
Abstract
Oxycodone is a semisynthetic opioid used for the treatment of moderate to severe pain. Two separate studies were conducted to assess the pharmacokinetic bioequivalence of a newly formulated oxycodone hydrochloride extended-release tablet to a marketed oxycodone product in Japan under fasting and fed conditions. Each study was a randomized, open-label, single-dose, single-center, two-period, two-way crossover study. Healthy male Japanese subjects received the oxycodone 10-mg products under fasting and fed conditions. Blood samples were collected at specified time intervals, and plasma concentrations of oxycodone were analyzed using a validated liquid chromatography tandem mass spectrometry assay method. The pharmacokinetic parameters were determined via non-compartmental analysis. Pharmacokinetic metrics used for bioequivalence assessment were the maximum observed plasma concentration (C max) and the area under the concentration–time curve up to the last sampling time (AUC t ). A total of 24 healthy subjects were enrolled in each study. One subject withdrew after completion of the first sequence under fed conditions. The ratios of geometric least square means for C max and AUC t under fasting conditions were 1.1110 (90% confidence interval [CI] 1.0562–1.1687) and 0.9946 (90% CI 0.9670–1.0231), respectively. The ratios of geometric least square means for C max and AUCt under fed conditions were 1.1417 (90% CI 1.0959–1.1895) and 1.0135 (90% CI 0.9810–1.0470), respectively. The 90% CIs were within the predefined range (0.80–1.25). Both treatments were well tolerated when taken without an opioid antagonist in healthy Japanese subjects. Pharmacokinetic bioequivalence between test and reference formulations under fasting and fed conditions was concluded in terms of both rate and extent of absorption.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
This bioequivalence study demonstrates that the pharmacokinetic properties of a newly formulated extended-release oxycodone 10-mg tablet are bioequivalent to a marketed reference product under fasting and fed conditions. |
Both treatments were well tolerated when taken without an opioid antagonist in healthy Japanese subjects. |
Hydrophilic matrix technology is one of the proper methods to manufacture an oxycodone extended-release formulation. |
1 Introduction
Oxycodone is a strong opioid that is commonly used as an alternative to morphine. After oral administration, oxycodone has a higher bioavailability (range 42–87%) than morphine (range 22–48%) [1]. Oxycodone is extensively metabolized in the liver, and less than 10% of the dose is excreted unchanged in urine [2]. First-pass metabolism most likely occurs in the liver and not in the gastrointestinal tract [3]. Cytochrome P450 (CYP)-3A4 and CYP2D6 are the main enzymes involved in its metabolism. Oxymorphone, a 3-O-demethylation metabolite of oxycodone catalyzed by CYP2D6, is a potent opioid with a μ-opioid receptor affinity that is three to five times higher than that of morphine. CYP3A4 and CYP3A5 displayed the highest activity for oxycodone N-demethylation to catalyze noroxycodone in human liver microsomes [3]. Both oxymorphone and noroxycodone are transformed to another possibly active metabolite, noroxymorphone, by CYP3A4 and CYP2D6, respectively. CYP2D6 genotypes caused expected differences in pharmacokinetics (PK), but they did not influence pain control in patients treated with oxycodone for cancer pain [4].
Opioid therapy frequently requires switching from one opioid to another to improve the response to analgesic therapy or to reduce adverse effects. There may be a benefit in switching to a different route of administration, drug, or formulation [5, 6]. Various oxycodone dosage forms and routes of administration, including immediate-release (IR) and extended-release (ER) oral formulations and intravenous and subcutaneous injections, allow a high degree of flexibility in switching from one route of administration to another and switching to oxycodone from other opioids. In Japan, oxycodone hydrochloride ER tablets and IR powder were approved in 2003 and 2007, respectively. The recommended daily dose of ER oxycodone in Japan is 10–80 mg divided into two oral doses. Four strengths (5, 10, 20, and 40 mg) of ER oxycodone have been used for the treatment of patients with cancer pain [7, 8]. Oxycodone hydrochloride injection formulation was developed as an alternative route for these patients with cancer pain and was approved in 2012. Population PK analysis indicated no PK differences for total body clearance between intravenous and subcutaneous infusion in Japanese patients with cancer pain [9].
