
Abstract
Background and Objective
Oral tramadol, an atypical opioid approved in the United States (US) since 1995 and a Schedule IV controlled substance, has less abuse liability compared to Schedule II conventional opioids. Intravenous (IV) tramadol is not available in the US, but has the potential to fill a gap between non-opioid medications and conventional opioids for treatment of acute pain. This study evaluates IV tramadol in the management of postoperative pain compared to placebo and standard-of-care active control.
Methods
A phase 3, multicenter, double-blind, three-arm, randomized, placebo- and active-controlled, multiple-dose, parallel-group study was conducted to evaluate the efficacy and safety of 50 mg IV tramadol versus placebo and 4 mg IV morphine over 48 h in patients with postoperative pain following abdominoplasty surgery.
Results
IV tramadol was statistically superior (p < 0.05) to placebo and comparable to IV morphine for the primary and all key secondary efficacy outcomes and demonstrated numerically lower rates for the incidence of most common treatment-emergent adverse events (TEAEs) compared to morphine. No unexpected findings were observed for TEAEs, laboratory tests, vital signs, or electrocardiograms (ECGs). Over 90% of patients completed the study.
Conclusion
The study demonstrated that IV tramadol 50 mg is highly effective in the management of postoperative pain following abdominoplasty. The consistency of effects between tramadol and morphine (as compared to placebo) for primary and key secondary endpoints validates the efficacy of tramadol observed. The study also provided direct evidence of improved tolerability of IV tramadol over a standard-of-care conventional Schedule II opioid. IV tramadol may become a useful option in patients where exposure to conventional opioids is not desired.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
IV tramadol 50 mg is effective in management of postoperative pain. |
It has improved tolerability over a Schedule II opioid. |
It is a treatment option in US patients where exposure to Schedule II opioids is not desired. |
1 Introduction
Clinicians in the United States (US) are currently limited in their choices of intravenous (IV) analgesics, which are widely used in the acute pain setting because of their bioavailability and inability of many patients to take medications orally following surgery. The only approved IV analgesics include three pharmacological classes: acetaminophen, non-steroidal anti-inflammatory drugs (NSAIDs), and conventional opioids. The lack of options contributes to the fact that IV conventional opioids are still used heavily in the acute pain setting. This is especially true if a patient has contraindications to one or more classes of non-opioid medications. Following IV opioids, physicians tend to transition patients to conventional oral opioids for outpatient pain management, some of which (including hydromorphone and oxycodone) have been shown to have a significant potential for abuse or misuse [1].
An often-overlooked analgesic for the treatment of acute pain in the hospital setting is tramadol, even though it is utilized around the world and has been shown to be effective for treating moderate to moderately severe levels of pain. Tramadol is a centrally acting atypical opioid with two mechanisms of action including weak activation of the mu opioid receptor by the parent drug, more potent activation by its primary metabolite (M1), and inhibition of the reuptake of serotonin and norepinephrine. These two distinct mechanisms serve to make tramadol an effective analgesic with a good tolerability profile. Tramadol is a member of the phenanthrene group of opium alkaloids, which includes morphine and codeine, and is structurally related to these opioids [2]. Like codeine, there is a substitution of the methyl group on the phenol ring that imparts a relatively weak affinity for opioid receptors. Therefore, the opioid component of tramadol comes primarily from the key metabolite M1, a stronger µ-agonist than the parent compound.
The primary advantage of tramadol, a Schedule IV controlled substance, over Schedule II opioids is that it carries less abuse liability [3,4,5], an important consideration in the context of the ongoing opioid epidemic in the US. There is a strong body of evidence that tramadol carries low but not zero abuse potential and that its abuse potential is lower than conventional opioid analgesic medications [6].
