
Abstract
Background
Genetic polymorphisms are known to influence outcomes with phenytoin yet effects in the Middle East and North Africa region are poorly understood.
Objectives
The objective of this systematic review was to evaluate the impact of genetic polymorphisms on phenytoin pharmacokinetics and clinical outcomes in populations originating from the Middle East and North Africa region, and to characterize genotypic and allelic frequencies within the region for genetic polymorphisms assessed.
Methods
MEDLINE (1946–3 May, 2017), EMBASE (1974–3 May, 2017), Pharmacogenomics Knowledge Base, and Public Health Genomics Knowledge Base online databases were searched. Studies were included if genotyping and analyses of phenytoin pharmacokinetics were performed in patients of the Middle East and North Africa region. Study quality was assessed using a National Institutes of Health assessment tool. A secondary search identified studies reporting genotypic and allelic frequencies of assessed genetic polymorphisms within the Middle East and North Africa region.
Results
Five studies met the inclusion criteria. CYP2C9, CYP2C19, and multidrug resistance protein 1 C3435T variants were evaluated. While CYP2C9*2 and *3 variants significantly reduced phenytoin metabolism, the impacts of CYP2C19*2 and *3 variants were unclear. The multidrug resistance protein 1 CC genotype was associated with drug-resistant epilepsy, but reported impacts on phenytoin pharmacokinetics were conflicting. Appreciable variability in minor allele frequencies existed both between and within countries of the Middle East and North Africa region.
Conclusions
CYP2C9 decrease-of-function alleles altered phenytoin pharmacokinetics in patients originating from the Middle East and North Africa region. The impacts of CYP2C19 and multidrug resistance protein 1 C3435T variants on phenytoin pharmacokinetic and clinical outcomes are unclear and require further investigation. Future research should focus on the clinical outcomes associated with phenytoin therapy.
PROSPERO 2017: CRD42017057850
Similar content being viewed by others

Avoid common mistakes on your manuscript.
In patients originating from the Middle East and North Africa region, phenytoin metabolism was significantly decreased by genetic polymorphisms of the cytochrome P450 (CYP) 2C9 enzyme; the impacts of CYP2C19 and multidrug resistance protein 1 C3435T polymorphisms were unclear. |
The impacts of genetic polymorphisms on clinical outcomes associated with phenytoin are not well described in the literature and warrant further investigation. |
1 Introduction
Since the discovery of its anticonvulsant properties in 1938 [1], phenytoin has been widely used for both the acute management and chronic prevention of seizures [2, 3]. Its primary mechanism in treating and preventing seizures is through preferential binding to inactivated sodium channels, which inhibits neuronal generation of sustained high-frequency bursts of action potentials [4]. Phenytoin has a narrow therapeutic index, with 40–80 µmol/L (10–20 mg/L) being the recommend therapeutic range; however, this is often difficult to achieve due to wide interpatient variability in dose response and concentrations achieved from a given dose, as well as the capacity-limited metabolism of phenytoin [5, 6].
Factors such as patient body size and composition, serum albumin levels, comorbid disease states, and drug–drug interactions have been shown to contribute to the interpatient variability of the effect of phenytoin. Additionally, genetic polymorphisms of enzymes and transporters targeting phenytoin pharmacokinetics have been recognized as a significant contributor to the exhibited interpatient variability [5,6,7]. Evidence linking the cytochrome P450 (CYP) 2C9 polymorphism to the concentration of phenytoin has led to the development of guidelines tailoring the initial dose of phenytoin based on the CYP2C9 genotype [7,8,9]. Though not as well studied, there is evidence to suggest that polymorphisms of CYP2C19 may also significantly impact the concentration of phenytoin and warrant dosage adjustments [7, 10]. Additional enzymes and drug transporters have been investigated, but the implications of these polymorphisms are not well established [7, 11].
Cited studies investigating the impact of genetic polymorphisms on the pharmacokinetics and pharmacodynamics of phenytoin have predominantly been conducted in patients of European, Indian, and East Asian origin; however, few studies have been conducted in patients originating from the Middle East and North Africa (MENA) region [11, 12]. As a result of migration and significant admixture of European, Asian, and African populations dating back over thousands of years, the MENA region has a unique genetic diversity, with great variability in minor allelic frequencies (MAF) of genetic polymorphisms [13,14,15]. Even within the MENA region itself, there is great diversity. For example, while populations of Arabic and Iranian origin have both European and African ancestry, those of Iranian origin also have a significant level of Asian admixture [15]. Furthermore, as a result of consanguinity, some countries within the MENA region may exhibit relatively less genetic diversity [15]. Therefore, results of previous studies conducted in patients of different ethnic origins are not readily generalizable, and a focused review of the literature addressing genetic polymorphisms and the associated implications regarding phenytoin in individuals originating from the MENA region is warranted.
The primary objective of this systematic review was to evaluate the impact of genetic polymorphisms on phenytoin pharmacokinetics, specifically in populations originating from the MENA region. Secondary objectives were to evaluate the impact of genetic polymorphisms on clinical outcomes associated with phenytoin therapy, as well as to characterize the genotypic and allelic frequencies of each polymorphism identified in the primary search within the MENA region.
2 Methods
2.1 Search Strategy
A comprehensive search of the MEDLINE (1946–May 2017), EMBASE (1974–May 2017), Pharmacogenomics Knowledge Base (PharmGKB), and Public Health Genomics Knowledge Base (PHGKB) online databases was conducted to identify existing literature addressing the primary research question. An example of the search terms and Boolean connectors used in MEDLINE is outlined in the Appendix, and is reflective of the search strategy used in EMBASE. References of pertinent studies were also searched manually to identify additional studies not identified in the electronic search. For each polymorphism assessed in the studies that met the inclusion criteria, a secondary search was conducted to identify the reported variant MAF, specifically within the MENA region.
2.2 Selection of Studies
Studies were eligible for inclusion if participants originated from the MENA region, received at least one dose of phenytoin with a subsequent pharmacokinetic analysis, and were genotyped for a polymorphism that may impact phenytoin pharmacokinetics. Although understanding the impact of genetic polymorphisms on clinical outcomes associated with phenytoin therapy was a secondary objective of this review, eligible studies were not required to report on clinical outcomes. Two reviewers (RD, KW) screened potentially relevant studies by title and abstract, and then by full text to exclude any studies that did not fulfill the inclusion criteria. For the review regarding variant MAF, studies were included if participants originated from the MENA region and were genotyped for one or more of the polymorphisms assessed in the primary review.
2.3 Data Extraction and Management
Data from each included study were extracted and tabulated. Extracted data included: author, year of publication, study population (country of origin, practice setting, inclusion and exclusion criteria), sample size, study design, genetic polymorphism(s) assessed, phenytoin dosing, assay methods for phenytoin serum concentrations and genotyping, MAF of the genetic polymorphism(s), as well as final study results. For studies also including participants from outside of the MENA region, only data regarding participants who originated from the MENA region were extracted.
Similarly, for studies included in the review of variant MAF, the following data were extracted: author, country of origin, sample size, genetic polymorphism(s) assessed, as well as the computed genotypic and allelic frequencies. If studies eligible for inclusion also reported data from participants originating outside of the MENA region, only data on participants who originated from the MENA region were extracted.
2.4 Quality Assessment
The quality of each study included in the primary review was assessed by two reviewers (RD, KW) using the Quality Assessment Tool for Observational Cohort and Cross-Sectional Studies, from the National Institutes of Health, National Heart, Lung, and Blood Institute [16].
2.5 Data Synthesis
Data extracted from the included studies were assessed descriptively. If two or more studies reported the genotypic or allelic frequency of a given polymorphism, data from all studies were reported. The highest and lowest frequencies extracted for each polymorphism were assessed to estimate variants’ MAF range within the MENA region.
3 Results
3.1 Study Selection
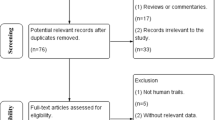
A total of five studies fulfilled the inclusion criteria for this review, as depicted in Fig. 1. Of the 30 articles screened for eligibility, ten were excluded for subjects not originating from the MENA region [17,18,19,20,21,22,23,24,25,26], seven were excluded for phenytoin concentrations not being assessed [27,28,29,30,31,32,33], five were excluded for both subjects not originating from the MENA region and phenytoin concentrations not being assessed [34,35,36,37,38], one was excluded for genetic polymorphisms not being assessed [39], and two were excluded for being poster abstracts [40, 41]. Between the five included studies, CYP2C9*1, *2, and *3; CYP2C19*1, *2, and *3; and multidrug resistance protein 1 [MDR1, also known as ATP-binding cassette sub-family B member 1 (ABCB1)] C3435T polymorphisms were evaluated. The study population, phenytoin regimen, and genetic polymorphism(s) assessed varied between studies; summaries of each study are provided in Table 1.

Preferred reporting items for systematic reviews and meta-analyses (PRISMA) flow chart of included studies [146]. MENA Middle East and North Africa, PHT phenytoin
Upon assessment, each of the five studies was rated to be of poor quality. The ratings of each study against the 14 quality assessment tool criteria are summarized in Table 2. Factors contributing to poor quality ratings included: unclear descriptions of study subject selection, introducing potential for selection bias; insufficient reporting of study subjects’ baseline characteristics; relatively small sample sizes, limiting the power to assess the impact of polymorphic genotypes with low frequencies; and absence of adjustments for potential confounders that could affect phenytoin pharmacokinetics (e.g., antiepileptic polytherapy, concomitant medications, comorbid medical conditions, or serum albumin levels) or clinical outcomes (e.g., seizure control at baseline, seizure etiology, or type of seizure).