ER opioid formulations can provide less fluctuation in plasma drug concentration than IR formulations and good patient compliance due to reduced dose frequency, which offers several clinical advantages to managing chronic pain as a longer-lasting analgesia. Hydrophilic matrix technology is a method for ER of drugs and has been widely used for oral controlled delivery of various drugs, including hydromorphone [10]. Given their hydrophilic nature, these polymers begin to swell upon contact with water, forming a gel layer. Drug release is controlled via diffusion through the layer and/or erosion mechanisms. The advantages of this technology are ease of formulation, the cost-effective manufacturing process, wide regulatory acceptance of the polymer systems, and flexibility in the control of the drug-release profiles [11]. A new oxycodone ER tablet with in vitro dissolution properties that were not affected by the pH of the test medium was manufactured with this technology. In addition, in vitro dissolution profiles, when evaluated with a bio-relevant method, were similar between the newly formulated ER tablet and a marketed oxycodone tablet [12].
The objective of this study was to investigate whether the newly formulated ER oxycodone tablet is bioequivalent to a marketed oxycodone tablet under fasting and fed conditions to provide an option for a generic oxycodone product in Japan.
2 Methods
This study was conducted in accordance with the ethical principles originating in or derived from the Declaration of Helsinki, the guidelines on Good Clinical Practice, and locally applicable laws and regulations. The study protocol, amendments, and the informed consent form were approved by the Institutional Review Boards of the study site. All subjects gave written informed consent prior to commencing the study. The study was conducted at Osaka Pharmacology Clinical Research Hospital (Osaka, Japan) and is registered on JapicCTI-163475.
2.1 Subjects
Healthy male Japanese subjects aged between 20 and 45 years, with a normal body mass index of ≥18.5 and <25.0 kg/m2 were eligible for inclusion. Subjects were excluded from this study if any of the following conditions existed: evidence of organ dysfunction or any clinically significant deviation from the normal range in a physical examination, vital signs (e.g., blood pressure, heart rate, and body temperature), 12-lead electrocardiogram (ECG), or clinical laboratory determinations; presence or history of severe adverse reaction to any medicine; a history of alcohol abuse or drug abuse; a positive result for hepatitis B, hepatitis C, or HIV; a positive result for drug urinalysis; donation of more than 200 ml of blood within 28 days before the screening test or more than 400 ml of blood within 84 days or more than 1200 ml of blood within a year, or apheresis within 14 days; receipt of an investigational agent within 120 days before the screening test; previous participation in an opioid study; refusal to use contraception; and inability to communicate satisfactorily with the investigator. Participants could withdraw from the study at their own request at any time, and reasons for withdrawal were recorded.
2.2 Study Design
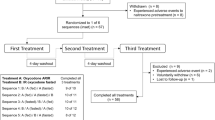
The studies were of randomized, open-label, single-dose, two-period, two-treatment, two-way cross-over design. Two separate studies were conducted to assess the bioequivalence of a new generic formulation of oxycodone hydrochloride ER tablets (test) to a marketed reference oxycodone product in Japan, OXYCONTIN® (reference), under fasting and fed conditions in healthy Japanese subjects.
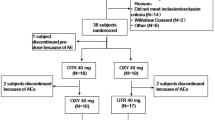
Each subject underwent screening assessments to confirm eligibility within 30 days before the first treatment period. Subjects were randomized in a 1:1 ratio to either test-reference or reference-test sequences. Treatment sequences were separated by a wash-out period of at least 6 days.
