
Abstract
Depression is one of the most frequent and burdensome non-motor symptoms in Parkinson’s disease (PD), across all stages. Even when its severity is mild, PD depression has a great impact on quality of life for these patients and their caregivers. Accordingly, accurate diagnosis, supported by validated scales, identification of risk factors, and recognition of motor and non-motor symptoms comorbid to depression are critical to understanding the neurobiology of depression, which in turn determines the effectiveness of dopaminergic drugs, antidepressants and non-pharmacological interventions. Recent advances using in vivo functional and structural imaging demonstrate that PD depression is underpinned by dysfunction of limbic networks and monoaminergic systems, depending on the stage of PD and its associated symptoms, including apathy, anxiety, rapid eye movement sleep behavior disorder (RBD), cognitive impairment and dementia. In particular, the evolution of serotonergic, noradrenergic, and dopaminergic dysfunction and abnormalities of limbic circuits across time, involving the anterior cingulate and orbitofrontal cortices, amygdala, thalamus and ventral striatum, help to delineate the variable expression of depression in patients with prodromal, early and advanced PD. Evidence is accumulating to support the use of dual serotonin and noradrenaline reuptake inhibitors (desipramine, nortriptyline, venlafaxine) in patients with PD and moderate to severe depression, while selective serotonin reuptake inhibitors, repetitive transcranial magnetic stimulation and cognitive behavioral therapy may also be considered. In all patients, recent findings advocate that optimization of dopamine replacement therapy and evaluation of deep brain stimulation of the subthalamic nucleus to improve motor symptoms represents an important first step, in addition to physical activity. Overall, this review indicates that increasing understanding of neurobiological changes help to implement a roadmap of tailored interventions for patients with PD and depression, depending on the stage and comorbid symptoms underlying PD subtypes and their prognosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Depression is highly prevalent in Parkinson’s disease (PD) across all stages of the disease, depending on dynamic neurobiological changes related to dysfunction of limbic networks and monoaminergic systems across time. |
Motor and non-motor symptoms (in particular apathy, anxiety, rapid eye movement sleep behavior disorder and cognitive impairment) comorbid to PD depression are critical to delineate the neurobiology of PD depression. |
In vivo functional imaging supports the effectiveness of dopaminergic and serotonergic treatments in PD depression and helps develop a roadmap for tailored therapeutic interventions according to PD subtypes and disease stage. |
1 Introduction
Non-motor symptoms are well-recognized as a major source of disability for patients with Parkinson’s disease (PD), which may exceed the burden related to motor impairment [1, 2]. Moreover, treatment of non-motor symptoms is often difficult and may either improve or worsen under dopaminergic replacement therapy used to alleviate motor symptoms. In particular, neuropsychiatric non-motor symptoms may severely alter the quality of life of both patients and their caregivers [3,4,5], and possibly even more during the coronavirus disease 2019 (COVID-19) pandemic [6]. Among these, depression is recognized as one of the most frequent and disabling conditions, alongside anxiety, fatigue and pain, affecting one-quarter of patients at PD onset [7], but which may occur at any stage of PD, including the prodromal stage [1, 4, 8, 9], and may shorten life expectancy [10].
Depression in patients with PD is mostly characterized by loss of pleasure/enjoyment, including dissatisfaction and loss of appetite, but less feelings of guilt, self-hate, and loss of libido [11], although fundamental supporting criteria are not different from those of depressive or anxiety disorders in the general population [12].
Symptoms related to depression were among the most frequent non-motor symptoms in patients with PD using the Non-Motor Symptoms Scale (NMSS) and highly correlated to lower quality of life across all stages [1, 9, 13]. The prevalence of depression in PD is high [14], but variable, depending on the stage of the disease and patients’ comorbidities, suggesting that depression may have different pathophysiological determinants and be related to specific PD subtypes [15]. Overall, the interaction of depression heralding PD and incidental depression after the diagnosis of PD is complex and a unified pathophysiological model of depression in PD is lacking. In addition, pathophysiological changes involved in the pathogenesis of PD (including neurotransmission abnormalities of the dopaminergic, serotonergic, noradrenergic or cholinergic systems, dysfunction of striato-thalamo-cortical circuits, frontal and limbic cortical structural and functional alterations, inflammation, and mitochondrial dysfunction) are also suspected to play a prominent role for depression in the general population, in addition to neuroendocrine changes, as reviewed previously [16, 17].
We followed a pragmatic literature search strategy using PubMed with the Medical Subject Heading (MeSH) terms ‘parkinson disease’ in the title and ‘depression’ combined with ‘prevalence’, ‘incidence’, ‘longitudinal’, ‘MRI’,‘SPECT’,‘PET’, ‘trial’, ‘therapy’ and ‘treatment’, yielding 1031 results. In addition, we queried the Cochrane library database of systematic reviews for treatments, yielding two relevant occurrences [18, 19] and the Movement Disorders Society Evidence-Based Medicine Reviews, with two relevant reviews [20, 21].
This review aims to help bridge the gap between novel hypotheses regarding the pathophysiology of depression in PD and the available and promising therapeutic interventions, and thereby promote accurate treatment of depression, which remains under-recognized and insufficiently treated.
2 Epidemiology and Clinical Characteristics of Depression in Parkinson’s Disease (PD)
2.1 Prevalence
Depression in PD is recognized as one of the most frequent and disabling non-motor symptoms. Prevalence estimates of depression are wide, ranging from 2.7 to 90% [14], depending on the evaluation and diagnostic criteria, patients’ characteristics and comorbidities, and stage of the disease. Overall, clinically significant depression is present in 35% [14, 22, 23], up to 40.4% for outpatients with PD and 54.3% for inpatients with PD, albeit with variable severity [24, 25]. Major depressive disorder in PD is found in about 20% [14, 26,27,28]. Risk of depression is considered four times higher than in the general population matched for age, sex and comorbidities [29].
Importantly, depression is found at any stage of PD. Recently, data obtained in a large population indicated that 38% of patients are treated or diagnosed for depression within the year following diagnosis [30], consistent with other studies in de novo patients [8, 31]. Depression is found in up to 70% of mid-to-advanced PD outpatients without dementia [4], and associated with other neuropsychiatric symptoms [32]. In addition, prevalence, severity and symptoms of depression were similar in patients with mid-to-advanced PD and in patients with essential tremor and (mostly focal) dystonia [33].
Importantly, depression in PD may be underdiagnosed and important manifestations, including fatigue, sleep disorders and weight changes, may be directly attributed to PD and not to depression complicating PD [34]. The reverse also stands true as initial PD motor (amimia, slowness) and non-motor symptoms may be interpreted as manifestations of depression, instead of PD, leading to erroneous treatment [35].
Furthermore, the sensitivity and specificity of recommended diagnostic tools remains variable [36,37,38,39,40], although the Geriatric Depression Scale (GDS-15), Beck Depression Inventory-I (BDI-I/1a), and Montgomery-Åsberg Depression Rating Scale (MADRS) are considered valid scales to measure depression severity [27], in line with previous guidelines [38].
In addition, screening of depression is a critical step and can also be performed using the BDI, MADRS and GDS thanks to their high-sensitivity in patients with PD [38]. In particular, the 15-item version of the GDS represents a time-efficient screening tool, as it is easily comprehensible and incorporates relatively few somatic-related items overlapping with the core symptoms of PD (sleep disturbances, alteration of facial expression, slowness), which may falsely inflate depression scores [38].
Overall, few symptoms are specific to PD depression in comparison with depression in the general population, and patients with PD may often experience clinically significant symptoms although they may not fulfill criteria for major depressive disorders [14, 27].
Overall, the burden of depression in PD related to both its prevalence and repercussions advocate for an ‘inclusive’ approach in which somatic symptoms are equally considered as part of PD depression for screening and severity scores to avoid underdiagnosis and undertreatment (see below) [41, 42]. However, although challenging, the distinction between depression and apathy has critical implications for treatment [21, 44], underpinned by specific limbic dysfunction [43, 45], and should be operated using dedicated scales [36, 38] such as the Lille Apathy Rating Scale (LARS) [46] or Starkstein Apathy Scale [39]. This is further emphasized in the Neurobiology and Treatment sections.
2.2 Incidence and Risk Factors
Longitudinal studies are particularly valuable to decipher the dynamics of depression across PD stages and identify clusters of patients with distinct demographic and clinical characteristics, which may inform the etiopathogenesis of PD-related motor and non-motor symptoms [47].
Data from clinical trials indicate that incidental depression occurs in about 16% of de novo patients newly diagnosed with PD and previously free of depression, in addition to the 13.8% of patients already suffering from depression at diagnosis [13, 48]. Overall, the incidence rate of depression in newly diagnosed patients is increased twofold in comparison with age-matched non-PD patients [49], and up to 10 times higher than in the general population aged over 50 years. Female sex is consistently regarded as a risk factor for depression in PD [23, 49,50,51] and in the general population [52]. Contrastingly, influence of age at onset is highly dependent on the setting [50, 53,54,55,56], although patients with depression after diagnosis may be older [57, 58]. Furthermore, non PD-specific risk factors (age, female sex, personal history of anxiety or depression, family history of depression, worse functioning on activities of daily living, and worse cognitive status) have been shown to be three times more influential for PD depression than PD-specific risk factors (longer disease duration, more severe motor symptoms and use of levodopa) [59, 60]. Moreover, personalized prediction of depression using machine learning confirms the influence of non-PD-specific predictors (depression and anxiety severity, presence of rapid eye movement sleep behavior disorder [RBD] and history of depression) in addition to PD-specific factors (age at onset and duration) [56]. In particular, evidence that RDB represents an important risk factor for depression in PD is accumulating [61].