A total of 82 studies were conducted within the MENA region and investigated the prevalence of CYP2C9, CYP2C19, and/or MDR1 variant alleles [27, 29, 42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121]. These studies were included in the secondary part of the review. Specific single nucleotide polymorphisms reviewed were: CYP2C9*1, *2, and *3; CYP2C19*1, *2, and *3; and MDR1 (ABCB1) C3435T. The study populations and computed genotypic and allelic frequencies for CYP2C9, CYP2C19, and MDR1 are summarized in Tables 3, 4, and 5, respectively.
3.2 CYP2C9
CYP2C9 accounts for 70–90% of phenytoin metabolism [122], with (S)-5-(p-hydroxyphenyl)-5-phenylhydantoin (p-HPPH) being the major resultant metabolite [5, 7]. Although up to 60 different CYP2C9 haplotypes have been identified, CYP2C9*2 and *3 are the two most common allelic variants [123, 124]. Both variants exhibit reduced functional activity, with losses of 29 and 94% enzymatic activity reported for CYP2C9*2 and *3, respectively, when compared with the wild-type genotype [125, 126]. Three of the included studies investigated CYP2C9 genotypes [42,43,44].
Aynacioglu et al. [42] genotyped all outpatients and healthy volunteers for CYP2C9, and phenotyped phenytoin through assessment of serum phenytoin and p-HPPH concentrations in a subset of 101 of the Turkish healthy volunteers. Twelve hours following a single dose of phenytoin, serum phenytoin concentrations differed significantly across groups with 0, 1, and 2 loss of function mutations (p < 0.001). Study results indicated serum phenytoin concentration in the CYP2C9*1/*1 wild-type genotype group was significantly lower when compared with CYP2C9*1/*2, CYP2C9*1/*3, and CYP2C9*2/*2 genotype groups, with the difference in means being −1.36 mg/L (p = 0.009), −1.49 mg/L (p = 0.001), and −2.42 mg/L (p = 0.02), respectively. Accordingly, results also indicated the ratio of serum p-HPPH-to-phenytoin concentration of the CYP2C9*1/*1 wild-type genotype group was significantly higher than that of CYP2C9*1/*2, CYP2C9*1/*3, and CYP2C9*2/*2 genotype groups, with the difference in means being 0.17 (p = 0.002), 0.22 (p < 0.0001), and 0.29 (p = 0.006), respectively. Differences between wild-type and CYP2C9*3/*3 genotypes were not compared statistically, as the latter was present in only one individual.
It was identified that data from the 96 healthy Turkish volunteers in the study conducted by Kerb et al. [43] were derived from the 101 volunteers of the Aynacioglu et al. study [42]. Thus, the results regarding influence of CYP2C9 genotype on measured phenytoin concentrations and the p-HPPH-to-phenytoin ratio are reflected in the aforementioned results [42, 43]. Using these data in a multiple linear regression analysis, Kerb et al. determined that the number of polymorphic CYP2C9 alleles accounted for 14.1% of variability in phenytoin serum concentrations [43]. The final regression equation, which also included the number of MDR1 C3435T alleles, had an r 2 = 0.154 (p = 0.0002).
Ozkaynakci et al. [44] reported data for 11 different combinations of CYP2C9 and CYP2C19 genotypes; therefore, the independent impact of the CYP2C9 genotype on serum phenytoin concentrations was difficult to discern. When compared with the CYP2C9 and CYP2C19 wild-type genotype group, CYP2C9*1/*3, CYP2C19*2/*3 was the only genotype group with a serum phenytoin trough that was significantly different, with a mean concentration that was 20.47 mg/L higher than the wild-type genotype group (p < 0.001). The mean serum phenytoin trough of the CYP2C9*1/*3, CYP2C19*2/*3 genotype group was also significantly higher than several other genotype groups (Table 1). Clinical outcomes associated with CYP2C9 polymorphisms were not reported and therefore could not be assessed.
3.3 CYP2C19
CYP2C19 accounts for 10–30% of phenytoin metabolism, with greater metabolic contributions as CYP2C9 becomes saturated [122, 127]. CYP2C19 primarily forms the (R)-p-HPPH metabolite [5, 7]. Up to 35 different CYP2C19 haplotypes have been identified, of which CYP2C19*2 and *3 are the two most common null alleles; presence of two null alleles results in a non-functional CYP2C19 enzyme [124, 125]. A third haplotype, CYP2C19*17, results in increased expression and activity of the enzyme [125]; however, this haplotype was not assessed in the included studies. Two of the included studies investigated CYP2C19 genotypes [43, 44].
In the study conducted by Kerb et al. [43], the CYP2C19 genotype did not significantly affect serum phenytoin concentration (p = 0.52) or p-HPPH-to-phenytoin ratio (p value not reported). As with CYP2C9, as a result of Ozkaynakci et al. [44] reporting the data for 11 different CYP2C9 and CYP2C19 genotype groups, it is difficult to distinguish the independent impact of CYP2C19 genotype on serum phenytoin concentrations; therefore, no inferences can be made. As with CYP2C9, clinical outcomes were not reported and therefore could not be assessed.
3.4 MDR1 (ABCB1) C3435T
The MDR1 (ABCB1) gene encodes for human P-glycoprotein, an efflux transporter of which phenytoin is a substrate [128, 129]. The MDR1 C3435T polymorphism is a silent mutation that decreases transporter expression and alters substrate specificity [130, 131]; therefore, it has the potential to affect substrate absorption and tissue distribution. The resultant impact of this polymorphism on pharmacokinetic and pharmacodynamic outcomes of phenytoin therapy is unclear [7, 130, 132]. Three of the included studies investigated the MDR1 C3435T polymorphism [43, 45, 46].
Kerb et al. [43] identified that the MDR1 C3435T genotype did not significantly impact serum phenytoin concentration overall (p = 0.064), and the number of MDR1 T alleles accounted for only 1.3% of the variability in serum phenytoin concentrations in their multiple regression analysis. As discussed previously, the final regression equation had an r 2 = 0.154 (p = 0.0002). When serum phenytoin concentrations were divided into quartiles (25th percentile: <3.79 mg/L, interquartile range: 3.79–5.79 mg/L, 75th percentile: >5.79 mg/L), the frequency distribution of MDR1 genotype indicated that the MDR1 CC genotype was more frequent in the 25th percentile group (p ≤ 0.001, χ 2 test). Moreover, while the presence of the MDR1 TT genotype reportedly had significant influence on p-HPPH-to-phenytoin ratio (p < 0.026), absolute values were not provided and there was considerable overlap in the depicted frequency distribution; therefore, clinical significance of this association cannot be inferred.
Alhazzani et al. [45] identified significant differences between the CC, CT, and TT genotypes (classified as extensive, intermediate, or poor metabolizers, respectively) for the pharmacokinetic parameters of maximum plasma concentration (p < 0.01), area under the plasma concentration–time curve from 0 to 12 h post-dose (p < 0.01), half-life (p < 0.01), and clearance (p < 0.01) using one-way analysis of variance. However, post-hoc analyses were not performed to identify which genotypes differed significantly for these parameters. There was no difference between genotype groups for the parameter of time to maximum plasma concentration (p > 0.05). The reported values for each genotype are presented in Table 1. Moreover, a significant association between the MDR1 C3435T genotype and phenytoin response was reported. Non-responders (i.e., phenytoin-resistant patients) had a higher frequency of the T allele (χ 2 = 9.76, p = 0.0017) and, accordingly, the CT or TT genotypes (χ 2 = 4.37, p = 0.036). The odds ratio (OR) calculated from the identified frequencies was significant for the allelic frequency [OR 3.14; 95% confidence interval (CI) 1.42–7.05], but not for the genotypic (OR 3.69; 95% CI 0.90–5.81).
Ebid et al. [46] did not identify a significant association between drug resistance or responsiveness and phenytoin concentrations overall, but reported associations between the MDR1 CC genotype and the likelihood of drug-resistant patients having serum phenytoin trough concentrations below or in the lower half of therapeutic range at the 3- and 6-month evaluations. Whether the MDR1 C3435T genotype influences the efficacy and toxicity of phenytoin in epilepsy management was also explored. Overall, patients who were drug resistant (i.e., had seizure recurrence in the preceding 3 months) were more likely to have the MDR1 CC genotype than the MDR1 TT genotype when compared with patients responsive to therapy (55.5 vs. 13.5%, χ 2 = 24.99; p < 0.0001). Moreover, as would be expected, within the drug-resistant group, seizures occurred at higher frequency in patients with serum phenytoin trough <10 mg/L, with average seizure frequencies of 4.4 and 3.7 at 3 and 6 months, respectively (p < 0.0001).
3.5 Genotypic and Allelic Frequencies Within the MENA Region
Variant MAF of CYP2C9, CYP2C19, and MDR1 (ABCB1) C3435T have been investigated in a variety of settings and study populations across the MENA region. Studies from 11 different countries were identified for both CYP2C9 and CYP2C19, and seven countries for MDR1 (ABCB1) C3435T; study populations and computed genotypic and allelic frequencies for each polymorphism are summarized in Tables 3, 4, and 5, respectively. Overall, there was appreciable variability in MAF both between and within countries of the MENA region for each of CYP2C9, CYP2C19, and MDR1 (ABCB1) C3435T. For example, in studies assessing subjects of Turkish origin, the reported CYP2C19*3 MAF ranged from 0 to 10.3% [43, 44, 87,88,89].