Each subject was confined to the study site from the day before administration through to completion of the 48-h post-dose procedures. In study 1 (fasting conditions), subjects fasted overnight (more than 10 h) and for 4 h after administration. Each dose was given with 200 ml of water, and water ad libitum was allowed 2 h after administration. In study 2 (fed conditions), the test and reference products were administered in a similar way after consumption of a high-fat high-calorie breakfast (a high-fat diet of ≥900 kcal and calories from fat >35% of total calories) as defined in the guidance [13]. The meal was to be eaten within 20 min and drugs administered within 10 min thereafter. The composition of the high-fat high-calorie breakfast was the same in both periods. The principal investigator assessed compliance with treatment administration by conducting a thorough examination of the oral cavity. Subjects were prohibited from resting in a supine position for 4 h after administration. Standardized meals were served at appropriate times through the study. Each subject was discharged from the study site on the morning of day 3 after completion of the 48-h post-dose assessment. The final follow-up assessment was carried out after 7 ± 1 days of administration.
2.3 Randomization
The order of investigational product administration was sequentially assigned via a computer-generated randomization scheme. The scheme remained unavailable to the bioanalytical contract research organization until completion of both clinical and analytical phases.
2.4 Drug Product
Oxycodone ER tablets and OXYCONTIN® containing oxycodone hydrochloride (10 mg) were obtained from Daiichi Sankyo Co., Ltd. (Tokyo, Japan), and Shionogi & Co., Ltd. (Osaka, Japan), respectively.
2.5 Blood Sampling
Blood (5 ml) was collected into Vacutainers containing EDTA-2K as an anticoagulant at the following times: pre-dose and at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 9, 12, 24, and 36 h after administration in study 1, and pre-dose and at 1, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 8, 12, 24, and 36 h after administration in study 2. Plasma was separated via centrifugation and stored at −20 °C until the assay.
2.6 Sample Analysis
Drug concentrations in human plasma were determined by validated sensitive and specific methods using solid-phase extraction of the analyte and deuterated internal standard (oxycodone-D6) from plasma, followed by reverse-phase high-pressure liquid chromatography with tandem mass spectrometry detection in the positive ionization mode (API-5000, AB SCIEX, Framingham, MA, USA). Briefly, plasma samples (50 μl) were combined with 250 μl of water and 20 μl of internal standard spiking solution, mixed and loaded onto a preconditioned solid-phase extraction plate (OASIS HLB μElution Plate; Waters Corp., Milford, MA, USA). Each sample was washed and eluted to a 96-well plate using 100 μl of acetonitrile/water (80:20, v/v). After addition of acetonitrile 100 μl to each sample, a 2-μl aliquot of the final solution was injected and analyzed using an Inertsil amide analytical column (3 μm, 100 mm × 2.1 mm id; GL Sciences, Tokyo, Japan). A gradient of 7.5 mM ammonium acetate in 0.2% formic acid and acetonitrile was used for the mobile phase, and the flow rate was 1.0 ml/min. The mass transitions were 316–241 (m/z) for oxycodone, and 322–247 (m/z) for oxycodone-D6, respectively. Two sets of low-, medium-, and high-quality control samples were evaluated with each run of clinical samples.
The lower limit of quantitation for the plasma oxycodone assay was 0.05 ng/ml, and the linear calibration ranges were 0.05–25.0 ng/ml, respectively. The coefficient of variation (CV) for the intra-day reproducibility of the plasma quality control samples for oxycodone was between 1.3 and 3.2%. The CV for the inter-day reproducibility of the plasma quality control samples was between 1.0 and 4.6%.
2.7 Pharmacokinetic Analysis
The PK parameters were calculated by non-compartmental analysis using the computer software Phoenix WinNonlin (version 6.2, Pharsight Corp., Sunnyvale, CA, USA) and SAS® (version 9.2, SAS Institute, Japan, Tokyo, Japan).
The maximum concentration (C max) and time to C max (t max) were obtained by observation. The apparent elimination half-life (t ½) was obtained by linear regression of three or more log-transformed data points in the terminal phase. The area under the concentration–time curve up to the last sampling time (AUC t ) was obtained by the linear trapezoidal method. The AUC values were extrapolated to infinity (AUCinf) using the last measurable concentration and the terminal elimination rate.