Oral tramadol has been approved by the Food and Drug Administration (FDA) since 1995, but IV tramadol, widely used outside the US for decades, has not been available in the US. A novel dosing regimen for IV tramadol was recently developed for the US, in which 50 mg is given for the first dose and repeated after 2 h, again at 4 h, and once every 4 h thereafter. Compared to oral tramadol 100 mg administered once every 6 h, IV tramadol reached initial peak serum concentration (Cmax) more rapidly, while resulting in similar overall steady-state Cmax and area under the curve (AUC). Importantly, IV tramadol results in less M1, the key opioid component, and a slower onset of exposure to M1 compared to the oral route, due to the avoidance of first-pass metabolism [7]. The reduced abuse potential of parenteral tramadol relative to conventional opioids was recognized by the Expert Committee on Drug Dependence convened by the World Health Organization (WHO) in 2018 [8].
Although parenteral tramadol has been widely used outside the US for decades and is well known to physicians outside the US, it has not been available in the US and is not familiar to US clinicians. Further, there is no current literature comparing the benefits and risks of IV tramadol versus placebo in an adequately powered and well-controlled clinical study, which is the gold standard in analgesic trials for approval in the US. Most studies involving parenteral tramadol have been limited in sample size, compared to a variety of comparators, and used different dosing strategies [9].
This study was designed to compare IV tramadol to both placebo and IV morphine in the management of postoperative pain. Comparing to placebo is important in all clinical trials aiming to demonstrate the efficacy of a new analgesic medication, to obtain a clear description of not only the efficacy, but, just as importantly, the safety and tolerability. Comparing to IV morphine, a standard-of-care IV opioid in post-surgical pain, provides assay sensitivity for the trial and provides a framework to understand a new drug’s efficacy and side effect profile. To our knowledge, this is the first high-quality, highly powered clinical trial that compares IV tramadol to an approximately equipotent amount of IV morphine.
2 Methods
2.1 Study Design and Ethics
This was a phase 3, multicenter, randomized, double-blind, three-arm study to evaluate the safety, tolerability, and efficacy of IV tramadol (tramadol hydrochloride) versus placebo and IV morphine (morphine sulfate) in the management of postoperative pain following abdominoplasty, a soft-tissue surgical model involving skin, muscle, and adipose tissue. All three study sites were located in the US. Patients were randomized in a 3:3:2 ratio to IV tramadol 50 mg, placebo, or IV morphine 4 mg. Study drugs were administered at baseline, hour 2, hour 4, and every 4 h thereafter through to hour 44.
Potentially equipotent doses of IV tramadol and IV morphine were chosen based on published data that support a 10:1 to 15:1 tramadol/morphine efficacy ratio [10,11,12,13,14,15,16,17]. The total amount of IV morphine administered was 28 mg on day 1 and 24 mg on day 2, similar to doses utilized in other comparable studies [18, 19].
In order to maintain the blind for study purposes, patients received both an IV infusion and an IV push at each dosing time point, with IV tramadol administered as a 15-min infusion and IV morphine 4 mg administered as an IV push, per usual clinical practice. Placebo IV infusion and IV push were also used to maintain the blind.
The study was performed in accordance with ethical principles that have their origin in the Declaration of Helsinki and are consistent with International Council for Harmonization (ICH)/Good Clinical Practice (GCP) guidelines, applicable regulatory requirements, and the sponsor or its delegate’s policy on bioethics. Aspire institutional review board (Santee, CA) reviewed the study protocol and informed consent forms. The study was registered at ClinicalTrials.gov (December 13, 2018; trial number NCT03774836), with the first patient enrolled on December 14, 2018 and the last patient completing the study on May 6, 2019.
The primary endpoint was the sum of pain intensity differences over 24 h (SPID24), and the key secondary outcomes were Patient Global Assessment (PGA) at 24 h, SPID48, and total rescue medication used through 24 h. Safety assessments included treatment-emergent adverse events (TEAEs), clinical laboratory tests, vital signs, and electrocardiograms (ECGs).