4 Discussion
The purpose of this systematic review was to evaluate the role of genetic polymorphisms on phenytoin pharmacokinetics and clinical outcomes specifically in populations originating from the MENA region, and to characterize assessed polymorphism variant MAF within the region. Recognizing and understanding factors that contribute to interpatient variability in pharmacokinetics, pharmacodynamics, and associated clinical outcomes is essential to providing patient-individualized pharmacotherapy and disease management. In the management of epilepsy and seizure disorders, the significant role of genetic polymorphisms as one such factor has received increasing recognition, with phenytoin being an antiepileptic drug of particular interest, owing to its unique pharmacokinetic and pharmacodynamic profile [5, 6, 17,18,19,20,21,22,23,24,25,26, 35, 38,39,40,41,42,43,44,45,46, 127, 133, 134]
4.1 CYP2C9
Findings of the studies included in this review indicate that the reduced functional capacity of CYP2C9 enzymes with CYP2C9*2 and CYP2C9*3 variant alleles results in significantly higher serum phenytoin concentrations [42,43,44]. This difference was significant after only a single dose of phenytoin in healthy volunteers included in the studies by Aynacioglu et al. [42] and Kerb et al. [43]. Although the absolute difference between the wild-type genotype and those with one or two reduced function alleles was only in the range of 1.32–2.42 mg/L, owing to the reduced functional capacity of enzymes with variant alleles, it is reasonable to surmise that the absolute difference in phenytoin concentrations would likely be of clinical significance with repeat dosing [5]. This notion is supported by an in-vitro study of CYP2C9 polymorphisms, wherein *2 and *3 allelic variants decreased maximal velocity by 29 and 79%, respectively, and while the *2 variant did not affect the Michaelis constant, the *3 variant increased it fourfold [126].
Ozkaynakci et al. [44] reported a statistically significant difference between the wild-type and CYP2C9*1/*3, CYP2C19*2/*3 genotype groups. However, five of the 11 reported genotype groups had frequencies of only 0.98–2.94%, which likely limited the power of the study to detect differences between the wild-type and other genotype groups. Moreover, patients were required to have been taking phenytoin for only 7 days. Therefore, it is possible that some patients were not yet at steady state when phenytoin trough concentrations were drawn, and that absolute differences in concentrations would be statistically and/or clinically significant over time [5]. This is especially noteworthy in those with CYP2C9*2 or *3 alleles, in whom reduced functional capacity may significantly prolong the time necessary to achieve steady-state concentrations [5, 46]. As well, patients were not on a fixed dose of phenytoin; therefore, knowing the mean dose within each genotype group or having a serum concentration adjusted for mean dose in each genotype group would allow for assessment of this as a potential confounder. Although the impact of polytherapy was explored, the study was underpowered to appropriately assess this outcome. Thus, these results do not provide a clear demonstration of the impact of CYP2C9*2 and *3 allelic variants on phenytoin pharmacokinetics.
The impact of CYP2C9 variants have been more clearly demonstrated in studies conducted in non-MENA populations. In a cohort of Japanese pediatric patients, when controlling for CYP2C19 polymorphisms, patients with the CYP2C9*1/*3 genotype required phenytoin maintenance doses that were 37.3% lower than those with the wild-type genotype to achieve target concentrations of 15–20 mg/L [10]. Similarly, in a sample of Dutch patients, those with at least one CYP2C9*2 or *3 allele required phenytoin maintenance doses that were 37% lower to achieve target concentrations of 10–20 mg/L [24]. A study of patients originating from India indicated that those with the CYP2C9*1/*3 genotype were more likely than the wild-type genotype to have serum phenytoin concentrations >20 mg/L (OR 4.8; 95% CI 1.89–12.17; p 0.001), despite receiving comparable maintenance doses of phenytoin (5.4 vs. 5.1 mg/kg/day; p = 0.16) [133]. In addition to the impact of CYP2C9 polymorphisms on phenytoin pharmacokinetics in non-MENA populations, it also had an effect on phenytoin safety outcomes. A case-control study of Thai patients demonstrated that, when on comparable maintenance phenytoin doses, patients with the CYP2C9*1/*3 genotype were more likely to develop phenytoin-induced severe cutaneous adverse reactions (i.e., Stevens–Johnson syndrome, toxic epidermal necrolysis, or drug reaction with eosinophilia and systemic symptoms) within 12 weeks of initiating therapy than those with the wild-type genotype (OR 14.52; 95% CI 1.18–∞; p 0.044) [134]. This association is supported by findings of a meta-analysis conducted in Asian populations, wherein the CYP2C9*3 allele was associated with an increased risk of phenytoin-induced severe cutaneous reactions (OR 11.0; 95% CI 6.2–18.0; p < 0.00001) [17]. Furthermore, in a case-control study of Tamilian patients, when adjusting for phenytoin maintenance dose, patients with the CYP2C9*1/*3 genotype were significantly more likely to experience phenytoin-induced neurological toxicity (e.g., ataxia, slurred speech, lethargy, nystagmus) when compared with wild-type genotype (adjusted OR 15.3; 95% CI 5.8–40.3; p 0.0001) [22].
Ultimately, the significant impact of CYP2C9*2 and *3 allelic variants on phenytoin pharmacokinetics and risk of adverse effects when standard doses are administered has led to development of genotype-specific recommendations. It is suggested that initial doses of phenytoin be reduced by 25% in patients with one variant allele (i.e., CYP2C9*1/*2 or *1/*3) and by 50% in patients with two variant alleles (i.e., CYP2C9*2/*2, *2/*3 or *3/*3) [8].
4.2 CYP2C19
Results of this review suggest that CYP1C19*2 and *3 allelic variants, on their own, did not significantly impact the pharmacokinetics of phenytoin. In the single-dose study by Kerb et al. [43], using the same rationale as discussed with CYP2C9, although CYP2C19*1/*2 and *2/*2 genotypes were not associated with significantly different phenytoin serum concentrations, owing to the non-functional nature of *2 alleles, it is possible that statistically and clinically differences would arise after repeat dosing [5].
For results regarding CYP2C19 reported by Ozkaynakci et al. [44], previously discussed limitations affecting interpretation of results regarding CYP2C9 also apply. Both studies included in this review were likely underpowered to assess the impact of CYP2C19 polymorphisms specifically. Moreover, because CYP2C19 accounts for only 10–30% of phenytoin metabolism [122, 127], decreased or loss of functional capacity may be less likely to significantly affect phenytoin serum concentrations when compared with CYP2C9 allelic variants with reduced function.
Evidence from studies conducted in non-MENA populations suggest that CYP2C19 allelic variants may significantly impact pharmacokinetics of phenytoin. In the previously described Japanese cohort, those with CYP2C9 wild-type genotype and one variant CYP2C19 allele (i.e., CYP2C19*1/*2 or *1/*3) required phenytoin doses 13.1% lower than patients with wild-type genotypes of both CYP2C9 and CYP2C19 to achieve target concentrations (p < 0.01), while those with two variant CYP2C19 alleles (i.e., CYP2C19*2/*2, *2/*3, or *3/*3) required doses 28.1% lower (p < 0.001) [10]. Regarding clinical outcomes, in the study conducted in Tamilian patients, when adjusting for maintenance dose, patients with CYP2C19*2/*2 genotype were significantly more likely to experience phenytoin-induced neurological toxicity when compared with wild-type genotype (adjusted OR 3.0; 95% CI 1.3–7.0; p 0.01) [22]. Evidence from studies assessing both CYP2C9 and CYP2C19 genotypes suggest additive impact on phenytoin concentrations when variant genotypes of both enzymes are present [20, 22]. Although official guidelines have not been developed for dosage adjustments in patients with CYP2C19 allelic variants, recommendations for both pediatric and adult patients have been suggested based on the combination of both CYP2C9 and CYP2C19 genotypes [10, 20].
4.3 MDR1 (ABCB1) C3435T
The reported impact of the MDR1 C3435T genotype on phenytoin pharmacokinetics in this review was inconsistent, and results in all three studies [43, 45, 46] may have been confounded because of aforementioned factors such as concomitant medications, comorbid conditions, and serum albumin levels, among other patient-specific factors.
Kerb et al. [43] reported no significant association between MDR1 genotype and serum phenytoin concentrations overall. Although post-hoc analysis identified a significant association between the MDR1 CC genotype and serum phenytoin concentrations in the 25th percentile, the association is likely of limited clinical significance because of the narrow concentration range defined by the three classes of low, medium, and high. Unlike CYP2C9 and CYP2C19, transporters encoded by MDR1 are not involved in phenytoin metabolism; therefore, it is unclear whether repeat dosing over time would significantly impact the observed degree of difference in phenytoin serum concentrations between genotype groups. Moreover, although presence of MDR1 T alleles accounted for 1.3% of variability in phenytoin serum concentrations, the clinical relevance of this is questionable, especially considering the final regression equation could account for only 15.4% of observed variability [43].
In contrast, results reported by Alhazzani et al. [45] indicate all pharmacokinetic parameters, with the exception of time to maximum plasma concentration, were significantly affected by the MDR1 C3435T genotype. In the absence of post-hoc analyses of the one-way analysis of variance, which genotypes significantly differed for these parameters cannot be confirmed; however, the results trend toward the MDR1 CC genotype group being different. Of note, the classification of extensive, intermediate, and poor metabolizer to describe the MDR1 C3435T genotype is misleading, as proteins encoded by MDR1 are involved in substrate absorption and distribution, not metabolism [129,130,131]. The trend of increased maximum plasma concentration, area under the plasma concentration–time curve from 0 to 12 h post-dose, half-life, and clearance in subjects with one or more T alleles holds face validity, as this variant is associated with lower levels of transporter expression [130], which, at the level of the intestine, would result in less phenytoin being effluxed from systemic circulation back into the intestinal lumen. The distributions of responders and non-responders across the extensive, intermediate, and poor metabolizer categories were not reported; therefore, associations between the MDR1 C3435T genotype, phenytoin pharmacokinetics, and resultant clinical outcomes could not be assessed.