2.8 Statistical Analysis
Ln-transformed AUC t and Cmax were analyzed using the general linear model procedure in SAS®. The statistical model included sequence, period, and treatment within sequence as fixed factors and subject within sequence as a random effect. The point estimate and the 90% geometric confidence interval (CI) for the test-to-reference geometric mean ratio were calculated for AUC t and C max. Bioequivalence of test compound relative to the reference compound was concluded if the 90% geometric CIs of the ratio of least-squares means of the test-to-reference product of ln-transformed parameter (AUC t and C max) were within the acceptance range of 0.80–1.25.
The sample size for this trial was derived from data characterizing the variability of oxycodone products. The intra-individual geometric CVs of the AUC t and C max of oxycodone are approximately 20%. Given this value, a sample size of 19 evaluable subjects was estimated to provide 90% power to demonstrate bioequivalence between two formulations. To allow for withdrawals, 24 subjects were enrolled into the study.
2.9 Safety Assessments
Safety and tolerability were assessed by physical examinations, vital signs, oxygen saturation, 12-lead ECGs, and laboratory measurements (including hematology, serum chemistry, and urinalysis). Adverse events (AEs) were monitored throughout the study. Subjects were questioned concerning their well-being. Investigators evaluated all clinical AEs in terms of intensity (mild, moderate, or severe), duration, severity, outcome, and relationship to the study drug.
3 Results
3.1 Subject Characteristics
In study 1, a total of 24 healthy subjects were enrolled and randomized, all of whom completed the study. At screening (N = 24), the mean ± standard deviation (SD) age was 26.5 ± 5.2 (range 20–38) years and mean body mass index was 21.7 ± 1.5 (range 18.7–24.0) kg/m2. In study 2, a total of 24 healthy subjects were enrolled, and one subject withdrew after completion of the first sequence because of nasopharyngitis that was unrelated to the study medication. At screening (N = 24), the mean ± SD age was 25.5 ± 5.1 (range 20–36) years and mean body mass index was 22.0 ± 1.9 (range 18.6–24.7) kg/m2.
3.2 Pharmacokinetics of Oxycodone
3.2.1 Study 1 (Fasting Conditions)
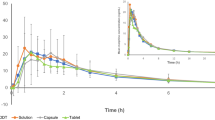
Figure 1 shows the mean plasma concentration–time profiles of oxycodone following administration of a single oral dose of either test or reference products under fasting conditions, and Table 1 summarizes the PK parameters. Quantifiable plasma concentrations were observed throughout the 36-h PK sampling period in all subjects who received the test or reference oxycodone under fasting conditions. No carry-over effect was observed in the plasma drug concentration analysis. The geometric mean C max for the test and reference were 11.6 and 10.5 ng/ml, respectively. The geometric mean AUCt for the test and reference were 123 and 124 ng·h/ml, respectively. The maximum concentrations of oxycodone were reached at about 3 h after administration, and the median t max was 3.00 and 2.25 h for the test and reference, respectively. The mean t ½ of oxycodone for the test and reference was 4.32 and 5.89 h, respectively. Table 2 shows the geometric mean ratios and 90% CIs for the primary endpoints. The ratio of geometric least-square means for C max was 1.1110 (90% CI 1.0562–1.1687). The ratio of geometric least-square means for AUCt was 0.9946 (90% CI 0.9670–1.0231). Both ratios of geometric least-square means and 90% CI were within the pre-specified bioequivalence range (0.80–1.25).

Mean plasma concentrations (±standard deviation) of oxycodone after a single 10-mg dose of test (filled circle) and reference (open circle) tablet under fasting conditions (study 1)
3.2.2 Study 2 (Fed Conditions)
Figure 2 shows the mean plasma concentration–time profiles of oxycodone following administration of a single oral dose of either test or reference products under fed conditions, and Table 3 summarizes the PK parameters. The geometric mean C max for the test and reference was 17.0 and 15.0 ng/ml, respectively. The geometric mean AUC t for the test and reference was 156 and 156 ng·h/ml, respectively. The median t max was 4.50 and 3.50 h for the test and reference, respectively. The mean t ½ for the test and reference was 4.26 and 5.45 h, respectively. Table 4 shows the geometric mean ratios and 90% CIs for the primary endpoints. The ratio of geometric least-square means for C max was 1.1417 (90% CI 1.0959–1.1895). The ratio of geometric least-square means for AUC t was 1.0135 (90% CI 0.9810–1.0470). Both ratios of geometric least-square means and 90% CI were within the pre-specified bioequivalence range (0.80–1.25).