2.2 Study Treatment and Eligibility
Male and female subjects between the ages of 18 and 75 years with an American Society of Anesthesiologists (ASA) classification of I or II undergoing abdominoplasty were enrolled. The surgical protocol consisted of a low transverse abdominal incision, infra-umbilical fascial plication, and unilateral or bilateral drain placement. Each site followed a study-specific surgical and anesthetic protocol to reduce variability. Patients were housed in a healthcare facility and were to receive parenteral analgesia for at least 48 h after surgery. Patients with clinically significant disease or conditions that might have created an unacceptable risk were excluded. Other exclusion criteria included known physical dependence on opioids and the use of any opioids (including tramadol) within the past 30 days prior to surgery. Post-surgical eligibility required patients report a score of moderate or severe on a 4-point categorical rating scale (with categories of none, mild, moderate, and severe) and have a Numerical Pain Rating Scale (NPRS) pain score of ≥ 5 (on a scale from 0 to 10, where higher scores indicate worse pain) within 3 h after the end of surgery and an NPRS score of ≥ 5 at baseline (T0). Each patient underwent screening, a pre-operative assessment within 24 h prior to surgery, the surgical/treatment visit, assessment through to hour 48, and assessment at the follow-up visit (day 7).
Pain intensity assessments were recorded immediately prior to the first dose (baseline, T0) and at frequent intervals through to 48 h after the first treatment. PGA was assessed by asking “How would you rate the study medication in terms of its effectiveness in controlling your pain?” (0 = poor; 1 = fair; 2 = good; 3 = very good; 4 = excellent).
2.3 Statistical Methods
A sample size of 360 patients (135 each in the tramadol and placebo groups and 90 in the morphine group) was planned, to provide over 90% power to detect an SPID24 difference of 15 between placebo and tramadol, assuming a common SPID24 standard deviation (SD) of 38 (40% effect size) and an alpha of 0.05 using a two-sided analysis of covariance (ANCOVA) testing mean differences. Ninety patients in the morphine group allowed for at least 80% power to detect a 20% absolute difference between tramadol and morphine in the incidence of each individual preferred term of opioid-related adverse events (ORAEs).
The full analysis set (FAS) population was defined as all randomized patients who received at least one dose of study medication. Patients were analyzed according to the treatment group they were randomized to. The safety population was defined as all patients who received study medication. Patients were analyzed according to the actual treatment they received. There was one patient randomized to placebo who received a single dose of IV tramadol 50 mg and thus was included in the tramadol 50 mg arm for the purposes of the safety assessment.
2.4 Assessment of Efficacy
All SPID calculations were performed using the standard trapezoidal rule:
where PIDi = the pain intensity difference (PID) at time i, and (Ti+1 − Ti) is the time difference in hours between time i and time i + 1. Multiple imputation (Rubin 1976) [30] was used to impute for missing pain scores. Specifically, for the primary endpoint (SPID24), 100 imputed datasets were created, with data imputation for missing values due to missingness at random as well as due to discontinuation due to adverse events (AEs) or lack of efficacy (missing, not at random) and to account for use of rescue medication (the last NPRS score prior to the use of any rescue medication was used to impute subsequent NPRS scores for the subsequent protocol-specified time points for measurement of pain intensity through to 4 h after the dosing of the rescue medication). The 100 imputed datasets were analyzed using an ANCOVA model with treatment as the main effect and study center, baseline body mass index (BMI) (< 30 kg/m2 vs ≥ 30 kg/m2), and baseline NPRS score as covariates (these covariates were prespecified for the model to account for any possible end-of-study imbalances among the treatment groups).
The PGA at hour 24 and hour 48 was assessed for treatment-group differences using an ANCOVA with pooled study center, BMI, and the baseline pain score as covariates. Total consumption of rescue medication (Advil 400 mg) was calculated as the total amount of rescue analgesia (milligrams) captured in the Rescue Medications electronic case report form (eCRF) and recorded as given to the patient between the first dose of study medication through to 24 h after the first dose. The total consumption of rescue analgesia was analyzed using the nonparametric Wilcoxon rank sum test to test each active versus placebo comparison separately.