The clinical significance of the association between serum phenytoin level and MDR1 C3435T genotype reported by Ebid et al. [46] is unclear. Specifically, at the 3-month evaluation, there was no significant association between phenytoin responsiveness and serum concentration overall, while at the 6-month evaluation, a ‘higher’ concentration in the drug-responsive group was not defined and whether or not weight adjusting the doses mitigated reported differences was not explored.
Reported associations between the MDR1 C3435T genotype and drug resistance reported in this review are conflicting [45, 46]. Historically, associations between drug-resistant epilepsy and overexpression of transporters encoded by MDR1 have been reported [135,136,137]. Despite this, several limitations must be considered when interpreting the clinical outcomes reported by Ebid et al. [46]. Foremost, distribution of seizure type within the drug-responsive and drug-resistant groups was not reported, nor adjusted for in the results. This is notable, as there is evidence to suggest phenytoin may aggravate generalized seizures [3]. Additional potential confounders, such as baseline seizure control and concomitant antiepileptic drugs, were also not adjusted for. It is unclear if appropriate corrections were made for multiple statistical analyses; thus, it is possible that identified associations were the result of chance.
The association of MDR1 C3435T genetic polymorphisms and drug-resistant epilepsy in general (as opposed to phenytoin specifically) was assessed within Iranian patients [33]. The MDR1 CC wild-type genotype was significantly more frequent in drug-resistant patients, and associations were also made with adult age and female sex; however, the latter two associations may have resulted from performing multiple statistical analyses. The investigators attempted to address confounders such as baseline etiology and seizure type through univariate analyses; however, because baseline characteristics were so different between study groups, residual confounding likely remained [33]. Two studies conducted in Turkish patients with seizure disorders did not find a significant association between the MDR1 C3435T genotype and drug resistance [29, 110]. More recently, a meta-analysis of studies conducted in Asian, Caucasian (under which populations originating from the MENA region were classified), and Indian populations investigated the role of MDR1 gene haplotypes in antiepileptic drug response [132]. Initial results indicated that the presence of the T variant of MDR1 C3435T may be associated with lower rates of drug resistance. Once corrected for multiple statistical analyses, there was no significant association overall, but a significant association remained in Caucasian patients (T vs. C allele, OR 0.83; 95% CI 0.71–0.96; p = 0.01) [132]. One hypothesis regarding inconsistency in positive associations found between MDR1 genotypes and responsiveness to antiepileptic drugs is that the gene falls within a significant block of high linkage disequilibrium [138]. Therefore, it is possible that the associations identified are not the result of the MDR1 polymorphism itself, but to an alternative gene within the linkage disequilibrium block [138].
4.4 Genotypic and Allelic Frequencies Within the MENA Region
Variability in genotypic and allelic frequencies of CYP2C9, CYP2C19, and MDR1 C3435T both between and within the MENA countries for which data were identified was not unexpected. Key factors contributing to this variability were likely small sample sizes and selective patient populations in whom many of the studies were conducted. The allelic frequencies of CYP2C9*2 and *3, as well as CYP2C19*2 and *3 were comparable to those reported for Middle Eastern populations [139, 140]. Similar population data for the MDR1 C3435T polymorphism were not available for comparison, nor were comparative data specific to North Africa populations.
Ethnicity significantly affects distributions of variants [124] and contributes to differences in MAF reported for populations outside of the MENA region for each of CYP2C9 [48, 50, 53, 57, 65, 67, 70, 139], CYP2C19 [27, 53, 57, 67, 140], and MDR1 C3435T [132, 141, 142]. This emphasizes the importance of population-specific studies assessing genetic polymorphisms.
4.5 Limitations
This review has several limitations that must be considered when interpreting the results. Foremost, in the primary analysis, only three different countries within the MENA region were represented by study populations, with three of five studies being in subjects of Turkish origin. The genotypic diversity across MENA countries as well as the small sample size of the included studies, therefore limit the generalizability of this review. Moreover, only three of the five studies were conducted in patients with seizure disorder, of which only one assessed clinical outcomes, thereby limiting ability to make clinical inferences. The two healthy volunteer studies (which used data from the same subjects) were based on a single dose of phenytoin; therefore, they did not reflect the potential impact of genetic polymorphisms on the steady-state pharmacokinetics of phenytoin or the influence of the non-linear capacity-limited metabolism of phenytoin. Furthermore, all five studies had poor methodological quality and were likely not powered to compare differences between genotypes and alleles of relatively low frequencies. Regarding the review of genotypic and allelic frequencies, the small select study populations and resultant variability in frequency estimates limit generalizability.
4.6 Future Directions
Identified limitations of this review highlight the need for future studies to investigate the impact of genetic polymorphisms on the pharmacokinetics of phenytoin and associated clinical outcomes, specifically in patients with epilepsy and seizure disorders. Although the impacts of CYP2C9*2 and *3 allelic variants on phenytoin metabolism and the risk of adverse effects have been recognized and dosage modification guidelines developed [8, 9], the potential impacts of CYP2C19*2 and *3 allelic variants have not been established and warrant further investigation. Moreover, owing to the limitations of individual studies, the association between the MDR1 C3435T genotype and drug-resistant epilepsy as well as phenytoin pharmacokinetics remains to be elucidated. To address these questions, future studies must be appropriately powered to assess differences between genotypes of low frequencies, and account for known confounding factors. Furthermore, studies should be designed to look at both pharmacokinetic and clinical outcomes once phenytoin concentrations have achieved a steady state. If possible, studies should be conducted in patient populations originating from countries of the MENA region that have not yet been assessed.
Future research may also focus on investigating the prevalence of the HLA-B*15:02 allele and its impact on the risk of phenytoin-induced severe cutaneous adverse reactions (i.e., Stevens–Johnson syndrome, toxic epidermal necrolysis, or drug reaction with eosinophilia and systemic symptoms) in populations originating within the MENA region. The presence of this allele has been associated with an increased risk of phenytoin-induced severe cutaneous adverse reactions [38, 143,144,145], resulting in pharmacogenetic guidelines recommending against the use of phenytoin in patients who are HLA-B*15:02 carriers [8].
5 Conclusion
Results of this review indicate the reduced functional capacities of CYP2C9*2 and *3 allelic variants significantly decrease phenytoin metabolism in patients originating from the MENA region. The impacts of CYP2C19 and MDR1 C3435T polymorphisms on phenytoin pharmacokinetics and clinical outcomes in this population are unclear and require further investigation. Future research should focus on better understanding the impact of genetic polymorphisms on clinical outcomes associated with phenytoin therapy.
References
Merritt HH, Putnam TJ. Sodium diphenyl hydantoinate in the treatment of convulsive disorders. JAMA. 1939;11:1068–73.
Glauser T, Shinnar S, Gloss D, Alldredge B, Arya R, Bainbridge J, et al. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: report of the guideline committee of the American Epilepsy Society. Epilepsy Curr. 2016;16:48–61.
Glauser T, Ben-Menachem E, Bourgeois B, Cnaan A, Guerreiro C, Kälviäinen R, et al. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2013;54:551–63.
Porter RJ, Meldrum BS. Antiseizure drugs. In: Katzung BG, Masters SB, Trevor AJ, editors. Basic and clinical pharmacology. 12th ed. London: The McGraw-Hill Companies, Inc.; 2012.
Winter ME, Tozer TN. Phenytoin. In: Burton ME, Shaw LM, Schentag JJ, Evans WE, editors. Applied pharmacokinetics and pharmacodynamics: principles of therapeutic drug monitoring. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2006.
Koch-Weser J. The serum level approach to individualization of drug dosage. Eur J Clin Pharmacol. 1975;9:1–8.
Thorn CF, Whirl-Carrillo M, Leeder JS, Klein TE, Altman RB. PharmGKB summary: phenytoin pathway. Pharmacogenetics Genom. 2012. https://www.pharmgkb.org/. Accessed 22 Feb 2017.
Caudle KE, Rettie AE, Whirl-carrillo M, Smith LH, Mintzer SE, Ta M, et al. Clinical pharmacogenetics implementation consortium guidelines for CYP2C9 and HLA-B genotype and phenytoin dosing. Clin Pharmacol Ther. 2014;96:542–8.
Swen JJ, Nijenhuis M, de Boer A, Grandia L, Maitland-van der Zee AH, Mulder H, et al. Pharmacogenetics: from bench to byte: an update of guidelines. Clin Pharmacol Ther. 2011;89:662–73.
Yamamoto Y, Takahashi Y, Imai K, Miyakawa K, Ikeda H, Ueda Y, et al. Individualized phenytoin therapy for Japanese pediatric patients with epilepsy based on CYP2C9 and CYP2C19 genotypes. Ther Drug Monit. 2015;37:229–35.
Public Health Genomics Knowledge Base. Phenytoin. 2016. https://phgkb.cdc.gov/. Accessed 25 Feb 2017.
PharmGKB. Phenytoin. 2017. https://www.pharmgkb.org/. Accessed 25 Feb 2017.