Mean plasma concentrations (±standard deviation) of oxycodone after a single 10-mg dose of test (filled circle) and reference (open circle) tablet under fed conditions (study 2)
3.3 Safety
AEs for test and reference oxycodone under fasting conditions were reported by three (12.5%) and four (16.7%) subjects, respectively. AEs for test and reference subjects under fed conditions were reported by five (21.7%) and five (20.8%) subjects, respectively. No severe-intensity or serious AEs were reported. One subject discontinued because of nasopharyngitis, which was not related to the study drug. Table 5 lists the drug-related treatment-emergent AEs (TEAEs). The most common drug-related TEAEs in both oxycodone formulations were somnolence and nausea. All TEAEs were relatively transient, of mild intensity, and resolved by the end of the study. Overall, clinical laboratory values and all other observations related to safety (vital signs and ECG) were normal, and no clinically relevant differences were observed between treatments.
4 Discussion
This study was undertaken to establish the bioequivalence of the oxycodone ER tablet and the marketed formulation to support development of new ER formulations of oxycodone in Japan. The results from this study show that the two oxycodone tablets at the dose strengths of 10 mg can be considered bioequivalent. Mean plasma concentrations and t max were similar under both fasting and fed conditions, and adjusted geometric mean ratios for the PK parameters were close to 1.00, with 90% CIs contained within pre-specified bioequivalence limits. Bioequivalence guidance in Japan recommends studies be conducted using a stronger dose than intended for regular consumption; however, to protect the safety of healthy subjects, these bioequivalence studies were conducted using a 10-mg tablet dose based on available published information concerning oxycodone administered without naltrexone, which has not been approved in Japan. The bioequivalence of the four strengths of the ER tablets has been demonstrated using in vitro dissolution tests.
The concomitant intake of alcoholic beverages together with ER opioid formulations poses a serious safety concern, since ethanol ingestion may modify the release characteristics and cause dose dumping, which can be a patient safety issue [14]. The in vitro dissolution profile in 40% ethanol of oxycodone from the ER tablets used in this study did not differ from that in water [12]. Hydrophilic polymers, which are insoluble in ethanol and expected to be unaffected when consumed together with alcohol, were used as excipients for the preparation of the ER oxycodone tablets. The dissolution profiles of the ER tablet with or without ethanol (5–40%) in test medium were evaluated using the USP apparatus 2 (paddle method). The in vitro dissolution rates of the ER tablet decreased slightly depending on the concentration of ethanol in the test medium (Fukui et al., unpublished data). In the medium with the highest ethanol concentration (40%), the dissolution rate decreased by approximately 20% compared with that in the ethanol-free medium. Therefore, the ER tablet would have a low potential for dose dumping when consumed with alcohol.
A delay of median t max by approximately 1 h and shorter mean terminal t ½ than the reference formulation was observed for the ER tablets in fasted and fed conditions, while bioequivalence was demonstrated in both conditions. As ER oxycodone is administered twice daily and the plasma concentration at 12 h after administration was similar for both formulations, the difference in t max and t ½ between the two formulations would not provide a clinically meaningful difference in the treatment of pain.
Although ER opioid formulations offer several clinical advantages, including consistent analgesia over the administration period, they are at particular risk for abuse via unintended routes because they contain higher amounts of the active drug than do IR formulations. The US FDA considers the development of abuse-deterrent opioid products a high public health priority and released guidance on the evaluation and labeling of abuse-deterrent opioids in 2015 [15]. Oxycodone products specifically intended to reduce the potential for abuse and misuse of the original oxycodone tablet formulation were developed and reformulated as OxyContin®; these became available in the USA in 2010. A tamper-resistant system was not actively provided for either the test or the reference tablets used in this study. No abuse-deterrent formulation of oxycodone is currently approved for use in Japan. In Japan, oxycodone is prescribed only to patients with cancer pain who require continuous around-the-clock opioid analgesics for an extended period of time. However, abuse-deterrent formulations will definitely ensure the safe and effective use of opioids for the relief of chronic pain, and the technology has been under consideration.