Overall, a systematic review identified that autonomic symptoms, motor fluctuations, severity and frequency of symptoms, staging of the disease, and PD onset and duration were associated with the presence of depression and anxiety in patients with PD [62]. However, one should not dismiss that personal and familial psychosocial and environmental factors also play an important role in the dynamics and repercussions of PD depression [42], which all influence the coping strategies of patients and their caregivers depending on age and stage [63].
2.3 Association Between Depression and Motor Symptoms
Greater motor and global impairment, closely related to worse quality of life, is a constant finding in depressed patients [28, 48, 64,65,66], although the impact of depression on motor impairment is modest [48, 67,68,69]. In addition, depression in early PD is regarded as a risk factor for worse motor and global prognosis [64, 70, 71]. This is also the case in patients with advanced PD and persistent depression [72]. This is consistent with previous studies confirming the association between depression and PD severity (PD duration, Hoehn and Yahr and Unified Parkinson’s Disease Rating Scale (UPDRS) scores, motor complications, duration of treatment) such as the DEPAR and DoPaMiP surveys in older and more advanced patients, including those with mild cognitive impairment [50, 73, 74]. Furthermore, worse motor prognosis was found in mid-stage patients with greater depression, apathy and anxiety as well as greater cognitive impairment [75]. Importantly, comorbid depression over time is not related to worse motor prognosis in prospective studies [64] but may accelerate the initiation of dopaminergic replacement therapy in early PD [48].
Higher depression scores were found in patients with motor fluctuations and/or dyskinesia [68, 76, 77], also in comparison with drug-naïve patients [78]. Greater dyskinesia, levodopa equivalent dose and impulse control disorders were also associated with depression [66, 79, 80]. In addition, greater severity of depression was the strongest predictor of impulse control disorders of all types, independently of treatment with dopamine agonists [81].
Association with specific motor subtypes is unclear and may reflect unstable classification in early PD [82]. Depression prevalence was similar in Postural Instability and Gait Disorder (PIGD) versus tremor-dominant (TD) patients in early [13], and across, PD stages [83] but its severity was greater in non-TD de novo patients [31] and early PIGD-dominant patients [84]. Furthermore, mild depression was significantly associated with gait impairment in early PD [85] and axial symptoms may be associated with greater prevalence and severity of depression across PD stages [28, 68, 69, 73, 86,87,88], especially in those with rapid progression with or without cognitive impairment [89]. This association is also found for anxiety [90]. Moreover, there is no definitive association between depression and the side of greatest motor impairment [59, 91, 92], similar to anxiety [90], although (mild) depression may be more severe in patients with bilateral symptoms [84].
2.4 Association Between Depression and Other Non-Motor Symptoms
The association of depression with other neuropsychiatric non-motor symptoms is particularly frequent in PD, which is considered as indirect evidence of co-occuring, and possibly intricately linked or synergistic pathophysiological mechanisms.
Anxiety is long recognized as one of the most frequent comorbid conditions accompanying depression [4, 93,94,95,96], although both conditions may exist on their own [4, 73, 90, 97]. As such, risk factors are partly distinct in depression with or without anxiety, and, in particular, anxiety may be more frequent in patients aged < 60 years at diagnosis, and be related to motor fluctuations [86, 90, 98]. Furthermore, the prevalence of depressive disorder is greater in patients with non-specific anxiety subtypes [98]. In addition, anxiety is an important risk factor when considering incident depression, besides insomnia [93]. Apathy is also frequently associated with depression, leading to consider a hypodopaminergic neuropsychiatric triad consisting of apathy, depression and anxiety in PD [99]. However, association of depression on one side, with apathy and anxiety on the other side, is complex, as symptoms most frequently co-exist [100], although they also occur in isolation [101,102,103], which may indicate overlapping and also distinct pathophysiological mechanisms related to distinct prognostic outcomes [44, 99].
Interestingly, earlier studies suggested a link between cognitive impairment and depression in older patients and those with long PD duration [23, 74, 76, 77, 104,105,106]. More precisely, many studies demonstrated a strong association between depression and anxiety and worse executive or multiple domain cognitive functioning [107,108,109,110,111]. In addition, lower education level was also associated with depression in PD [68, 112, 113]. Psychosis in patients with early PD also predicted greater prevalence of depression and anxiety, in addition to greater cognitive impairment, and worse prognosis over 2 years [114]. Overall, depression and cognitive impairment share many common risk factors [31, 115].
Recent studies show that depression may herald dementia in PD and across other neurodegenerative disorders [116]. Depression and anxiety were found to be the strongest predictors of the longitudinal decline in verbal and visual learning performance in PD [117], as well as in old-age depression in the general population [118]. Analysis in the Parkinson’s Progression Markers Initiative (PPMI) cohort showed that patients with early PD and depression had an almost twofold risk to develop mild cognitive impairment within 4 years [119]. In addition, patients with the more severe depressive symptoms experience greater and more rapid decrease of performance in critical cognitive tasks (processing speed, verbal learning and verbal delayed recall) [120]. Interestingly, poorer multi-domain cognitive performance predicts increased incidence of depression and anxiety in early PD [121, 122]. In addition, cognitive decline is also related to incidental depression in mid-stage patients [123]. Moreover, specific association of apathy with subsequent cognitive impairment or dementia was also demonstrated in non-depressed patients [124]. As such, depression, apathy and anxiety, represent risk factors of cognitive impairment and dementia, whereas risk and domain-specific cognitive impairment restricted to one of these three neuropsychiatric conditions remains inconclusive [109, 117, 120, 125, 126]. Overall, evidence is strong to support that depression related to cognitive impairment and dementia represents a different nosologic entity from depression in de novo and prodromal patients with PD, and is underpinned by distinct, possibly more widespread, pathological burden.
Most interestingly, it was recently pointed out that patients with RBD in early PD had greater worsening of depression over time, also associated with worse motor and non-motor prognosis and death, in a large cohort [127]. Overall, patients with RBD have more severe depression and may represent a PD subtype with faster motor progression and cognitive decline [128], possibly related to the extent of amyloid pathology [129].
Recent mediation analyses indicate that longitudinal changes in autonomic dysfunction and depressive symptoms are linked [130, 131], with greater depression in those with more severe autonomic dysfunction [132]. Association of constipation and depression was also found [66, 133]. Interestingly, earlier development of autonomic dysfunction was demonstrated to predict more rapid progression in PD and death, although neuropathological findings assessing the burden of Lewy bodies were similar [134]. Depression is also more severe in patients with pain [135, 136]. Furthermore, greater severity of depression was found to better predict dysphonia than global motor impairment [137]. Depression, as well as anxiety, also predicts greater insomnia in early PD [138].
2.5 Course of Depression
Longitudinal follow-up in prospective studies and the PPMI cohort provide new insight in the variable course of depression, in particular in early PD [28, 104, 139]. Importantly, in early PD, depressive symptoms resolved in one-third [28, 123] to one-half of patients, especially for younger patients and those with milder symptoms [139]. Similarly, in de novo patients with PD, depression was half less frequent 2 years after diagnosis, following the initiation of dopamine replacement therapy [140]. However, one has to keep in mind that the role of dopaminergic and antidepressant treatments remains complex to disentangle in observational studies, and that clinical factors may both influence the natural history of PD depression and the response to treatments. In particular, risk factors for depression (female sex, age and PD duration) are associated with a lower likelihood that depressive symptoms resolve [123, 139]. In addition, greater cognitive decline and worsening of UPDRS part II were associated with the worsening of depression score over 30 months in mid-stage patients [123]. However, further research is needed to identify the factors that predict favorable evolution and good response to treatments of PD depression at the individual level and may be applicable in the clinical context. Notably, longitudinal positron emission tomography (PET) imaging studies have the potential to identify compensatory mechanisms, possibly involving the serotonergic system [141] and related to the improvement of apathy in early PD [43].
2.6 Depression in Prodromal PD
Depression represents one of the earliest and most frequent alerting symptom across age-related neurodegenerative disorders. Prospective historical cohort studies using registry and medical records showed that history of depression is about twice more frequent at the time of PD diagnosis compared with age-matched controls (9.2% vs. 4%) [142,143,144] and multiplies the risk to be diagnosed with PD by at least twofold [145, 146]. Meta-analysis regarding risk factors for PD confirmed that the odds ratio (OR) for mood disorders was 1.86 (95 % confidence interval [CI] 1.64–2.11], behind familial history of PD, constipation and absence of smoking [147]. Interestingly, association of depression and anxiety was associated with the highest risk (OR 2.4, 95 % CI 1.2–4.8), and lag time might be higher for anxiety [144]. Overall, depression may precede motor symptoms by 5 years or more [148] and the risk for depression in premotor patients is regularly increasing until PD diagnosis [142, 144, 148]. Interestingly, all patients with major depressive disorder who developed PD within 10 years were characterized by the triad consisting in mild asymmetric motor slowing, idiopathic hyposmia and substantia nigra hyperechogenicity [149]. Moreover, it was recently demonstrated that patients with depression following PD diagnosis were older and had a twofold risk of dementia in comparison with patients with prediagnostic depression [57].