2.5 Alpha Control
All inferential assessments were two-sided tests performed at the 0.05 alpha level unadjusted for multiple comparisons. A hierarchical alpha testing strategy was utilized to control for the overall experiment-wise alpha for the tramadol versus placebo comparison (the comparisons to morphine were exploratory only, and thus no adjustments were made for those comparisons). As there were multiple tests being performed (the single primary efficacy variable pairwise test and the three key secondary efficacy tests), the following strategy was applied. If the primary efficacy endpoint, SPID24, was significant for the tramadol versus placebo comparison (in favor of the tramadol arm), then statistical testing was to proceed to the tramadol key secondary endpoints, to be tested in the following order:
-
PGA at hour 24
-
SPID48
-
Total consumption of rescue medication through to hour 24.
If the statistical test was a significant comparison at the nominal 0.05 level and two-sided (in favor of the tramadol arm) for the first endpoint, then testing was to proceed to the next endpoint in the list, and so on. Once a non-significant test occurred, endpoints lower in the list were to be considered not statistically significant. All analyses were performed using the SAS System® version 9.4.
2.6 Assessment of Safety
The incidence of TEAEs was summarized for each treatment group by Medical Dictionary for Regulatory Authorities (MedDRA) system organ class (SOC) and preferred term.
This study was designed to carefully assess the relative safety and tolerability of IV tramadol versus morphine, with specific pre-defined safety outcomes for ORAEs (bradypnea, constipation, dizziness, dizziness postural, hypoxia, respiratory disorder, nausea, somnolence, sedation, vomiting, pruritus, and pruritus generalized), TEAEs potentially related to substance abuse (defined as indicated in the FDA guidance “Assessment of Abuse Potential of Drugs Guidance for Industry: January 2017”), gastrointestinal TEAEs (that are often associated with opioid treatment, including nausea, grade 2 nausea, vomiting, and use of antiemetics for nausea). Local tolerability of the infusion site was also assessed. Respiratory impairment (RI) was defined as a clinically relevant worsening of respiratory status—taking into account selected safety parameters such as respiratory rate, oxygen saturation, and somnolence/sedation.
3 Results
3.1 Patient Disposition
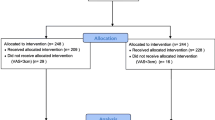
This study was conducted between December 14, 2018 and May 6, 2019. Patients were enrolled from three investigational study centers, with each center contributing at least 23.9% of the total number of randomized patients. No center contributed more than 38.4% of the total randomized patients. Over 90% of total patients completed the study (Fig. 1).

Study consort diagram of patient disposition from screening to study completion. DC discontinued, TEAE treatment-emergent adverse event
3.2 Demographics
The mean (SD) qualifying NPRS score overall was 6.5 (1.45), and the majority of patients (73.2%) reported moderate pain at the time they qualified for study drug. The treatment groups were similar with respect to demographic and baseline characteristics (Table 1).
3.3 Primary and Secondary Efficacy
The outcomes demonstrated that IV tramadol 50 mg was statistically significantly superior to placebo in the management of postoperative pain following abdominoplasty (Table 2), in accordance with the pre-defined hierarchical testing strategy.
The PID scores over time demonstrated similar profiles of pain relief during the 48-h treatment period (Fig. 2). The magnitude of the differences in the primary and key secondary efficacy outcomes as well as most of the tertiary efficacy outcomes were similar between tramadol and morphine, including the SPID24 and SPID48 (Fig. 3). The only notable differences were that patients in the morphine group utilized less rescue medication over the first 24 h and showed earlier time to onset of pain relief.

Least squares means (± standard errors of the means) of pain intensity differences over the 48-h treatment period

LS mean (SE) summary of pain intensity differences: SPID24 and SPID48 comparisons across treatment groups. ANCOVA analysis of covariance, LS least squares, NPRS Numerical Pain Rating Scale, SE standard error, SPID24 sum of pain intensity differences over 24 h, SPID48 sum of pain intensity differences over 48 h
3.4 Safety and Tolerability
The incidence of patients with at least one TEAE was highest in the morphine group (92.5%), followed by the IV tramadol group (85.9%), and then the placebo group (57.0%) (Table 3).
The incidence of patients with at least one TEAE considered to be at least possibly related to study drug was greatest in the morphine group (72.0%), followed by the tramadol 50 mg group (66.2%), and placebo group (34.8%) (Fig. 4).