Henn BM, Botigué LR, Gravel S, Wang W, Brisbin A, Byrnes JK, et al. Genomic ancestry of North Africans supports back-to-Africa migrations. PLoS Genet. 2012;8:e1002397.
Yang Y, Peter I, Scott SA. Pharmacogenetics in Jewish populations. Drug Metabol Drug Interact. 2014;29:221–33.
Yang X, Al-Bustan S, Feng Q, Guo W, Ma Z, Marafie M, et al. The influence of admixture and consanguinity on population genetic diversity in Middle East. J Hum Genet. 2014;59:615–22.
NIH National Heart, Lung, and Blood Institute. Quality assessment tool for observational cohort and cross-sectional studies. 2014. https://www.nhlbi.nih.gov/. Accessed 1 Feb 2017.
Chung WH, Chang WC, Lee YS, Wu YY, Yang CH, Ho HC, et al. Genetic variants associated with phenytoin-related severe cutaneous adverse reactions. JAMA. 2014;312:525–34.
Depondt C, Godard P, Espel RS, da Cruz AL, Lienard P, Pandolfo M. A candidate gene study of antiepileptic drug tolerability and efficacy identifies an association of CYP2C9 variants with phenytoin toxicity. Eur J Neurol. 2011;18:1159–64.
Huang Y, Yang JF, Qi XL, Wang YQ, Wang WZ, Chen B. Association between genetic polymorphisms of CYP2C19 and CYP2C9 and phenytoin serum concentration. Zhongua Yi Xue Za Zhi. 2004;84:1686–9.
Hung CC, Lin CJ, Chen CC, Chang CJ, Liou HH. Dosage recommendation of phenytoin for patients with epilepsy with different CYP2C9/CYP2C19 polymorphisms. Ther Drug Monit. 2004;26:534–40.
Kousar S, Wafai ZA, Wani MA, Jan TR, Andrabi KI. Clinical relevance of genetic polymorphism in the CYP2C9 gene to pharmacodynamics and pharmacokinetics of phenytoin in epileptic patients: validatory pharmacogenomic approach to pharmacovigilance. Int J Clin Pharmacol Ther. 2015;53:504–16.
Kesavan R, Narayan SK, Adithan C. Influence of CYP2C9 and CYP2C19 genetic polymorphisms on phenytoin-induced neurological toxicity in Indian epileptic patients. Eur J Clin Pharmacol. 2010;66:689–96.
Sun F, Cao BQ, Wang B, Wu SQ, Jiang DH. Association between ABCB1 genetic polymorphism and the effect on epilepsy following phenytoin treatment. Exp Ther Med. 2016;12:1780–4.
van der Weide J, Steijns LS, van Weelden MJ, de Haan K. The effect of genetic polymorphism of cytochrome P450 CYP2C9 on phenytoin dose requirement. Pharmacogenetics. 2001;11:287–91.
Ponnala S, Chaudhari JR, Jaleel MA, Bhiladvala D, Kaipa PR, Das UN. Role of MDR1 C3435T and GABRG2 C588T gene polymorphisms in seizure occurrence and MDR1 effect on anti-epileptic drug (phenytoin) absorption. Genet Test Mol Biomark. 2012;16:550–7.
Thaker SJ, Gandhe PP, Godbole CJ, Bendkhale SR, Mali NB, Thatte UM, et al. A prospective study to assess the association between genotype, phenotype and Prakriti in individuals on phenytoin monotherapy. J Ayurveda Integr Med. 2017;8:37–41.
Tabari RG, Naseri F, Marjani A. Genetic polymorphism of CYPP450 (2C19) enzyme in Iranian Baluch ethnic group. Open Biochem J. 2015;9:37–41.
Seven M, Batar B, Unal S, Yesil G, Yuksel A, Guven M. The effect of genetic polymorphisms of cytochrome P450 CYP2C9, CYP2C19, and CYP2D6 on drug-resistant epilepsy in Turkish children. Mol Diagn Ther. 2014;18:229–36.
Seven M, Batar B, Unal S, Yesil G, Yuksel A, Guven M. The drug-transporter gene MDR1 C3435T and G2677T/A polymorphisms and the risk of multidrug-resistant epilepsy in Turkish children. Mol Biol Rep. 2014;41:331–6.
Britzi M, Bialer M, Arcavi L, Shachbari A, Kapitulnik T, Soback S. Genetic polymorphism of CYP2D6 and CYP2C19 metabolism determined by phenotyping Israeli ethnic groups. Ther Drug Monit. 2000;22:510–6.
Sviri S, Shpizen S, Leitersdorf E, Levy M, Caraco Y. Phenotypic-genotypic analysis of CYP2C19 in the Jewish Israeli population. Clin Pharmacol Ther. 1999;65:275–82.
Maleki M, Sayyah M, Kamgarpour F, Karimipoor M, Arab A, Rajabi A, et al. Association between ABCB1-T1236C polymorphism and drug-resistant epilepsy in Iranian female patients. Iran Biomed J. 2010;14:89–96.
Sayyah M, Kamgarpour F, Maleki M, Karimipoor M, Gharagozli K, Shamshiri AR. Association analysis of intractable epilepsy with C3435T and G2677T/A ABCB1 gene polymorphisms in Iranian patients. Epileptic Disord. 2011;13:155–65.
Hennessy S, Leonard CE, Freeman CP, Metlay JP, Chu X, Strom BL, Bilker WB. CYP2C9, CYP2C19 and ABCB1 genotype and hospitalization for phenytoin toxicity. Clin Lymphoma. 2010;9:19–22.
Lai ML, Tien YE, Huang YS, Huang JD. Studies on pharmacokinetic mechanism of phenytoin resistance in refractory epilepsy. J Pharm Sci. 2013;102:3189–95.
Shaheen U, Prasad DKV, Sharma V, Suryaprabha T, Ahuja YR, Jyothy A, et al. Significance of MDR1 gene polymorphism C3435T in predicting drug response in epilepsy. Epilepsy Res. 2014;108:251–6.
Grover S, Gourie-Devi M, Baghel R, Sharma S, Bala K, Gupta M, et al. Genetic profile of patients with epilepsy on first-line antiepileptic drugs and potential directions for personalized treatment. Pharmacogenomics. 2010;11:927–41.
Yampayon K, Sukasem C, Limwongse C, Chinvarun Y, Tempark T, Rerkpattanapipat T, et al. Influence of genetic and non-genetic factors on phenytoin-induced severe cutaneous adverse drug reactions. Eur J Clin Pharmacol. 2017;73:855–65.
Salehifar E, Zohrabi M, Eshghi S, Saeedi M, Ebrahimi P. Different pharmacokinetic parameters of phenytoin in Iranian outpatients: need to optimize the current dosage regimen. IJPR. 2009;8:37–45.
Alhazzani AA, Munisamy M, Karunakaran G. Pharmacogenetics of ATP binding cassette transporter—MDR1 gene polymorphism (MDR1—1236C>T polymorphism) and response to antiepileptic drug phenytoin pharmacokinetics in epilepsy. Neurology. 2016;86(Suppl. 16):P2.046.
Alhazzani A, Al-Gahtany M, Munisamy M, Karunakaran G. Pharmacogenetics of ATP binding cassette transporter-MDR1 gene polymorphism (C3435T) and response to antiepileptic drug phenytoin pharmacokinetics in epilepsy. J Neurol Sci. 2015;357:e142–60.
Aynacioglu AS, Brockmöller J, Bauer S, Sachse C, Guzelbey P, Ongen Z, et al. Frequency of cytochrome P450 CYP2C9 variants in a Turkish population and functional relevance for phenytoin. Br J Clin Pharmacol. 1999;48:409–15.
Kerb R, Aynacioglu AS, Brockmöller J, Schlagenhaufer R, Bauer S, Szekeres T, et al. The predictive value of MDR1, CYP2C9, and CYP2C19 polymorphisms for phenytoin plasma levels. Pharmacogenom J. 2001;1:204–10.
Ozkaynakci A, Gulcebi MI, Ergeç D, Ulucan K, Uzan M, Ozkara C, et al. The effect of polymorphic metabolism enzymes on serum phenytoin level. Neurol Sci. 2015;36:397–401.
Alhazzani AA, Munisamy M, Karunakaran G. MDR1 gene polymorphism and phenytoin pharmacokinetics in epilepsy. Bahrain Med Bull. 2017;39:29–32.
Ebid AH, Ahmed MM, Mohammed SA. Therapeutic drug monitoring and clinical outcomes in epileptic Egyptian patients: a gene polymorphism perspective study. Ther Drug Monit. 2007;29:305–12.
Hamdy SI, Hiratsuka M, Narahara K, El-Enany M, Moursi N, Ahmed MS, et al. Allele and genotype frequencies of polymorphic cytochromes P450 (CYP2C9, CYP2C19, and CYP2E1) and dihydropyrimidine dehydrogenase (DPYD) in the Egyptian population. Br J Clin Pharmacol. 2002;52:596–603.
El Din MS, Amin DG, Ragab SB, Ashour EE, Mohamed MH, Mohamed AM. Frequency of VKORC1 (C1173T) and CYP2C9 genetic polymorphisms in Egyptians and their influence on warfarin maintenance dose: proposal for a new dosing regimen. Int J Lab Hematol. 2012;34:517–24.
Azzam H, Elwakeel H, Awad I, El-Farahaty R, El-Gilany AH, El-Sharawy S. VKORC1 and CYP2C9 genotypes in Egyptian patients with warfarin resistance. Blood Coagul Fibrinolysis. 2016;27:121–6.
