need to note that this study has certain limitations. During phase I clinical trials, the primary objective is to assess the PK data and preliminary safety of drugs. Healthy individuals are commonly selected as participants due to their absence of underlying diseases or concomitant medications, which could potentially influence the outcomes. This approach enables a more precise evaluation of the drug’s effects. To minimize variability between subjects, a crossover design was implemented [31]. However, the strictly controlled conditions in this study, such as including only healthy volunteer participation and serving a standardized high-fat meal, may differ from a real-word setting. Therefore, further studies are needed to estimate the safety and PK/PD profiles of the T formulation between patients with major depressive disorders. In addition, the sample size is relatively small and may not be fully representative of the entire population. Furthermore, venlafaxine is well acknowledged to be mainly metabolized to its active metabolite, ODV, by the cytochrome P450 2D6 enzyme (CYP2D6) and 2C19 enzyme (CYP2C19) [32]. Nevertheless, these members of the CYP superfamily exhibit many allele variants [33, 34], resulting in a wide range of metabolic activity phenotypes, such as ultra-rapid metabolizer (UM), normal metabolizer (NM), intermediate metabolizer (IM), and poor metabolizer (PM) [35]. The different metabolizer phenotypes are a reliable possibility of inter-individual variability in exposure and thus are likely to affect the PK parameters of venlafaxine [36]. However, we did not investigate the influence of the T formulation on cytochrome P450s (CYPs), including CYP2C19 and CYP2D6.
5 Conclusion
In this single-dose study conducted under fed conditions with healthy volunteers, the test and reference extended-release venlafaxine 150-mg capsules were determined to meet the bioequivalence criteria set by Chinese standards. Additionally, both the test and reference formulations exhibited mild adverse reactions and demonstrated good safety in Chinese healthy subjects. However, it is important to note that the active metabolite of venlafaxine, ODV, displayed variations in t½ and Tmax between the test and reference formulations.
References
Disease GBD, Injury I, Prevalence C. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1545–602. Available from: https://www.ncbi.nlm.nih.gov/pubmed/27733282.
Korte SM, Prins J, Krajnc AM, et al. The many different faces of major depression: it is time for personalized medicine. Eur J Pharmacol. 2015;753:88–104. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25592320.
Effexor XR (venlafaxine hydrochloride) extended-release capsules [package insert].
Troy SM, Dilea C, Martin PT, et al. Pharmacokinetics of once-daily venlafaxine extended release in healthy volunteers. Current Therapeutic Research. 1997;58(8):504–14. Available from: https://www.sciencedirect.com/science/article/pii/S0011393X97800422.
Troy SM, Parker VP, Hicks DR, et al. Pharmacokinetics and effect of food on the bioavailability of orally administered venlafaxine. J Clin Pharmacol. 1997;37(10):954–61. Available from: https://www.ncbi.nlm.nih.gov/pubmed/9505987.
Karlsson L, Zackrisson AL, Josefsson M, et al. Influence of CYP2D6 and CYP2C19 genotypes on venlafaxine metabolic ratios and stereoselective metabolism in forensic autopsy cases. Pharmacogenomics J. 2015;15(2):165–71. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25245581.
Howell SR, Husbands GE, Scatina JA, et al. Metabolic disposition of 14C-venlafaxine in mouse, rat, dog, rhesus monkey and man. Xenobiotica. 1993;23(4):349–59. Available from: https://www.ncbi.nlm.nih.gov/pubmed/8337893.
Horst WD, Preskorn SH. Mechanisms of action and clinical characteristics of three atypical antidepressants: venlafaxine, nefazodone, bupropion. J Affect Disord. 1998;51(3):237–54. Available from: https://www.ncbi.nlm.nih.gov/pubmed/10333980.
U.S. FOOD AND DRUG ADMINISTRATION (FDA). Draft guidance on venlafaxine hydrochloride [EB/OL]. Silver Spring: FDA. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Venlafaxine_HCl_ERcap_20699_RC2-06.pdf.
Klamerus KJ, Maloney K, Rudolph RL, et al. Introduction of a composite parameter to the pharmacokinetics of venlafaxine and its active O-desmethyl metabolite. J Clin Pharmacol. 1992;32(8):716–24. Available from: https://www.ncbi.nlm.nih.gov/pubmed/1487561.
Bhatt J, Jangid A, Venkatesh G, et al. Liquid chromatography-tandem mass spectrometry (LC-MS-MS) method for simultaneous determination of venlafaxine and its active metabolite O-desmethyl venlafaxine in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;829(1-2):75–81. Available from: https://www.ncbi.nlm.nih.gov/pubmed/16249126.