2.7 Depression in Genetic PD
Whether genetic forms of PD predispose to depression remains an open question, although monogenic forms of PD resembling idiopathic PD with Lewy-body synucleinopathy represent critical etiopathogenic models.
Depression is frequent in patients with genetic PD [150,151,152], although depression was not related to mutations of parkin, leucine-rich repeat kinase 2 (LRRK2) and apolipoprotein E (APOE) status in a large study gathering 632 families [153]. In addition, a recent study showed that depression severity was similar to healthy controls in both LRRK2 and glucocerebrosidase (GBA) mutation carriers at risk of PD as well as RBD and impulse control disorders [154]. Nevertheless, prevalence of depression is close to 30% in LRRK2 patients with manifest PD and may predate motor symptoms, similar to patients with idiopathic PD [155, 156], whereas prevalence of dementia might be lower [156,157,158]. Moreover, depression severity increased over 2 years in heterozygous carriers of GBA mutations, who are at high risk of PD, also associated with greater olfactory and cognitive impairment [159], and depending on the alleles [157].
Several studies indicate that there are no associations between depression in PD and known genetic polymorphisms of the serotonin and dopamine transporter genes [160, 161]. However, other studies are positive for the short allele of the serotonin transporter (SERT) gene-linked polymorphic region [162, 163], but not for the monoamine oxidase (MAO)-A gene promoter-associated polymorphism [163]. Interestingly, PD depression is associated with polymorphism of the cannabinoid 1 receptor (CB1) gene [164], which is of interest since cannabinoid CB1 receptors likely modulate the excitability of dorsal raphe neurons [165].
Overall, these robust statistical associations and temporal precedence suggest that depression and PD may share common pathophysiological mechanisms (Table 1), and that association with specific comorbidities and risks factors may reflect different pathological routes [15, 166, 167].
3 Neurobiology of Depression
Understanding the pathophysiological mechanisms and the respective importance of dopaminergic versus non-dopaminergic pathology underlying depression in patients with PD is critical to address the heterogeneity of PD depression and develop tailored interventions to treat depression at all stages. In vivo structural and functional imaging have greatly improved our understanding of changes related to PD depression and dysfunction of corticostriatal circuits [44].
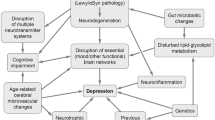
In particular, the role of monoamine deficits related to depression has long been investigated in PD, which is characterized by multiple and progressive monoaminergic and catecholaminergic dysfunction, largely extending beyond the core nigrostriatal dopaminergic degeneration. Indeed, it is now established that serotonergic and noradrenergic deficits occur in prodromal patients with RBD [168, 169] and in A53T mutation carriers of the alpha-synuclein (SNCA) gene [170], and are prominent in de novo patients with idiopathic PD [171]. Moreover, postmortem histopathological staging suggests that monoaminergic deficit might be sequential, starting with noradrenergic and serotonergic dysfunction heralding nigrostriatal dopaminergic dysfunction in prodromal patients, and progressively extending to cholinergic and neocortical dysfunction, based on the topography of Lewy-body synucleinopathy [172], although this may apply to a subset of patients and remains controversial [173,174,175,176]. In addition, depression is more severe in patients with akinetic-rigid, bilateral and axial symptoms (see Sect. 2.3) and correlated to bradykinesia [28, 69, 177, 178], suggesting greater dopaminergic injury possibly involving the motor nigrostriatal system, but also more widespread, and extrastriatal pathology associated with axial symptoms. Overall, evidence accumulates to support that PD depression is causally related to pathological mechanisms starting in the prodromal state and that progression of dopaminergic and non-dopaminergic pathology over the PD course may explain the variable expression and outcome of depression over time (Fig. 1).

Structural and functional abnormalities related to depression in Parkinson’s disease (red: striatum; light green: prefrontal cortex and insula; green: brainstem; light blue: cingulate cortex; blue: thalamus; violet: amygdala and hippocampus). ACC anterior cingulate cortex, sgACC subgenual ACC, Amyg amygdala, CN caudate nucleus, GP globus pallidus, GR gyrus rectus, Hipp hippocampus, HypoT hypothalamus, Ins insula, LC locus coeruleus, MTG medial temporal gyrus, OFC orbito-frontal cortex, PCC posterior cingulate cortex, PFC prefrontal cortex, Put putamen, VS ventral striatum
3.1 Neurotransmission Abnormalities
3.1.1 Dopamine Imaging
The increased prevalence and incidence of depression in PD and its association with greater motor impairment and bradykinesia suggest that dopaminergic denervation may play an important role for depression. In particular, dysfunction of the presynaptic terminals of the mesolimbic dopaminergic system has been demonstrated using single photon emission computed tomography (SPECT) and PET imaging across PD stages (Fig. 1).
Indeed, decreased binding of [11C]RTI-32, a PET ligand for the dopamine (DAT) and noradrenaline (NERT) transporters, was demonstrated in the (left) ventral striatum in patients with PD and more severe depression, next to noradrenergic dysfunction in other limbic structures [179]. In addition, decreased DAT correlating with depression severity was found in the left anterior putamen using (99m)Tc-TRODAT-1 [180], and in the bilateral striatum and putamen [181], thalamus [182, 183], right caudate [184] and left cingulate cortex [185] using [123I]FP-CIT-SPECT. Interestingly, decreased DAT in the left caudate in PD and left putamen in primary dystonia correlated with greater depression [186]. Decreased dopaminergic metabolism was also demonstrated in the bilateral putamen and caudate nucleus proportional to PD depression severity [187]. Furthermore, striatal decrease of DAT labeling is also found in de novo apathetic patients with PD [188] and in non-PD patients with major depressive disorder [189].
On the contrary, other studies have rather found greater DAT labeling in the bilateral striatum, left caudate and right putamen in depressed patients in comparison with non-depressed patients with PD, including patients taking antidepressants [190, 191], or no dopaminergic striatal change related to depression using [18F]FP-CIT PET [192].
Overall, DAT imaging provides evidence for presynaptic dopaminergic system dysfunction related to PD depression, either via a reduced (i.e greater degeneration) or increased availability (possibly leading to abnormal dopamine clearance) of such a transporter.
In addition, disequilibrium of D3 versus D2 receptors has been shown to be related to not only depression severity but also to motor impairment in the ventral striatum and pallidum [193]. Indeed, a lower [11C]-(+)-PHNO (D3-specific) to [11C]raclopride (D2/3 non-specific) BPND ratio was observed in the striatum in depressed drug-naïve patients, possibly as a result of preferential downregulation of postsynaptic D3 receptors.
Overall, these studies pinpoint the role of mesolimbic dopaminergic denervation in PD depression, although small sample sizes in PET and SPECT studies, use of different tracers, and custom methods of analysis may limit the comparison of these findings.
3.1.2 Serotonin Imaging
SPECT studies suggested a relationship between depression in PD and decreased [123I]FP-CIT binding in the dorsal midbrain, classically considered to reflect binding to the SERT [182, 194]; however such association was not found in the large PPMI cohort [195]. Using [11C]DASB, a highly specific PET radiotracer for SERT, it was demonstrated that depression was associated with an increased binding in the caudal raphe nuclei, amygdala, hypothalamus, and posterior cingulate cortex [196]. Furthermore, such an increase positively correlated to depression severity in the prefrontal and cingulate cortex, insula and putamen in antidepressant-naive patients with PD [197], possibly reflecting upregulation of SERT, in turn resulting in greater serotonin clearance. Contrastingly, depression in de novo, drug-naïve patients with apathy, depression and anxiety was associated with decreased [11C]DASB in the limbic system and greater severity specifically correlated to greater serotonergic dysfunction in the bilateral subgenual anterior cingulate cortex (ACC) [171]. Moreover, greater serotonergic pathology related to depression was recently demonstrated across PD stages, underlying the major influence of serotonergic dysfunction in limbic corticostriatal circuits [198], which may specifically underpin microstructural alterations in the caudate nucleus [45]. Regarding postsynaptic dysfunction, patients with PD depression had lower binding of [18F]MPPF to the 5HT1A receptor in the left hippocampus, right insula, left superior temporal cortex, and the orbitofrontal cortex [199]. Decreased echogenicity of the basal limbic midbrain was also found in patients with PD depression using transcranial sonography, as well as alterations of signal intensity and relaxation time of the pontomesencephalic midline structures using T2-weighted magnetic resonance imaging (MRI) [200,201,202].
3.1.3 Noradrenaline Imaging
Studies investigating noradrenergic involvement in PD depression are scare. However, using [11C]RTI-32 PET imaging, an in vivo marker of both the dopamine and noradrenaline transporters, noradrenergic dysfunction related to PD depression and anxiety was found in the bilateral locus coeruleus and thalamus as well as the right amygdala [179]. Overall, widespread noradrenergic dysfunction was recently evidenced in PD using [11C]-MeNER PET imaging, in association with decreased neuromelanin in the locus coeruleus [203]. Most interestingly, noradrenergic injury was greater in parkinsonian patients with RBD, who also had greater cognitive impairment, slowed EEG activity, and orthostatic hypotension in comparison with patients without RBD [203], which may be related to depression [61, 93].