Bazan NS, Sabry NA, Rizk A, Mokhtar S, Badary OA. Factors affecting warfarin dose requirements and quality of anticoagulation in adult Egyptian patients: role of gene polymorphism. Ir J Med Sci. 2014;183:161–72.
Ekladious SM, Issac MS, Sharaf SA, Abou-Youssef HS. Validation of a proposed warfarin dosing algorithm based on the genetic make-up of Egyptian patients. Mol Diagn Ther. 2013;17:381–90.
Shahin MH, Khalifa SI, Gong Y, Hammad LN, Sallam MT, Shafey ME, et al. Genetic and nongenetic factors associated with warfarin dose requirements in Egyptian patients. Pharmacogenet Genom. 2011;21:130–5.
Zand N, Tajik N, Moghaddam AS, Milanian I. Genetic polymorphisms of cytochrome P450 enzymes 2C9 and 2C19 in a healthy Iranian population. Clin Exp Pharmacol Physiol. 2007;34:102–5.
Amini-Shirazi N, Ghahremani MH, Ahmadkhaniha R, Mandegary A, Dadgar A, Abdollahi M, et al. Influence of CYP2C9 polymorphism on metabolism of valproate and its hepatotoxin metabolite in Iranian patients. Toxicol Mech Methods. 2010;20(8):452–7.
Ghadam P, Sharifian R, Farsangi ZJ, Kianmehr Z, Lak M. CYP2C9 gene analysis of some Iranian hypersensitive patients to warfarin. Pak J Biol Sci. 2009;12(16):1160–3.
Namazi S, Azarpira N, Hendijani F, Khorshid MB, Vessal G, Mehdipour AR. The impact of genetic polymorphisms and patient characteristics on warfarin dose requirements: a cross-sectional study in Iran. Clin Ther. 2010;32(6):1050–60.
Yousef A-M, Bulatova NR, Newman W, Hakooz N, Ismail S, Qusa H, et al. Allele and genotype frequencies of the polymorphic cytochrome P450 genes (CYP1A1, CYP3A4, CYP3A5, CYP2C9 and CYP2C19) in the Jordanian population. Mol Biol Rep. 2012;39(10):9423–33.
Alrashid MH, Al-Serri A, Alshemmari SH, Koshi P, Al-Bustan SA. Association of genetic polymorphisms in the VKORC1 and CYP2C9 genes with warfarin dosage in a group of Kuwaiti individuals. Mol Diagn Ther. 2016;20(2):183–90.
Djaffar-Jureidini I, Chamseddine N, Keleshian S, Naoufal R, Zahed L, Hakime N. Pharmacogenomics of coumarin dosing: prevalence of CYP2C9 and VKORC1 polymorphisms in the Lebanese population. Genet Test Mol Biomark. 2011;15(11):827–30.
Esmerian MO, Mitri Z, Habbal MZ, Geryess E, Zaatari G, Alam S, et al. Influence of CYP2C9 and VKORC1 polymorphisms on warfarin and acenocoumarol in a sample of Lebanese people. J Clin Pharmacol. 2011;51(10):1418–28.
Smires FZ, Habbal R, Moreau C, Assaidi A, Loriot MA, Nadifi S. Effect of different genetics variants: CYP2C9*2, CYP2C9*3 of cytochrome P450 CYP2C9 and 1639G>A of the VKORC1 gene; on acenocoumarol requirement in Moroccan patients. Pathol Biol. 2013;61(3):88–92.
Pathare A, Khabori MA, Alkindi S, Zadjali SA, Misquith R, Khan H, et al. Warfarin pharmacogenetics: development of a dosing algorithm for Omani patients. J Hum Genet. 2012;57(10):665–9.
Tanira MO, Al-Mukhaini MK, Al-Hinai AT, Al Balushi KA, Ahmed IS. Frequency of CYP2C9 genotypes among Omani patients receiving warfarin and its correlation with warfarin dose. Community Genet. 2006;10(1):32–7.
Alzahrani AM, Ragia G, Hanieh H, Manolopoulos VG. Genotyping of CYP2C9 and VKORC1 in the Arabic population of Al-Ahsa, Saudi Arabia. Biomed Res Int. 2013;2013:3–8.
Mirghani RA, Chowdhary G, Elghazali G. Distribution of the major cytochrome P450 (CYP) 2C9 genetic variants in a Saudi Population. Basic Clin Pharmacol Toxicol. 2011;109(2):111–4.
Shrif NE, Won HH, Lee ST, Park JH, Kim KK, Kim MJ, et al. Evaluation of the effects of VKORC1 polymorphisms and haplotypes, CYP2C9 genotypes, and clinical factors on warfarin response in Sudanese patients. Eur J Clin Pharmacol. 2011;67(11):1119–30.
Abdelhedi R, Bouayed NA, Alfadhli S, Abid L, Rebai A, Kharrat N. Characterization of drug-metabolizing enzymes CYP2C9, CYP2C19 polymorphisms in Tunisian, Kuwaiti and Bahraini populations. J Genet. 2015;94(4):765–70.
Yilmaz N, Erbağci AB, Aynacioğlu AŞ. Cytochrome P4502C9 genotype in Southeast Anatolia and possible relation with some serum tumour markers and cytokines. Acta Biochim Pol. 2001;48(3):775–82.
Babaoglu MO, Yasar U, Sandberg M, Eliasson E, Dahl ML, Kayaalp SO, et al. CYP2C9 genetic variants and losartan oxidation in a Turkish population. Eur J Clin Pharmacol. 2004;60(5):337–42.
Ozgon GO, Langaee TY, Feng H, Buyru N, Ulutin T, Hatemi AC, et al. VKORC1 and CYP2C9 polymorphisms are associated with warfarin dose requirements in Turkish patients. Eur J Clin Pharmacol. 2008;64(9):889–94.
Ozer N, Cam N, Tangurek B, Ozer S, Uyarel H, Oz D, et al. The impact of CYP2C9 and VKORC1 genetic polymorphism and patient characteristics upon warfarin dose requirements in an adult Turkish population. Heart Vessels. 2010;25(2):155–62.
Silan C, Dogan OT, Silan F, Kukulguven FM, Asgun HF, Ozdemir S, et al. The prevalence of VKORC1 1639 G>A and CYP2C9*2*3 genotypes in patients that requiring anticoagulant therapy in Turkish population. Mol Biol Rep. 2012;39:11017–22.
Ellison CA, Abou El-Ella SS, Tawfik M, Lein PJ, Olson JR. Allele and genotype frequencies of CYP2B6 and CYP2C19 polymorphisms in Egyptian agricultural workers. J Toxicol Environ Health A. 2012;75(4):232–41.
Sameer AE, Amany GM, Abdela AA, Fadel SA. CYP2C19 genotypes in a population of healthy volunteers and in children with hematological malignancies in Gaza Strip. Can J Clin Pharmacol. 2009;16(1):156–62.
Payan M, Rouini MR, Tajik N, Ghahremani MH, Tahvilian R. Hydroxylation index of omeprazole in relation to CYP2C19 polymorphism and sex in a healthy Iranian population. Daru. 2014;22:81.
Zendehdel N, Biramijamal F, Hossein-Nezhad A, Zendehdel N, Sarie H, Doughaiemoghaddam M, et al. Role of cytochrome P450 2C19 genetic polymorphisms in the therapeutic efficacy of omeprazole in iranian patients with erosive reflux esophagitis. Arch Iran Med. 2010;13(5):406–12.
Namazi S, Kojuri J, Khalili A, Azarpira N. The impact of genetic polymorphisms of P2Y12, CYP3A5 and CYP2C19 on clopidogrel response variability in Iranian patients. Biochem Pharmacol. 2012;83:903–8.
Akhlaghi A, Shirani S, Ziaie N, Pirhaji O, Yaran M, Shahverdi G, et al. Cytochrome P450 2C19 polymorphism in Iranian patients with coronary artery disease. ARYA Atheroscler. 2011;7(3):106–10.
Nozari Y, Vosooghi S, Boroumand M, Poorhosseini H, Nematipour E, Salarifar M, et al. The impact of cytochrome P450 2C19 polymorphism on the occurrence of one-year in-stent restenosis in patients who underwent percutaneous coronary intervention: a case-match study. Anatol J Cardiol. 2015;15:348–53.
Sviri S, Shpizen S, Leitersdorf E, Levy M, Caraco Y. Phenotypic-genotypic analysis of CYP2C19 in the Jewish Israeli population. Clin Pharmacol Ther. 1999;65(3):275–82.
Luo HR, Aloumanis V, Lin KM, Gurwitz D, Wan YJ. Polymorphisms of CYP2C19 and CYP2D6 in Israeli ethnic groups. Am J Pharmacogenom. 2004;4(6):395–401.
Zalloum I, Hakooz N, Arafat T. Genetic polymorphism of CYP2C19 in a Jordanian population: influence of allele frequencies of CYP2C19*1 and CYP2C19*2 on the pharmacokinetic profile of lansoprazole. Mol Biol Rep. 2012;39(4):4195–200.
Jureidini ID, Chamseddine N, Keleshian S, Naoufal R, Zahed L, Hakime N. Prevalence of CYP2C19 polymorphisms in the Lebanese population. Mol Biol Rep. 2011;38(8):5449–52.
Saeed LH, Mayet AY. Genotype-phenotype analysis of CYP2C19 in healthy Saudi individuals and its potential clinical implication in drug therapy. Int J Med Sci. 2013;10(11):1497–502.