Patient Global Assessment of treatment at hour 24 and hour 48: tramadol 50 mg vs placebo. PGA Patient Global Assessment
There were three serious adverse events (SAEs) reported during this study, including two post-procedural hematomas, judged by the investigator to be unrelated to study treatment, in tramadol-treated patients. A third SAE (acute cholecystitis), also in the tramadol group, was reported post-treatment (over 24 h after the last dose of study medication) and was judged by the investigator as not related to study drug. There was one grade 3 TEAE reported (one of the post-procedural hematoma SAEs); the remaining TEAEs reported in this study were of mild to moderate intensity.
This study was designed to carefully assess the relative safety and tolerability of IV tramadol versus morphine, with specific pre-defined safety outcomes for ORAEs, TEAEs potentially related to substance abuse, gastrointestinal TEAEs (that are often associated with opioid treatment, including nausea, grade 2 nausea, vomiting, and use of antiemetics for nausea), and local tolerability of the infusion site. In each of these types of events, the morphine group demonstrated a numerically higher incidence than that of the tramadol group. The placebo group generally had the lowest incidence for these types of events, with the exception of local tolerability events at the infusion site. The tramadol group had the lowest incidence of local tolerability events at the infusion site.
The incidence of RI was 0% in the placebo arm, 6.3% in the tramadol arm and 4.3% in the morphine arm. All these events occurred at one site, driven by hypoxemia defined on pulse oximetry. There were no reports of shortness of breath or clinical symptoms linked to a depression of respiratory function.
The incidence of at least one TEAE related to potential risk of substance abuse was 8.1% in the placebo group, 16.2% in the tramadol group, and 22.6% in the morphine group. Dizziness was the most frequently reported TEAE of this type, reported in 6.7% of placebo patients, 12.7% of tramadol patients, and 18.3% of morphine patients. No dizziness, somnolence, or sedation occurred in conjunction with euphoria (euphoria was not reported at all). The incidence of the individual preferred terms was low for each treatment group, with similar incidences among the treatment groups.
The most common TEAEs, occurring in at least 10% of total patients, were nausea, vomiting, headache, and dizziness. The incidence of each of these TEAEs was slightly higher for the morphine group as compared to the tramadol group, while the incidence was lowest for each of these TEAEs in the placebo group (Table 4).
4 Discussion
While there is always a need for new options for the treatment of postoperative pain, this need is more acute in the context of the ongoing opioid crisis, which has put additional pressure on clinicians to minimize the use of conventional opioids. Once a patient’s pain cannot be adequately treated with non-opioid medications even when multi-modal analgesia is used, the clinician will have no option but to turn to a conventional opioid at the present time. IV tramadol fills in a gap between non-opioid medicine and conventional opioids and can be effectively used in combination with non-opioid medications, based on differing mechanisms of action. The primary advantage of tramadol (a Schedule IV controlled substance) over Schedule II conventional opioids is that it carries less abuse liability, and this study answers the important question of how it compares to conventional opioids for both efficacy and safety.
IV tramadol 50 mg was statistically significantly superior to placebo for the primary efficacy endpoint (SPID24), and all three key secondary efficacy endpoints (PGA at 24 h, SPID48, and total rescue medication used through to 24 h). In addition, IV tramadol demonstrated a similar efficacy profile for each of these pre-defined endpoints to that of IV morphine 4 mg, which was included as a standard-of-care active comparator in this study.
Comparison of the safety outcomes were compared to other published studies of similar design. Erolcay and Yüceyar [20] compared tramadol with morphine using IV patient-controlled analgesia during the first 24 h after thoracotomy in 44 patients and found that postoperative analgesia and side effects in the tramadol arm were similar to those of the morphine arm. In particular, 26% of patients in the tramadol group and 33% in the morphine group had nausea.
Similarly, Houmes et al. [21] compared tramadol and morphine in a double-blind, randomized study of 150 female patients after gynecologic surgery and found that both drugs produced acceptable analgesia. AEs (nausea, vomiting, dizziness, drowsiness, etc.) were reported by 23% of tramadol patients versus 27% of morphine patients. While the absolute incidence of events in these prior studies varied, the finding of a slightly higher incidence of events in the morphine arm was consistent.