3.1.4 Cholinergic Imaging
The role of cholinergic cortical deficit in PD dementia is well established [204]. Furthermore, striatal and limbic cholinergic dysfunction were also demonstrated in patients with axial or gait disorders [205] and RBD [206]. As such, cortical cholinergic loss was also related to depression score in mid-stage demented and non-demented male patients with PD [207]. In addition, decrease of 2-[18F]FA-85380 binding to α4β2-nicotinic receptors was prominent in the anterior cingulate cortex and frontoparietal cortex related to PD depression, in addition to the left putamen, left midbrain, and occipital cortex [208], partly overlapping with cognitive impairment. Interestingly, progression of cholinergic deficit has been found to spare the prefrontal cortex in early PD, and subsequently follow an anterior-to-posterior prefrontal degeneration gradient, which may relate to progressive cognitive impairment and comorbid depression [209].
3.2 Structural and Metabolic Abnormalities
Limbic and striatal atrophy were consistently demonstrated in PD depression, associated with widespread dysfunction of the limbic network. In particular, depression severity was correlated to lower grey matter density in the bilateral orbitofrontal, bilateral rectal gyrus, right medial temporal gyrus, anterior and medial cingular gyrus, and parahippocampal gyrus [210], left hippocampus and right parahippocampal gyrus [211], in addition to white matter loss in the anterior cingulate bundle and the inferior orbitofrontal region [212]. This is consistent with earlier studies indicating hypometabolism in the anterior cingulate cortex and medial frontal and orbitofrontal cortex [213,214,215], whereas hypermetabolism is present in the amygdala [216], which is preserved from atrophy [217].
In addition, recent diffusion-weighted imaging studies showed decreased fractional anisotropy in the anterior cingular bundles [218] and other limbic association tracts in the left [219, 220] and right [45] hemispheres. Specifically, microstructural alterations of the inferior longitudinal fasciculus were consistently associated with PD depression and cognitive impairment, as well as RBD, olfactory impairment and systemic inflammation [221]. In addition, functional connectivity between the striatum, thalamus and limbic frontotemporal regions [222, 223] is altered in depressed patients as well as between the amygdala and frontoparietal regions [217]. Notably, functional connectivity in the posterior cingulate cortex was increased in patients with PD depression and was negatively correlated to the severity of depression [224], in line with recent findings [225]. As such, differences mainly involving the functional connectivity of the posterior cingulate cortex and insula with the amygdala, hippocampus, precuneus and frontal cortex may help to classify patients with or without PD depression diagnosed with Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) criteria [225], although further research is needed to validate imaging biomarkers of PD depression in independent and larger cohorts. Overall, the importance of the anterior and subgenual cingulate cortex is paramount, as previously evidenced [226]. Moreover, greater white matter injury is found in PD depression and is associated with cognitive impairment and gait disorders [227]. Finally, patients with Lewy body disorders and depression had greater synucleinopathy in the substantia nigra, ventral tegmental area and nucleus accumbens, whereas beta-amyloid and hyperphosphorylated tau burden was similar in patients with or without depression [228].
3.3 Inflammation
Inflammation is one of the mechanisms of neurodegeneration, first suggested by the presence of activated microglial cells in the substantia nigra pars compacta and elevated levels of proinflammatory cytokines in the brain and blood of patients with PD [229]. Regarding PD depression, elevated levels of the soluble interleukin-2 receptor (sIL-2R) and tumor necrosis factor-alpha (TNFα) [230], as well as C-reactive protein (CRP) [231], were reported. Most importantly, elevated CRP was associated with decreased connectivity between the ventral striatum and ventromedial prefrontal cortex, correlated to greater anhedonia in non-PD patients with major depressive disorders [232]. In addition, dysregulation of genes involved in signaling pathways related to the immune system and metabolism present in patients with depression, including the nicotinamide phosphoribosyltransferase (NAMPT) for lipid and glucose regulation, was recently demonstrated in early untreated patients with PD, using transcriptomic meta-analysis and network analysis of blood microarrays [233]. Furthermore, inflammation is critical in changes of the microbiome, which may also be involved in PD depression [234].
4 Treatment
The consequences of depression, even when its severity is mild, are dramatic and overwhelming, acting negatively on global well-being, quality of life, physical activity, healthcare burden and family equilibrium [60, 235]. Notably, the evidence base for treating depression has grown substantially in the latest decade, including randomized clinical trials of non-pharmacological interventions and meta-analysis of pharmacological treatments [18, 21, 236,237,238]. Randomized placebo-controlled trials of serotonin and noradrenaline reuptake inhibitors are presented in Table 2, in addition to the most recent studies not previously reviewed [18, 21]. Importantly, several factors may explain that treatment of depression in PD may still be delayed, insufficient, or even absent, especially if severity is mild [48, 73, 83, 239]. Indeed, underrecognition of depression in PD remains problematic, as manifestations of depression are intermingled with multiple comorbid motor and non-motor symptoms. Moreover, depression may resolve over time in up to one-third of patients with PD, in line with a strong placebo effect in randomized clinical trials [240, 241], where intensive medical follow-up and coping strategies may help, as observed for apathy [242]. Retrospective analysis of registry data shows that trend of diagnosis and treatment of depression in the first year following PD diagnosis slightly decreased from the years 2000 to 2015 [30]. Citalopram, amitriptyline, fluoxetine and mirtazapine were the most frequent pharmacological treatments used in recent years [30]. However, use of pharmaceutical and non-pharmaceutical interventions is increasingly supported by the pathophysiological mechanisms described in Sect. 3 of this review and help develop a roadmap of tailored interventions for patients with PD and depression, depending on the stage and comorbid symptoms (Fig. 2).

Suggested roadmap for the diagnosis and treatment of depression in patients with PD. Treatments in bold are those determined as efficacious or likely efficacious in the MDS evidence-based medicine review for the treatment of non-motor symptoms in PD [21]. CBT cognitive behavioral therapy, DBS deep brain stimulation, ECG electrocardiogram, ECT electroconvulsive therapy, PD Parkinson’s disease, rTMS repetitive transcranial magnetic stimulation, SNRI serotonin and noradrenaline reuptake inhibitor, TCA tricyclic antidepressant
4.1 Dopaminergic Treatments
Optimal motor control is critical and should be finely tuned with oral dopamine replacement therapy using levodopa or dopamine agonists as a first step (Fig. 2) [21]. Importantly, a positive effect on PD depression was demonstrated for levodopa (DoPaMiP [73]) and the dopamine agonist pramipexole [243], while ropinirole [244, 245], rotigotine [246] and MAO-B inhibitors deserve further investigation [20, 21, 247]. In particular, a direct antidepressant effect (rather than mediated by the improvement of motor symptoms) of pramipexole (a highly potent D3-agonist) on depressive symptoms across PD stages was demonstrated in a multicentric, large-scale, randomized, placebo-controlled trial over 12 weeks [243], in line with earlier studies [248]. Notably, 25% of patients also received antidepressant drugs. In addition, a randomized clinical trial indicated significantly higher recovery (60.6% vs. 27.3%) for pramipexole versus sertraline (50 mg/day) [249], although sertraline was also effective [250].
In addition, piribedil, a potent D2/3 agonist, highly effective to treat apathy following subthalamic nucleus deep brain stimulation (STN-DBS) was also shown to improve depression [251]. Contrastingly, trait anxiety improved in de novo patients using low-dose rotigotine compared with placebo, whereas apathy and depression improved but similarly in the rotigotine and placebo groups, stressing the importance of care, follow-up and placebo effects [242].
Furthermore, enhancement of dopaminergic transmission using the selective MAO-B inhibitor rasagiline was not found to be superior to placebo on PD depression in a randomized controlled trial [252], although previous evidence indicated that rasagiline may improve mood [253] and depression in combination with antidepressants [254], with a low risk for adverse serotonin syndrome. However, safinamide, a reversible MAO-B inhibitor and GABAergic modulator, improved depressive symptoms in addition to the expected improvement of motor fluctuations [255]. Moreover, there is growing evidence that continuous infusion therapies using levodopa/carbidopa intestinal gel or subcutaneous apomorphine may also improve depressive symptoms [256,257,258], in addition to their important benefits on motor symptoms and quality of life [259,260,261,262]. Moreover, results from the open-label, randomized INSIGHTS study evaluating the efficacy of levodopa infusion on non-motor symptoms are awaited [261]. Overall, treatment should be optimized to achieve the best control of motor and non-motor fluctuations in the first instance [21, 258], considering STN-DBS or infusion therapies as appropriate (Fig. 2).
4.2 Serotonergic, Noradrenergic, and Cholinergic Treatments
As in non-PD depression, selective serotonin reuptake inhibitors (SSRIs) or tricyclic antidepressants (TCAs) are considered effective and well tolerated (Table 2). Effectiveness of antidepressants in PD depression was confirmed in the three largest randomized clinical trials with 48, 52 and 115 non-demented patients with PD and major depressive disorder, respectively [263,264,265]. These trials demonstrated the short-term efficacy of both desipramine and citalopram within 4 weeks [263], of nortryptiline versus paroxetine and placebo within 8 weeks [264], and of both paroxetine and venlafaxine versus placebo over 12 weeks [265]. Moreover, superiority of dual (nortryptiline, amitriptyline, venlafaxine) or predominantly noradrenergic (desipramine) reuptake inhibitors was suggested, in line with earlier trials [250, 266,267,268] and confirmed using network analysis [269]. In particular, desipramine had a more rapid efficacy compared with citalopram, possibly mediated by the effect on lassitude, inability to feel, apparent sadness, and concentration difficulties, although mild adverse effects were twice as frequent [263], related to anticholinergic effects in particular. Overall, initiation of TCAs requires special attention and should be carefully evaluated in patients with orthostatic hypotension and those with cardiovascular diseases or family history of sudden death, cardiac dysrhythmia or conduction defects. To date, insufficient evidence is available to support the clinical use of other noradrenergic agents, including reboxetine [270], atomoxetine [271] and duloxetine [272]. Administration of 5-hydroxytryptophan (50 mg) over 4 weeks, the precursor of serotonin synthesis, may also ameliorate PD depression, but not apathy, as demonstrated in a recent single-center, randomized, placebo-controlled crossover trial in 25 patients [273]. One has to keep in mind that contrary to dopaminergic treatments, SSRIs may, in rare cases, aggravate motor symptoms [274], notably tremor [263]. A pilot randomized trial suggested that 5HT2A antagonism by nefazodone may improve motor symptoms while being equally effective on depressive symptoms as citalopram [275], consistent with the improvement of depression by pimavanserin (34 mg) administered as monotherapy or adjunctive therapy over 8 weeks in a recent single-arm, open-label, phase II study [276]. Interestingly, decreased brainstem (raphe) echogenicity predicted response to SSRIs [202].