The most common oxycodone-related AEs are similar to those experienced with other opioids, e.g., constipation, sedation, and nausea. Although oxycodone-related AEs are similar to those of morphine, the lack of opioid-induced hallucinations with oxycodone means it could be superior to morphine [1]. Opioid-induced bowel dysfunction, which includes constipation, hard dry stools, incomplete evacuation, bloating, abdominal cramping, and increased gastric reflux, is the most distressing symptom, and the frequency of symptoms ranges from 26 to 49% in patients receiving oxycodone [16]. An ER tablet comprising oxycodone and naloxone was developed, and analgesic efficacy was reported as comparable with a significant and clinically relevant improvement in opioid-induced constipation in various types of pain even after long-term treatment [17, 18]. Both test and reference products in the present study were well tolerated, with no severe AEs experienced. The AEs reported and observed in this study were similar and consistent with the known safety profiles of oxycodone. In a phase I study, naltrexone, an opioid antagonist, was used to reduce the likelihood of these AEs. However, naltrexone potentially altered the PK profile because it reverses the slowing of gastric transit associated with opioids [19]. In addition, naltrexone is capable of producing nausea and vomiting [20, 21]. Although blockade with naltrexone was not used in this study, all AEs were of mild and moderate severity and there were no clinically meaningful changes in the safety laboratory values, ECGs, or vital signs in any subjects.
5 Conclusion
This study demonstrated that a newly formulated oxycodone tablet using hydrophilic matrix technology is bioequivalent to a marketed reference product in Japan when administered to healthy male Japanese subjects under fasting and fed conditions. Both oxycodone formulations were well tolerated by these subjects.
References
Riley J, Eisenberg E, Müller-Schwefe G, Drewes AM, Arendt-Nielsen L. Oxycodone: a review of its use in the management of pain. Curr Med Res Opin. 2008;24:175–92.
Pöyhiä R, Seppälä T, Olkkola KT, Kalso E. The pharmacokinetics and metabolism of oxycodone after intramuscular and oral administration to healthy subjects. Br J Clin Pharmacol. 1992;33:617–21.
Lalovic B, Phillips B, Risler LL, Howald W, Shen DD. Quantitative contribution of CYP2D6 and CYP3A to oxycodone metabolism in human liver and intestinal microsomes. Drug Metab Dispos. 2004;32(4):447–54.
Andreassen TN, Eftedal I, Klepstad P, Davies A, Bjordal K, Lundström S, Kaasa S, Dale O. Do CYP2D6 genotypes reflect oxycodone requirements for cancer patients treated for cancer pain? A cross-sectional multicentre study. Eur J Clin Pharmacol. 2012;68(1):55–64.
Bruera E, Pereira J, Watanabe S, Belzile M, Kuehn N, Hanson J. Opioid rotation in patients with cancer pain. A retrospective comparison of dose ratios between methadone, hydromorphone, and morphine. Cancer. 1996;78(4):852–7.
Knotkova H, Fine PG, Portenoy RK. Opioid rotation: the science and the limitations of the equianalgesic dose table. J Pain Symptom Manag. 2009;38(3):426–39.
Koizumi W, Toma H, Watanabe K, Katayama K, Kawahara M, Matsui K, et al. Efficacy and tolerability of cancer pain management with controlled-release oxycodone tablets in opioid-naive cancer pain patients, starting 5 mg tablets. Jpn J Clin Oncol. 2004;34:608–14.
Narabayashi M, Saijo Y, Takenoshita S, Chida M, Shimoyama N, Miura T, et al. Opioid rotation from oral morphine to oral oxycodone in cancer patients with intolerable adverse effects: an open-label trial. Jpn J Clin Oncol. 2008;38:296–304.