While the study is not powered to formally compare the efficacy of IV tramadol and IV morphine, the sample size was adequate to provide assay sensitivity and comparative data. The magnitude of the differences in the primary and key secondary efficacy outcomes as well as most of the tertiary efficacy outcomes was similar between tramadol and morphine, including the SPID24, SPID48, PGA at 24 and at 48 h, and total rescue mediation consumption through 48 h. The consistency of effects between IV tramadol and IV morphine (as compared to placebo) for the primary and key secondary endpoints provide assay sensitivity and validation of the observed efficacy of tramadol. Notable differences were that the morphine group utilized less rescue medication early in the study and showed earlier onset of relief. Tramadol is an atypical opioid with two mechanisms of action including weak activation of the μ-opioid receptor by the parent drug, more potent activation by its primary metabolite, and inhibition of the reuptake of serotonin and norepinephrine. Based on this pharmacology, it was anticipated that morphine, as a pure μ-opioid receptor agonist, will have an effect more noticeable at very early time points. Onset difference may also be due to administration of the morphine full dose as an injection rather than 15-min infusion. This difference may not be clinically relevant for two reasons: (1) the study did not use a multimodal analgesic approach that is common in clinical practice; and (2) the study required patients’ pain levels to rise to a certain threshold before dosing was allowed. In an actual clinical setting, IV tramadol will likely be used with non-opioid analgesics and administered before anesthetics are worn off instead of waiting for pain levels to rise. Therefore, the slower onset versus IV morphine may not be important in practice.
Treatment with IV tramadol 50 mg was well-tolerated in this sample of patients with postoperative pain. There were no unexpected findings in the assessment of TEAEs, laboratory tests, vital signs, or ECGs. The incidence of opioid-related TEAEs (nausea, vomiting) and use of antiemetics associated with treatment-emergent nausea were numerically higher in the IV morphine group than in the IV tramadol group; the incidence of TEAEs related to potential risk of substance abuse and the incidence of local infusion site TEAEs were also higher in the IV morphine group than in the IV tramadol group.
It is important to note that these findings are consistent with the literature on IV tramadol, in which clinical studies conducted in Europe reported similar effectiveness and lower or similar AE rates among patients receiving tramadol relative to patients receiving comparator opioid products [12, 15, 22,23,24,25,26,27,28,29].
This study did not assess patients’ CYP2D6 genotype status (even though the function of the enzyme is directly linked to the production of M1) for two reasons: (1) the goal of the study was to assess the efficacy and safety of IV tramadol as compared to placebo and IV morphine regardless of the status of CYP2D6; and (2) clinicians in the US do not routinely test for it or use it to make clinical decisions in the acute pain setting.
A characteristic of the study population is that very few men were enrolled, reflecting the patient population who elect to undergo abdominoplasty (who tend to be female). This single, standardized procedure was chosen for the purpose of maximizing signal detection for both efficacy and safety. Further, the study design required a fixed dose and dosing interval for morphine to allow for maintenance of the blind and reliability of comparisons at each specific protocol-scheduled time point (and thus also to maximize signal detection).
5 Conclusions
IV tramadol 50 mg is highly effective and well-tolerated in patients treated for pain post-abdominoplasty surgery. Further, it was similarly efficacious to IV morphine, the standard-of-care control arm employed in this study, and somewhat better tolerated than the morphine arm. IV tramadol may become a useful option in patients where exposure to conventional opioids is not desired.
Availability of Data and Materials
Data may be available upon consent of the funding Sponsor.
References
Brat GA, Agniel D, Beam A, et al. Postsurgical prescriptions for opioid naïve patients and association with overdose and misuse: retrospective cohort study. BMJ. 2018;360:j5790.
Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43(879):923.
World Health Organization (WHO). Expert Committee on Drug Dependence. Tramadol Update Review Report; 2014. https://www.who.int/medicines/areas/quality_safety/6_1_Update.pdf. Accessed Apr 13, 2019.