Despite evidence that cholinergic deficit is involved in PD depression and dementia, well-conducted clinical trials remain scarce. Although effective on cognition in PD dementia and dementia with Lewy bodies, the effectiveness of donepezil (5–10 mg) was modest on depressive symptoms in an open-label study over 20 weeks [277]. Notably, weekly subcutaneous injection of exanetide (2 mg), an agonist of the GLP-1 receptor abundant in limbic structures, also contributed to improvement of depressive symptoms sustained over 48 weeks, in a recent single-center, randomized, double-blind, placebo-controlled trial that evaluated 62 patients with PD to investigate neuroprotection [278].
4.3 Non-Pharmacological Interventions
STN-DBS is highly effective for the control of motor complications of PD, with a generally favorable effect on depression, impulse control disorders and non-motor fluctuations [279,280,281], contrasting with possible worsening of apathy following STN-DBS [282], partly mediated by dopaminergic withdrawal and reversed by dopamine agonists [251, 283]. Better non-motor outcome is classically related to a more anterior, medial, and ventral stimulation site, mapping with limbic connections of the STN [284], whereas thalamic and pallidal stimulation are regarded as more neutral for mood. However, postoperative depression is regarded as the main risk factor for suicide after STN-DBS [285]. Some studies have suggested a greater risk of suicide attempt after STN-DBS than in the general population within the first 3 years after surgery [286], but others did not support this finding [287].
The use of repetitive transcranial magnetic stimulation over the dorsolateral prefrontal cortex in PD depression could provide some, but modest, improvement [21, 288]. Contrastingly, meta-analyses confirmed that electroconvulsive therapy had important beneficial effects for both motor symptoms and severe depression in PD [289, 290].
In addition, randomized clinical trials consistently demonstrated the effectiveness of cognitive behavioral therapy in depressed patients with PD [21, 288]. Interestingly, use of telemedicine with video-to-home cognitive behavioral therapy was recently demonstrated to be an effective treatment in randomized controlled clinical trials in comparison with usual clinic-based treatment [291], in addition to the efficacy of telephone-based cognitive behavioral therapy [292, 293], confirming an earlier randomized controlled trial in depressed PD outpatients [294].
Finally, physical activity in PD [295] and other neurodegenerative disorders [296] may have a beneficial effect to treat depression, in addition to global and motor benefits, in particular for aerobic training exercise, Qiqong, dance, and weekly or biweekly yoga. Indeed, exercise was demonstrated to enhance mesolimbic functioning and increase dopaminergic release in the caudate nucleus [297], and may promote the anti-inflammatory response [298, 299]. Efficacy of expression therapy also suggested that ‘laughter was the best medicine’ [300]. In addition, in a randomized controlled trial, bright-light therapy was found to be effective for depression in PD [301].
5 Conclusions and Future Developments
Enhancement of SSRI efficacy could be evaluated, especially in non-responders. This relates to the use of S-adenosyl methionine in monotherapy [302] or add-on therapy [303]. This component is involved in catechol-O-methyltransferase (COMT)-dependent metabolism of catecholamines and serotonin [304]. Combined therapy using mirtazapine and fluoxetine may also prove effective [305] but remains untested in PD. In addition, no definitive evidence is available regarding the antidepressant efficacy of bupropion in PD [306,307,308], presenting a favorable dual norepinephrine and dopamine reuptake inhibition profile. Triple reuptake inhibitors may also have a favorable outcome in PD to alleviate both motor and non-motor symptoms [309,310,311]. Overall, accurate diagnosis of depression, even when its severity is considered mild, and recognition of comorbid motor and non-motor symptoms in PD, at each stage, is critical to offer effective relief for the patient and improve quality of life. Undoubtedly, further research is needed to improve the performance of screening tools in the clinical context, and simplify the use and enhance the specificity of clinical scales for depression, apathy and anxiety in patients with PD [38, 312]. These developments will prove instrumental for the recognition of imaging biomarkers specific to PD depression, which may in turn serve as enrichment biomarkers in clinical trials [313].
However, increasing understanding of pathophysiological mechanisms already help to implement tailored interventions in PD depression and related comorbid symptoms by targeting the neurodegenerative processes that underpin PD subtypes and their prognosis. Furthermore, it is expected that the occurrence and course of PD depression will change with the development of disease-modifying treatments and interventions to promote compensatory plasticity, in particular in patients with prodromal and early PD.
Change history
03 September 2022
Missing Open Access funding information has been added in the Funding Note.
References
Gallagher DA, Lees AJ, Schrag A. What are the most important nonmotor symptoms in patients with Parkinson’s disease and are we missing them? Mov Disord. 2010;25:2493–500.
The Global Parkinson’s Disease Survey (GPDS) Steering Committee. Factors impacting on quality of life in Parkinson’s disease: Results from an international survey. Mov Disord. 2002;17:60–7.
Carter JH, Stewart BJ, Lyons KS, Archbold PG. Do motor and nonmotor symptoms in PD patients predict caregiver strain and depression? Mov Disord. 2008;23:1211–6.
Kulisevsky J, Pagonabarraga J, Pascual-Sedano B, García-Sánchez C, Gironell A, Trapecio GS. Prevalence and correlates of neuropsychiatric symptoms in Parkinson’s disease without dementia. Mov Disord. 2008;23:1889–96.
Weintraub D, Burn DJ. Parkinson’s disease: the quintessential neuropsychiatric disorder. Mov Disord. 2011;26:1022–31.
Montanaro E, Artusi CA, Rosano C, Boschetto C, Imbalzano G, Romagnolo A, et al. Anxiety, depression, and worries in advanced Parkinson disease during COVID-19 pandemic. Neurol Sci. 2022;43:341–8.
Barone P, Antonini A, Colosimo C, Marconi R, Morgante L, Avarello TP, et al. The PRIAMO study: a multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson’s disease. Mov Disord. 2009;24:1641–9.
Aarsland D, Brønnick K, Alves G, Tysnes OB, Pedersen KF, Ehrt U, et al. The spectrum of neuropsychiatric symptoms in patients with early untreated Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2009;80:928–30.
Martinez-Martin P, Rodriguez-Blazquez C, Kurtis MM, Chaudhuri KR, on B of the NVG. The impact of non-motor symptoms on health-related quality of life of patients with Parkinson’s disease. Mov Disord 2011;26:399–406.
Hughes TA, Ross HF, Mindham RHS, Spokes EGS. Mortality in Parkinson’s disease and its association with dementia and depression. Acta Neurol Scand. 2004;110:118–23.
Kritzinger C, Vollstedt E-J, Hückelheim K, Lorwin A, Graf J, Tunc S, et al. Qualitative characteristics of depression in Parkinson’s patients and controls. Behav Neurol 2015 ;2015:e961372.
Schrag A, Taddei RN. Chapter twenty—depression and anxiety in Parkinson’s disease. In: Chaudhuri KR, Titova N, editors. Int Rev Neurobiol. Academic Press; 2017. p. 623–55.
Duncan GW, Khoo TK, Yarnall AJ, O’Brien JT, Coleman SY, Brooks DJ, et al. Health-related quality of life in early Parkinson’s disease: the impact of nonmotor symptoms. Mov Disord. 2014;29:195–202.
Reijnders JSAM, Ehrt U, Weber WEJ, Aarsland D, Leentjens AFG. A systematic review of prevalence studies of depression in Parkinson’s disease: the prevalence of depression in PD. Mov Disord. 2008;23:183–9.
Chaudhuri KR, Sauerbier A. Parkinson disease. Unravelling the nonmotor mysteries of Parkinson disease. Nat Rev Neurol 2016;12:10–1.
Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902.
Ferrari F, Villa RF. The neurobiology of depression: an integrated overview from biological theories to clinical evidence. Mol Neurobiol. 2017;54:4847–65.
Ghazi-Noori S, Chung TH, Deane K, Rickards HE, Clarke CE. Therapies for depression in Parkinson’s disease. Cochrane Database Syst Rev. 2003;3:CD003465.
Elbers RG, Verhoef J, van Wegen EE, Berendse HW, Kwakkel G. Interventions for fatigue in Parkinson’s disease. Cochrane Database Syst Rev. 2015;10:CD010925.
Fox SH, Katzenschlager R, Lim S-Y, Barton B, de Bie RMA, Seppi K, et al. International Parkinson and movement disorder society evidence-based medicine review: update on treatments for the motor symptoms of Parkinson’s disease: treatment of motor symptoms in PD. Mov Disord. 2018;33:1248–66.