Goldstein JA, Ishizaki T, Chiba K, de Morais SM, Bell D, Krahn PM, et al. Frequencies of defective CYP2C19 alleles responsible for the mephenytoin poor metabolizer phenotype in various Oriental, Caucasian, Saudi Arabian and American black populations. Pharmacogenetics. 1997;7:59–64.
Khalaf H, Al Meman AR, Rasool S. Impact of cytochrome P450 2C19*2 and *3 on clopidogrel loading dose in Saudi patients with acute coronary syndrome. Drug Metab Lett. 2016;10:65–70.
Aynacioglu AS, Sachse C, Bozkurt A, Kortunay S, Nacak M, Schroder T, et al. Low frequency of defective alleles of cytochrome P450 enzymes 2C19 and 2D6 in the Turkish population. Clin Pharmacol Ther. 1999;66(2):185–92.
Gumus E, Karaca O, Babaoglu MO, Baysoy G, Balamtekin N, Demir H, et al. Evaluation of lansoprazole as a probe for assessing cytochrome P450 2C19 activity and genotype-phenotype correlation in childhood. Eur J Clin Pharmacol. 2012;68(5):629–36.
Sen HM, Silan F, Silan C, Degirmenci Y, Ozisik Kamaran HI. Effects of CYP2C19 and P2Y12 gene polymorphisms on clinical results of patients using clopidogrel after acute ischemic cerebrovascular disease. Balkan J Med Genet. 2014;17(2):37–41.
Khalil BM, Shahin MH, Solayman MHF, Gong Y, Hammad LN, Al-Mesallamy HO, et al. Genetic and nongenetic factors affecting clopidogrel response in the Egyptian population. Clin Transl Sci. 2016;9(1):23–8.
Issac MS, El-Nahid MS, Wissa MY. Is there a role for MDR1, EPHX1 and Protein Z gene variants in modulation of warfarin dosage? A study on a cohort of the Egyptian population. Mol Diagn Ther. 2014;18(1):73–83.
Shabana MF, Mishriki AA, Issac MS, Bakhoum SW. Do MDR1 and SLCO1B1 polymorphisms influence the therapeutic response to atorvastatin? A study on a cohort of Egyptian patients with hypercholesterolemia. Mol Diagn Ther. 2013;17(5):299–309.
Ali MA, Elsalakawy WA. ABCB1 haplotypes but not individual SNPs predict for optimal response/failure in Egyptian patients with chronic-phase chronic myeloid leukemia receiving imatinib mesylate. Med Oncol. 2014;31(11):279.
El-Beblawy NM, Elbarbary NS, Kamal TM, Mahmoud PM. A study of human killer cell immunoglobulin-like receptor and multidrug resistance gene polymorphisms in children with immune thrombocytopenia. Clin Appl Thromb Hemost. 2016;22(5):429–40.
Saidijam M, Mahjub H, Shabab N, Yadegarazari R. Simultaneous analysis of multidrug resistance 1 (MDR1) C3435T, G2677T/A, and C1236T genotypes in Hamadan city population, West of Iran. Iran Biomed J. 2015;19(1):57–62.
Sayyah M, Kamgarpour F, Maleki M, Karimipoor M, Gharagozli K, Shamshiri AR. Association analysis of intractable epilepsy with C3435T and G2677T/A ABCB1 gene polymorphisms in Iranian patients. Epileptic Disord. 2011;13(2):155–65.
Bonyadi M, Gholizadeh M, Soltan-Ali M. MDR1 C3435T polymorphism associated with the development of clinical features in Behçet’s disease in Iranian Azeri Turkish patients. Int J Dermatol. 2014;53:1235–40.
Ghafouri H, Ghaderi B, Amini S, Nikkhoo B, Abdi M, Hoseini A. Association of ABCB1 and ABCG2 single nucleotide polymorphisms with clinical findings and response to chemotherapy treatments in Kurdish patients with breast cancer. Tumor Biol. 2016;37(6):7901–6.
Taheri M, Mahjoubi F, Omranipour R. Effect of MDR1 polymorphism on multidrug resistance expression in breast cancer patients. Genet Mol Res. 2010;9(1):34–40.
Tatari F, Salek R, Mosaffa F, Khedri A, Behravan J. Association of C3435T single-nucleotide polymorphism of MDR1 gene with breast cancer in an Iranian population. DNA Cell Biol. 2009;28(5):259–63.
Khabour OF, Alzoubi KH, Al-Azzam SI, Mhaidat NM. Frequency of MDR1 single nucleotide polymorphisms in a Jordanian population, including a novel variant. Genet Mol Res. 2013;12(1):801–8.
Alzoubi KH, Khabour OF, Al-Azzam SI, Mayyas F. The role of multidrug resistance-1 (MDR1) variants in response to fexofenadine among Jordanians. Int J Clin Pharmacol Ther. 2013;51(11):880–7.
Samara SA, Irshaid YM, Mustafa KN. Association of MDR1 C3435T and RFC1 G80A polymorphisms with methotrexate toxicity and response in Jordanian rheumatoid arthritis patients. Int J Clin Pharmacol Ther. 2014;52(9):746–55.
Abuhaliema AM, Yousef AM, El-Madany NN, Bulatova NR, Awwad NM, Yousef MA, et al. Influence of genotype and haplotype of MDR1 (C3435T, G2677A/T, C1236T) on the incidence of breast cancer: a case-control study in Jordan. Asian Pacific J Cancer Prev. 2016;17(1):261–6.
Zgheib NK, Akra-Ismail M, Aridi C, Mahfouz R, Abboud MR, Solh H, et al. Genetic polymorphisms in candidate genes predict increased toxicity with methotrexate therapy in Lebanese children with acute lymphoblastic leukemia. Pharmacogenet Genomics. 2014;24(8):387–96.
Kassogue Y, Dehbi H, Nassereddine S, Quachouh M, Nadifi S. Genotype variability and haplotype frequency of MDR1 (ABCB1) gene polymorphism in Morocco. DNA Cell Biol. 2013;32(10):582–8.
Al-Mohizea AM, Alkharfy KM, Bagulb KM, Alghamdi AM, Al-Jenoobi FI, Al-Muhsen S, et al. Genetic variability and haplotype profile of MDR1 in Saudi Arabian males. Mol Biol Rep. 2012;39:10294–301.
Alsaif AA, Hasan TN, Shafi G, Syed NA, Alsaif MA, Al-Assaf A, et al. Association of multiple drug resistance-1 gene polymorphism with multiple drug resistance in breast cancer patients from an ethnic Saudi Arabian population. Cancer Epidemiol. 2013;37(5):762–6.
Gümüş-Akay G, Rüstemoğlu A, Karadağ A, Sunguroğlu A. Haplotype-based analysis of MDR1/ABCB1 gene polymorphisms in a Turkish population. DNA Cell Biol. 2010;29(2):83–90.
Ozgon GO, Bebek N, Gul G, Cine N. Association of MDR1 (C3435T) polymorphism and resistance to carbamazepine in epileptic patients from Turkey. Eur Neurol. 2008;59(1–2):67–70.
Kesimci E, Engin AB, Kanbak O, Karahalil B. Association between ABCB1 gene polymorphisms and fentanyl’s adverse effects in Turkish patients undergoing spinal anesthesia. Gene. 2012;493(2):273–7.
Ayaz G, Batar B, Kanigur G, Guven M, Onaran I, Karadag B, et al. The association of MDR1 C3435T and G2677T/A polymorphisms with plasma platelet-activating factor levels and coronary artery disease risk in Turkish population. Gene. 2013;527(1):301–5.
Aydos SE, Karadağ A, Özkan T, Altınok B, Bunsuz M, Heidargholizadeh S, et al. Association of MDR1 C3435T and C1236T single nucleotide polymorphisms with male factor infertility. Genet Mol Res. 2015;14(2):6330–9.
Rustemoglu A, Gumus-Akay G, Yigit S, Tasliyurt T. Analysis of common MDR1 (ABCB1) gene C1236T and C3435T polymorphisms in Turkish patients with familial mediterranean fever. Genet Mol Res. 2011;10(4):3411–20.
Rustemoglu A, Gumus-Akay G, Karakus N, Yigit S, Sahin S, Tasliyurt T. Association analysis of three ABCB1 (MDR1) gene variants (C1236T, G2677A/T and C3435T) and their genotype/haplotype combinations with the familial Mediterranean fever. Xenobiotica. 2014;44(10):933–40.
Dogruer D, Tug E, Bes C, Soy M. Lack of an effect of CYP3A4 and MDR1 gene polymorphisms on colchicine pharmacogenetics in the treatment of familial Mediterranean fever. Genet Mol Res. 2013;12(3):3521–8.
Akbas SH, Bilgen T, Keser I, Tuncer M, Yucetin L, Tosun O, et al. The effect of MDR1 (ABCB1) polymorphism on the pharmacokinetic of tacrolimus in Turkish renal transplant recipients. Transplant Proc. 2006;38(5):1290–2.
Kazancioglu HO, Ak G, Turkmen A, Ozbek U, Tuncer FN, Karabulut A. The role of MDR1 C3435T gene polymorphism on gingival hyperplasia in Turkish renal transplant patients treated with cyclosporine in the absence of calcium channel blockers. Transplant Proc. 2013;45(6):2233–7.
Yağcioğlu AE, Yoca G, Ayhan Y, Karaca RÖ, Çevik L, Müderrisoğlu A, et al. Relation of the allelic variants of multidrug resistance gene to agranulocytosis associated with clozapine. J Clin Psychopharmacol. 2016;36(3):257–61.