Wei Z, Bing-Ren X, Cai-Yun W. Liquid chromatography-mass spectrometry method for the determination of venlafaxine in human plasma and application to a pharmacokinetic study. Biomed Chromatogr. 2007;21(3):266–72. Available from: https://www.ncbi.nlm.nih.gov/pubmed/17230450.
Wang Z, Li L, Huang S, et al. Joint population pharmacokinetic modeling of venlafaxine and O-desmethyl venlafaxine in healthy volunteers and patients to evaluate the impact of morbidity and concomitant medication. Front Pharmacol. 2022;13:978202. Available from: https://www.ncbi.nlm.nih.gov/pubmed/36569310.
Troy SM, Parker VD, Fruncillo RJ, et al. The pharmacokinetics of venlafaxine when given in a twice-daily regimen. J Clin Pharmacol. 1995;35(4):404–9. Available from: https://www.ncbi.nlm.nih.gov/pubmed/7650231.
Hicks DR, Wolaniuk D, Russell A, et al. A high-performance liquid chromatographic method for the simultaneous determination of venlafaxine and O-desmethylvenlafaxine in biological fluids. Ther Drug Monit. 1994;16(1):100–7. Available from: https://www.ncbi.nlm.nih.gov/pubmed/8160247.
Furukawa TA, Cipriani A, Cowen PJ, et al. Optimal dose of selective serotonin reuptake inhibitors, venlafaxine, and mirtazapine in major depression: a systematic review and dose-response meta-analysis. Lancet Psychiatry. 2019;6(7):601–9. Available from: https://www.ncbi.nlm.nih.gov/pubmed/31178367.
Montgomery SA. Rapid onset of action of venlafaxine. Int Clin Psychopharmacol. 1995;10(Suppl 2):21–7. Available from: https://www.ncbi.nlm.nih.gov/pubmed/7622814.
Higuchi T, Kamijima K, Nakagome K, et al. A randomized, double-blinded, placebo-controlled study to evaluate the efficacy and safety of venlafaxine extended release and a long-term extension study for patients with major depressive disorder in Japan. Int Clin Psychopharmacol. 2016;31(1):8–19. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26513202.
Lense XM, Hiemke C, Funk CSM, et al. Venlafaxine's therapeutic reference range in the treatment of depression revised: a systematic review and meta-analysis. Psychopharmacology (Berl). 2024;241(2):275–89. Available from: https://www.ncbi.nlm.nih.gov/pubmed/37857898.
Lyndon GJ, Prieto R, Wajsbrot DB, et al. Efficacy of venlafaxine extended release in major depressive disorder patients: effect of baseline anxiety symptom severity. Int Clin Psychopharmacol. 2019;34(3):110–8. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30870236.
Wright CW, Aikman MS, Werts E, et al. Bioequivalence of single and multiple doses of venlafaxine extended-release tablets and capsules in the fasted and fed states: four open-label, randomized crossover trials in healthy volunteers. Clin Ther. 2009;31(11):2722–34. Available from: https://www.ncbi.nlm.nih.gov/pubmed/20110014.
Karch FE, Lasagna L. Toward the operational identification of adverse drug reactions. Clin Pharmacol Ther. 1977;21(3):247–54. Available from: https://www.ncbi.nlm.nih.gov/pubmed/837643.
Jiang F, Kim HD, Na HS, et al. The influences of CYP2D6 genotypes and drug interactions on the pharmacokinetics of venlafaxine: exploring predictive biomarkers for treatment outcomes. Psychopharmacology (Berl). 2015;232(11):1899–909. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25510856.
Practice guideline for the treatment of patients with major depressive disorder (revision). American Psychiatric Association. Am J Psychiatry. 2000;157(4 Suppl):1–45. Available from: https://www.ncbi.nlm.nih.gov/pubmed/10767867.
Depression in adults with a chronic physical health problem: Evidence Update March 2012: a summary of selected new evidence relevant to NICE clinical guideline 91 ‘Depression in adults with a chronic physical health problem: treatment and management’ (2009). London 2012.
Melichar JK, Haida A, Rhodes C, et al. Venlafaxine occupation at the noradrenaline reuptake site: in-vivo determination in healthy volunteers. J Psychopharmacol. 2001;15(1):9–12. Available from: https://www.ncbi.nlm.nih.gov/pubmed/11277612.
Strawn JR, Geracioti L, Rajdev N, et al. Pharmacotherapy for generalized anxiety disorder in adult and pediatric patients: an evidence-based treatment review. Expert Opin Pharmacother. 2018;19(10):1057–70. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30056792.