Seppi K, Ray Chaudhuri K, Coelho M, Fox SH, Katzenschlager R, Perez Lloret S, et al. Update on treatments for nonmotor symptoms of Parkinson’s disease—an evidence-based medicine review. Mov Disord. 2019;34:180–98.
Riedel O, Bitters D, Amann U, Garbe E, Langner I. Estimating the prevalence of Parkinson’s disease (PD) and proportions of patients with associated dementia and depression among the older adults based on secondary claims data. Int J Geriatr Psychiatry. 2016;31:938–43.
Riedel O, Heuser I, Klotsche J, Dodel R, Wittchen H-U, GEPAD Study Group. Occurrence risk and structure of depression in Parkinson disease with and without dementia: results from the GEPAD Study. J Geriatr Psychiatry Neurol. 2010;23:27–34.
Hantz P, Caradoc-Davies G, Caradoc-Davies T, Weatherall M, Dixon G. Depression in Parkinson’s disease. Am J Psychiatry. 1994;151:1010–4.
Tandberg E, Larsen JP, Aarsland D, Cummings JL. The occurrence of depression in Parkinson’s disease. A community-based study. Arch Neurol. 1996;53:175–9.
Ehmann TS, Beninger RJ, Gawel MJ, Riopelle RJ. Depressive symptoms in Parkinson’s disease: a comparison with disabled control subjects. J Geriatr Psychiatry Neurol. 1990;3:3–9.
Goodarzi Z, Mrklas KJ, Roberts DJ, Jette N, Pringsheim T, Holroyd-Leduc J. Detecting depression in Parkinson disease. Neurology. 2016;87:426–37.
Rojo A, Aguilar M, Garolera MT, Cubo E, Navas I, Quintana S. Depression in Parkinson’s disease: clinical correlates and outcome. Parkinsonism Relat Disord. 2003;10:23–8.
Hsu Y-T, Liao C-C, Chang S-N, Yang Y-W, Tsai C-H, Chen T-L, et al. Increased risk of depression in patients with Parkinson disease: a nationwide cohort study. Am J Geriatr Psychiatry. 2015;23:934–40.
Orayj K, Almeleebia T, Vigneshwaran E, Alshahrani S, Alavudeen-Sirajudeen S, Alghamdi W. Trend of recognizing depression symptoms and antidepressants use in newly diagnosed Parkinson’s disease: population-based study. Brain Behav. 2021;11:e2228.
Weintraub D, Simuni T, Caspell-Garcia C, Coffey C, Lasch S, Siderowf A, et al. Cognitive performance and neuropsychiatric symptoms in early, untreated Parkinson’s disease. Mov Disord. 2015;30:919–27.
Hommel ALAJ, Meinders MJ, Lorenzl S, Dodel R, Coelho M, Ferreira JJ, et al. The prevalence and determinants of neuropsychiatric symptoms in late-stage Parkinsonism. Mov Disord Clin Pract. 2020;7:531–42.
Miller KM, Okun MS, Fernandez HF, Jacobson CE, Rodriguez RL, Bowers D. Depression symptoms in movement disorders: comparing Parkinson’s disease, dystonia, and essential tremor. Mov Disord. 2007;22:666–72.
Shulman LM, Taback RL, Rabinstein AA, Weiner WJ. Non-recognition of depression and other non-motor symptoms in Parkinson’s disease. Parkinsonism Relat Disord. 2002;8:193–7.
Marinus J, Zhu K, Marras C, Aarsland D, van Hilten JJ. Risk factors for non-motor symptoms in Parkinson’s disease. Lancet Neurol. 2018;17:559–68.
Leentjens AFG, Dujardin K, Marsh L, Martinez-Martin P, Richard IH, Starkstein SE, et al. Apathy and anhedonia rating scales in Parkinson’s disease: critique and recommendations. Mov Disord. 2008;23:2004–14.
Martinez-Martin P, Leentjens AFG, de Pedro-Cuesta J, Chaudhuri KR, Schrag AE, Weintraub D. Accuracy of screening instruments for detection of neuropsychiatric syndromes in Parkinson’s disease. Mov Disord. 2016;31:270–9.
Schrag A, Barone P, Brown RG, Leentjens AFG, McDonald WM, Starkstein S, et al. Depression rating scales in Parkinson’s disease: critique and recommendations. Mov Disord. 2007;22:1077–92.
Starkstein SE, Leentjens AFG. The nosological position of apathy in clinical practice. J Neurol Neurosurg Psychiatry. 2008;79:1088–92.
Visser M, Leentjens AFG, Marinus J, Stiggelbout AM, van Hilten JJ. Reliability and validity of the Beck depression inventory in patients with Parkinson’s disease. Mov Disord. 2006;21:668–72.
Marsh L, McDonald WM, Cummings J, Ravina B, NINDS/NIMH Work Group on Depression and Parkinson’s Disease. Provisional diagnostic criteria for depression in Parkinson’s disease: Report of an NINDS/NIMH Work Group. Mov Disord. 2006;21:148–58.
Aarsland D, Påhlhagen S, Ballard CG, Ehrt U, Svenningsson P. Depression in Parkinson disease—epidemiology, mechanisms and management. Nat Rev Neurol. 2012;8:35–47.
Prange S, Metereau E, Maillet A, Klinger H, Schmitt E, Lhommée E, et al. Limbic serotonergic plasticity contributes to the compensation of apathy in early Parkinson’s disease. Mov Disord. https://doi.org/10.1002/mds.28971. (Epub 3 Mar 2022).
Thobois S, Prange S, Sgambato-Faure V, Tremblay L, Broussolle E. Imaging the etiology of apathy, anxiety, and depression in Parkinson’s disease: implication for treatment. Curr Neurol Neurosci Rep. 2017;17:76.
Prange S, Metereau E, Maillet A, Lhommée E, Klinger H, Pelissier P, et al. Early limbic microstructural alterations in apathy and depression in de novo Parkinson’s disease. Mov Disord. 2019;34:1644–54.
Sockeel P, Dujardin K, Devos D, Denève C, Destée A, Defebvre L. The Lille apathy rating scale (LARS), a new instrument for detecting and quantifying apathy: validation in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2006;77:579–84.
Prange S, Danaila T, Laurencin C, Caire C, Metereau E, Merle H, et al. Age and time course of long-term motor and nonmotor complications in Parkinson disease. Neurology. 2019;92: e148.
Ravina B, Camicioli R, Como PG, Marsh L, Jankovic J, Weintraub D, et al. The impact of depressive symptoms in early Parkinson disease. Neurology. 2007;69:342–7.
Becker C, Brobert GP, Johansson S, Jick SS, Meier CR. Risk of incident depression in patients with Parkinson disease in the UK: depression in Parkinson disease. Eur J Neurol. 2011;18:448–53.
Davous P, Auquier P, Grignon S, Neukirch HC. A prospective study of depression in French patients with Parkinson’s disease. The Depar study. Eur J Neurol. 1995;2:455–61.
Riedel O, Klotsche J, Spottke A, Deuschl G, Förstl H, Henn F, et al. Frequency of dementia, depression, and other neuropsychiatric symptoms in 1,449 outpatients with Parkinson’s disease. J Neurol. 2010;257:1073–82.
Murphy JM, Olivier DC, Monson RR, Sobol AM, Leighton AH. Incidence of depression and anxiety: the Stirling County Study. Am J Public Health. 1988;78:534–40.
Bertucci Filho D, Teive HAG, Werneck LC. Early-onset Parkinson’s disease and depression. Arq Neuropsiquiatr. 2007;65:5–10.
Dissanayaka NN, O’Sullivan JD, Silburn PA, Mellick GD. Assessment methods and factors associated with depression in Parkinson’s disease. J Neurol Sci. 2011;310:208–10.
Farabaugh AH, Locascio JJ, Yap L, Fava M, Bitran S, Sousa JL, et al. Assessing depression and factors possibly associated with depression during the course of Parkinson’s disease. Ann Clin Psychiatry. 2011;23:171–7.
Gu S-C, Zhou J, Yuan C-X, Ye Q. Personalized prediction of depression in patients with newly diagnosed Parkinson’s disease: a prospective cohort study. J Affect Disord. 2020;268:118–26.
Wu Y-H, Chen Y-H, Chang M-H, Lin C-H. Depression in Parkinson’s disease: a case-control study. PLoS ONE. 2018;13: e0192050.
Yapici Eser H, Bora HA, Kuruoğlu A. Depression and Parkinson disease: prevalence, temporal relationship, and determinants. Turk J Med Sci. 2017;47:499–503.
Leentjens AFG, Lousberg R, Verhey FRJ. Markers for depression in Parkinson’s disease. Acta Psychiatr Scand. 2002;106:196–201.
Leentjens AFG, Moonen AJH, Dujardin K, Marsh L, Martinez-Martin P, Richard IH, et al. Modeling depression in Parkinson disease: Disease-specific and nonspecific risk factors. Neurology. 2013;81:1036–43.
Neikrug AB, Avanzino JA, Liu L, Maglione JE, Natarajan L, Corey-Bloom J, et al. Parkinson’s disease and REM sleep behavior disorder result in increased non-motor symptoms. Sleep Med. 2014;15:959–66.
Sagna A, Gallo JJ, Pontone GM. Systematic review of factors associated with depression and anxiety disorders among older adults with Parkinson’s disease. Parkinsonism Relat Disord. 2014;20:708–15.