Dogu GG, Kargi A, Turgut S, Ayada C, Taskoylu BY, Demiray G, et al. MDR1 single nucleotide polymorphism C3435T in Turkish patients with non-small-cell lung cancer. Gene. 2012;506(2):404–7.
Özhan G, Kara M, Sari FM, Yanar HT, Ercan G, Alpertunga B. Associations between the functional polymorphisms in the ABCB1 transporter gene and colorectal cancer risk: a case-control study in Turkish population. Toxicol Mech Methods. 2013;23(4):235–9.
Saruwatari J, Ishitsu T, Nakagawa K. Update on the genetic polymorphisms of drug-metabolizing enzymes in antiepileptic drug therapy. Pharmaceuticals. 2010;3:2709–32.
PharmGKB. CYP2C9. 2017. https://www.pharmgkb.org/. Accessed 25 Feb 2017.
Ma Q, Lu AY. Pharmacogenetics, pharmacogenomics, and individualized medicine. Pharmacol Rev. 2011;63:437–59.
Zanger UM, Turpeinen M, Klein K, Schwab M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal Bioanal Chem. 2008;392:1093–108.
Rettie AE, Haining RL, Bajpai M, Levy RH. A common genetic basis for idiosyncratic toxicity of warfarin and phenytoin. Epilepsy Res. 1999;35:253–5.
Bajpai M, Roskos L, Shen D, Levy R. Roles of cytochrome P4502C9 and cytochrome P4502C19 in the stereoselective metabolism of phenytoin to its major metabolite. Drug Metab Dispos. 1996;24(12):1401–3.
Luna-Tortós C, Fedrowitz M, Löscher W. Several major antiepileptic drugs are substrates for human P-glycoprotein. Neuropharmacology. 2008;55:1364–75.
Zhang C, Kwan P, Zuo Z, Baum L. In vitro concentration dependent transport of phenytoin and phenobarbital, but not ethosuximide, by human P-glycoprotein. Life Sci. 2010;86:899–905.
Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmöller J, Johne A, et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA. 2000;97:3473–8.
Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–7.
Li H, Wang B, Chang C, Wu M, Xu Y, Jiang Y. The roles of variants in human multidrug resistance (MDR1) gene and their haplotypes on antiepileptic drugs response: a meta-analysis of 57 studies. PLoS One. 2015;10:1–16.
Thakkar AN, Bendkhale SR, Taur SR, Gogtay NJ, Thatte UM. Association of CYP2C9 polymorphisms with phenytoin toxicity in Indian patients. Neurol India. 2012;60:577–80.
Suvichapanic S, Jittikoon J, Wichukchinda N, Kamchaisatian W, Visudtbhan A, Benjapopitak S, et al. Association analysis of CYP2C9*3 and phenytoin-induced severe cutaneous adverse reactions (SCARs) in Thai epilepsy children. J Hum Genet. 2015;60:413–7.
Loscher W, Potschka H. Role of multidrug transporters in pharmacoresistance to antiepileptic drugs. J Pharmacol Exp Ther. 2002;301:7–14.
Sisodiya SM, Lin WR, Harding BN, Squier MV, Thom M. Drug resistance in epilepsy: expression of drug resistance proteins in common causes of refractory epilepsy. Brain. 2002;125:22–31.
Tishler DM, Weinberg KI, Hinton DR, Barbaro N, Annett GM, Raffel C. MDR1 gene expression in brain of patients with medically intractable epilepsy. Epilepsia. 1995;36:1–6.
Siddiqui A, Kerb R, Weale ME, Brinkmann U, Smith A, Goldstein DB, et al. Association of multidrug resistance in epilepsy with a polymorphism in the drug-transporter gene ABCB1. N Engl J Med. 2003;348:1442–8.
PharmGKB. Gene-specific information tables for CYP2C9. 2017. https://www.pharmgkb.org/. Accessed 10 Mar 2017.
PharmGKB. Gene-specific information tables for CYP2C19. 2017. https://www.pharmgkb.org/. Accessed 10 Mar 2017.
Kassogue Y, Dehbi H, Nassereddine S, Quachouh M, Nadifi S. Genotype variability and haplotype frequency of MDR1 (ABCB1) gene polymorphism in Morocco. DNA Cell Biol. 2013;32:582–8.
Bournissen FG, Moretti ME, Juurlink DN, Koren G, Walker M, Finkelstein Y. Polymorphism of the MDR1/ABCB1 C3435T drug-transporter and resistance to anticonvulsant drugs: a meta-analysis. Epilepsia. 2009;50:898–903.
Chang CC, Ng CC, Too CL, Choon SE, Lee CK, Chung WH, et al. Association of HLA-B*15:13 and HLA-B*15:02 with phenytoin-induced severe cutaneous adverse reactions in a Malay population. Pharmacogenom J. 2017;17:170–3.
Cheung YK, Cheng SH, Chan EJ, Lo SV, Ng MH, Kwan P. HLA-B alleles associated with severe cutaneous reactions to antiepileptic drugs in Han Chinese. Epilepsia. 2013;54:1307–14.
Celeste BL, Kwan P, Baum L, Yu E, Lau KM, Cheng AS, et al. Association between HLA-B*1502 allele and antiepileptic drug-induced cutaneous reactions in Han Chinese. Epilepsia. 2007;48:1015–8.
Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097. doi:10.1371/journal.pmed1000097.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the preparation of this article.
Conflict of interest
Renée Dagenais, Kyle John Wilby, Hazem Elewa, and Mary H.H. Ensom have no conflicts of interest directly relevant to the content of this article.
Appendix: MEDLINE search strategy
Appendix: MEDLINE search strategy
-
1.
MENA.mp.
-
2.
exp Africa, Northern/ or “Middle East and North Africa”.mp. or exp Middle East/
-
3.
Afghanistan.mp. or exp Afghanistan/
-
4.
Bahrain.mp. or exp Bahrain/
-
5.
Djibouti.mp. or exp Djibouti/
-
6.
Egypt.mp. or exp Egypt/
-
7.
Algeria.mp. or exp Algeria/
-
8.
Iran.mp. or exp Iran/
-
9.
exp Iraq/ or Iraq.mp.
-
10.
Israel.mp. or exp Israel/
-
11.
Jordan.mp. or exp Jordan/
-
12.
Kuwait.mp. or exp Kuwait/
-
13.
Lebanon.mp. or exp Lebanon/
-
14.
Libya.mp. or exp Libya/
-
15.
Morocco.mp. or exp Morocco/
-
16.
Oman.mp. or exp Oman/
-
17.
Palestine.mp.
-
18.
West Bank.mp.
-
19.
Gaza.mp.
-
20.
Qatar.mp. or exp Qatar/
-
21.
Saudi Arabia.mp. or exp Saudi Arabia/
-
22.
Somalia.mp. or exp Somalia/
-
23.
exp South Sudan/ or exp Sudan/ or Sudan.mp.
-
24.
Syria.mp. or exp Syria/
-
25.
Tunisia.mp. or exp Tunisia/
-
26.
exp Turkey/ or Turkey.mp.
-
27.
United Arab Emirates.mp. or exp United Arab Emirates/
-
28.
Yemen.mp. or exp Yemen/
-
29.
1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27 or 28
-
30.
Polymorphism.mp. or exp Polymorphism, Genetic/ or exp Polymorphism, Single Nucleotide/
-
31.
Pharmacogenetics.mp. or exp Pharmacogenetics/
-
32.
Pharmacogenomics.mp.
-
33.
cytochrome p450.mp. or exp Cytochrome P-450 Enzyme System/
-
34.
CYP2C9.mp. or exp Cytochrome P-450 CYP2C9/
-
35.
CYP2C19.mp. or exp Cytochrome P-450 CYP2C19/
-
36.
CYP3A4.mp. or exp Cytochrome P-450 CYP3A/
-
37.
CYP2B6.mp. or exp Cytochrome P-450 CYP2B6/
-
38.
CYP1A2.mp. or exp Cytochrome P-450 CYP1A2/
-
39.
CYP2E1.mp. or exp Cytochrome P-450 CYP2E1/
-
40.
CYP2C8.mp. or exp Cytochrome P-450 CYP2C8/
-
41.
exp Cytochrome P-450 CYP2A6/ or CYP2A6.mp.
-
42.
exp Glucuronosyltransferase/ or UGT.mp.
-
43.
UGT1A1.mp.
-
44.
UGT1A4.mp.
-
45.
UGT1A6.mp.
-
46.
UGT1A9.mp.
-
47.
P-glycoprotein.mp. or exp P-Glycoprotein/
-
48.
multi drug resistance protein.mp. or exp Multidrug Resistance-Associated Proteins/ or exp ATP-Binding Cassette Transporters/
-
49.
exp P-Glycoproteins/ or exp P-Glycoprotein/ or MDR1.mp.
-
50.
ABCB1.mp.
-
51.
30 or 31 or 32 or 33 or 34 or 35 or 36 or 37 or 38 or 39 or 40 or 41 or 42 or 43 or 44 or 45 or 46 or 47 or 48 or 49 or 50
-
52.
phenytoin.mp. or exp Phenytoin/
-
53.
antiepileptic.mp. or exp Anticonvulsants/
-
54.
exp Epilepsy/ or epilepsy.mp.
-
55.
52 or 53 or 54
-
56.
29 and 51 and 55
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dagenais, R., Wilby, K.J., Elewa, H. et al. Impact of Genetic Polymorphisms on Phenytoin Pharmacokinetics and Clinical Outcomes in the Middle East and North Africa Region. Drugs R D 17, 341–361 (2017). https://doi.org/10.1007/s40268-017-0195-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-017-0195-7