Kokubun H, Yoshimoto T, Hojo M, Fukumura K, Matoba M. Pharmacokinetics of oxycodone after intravenous and subcutaneous administration in Japanese patients with cancer pain. J Pain Palliat Care Pharmacother. 2014;28:338–50.
Toyama K, Uchida N, Ishizuka H, Sambe T, Kobayashi S. Single-dose evaluation of safety, tolerability and pharmacokinetics of newly formulated hydromorphone immediate-release and hydrophilic matrix extended-release tablets in healthy Japanese subjects without co-administration of an opioid antagonist. J Clin Pharmacol. 2015;55(9):975–84.
Mehta RY, Missaghi S, Tiwari SB, Rajabi-Siahboomi AR. Application of ethylcellulose coating to hydrophilic matrices: a strategy to modulate drug release profile and reduce drug release variability. AAPS PharmSciTech. 2014;15(5):1049–59.
Fukui S, Yano H, Yada S, Mikkaichi T, Minami H. Design and evaluation of an extended-release matrix tablet formulation; the combination of hypromellose acetate succinate and hydroxypropylcellulose. Asian J Pharm Sci. 2017;12(2):149–56. doi:10.1016/j.ajps.2016.11.002.
Pharmaceutical and Food Safety Bureau of Japan. Guideline for bioequivalence studies of generic products. 2012. http://www.nihs.go.jp/drug/be-guide(e)/Generic/GL-E_120229_BE.pdf. Accessed 1 Feb 2017.
Jedinger N, Khinast J, Roblegg E. The design of controlled-release formulations resistant to alcohol-induced dose dumping: a review. Eur J Pharm Biopharm. 2014;87(2):217–26.
US Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Abuse-deterrent opioids—evaluation and labeling. Guidance for industry. 2015. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM334743.pdf. Accessed 1 Feb 2017.
Lazzari M, Greco MT, Marcassa C, Finocchi S, Caldarulo C, Corli O. Efficacy and tolerability of oral oxycodone and oxycodone/naloxone combination in opioid-naïve cancer patients: a propensity analysis. Drug Des Devel Ther. 2015;9:5863–72.
Meissner W, Leyendecker P, Mueller-Lissner S, Nadstawek J, Hopp M, Ruckes C, et al. A randomised controlled trial with prolonged-release oral oxycodone and naloxone to prevent and reverse opioid-induced constipation. Eur J Pain. 2009;13(1):56–64.
Leppert W. Emerging therapies for patients with symptoms of opioid-induced bowel dysfunction. Drug Des Dev Ther. 2015;9:2215–31.
Murphy DB, Sutton JA, Prescott LF, Murphy MB. Opioid-induced delay in gastric emptying: a peripheral mechanism in humans. Anesthesiology. 1997;87(4):765–70.
Bigelow GE, Preston KL, Schmittner J, Dong Q, Gastfriend DR. Opioid challenge evaluation of blockade by extended-release naltrexone in opioid-abusing adults: dose-effects and time-course. Drug Alcohol Depend. 2012;123(1–3):57–65.
Garbutt JC. Efficacy and tolerability of naltrexone in the management of alcohol dependence. Curr Pharm Des. 2010;16(19):2091–7.
Acknowledgements
The authors thank Tsunenori Nakazawa for the bioanalytical work and gratefully acknowledge the contributions of Sachiko Fukui and Hideki Yano and their valuable technical comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Daiichi Sankyo Co., Ltd.
Conflicts of interest
Hidetoshi Furuie has no conflicts of interest. Kaoru Toyama, Kana Kuroda, and Hitoshi Ishizuka are employees of Daiichi Sankyo Co., Ltd.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Toyama, K., Furuie, H., Kuroda, K. et al. Pharmacokinetic Bioequivalence Studies of an Extended-Release Oxycodone Hydrochloride Tablet in Healthy Japanese Subjects Under Fasting and Fed Conditions Without an Opioid Antagonist. Drugs R D 17, 363–370 (2017). https://doi.org/10.1007/s40268-017-0184-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-017-0184-x