Grünenthal GmBH Application for inclusion of tramadol into the WHO Model List of Essential medicines (EML); 2017. World Health Organization 2017. https://www.who.int/selection_medicines/committees/expert/21/applications/Grunethal_tramadol.pdf. Accessed Apr 13, 2019.
Schneider MF, Bailey JE, Cicero TJ, et al. Integrating nine prescription opioid analgesics and/or four signal detection systems to summarize statewide prescription drug abuse in the United States in 2007. Pharmacoepidemiol Drug Saf. 2009;18(9):778–90.
Dunn KE, Bergeria CL, Huhn AS, Strain EC. A systematic review of laboratory evidence for the abuse potential of tramadol in humans. Front Psychiatry. 2019;10:704.
Lu L, Ryan M, Harnett M, Atiee GJ Reines SA. Comparing the pharmacokinetics of 2 novel intravenous tramadol dosing regimens to oral tramadol: a randomized 3-arm crossover study. Clin Pharmacol Drug Dev. 2019.
WHO Expert Committee on Drug Dependence. Critical review report: tramadol. 41st ECDD (2018).
Scott LJ, Perry CM. Tramadol: a review of its use in perioperative pain. Drugs. 2000;60(1):139–76.
Hadi M, Kamaruljan S, Saedah A, et al. A comparative study of intravenous patient-controlled analgesia morphine and tramadol in patients undergoing major operation. Med J Malaysia. 2006;61(5):570–6.
Houmes RJ, Voets M, Verkaaik, et al. Efficacy and safety of tramadol versus morphine for moderate and severe postoperative pain with special regard to respiratory depression. Anesth Analg. 1992;74:510–4.
Naguib M, Seraj M, Attia M, et al. Perioperative anti-nociceptive effects of tramadol. A prospective, randomized, double-blind comparison with morphine. Can J Anaesth. 1998;45(12):1168–75.
Ng K, Tsui S, Yang J et al. Increased nausea and dizziness when using tramadol for post- operative patient-controlled analgesia (PCA) compared with morphine after intraoperative loading with morphine. Eur J Anaesthesiol (EJA); 1998. https://journals.lww.com/ejanaesthesiology/Fulltext/1998/09000/Increased_nausea_and_dizziness_when_using_tramadol.10.aspx.
Ozalevli M, Unlugenc H, Tuncer U, et al. Comparison of morphine and tramadol by patient-controlled analgesia for postoperative analgesia after tonsillectomy in children. Pediatr Anesth. 2005;15:979–84.
Silvasti M, Svartling N, Pitkanen N, et al. Comparison of intravenous patient-controlled analgesia with tramadol versus morphine after microvascular breast reconstruction. Eur J Anaesthesiol. 2000;17(7):448–55.
Stamer U, Maier C, Grond S, et al. Tramadol in the management of post-operative pain: a double-blind, placebo- and active drug-controlled study. Eur J Anaesthesiol (EJA). 1997;14(6):646–54.
Vergnion M, Degesves S, Garcet L, et al. Tramadol, an alternative to morphine for treating posttraumatic pain in the prehospital situation. Anesth Analg. 2001;92:1543–6.
Singla N, Minkowitz H, Soergel D, Burt D, et al. A randomized, phase IIb study investigating oliceridine (TRV130), a novel µ-receptor G-protein pathway selective (μ-GPS) modulator, for the management of moderate to severe acute pain following abdominoplasty. J Pain Res. 2017;10:2413–24.
Singla N, Skobieranda F, Soergel D, Salamea M, et al. APOLLO-2: a randomized, placebo and active-controlled phase III study investigating oliceridine (TRV130), a G protein–biased ligand at the μ-opioid receptor, for management of moderate to severe acute pain following abdominoplasty. Pain Pract. 2019;19(7):715–31.
Erolcay H, Yüceyar L. Intravenous patient-controlled analgesia after thoracotomy: a comparison of morphine with tramadol. Eur J Anaesthesiol. 2003;20(2):141–6.