Brown R, Jahanshahi M. Depression in Parkinson’s disease: a psychosocial viewpoint. Adv Neurol. 1995;65:61–84.
Bega D, Luo S, Fernandez H, Chou K, Aminoff M, Parashos S, et al. Impact of depression on progression of impairment and disability in early Parkinson’s disease. Mov Disord Clin Pract. 2015;2:371–8.
Holroyd S, Currie LJ, Wooten GF. Depression is associated with impairment of ADL, not motor function in Parkinson disease. Neurology. 2005;64:2134–5.
Lubomski M, Davis RL, Sue CM. Depression in Parkinson’s disease: Perspectives from an Australian cohort. J Affect Disord. 2020;277:1038–44.
Alves G, Wentzel-Larsen T, Aarsland D, Larsen JP. Progression of motor impairment and disability in Parkinson disease: a population-based study. Neurology. 2005;65:1436–41.
Dissanayaka NNW, Sellbach A, Silburn PA, O’Sullivan JD, Marsh R, Mellick GD. Factors associated with depression in Parkinson’s disease. J Affect Disord. 2011;132:82–8.
Papapetropoulos S, Ellul J, Argyriou AA, Chroni E, Lekka NP. The effect of depression on motor function and disease severity of Parkinson’s disease. Clin Neurol Neurosurg. 2006;108:465–9.
Marras C, McDermott MP, Rochon PA, Tanner CM, Naglie G, Lang AE, et al. Predictors of deterioration in health-related quality of life in Parkinson’s disease: Results from the DATATOP trial. Mov Disord. 2008;23:653–9.
Post B, Muslimovic D, van Geloven N, Speelman JD, Schmand B, de Haan RJ, et al. Progression and prognostic factors of motor impairment, disability and quality of life in newly diagnosed Parkinson’s disease. Mov Disord. 2011;26:449–56.
Pontone GM, Bakker CC, Chen S, Mari Z, Marsh L, Rabins PV, et al. The longitudinal impact of depression on disability in Parkinson disease. Int J Geriatr Psychiatry. 2016;31:458–65.
Nègre-Pagès L, Grandjean H, Lapeyre-Mestre M, Montastruc JL, Fourrier A, Lépine JP, et al. Anxious and depressive symptoms in Parkinson’s disease: the French cross-sectionnal DoPaMiP study: anxiety in Parkinson’s disease. Mov Disord. 2010;25:157–66.
Starkstein SE, Mayberg HS, Leiguarda R, Preziosi TJ, Robinson RG. A prospective longitudinal study of depression, cognitive decline, and physical impairments in patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1992;55:377–82.
Ng A, Chander RJ, Tan LCS, Kandiah N. Influence of depression in mild Parkinson’s disease on longitudinal motor and cognitive function. Parkinsonism Relat Disord. 2015;21:1056–60.
Tandberg E, Larsen JP, Aarsland D, Laake K, Cummings JL. Risk factors for depression in Parkinson disease. Arch Neurol. 1997;54:625–30.
Wichowicz HM, Sławek J, Derejko M, Cubała WJ. Factors associated with depression in Parkinson’s disease: a cross-sectional study in a Polish population. Eur Psychiatry J. 2006;21:516–20.
Canesi M, Lavolpe S, Cereda V, Ranghetti A, Maestri R, Pezzoli G, et al. Hypomania, depression, euthymia: new evidence in Parkinson’s disease. Behav Neurol. 2020;2020: e5139237.
Callesen MB, Weintraub D, Damholdt MF, Møller A. Impulsive and compulsive behaviors among Danish patients with Parkinson’s disease: prevalence, depression, and personality. Parkinsonism Relat Disord. 2014;20:22–6.
Voon V, Sohr M, Lang AE, Potenza MN, Siderowf AD, Whetteckey J, et al. Impulse control disorders in parkinson disease: a multicenter case–control study. Ann Neurol. 2011;69:986–96.
Joutsa J, Martikainen K, Vahlberg T, Voon V, Kaasinen V. Impulse control disorders and depression in Finnish patients with Parkinson’s disease. Parkinsonism Relat Disord. 2012;18:155–60.
Nutt JG. Motor subtype in Parkinson’s disease: different disorders or different stages of disease? Mov Disord. 2016;31:957–61.
van der Hoek TC, Bus BAA, Matui P, van der Marck MA, Esselink RA, Tendolkar I. Prevalence of depression in Parkinson’s disease: effects of disease stage, motor subtype and gender. J Neurol Sci. 2011;310:220–4.
Jankovic J, McDermott M, Carter J, Gauthier S, Goetz C, Golbe L, et al. Variable expression of Parkinson’s disease A base-line analysis of the DAT ATOP cohort. Neurology. 1990;40:1529–1529.
Lord S, Galna B, Coleman S, Burn D, Rochester L. Mild depressive symptoms are associated with gait impairment in early Parkinson’s disease. Mov Disord. 2013;28:634–9.
Burn DJ, Landau S, Hindle JV, Samuel M, Wilson KC, Hurt CS, et al. Parkinson’s disease motor subtypes and mood: Parkinson’s disease motor subtypes and mood. Mov Disord. 2012;27:379–86.
Factor SA, Steenland NK, Higgins DS, Molho ES, Kay DM, Montimurro J, et al. Postural instability/gait disturbance in Parkinson’s disease has distinct subtypes: an exploratory analysis. J Neurol Neurosurg Psychiatry. 2011;82:564–8.
Reijnders JSAM, Ehrt U, Lousberg R, Aarsland D, Leentjens AFG. The association between motor subtypes and psychopathology in Parkinson’s disease. Parkinsonism Relat Disord. 2009;15:379–82.
Lewis SJG, Foltynie T, Blackwell AD, Robbins TW, Owen AM, Barker RA. Heterogeneity of Parkinson’s disease in the early clinical stages using a data driven approach. J Neurol Neurosurg Psychiatry. 2005;76:343–8.
Dissanayaka NNW, Sellbach A, Matheson S, O’Sullivan JD, Silburn PA, Byrne GJ, et al. Anxiety disorders in Parkinson’s disease: Prevalence and risk factors. Mov Disord. 2010;25:838–45.
Barber J, Tomer R, Sroka H, Myslobodsky MS. Does unilateral dopamine deficit contribute to depression? Psychiatry Res. 1985;15:17–24.
Fleminger S. Left-sided Parkinson’s disease is associated with greater anxiety and depression. Psychol Med. 1991;21:629–38.
Chang Y-P, Lee M-S, Wu D-W, Tsai J-H, Ho P-S, Lin C-HR, et al. Risk factors for depression in patients with Parkinson’s disease: A nationwide nested case-control study. PLOS One. 2020;15:e0236443.
Marinus J, Leentjens AFG, Visser M, Stiggelbout AM, van Hilten JJ. Evaluation of the hospital anxiety and depression scale in patients with Parkinson’s disease. Clin Neuropharmacol. 2002;25:318–24.
Menza MA, Robertson-Hoffman DE, Bonapace AS. Parkinson’s disease and anxiety: comorbidity with depression. Biol Psychiatry. 1993;34:465–70.
Qureshi SU, Amspoker AB, Calleo JS, Kunik ME, Marsh L. Anxiety disorders, physical illnesses, and health care utilization in older male veterans with Parkinson disease and comorbid depression. J Geriatr Psychiatry Neurol. 2012;25:233–9.
Wee N, Kandiah N, Acharyya S, Chander RJ, Ng A, Au WL, et al. Depression and anxiety are co-morbid but dissociable in mild Parkinson’s disease: a prospective longitudinal study of patterns and predictors. Parkinsonism Relat Disord. 2016;23:50–6.
Pontone GM, Williams JR, Anderson KE, Chase G, Goldstein SA, Grill S, et al. Prevalence of anxiety disorders and anxiety subtypes in patients with Parkinson’s disease. Mov Disord. 2009;24:1333–8.
Pagonabarraga J, Kulisevsky J, Strafella AP, Krack P. Apathy in Parkinson’s disease: clinical features, neural substrates, diagnosis, and treatment. Lancet Neurol. 2015;14:518–31.
Oguru M, Tachibana H, Toda K, Okuda B, Oka N. Apathy and depression in Parkinson disease. J Geriatr Psychiatry Neurol. 2010;23:35–41.
Kirsch-Darrow L, Fernandez HF, Marsiske M, Okun MS, Bowers D. Dissociating apathy and depression in Parkinson disease. Neurology. 2006;67:33–8.
Kirsch-Darrow L, Marsiske M, Okun MS, Bauer R, Bowers D. Apathy and depression: separate factors in Parkinson’s disease. J Int Neuropsychol Soc. 2011;17:1058–66.
Ziropadja L, Stefanova E, Petrovic M, Stojkovic T, Kostic VS. Apathy and depression in Parkinson’s disease: the Belgrade PD study report. Parkinsonism Relat Disord. 2012;18:339–42.
Brown RG, MacCarthy B, Gotham AM, Der GJ, Marsden CD. Depression and disability in Parkinson’s disease: a follow-up of 132 cases. Psychol Med. 1988;18:49–55.
Mayeux R, Stern Y, Rosen J, Leventhal J. Depression, intellectual impairment, and Parkinson disease. Neurology. 1981;31:645–50.
Sano M, Stern Y, Williams J, Coté L, Rosenstein R, Mayeux R. Coexisting dementia and depression in Parkinson’s disease. Arch Neurol. 1989;46:1284–6.