Feighner JP. Mechanism of action of antidepressant medications. J Clin Psychiatry. 1999;60 (Suppl 4):4–11; (discussion 2-3). Available from: https://www.ncbi.nlm.nih.gov/pubmed/10086478.
Fagiolini A, Albert U, Ferrando L, et al. A randomized, double-blind study comparing the efficacy and safety of trazodone once-a-day and venlafaxine extended-release for the treatment of patients with major depressive disorder. Int Clin Psychopharmacol. 2020;35(3):137–46. Available from: https://www.ncbi.nlm.nih.gov/pubmed/31972628.
Kishi T, Ikuta T, Sakuma K, et al. Antidepressants for the treatment of adults with major depressive disorder in the maintenance phase: a systematic review and network meta-analysis. Mol Psychiatry. 2023;28(1):402–9. Available from: https://www.ncbi.nlm.nih.gov/pubmed/36253442.
Krishna R, Kesisoglou F. Clinical endpoint bioequivalence studies are needed: a perspective from brand drugs. Clin Pharmacol Ther. 2019;105(2):298–300. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30456848.
Hiemke C, Bergemann N, Clement HW, et al. Consensus guidelines for therapeutic drug monitoring in neuropsychopharmacology: update 2017. Pharmacopsychiatry. 2018;51(1-02):e1. Available from: https://www.ncbi.nlm.nih.gov/pubmed/29390205.
Gaedigk A, Sangkuhl K, Whirl-Carrillo M, et al. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med. 2017;19(1):69–76. Available from: https://www.ncbi.nlm.nih.gov/pubmed/27388693.
Fricke-Galindo I, Cespedes-Garro C, Rodrigues-Soares F, et al. Interethnic variation of CYP2C19 alleles, ‘predicted’ phenotypes and ‘measured’ metabolic phenotypes across world populations. Pharmacogenomics J. 2016;16(2):113–23. Available from: https://www.ncbi.nlm.nih.gov/pubmed/26503820.
Lin XQ, Wang P, Cai WK, et al. The associations between CYP2D6 metabolizer status and pharmacokinetics and clinical outcomes of venlafaxine: a systematic review and meta-analysis. Pharmacopsychiatry. 2019;52(5):222–31. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30485867.
Milosavljevic F, Bukvic N, Pavlovic Z, et al. Association of CYP2C19 and CYP2D6 poor and intermediate metabolizer status with antidepressant and antipsychotic exposure: a systematic review and meta-analysis. JAMA Psychiatry. 2021;78(3):270–80. Available from: https://www.ncbi.nlm.nih.gov/pubmed/33237321.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This study was funded by Hubei Duorui Pharmaceutical Company.
Conflicts of Interest
All authors declare no conflicts of interest in this study.
Ethics Approval
All the protocol and documents were reviewed and approved by the Ethics Committee of Wuhan Pulmonary Hospital (NO.2021004).
Consent to Participate
All study participants provided written informed consent.
Consent for Publication
All researchers, participants, institutions, and sponsors consented to the submission of this report to the journal.
Author Contributions
The principal investigator for this study was Guan Liu. Patient recruitment and data collection were undertaken by Fang Yao and Pan Lu. Vital signs monitoring was performed by Yafang Xie, Xiuwen Li, Qiangwei Liu, and Yang Liu. Statistical analysis was conducted by Dan Cao and Jun Liang. The manuscript was written with contributions from Yingxia He, Jie Wang, and Dan Tian.
Data and Code Availability
Datasets used in the current analysis are available from Yingxia He on reasonable request.
Supplementary Information
Below is the link to the electronic supplementary material.
Fig. S1
Bioequivalence study design of venlafaxine extended-release capsule; Fig. S2 Boxplot of Cmax, Tmax and AUC0–t for venlafaxine (left) and O-desmethylvenlafaxine (right) in the fed state. Pharmacokinetic parameters: Cmax the maximum observed concentration, Tmax, the time to peak drug concentration, AUC0–t the area under the plasma concentration-time curve from time zero to the last sampling time. (PDF 367 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
He, Y., Wang, J., Yao, F. et al. Pharmacokinetic Bioequivalence and Safety Assessment of Two Venlafaxine Hydrochloride Extended-Release Capsules in Healthy Chinese Subjects Under Fed Conditions: A Randomized, Open-Label, Single-Dose, Crossover Study. Drugs R D 24, 275–283 (2024). https://doi.org/10.1007/s40268-024-00470-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-024-00470-w