Houmes R-J, Voets MA, Verkaaik A, Erdmann W, Lachmann B. Efficacy and safety of tramadol versus morphine for moderate and severe postoperative pain with special regard to respiratory depression. Anesth Analg. 1992;74(4):510–4.
Broome IJ, Robb HM, Raj N, Girgis Y, Wardall GJ. The use of tramadol following day-case oral surgery. Anaesthesia. 1999;54(3):289–92.
Hadi M, Kamaruljan HS, Saedah A, Abdullah N. A comparative study of intravenous patient-controlled analgesia morphine and tramadol in patients undergoing major operation. The Medical journal of Malaysia. 2006;61(5):570–6.
Langford RM, Bakhshi KN, Moylan S, Foster JM. Hypoxaemia after lower abdominal surgery: comparison of tramadol and morphine. Acute Pain. 1998;1(2):7–12.
Ng K, Tsui S, Yang J, Ho E. Increased nausea and dizziness when using tramadol for post-operative patient-controlled analgesia (PCA) compared with morphine after intraoperative loading with morphine. Eur J Anaesthesiol. 1998;15(5):565–70.
Ng KF, Yuen TS, Ng VM. A comparison of postoperative cognitive function and pain relief with fentanyl or tramadol patient-controlled analgesia. J Clin Anesth. 2006;18(3):205–10.
Pang WW, Mok MS, Lin CH, Yang TF, Huang MH. Comparison of patient-controlled analgesia (PCA) with tramadol or morphine. Can J Anaesth. 1999;46(11):1030–5.
Shamim F, Hoda MQ, Samad K, Sabir S. Comparison between tramadol and pethidine in patient controlled intravenous analgesia. J Pak Med Assoc. 2006;56(10):433.
Silvasti M, Tarkkila P, Tuominen M, Svartling N, Rosenberg PH. Efficacy and side effects of tramadol versus oxycodone for patient-controlled analgesia after maxillofacial surgery. Eur J Anaesthesiol. 1999;16(12):834–9.
Rubin DB. Inference and missing data. Biometrika. 1976;63:581–92.
Acknowledgements
This study was funded by Avenue Therapeutics. Special thanks to Robert Criscola and Amy Landry Wheeler for support and efforts during the performance of this clinical study.
Author information
Authors and Affiliations
Contributions
Dr. Minkowitz was a coordinating investigator. Dr. Salazar was a principle investigator. Dr. Leiman was a coordinating investigator. Dr. Solanki was a principle investigator. Dr. Lu participated in the design conceptualization, planning, and conduct of the study. Dr. Reines participated in the design conceptualization, planning, medical oversight, and analysis interpretation. Mr. Ryan participated in the design conceptualization, planning, conduct, and data acquisition of the study. Mr. Harnett participated in the statistical analysis plan, analysis interpretation, and medical writing. Dr. Singla participated in the design conceptualization and planning of the study and was a coordinating investigator.
Corresponding author
Ethics declarations
Funding
This study was funded in full by Avenue Therapeutics, Inc.
Conflict of interest
All authors were either compensated directly (via salary or consulting fees) or indirectly (through employment within an organization that was paid directly by the Sponsor).
Ethics approval and consent to participate
The study was performed in accordance with ethical principles that have their origin in the Declaration of Helsinki and are consistent with International Council for Harmonization (ICH)/Good Clinical Practice (GCP) guidelines, applicable regulatory requirements, and the Sponsor or its delegate’s policy on Bioethics. Aspire institutional review board (Santee, CA) performed a review of the study protocol and informed consent forms. All patients provided signed informed consent prior to participation.
Study registration
The study was registered at ClinicalTrials.gov (December 13, 2018; trial number NCT03774836), with the first patient enrolled on December 14, 2018 and the last patient completing the study on May 6, 2019.
Consent for publication
All authors consent for this manuscript to be published.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Minkowitz, H., Salazar, H., Leiman, D. et al. Intravenous Tramadol is Effective in the Management of Postoperative Pain Following Abdominoplasty: A Three-Arm Randomized Placebo- and Active-Controlled Trial. Drugs R D 20, 225–236 (2020). https://doi.org/10.1007/s40268-020-00309-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-020-00309-0