Ehgoetz Martens KA, Silveira CRA, Intzandt BN, Almeida QJ. State anxiety predicts cognitive performance in patients with Parkinson’s disease. Neuropsychology. 2018;32:950–7.
Fernandez HH, See RH, Gary MF, Bowers D, Rodriguez RL, Jacobson C, et al. Depressive symptoms in Parkinson disease correlate with impaired global and specific cognitive performance. J Geriatr Psychiatry Neurol. 2009;22:223–7.
Jones JD, Mangal P, Lafo J, Okun MS, Bowers D. Mood differences among Parkinson’s disease patients with mild cognitive impairment. J Neuropsychiatry Clin Neurosci. 2016;28:211–6.
Petkus AJ, Filoteo JV, Schiehser DM, Gomez ME, Hui JS, Jarrahi B, et al. Mild cognitive impairment, psychiatric symptoms, and executive functioning in patients with Parkinson’s disease. Int J Geriatr Psychiatry. 2020;35:396–404.
Reynolds GO, Hanna KK, Neargarder S, Cronin-Golomb A. The relation of anxiety and cognition in Parkinson’s disease. Neuropsychology. 2017;31:596–604.
Nation DA, Katzen HL, Papapetropoulos S, Scanlon BK, Levin BE. Subthreshold depression in Parkinson’s disease. Int J Geriatr Psychiatry. 2009;24:937–43.
Rodríguez-Violante M, Cervantes-Arriaga A, Berlanga-Flores C, Ruiz-Chow A. Prevalence and determinants of depression in Mexican patients with Parkinson’s disease. Clin Neurol Neurosurg. 2012;114:1293–6.
Morgante L, Colosimo C, Antonini A, Marconi R, Meco G, Pederzoli M, et al. Psychosis associated to Parkinson’s disease in the early stages: relevance of cognitive decline and depression. J Neurol Neurosurg Psychiatry. 2012;83:76–82.
Hu MTM, Szewczyk-Królikowski K, Tomlinson P, Nithi K, Rolinski M, Murray C, et al. Predictors of cognitive impairment in an early stage Parkinson’s disease cohort. Mov Disord. 2014;29:351–9.
Ismail Z, Smith EE, Geda Y, Sultzer D, Brodaty H, Smith G, et al. Neuropsychiatric symptoms as early manifestations of emergent dementia: provisional diagnostic criteria for mild behavioral impairment. Alzheimers Dement. 2016;12:195–202.
Pirogovsky-Turk E, Moore RC, Filoteo JV, Litvan I, Song DD, Lessig SL, et al. Neuropsychiatric predictors of cognitive decline in Parkinson disease: a longitudinal study. Am J Geriatr Psychiatry. 2017;25:279–89.
Pantzar A, Atti AR, Fratiglioni L, Fastbom J, Bäckman L, Laukka EJ. Cognitive performance in unipolar old-age depression: a longitudinal study. Int J Geriatr Psychiatry. 2017;32:675–84.
Park JH, Lee SH, Kim Y, Park S-W, Byeon GH, Jang J-W, et al. Depressive symptoms are associated with worse cognitive prognosis in patients with newly diagnosed idiopathic Parkinson disease. Psychogeriatr. 2020;20:880–90.
Jones JD, Kurniadi NE, Kuhn TP, Szymkowicz SM, Bunch J, Rahmani E. Depressive symptoms precede cognitive impairment in de novo Parkinson’s disease patients: analysis of the PPMI cohort. Neuropsychology. 2019;33:1111–20.
Petkus AJ, Filoteo JV, Schiehser DM, Gomez ME, Petzinger G. Worse cognitive performance predicts increased anxiety and depressive symptoms in patients with Parkinson’s disease: a bidirectional analysis. Neuropsychology. 2019;33:35–46.
Rutten S, van der Ven PM, Weintraub D, Pontone GM, Leentjens AFG, Berendse HW, et al. Predictors of anxiety in early-stage Parkinson’s disease—results from the first two years of a prospective cohort study. Parkinsonism Relat Disord. 2017;43:49–55.
Xu Y-Y, Kuo S-H, Liang Z, Xu H, Feng W-R, Yu C-Y, et al. The natural history of depression in Parkinson’s disease within 30-month follow-up. Park Dis. 2015;2015: 362892.
Dujardin K, Sockeel P, Delliaux M, Destée A, Defebvre L. Apathy may herald cognitive decline and dementia in Parkinson’s disease: Apathy in Parkinson’s Disease. Mov Disord. 2009;24:2391–7.
Butterfield LC, Cimino CR, Oelke LE, Hauser RA, Sanchez-Ramos J. The independent influence of apathy and depression on cognitive functioning in Parkinson’s disease. Neuropsychology. 2010;24:721–30.
Varanese S, Perfetti B, Ghilardi MF, Di Rocco A. Apathy, but Not depression, reflects inefficient cognitive strategies in parkinson’s disease. Aleman A (ed). PLoS One. 2011;6:e17846.
Liu Y, Lawton MA, Lo C, Bowring F, Klein JC, Querejeta-Coma A, et al. Longitudinal changes in parkinson’s disease symptoms with and without rapid eye movement sleep behavior disorder: the Oxford Discovery Cohort Study. Mov Disord. 2021;36(12):2821–32.
Duarte Folle A, Paul KC, Bronstein JM, Keener AM, Ritz B. Clinical progression in Parkinson’s disease with features of REM sleep behavior disorder: a population-based longitudinal study. Parkinsonism Relat Disord. 2019;62:105–11.
Pagano G, De Micco R, Yousaf T, Wilson H, Chandra A, Politis M. REM behavior disorder predicts motor progression and cognitive decline in Parkinson disease. Neurology. 2018;91:e894-905.
Cui J, Qin Y, Tian Y, Ge X, Han H, Yang Z, et al. Activities of daily living as a longitudinal moderator of the effect of autonomic dysfunction on anxiety and depression of Parkinson’s patients. Brain Behav. 2021;11: e2297.
Sklerov M, Shih C-H, Browner N, Palma J-A, Styner M, Dayan E. Longitudinal change in autonomic symptoms predicts activities of daily living and depression in Parkinson’s disease. Clin Auton Res. 2020;30:223–30.
Matsubara T, Suzuki K, Fujita H, Watanabe Y, Sakuramoto H, Matsubara M, et al. Autonomic symptoms correlate with non-autonomic non-motor symptoms and sleep problems in patients with Parkinson’s disease. Eur Neurol. 2018;80:193–9.
Xiao-ling Q, Gang C, Bo L, Zai-li L, Xue-kui L, Xue L, et al. Depression is associated with constipation in patients with Parkinson’s disease. Front Neurol. 2020;11: 567574.
Pablo-Fernandez ED, Tur C, Revesz T, Lees AJ, Holton JL, Warner TT. Association of autonomic dysfunction with disease progression and survival in Parkinson disease. JAMA Neurol. 2017;74:970–6.
Ehrt U, Larsen JP, Aarsland D. Pain and its relationship to depression in Parkinson disease. Am J Geriatr Psychiatry. 2009;17:269–75.
Valkovic P, Minar M, Singliarova H, Harsany J, Hanakova M, Martinkova J, et al. Pain in Parkinson’s disease: a cross-sectional study of its prevalence, types, and relationship to depression and quality of life. PLoS ONE. 2015;10: e0136541.
Sunwoo MK, Hong JY, Lee JE, Lee HS, Lee PH, Sohn YH. Depression and voice handicap in Parkinson disease. J Neurol Sci. 2014;346:112–5.
Rutten S, Vriend C, van der Werf YD, Berendse HW, Weintraub D, van den Heuvel OA. The bidirectional longitudinal relationship between insomnia, depression and anxiety in patients with early-stage, medication-naïve Parkinson’s disease. Parkinsonism Relat Disord. 2017;39:31–6.
Ravina B, Elm J, Camicioli R, Como PG, Marsh L, Jankovic J, et al. The course of depressive symptoms in early Parkinson’s disease. Mov Disord. 2009;24:1306–11.
Erro R, Picillo M, Vitale C, Amboni M, Moccia M, Longo K, et al. Non-motor symptoms in early Parkinson’s disease: a 2-year follow-up study on previously untreated patients. J Neurol Neurosurg Psychiatry. 2013;84:14–7.
Wile DJ, Agarwal PA, Schulzer M, Mak E, Dinelle K, Shahinfard E, et al. Serotonin and dopamine transporter PET changes in the premotor phase of LRRK2 parkinsonism: cross-sectional studies. Lancet Neurol. 2017;16:351–9.
Fang F, Xu Q, Park Y, Huang X, Hollenbeck A, Blair A, et al. Depression and the subsequent risk of Parkinson’s disease in the NIH-AARP Diet and Health Study. Mov Disord. 2010;25:1157–62.
Leentjens AFG, den Akker MV, Metsemakers JFM, Lousberg R, Verhey FRJ. Higher incidence of depression preceding the onset of Parkinson’s disease: a register study. Mov Disord. 2003;18:414–8.
Shiba M, Bower JH, Maraganore DM, McDonnell SK, Peterson BJ, Ahlskog JE, et al. Anxiety disorders and depressive disorders preceding Parkinson’s disease: a case-control study. Mov Disord. 2000;15:9.
Nilsson FM, Kessing LV, Bolwig TG. Increased risk of developing Parkinson’s disease for patients with major affective disorder: a register study. Acta Psychiatr Scand. 2001;104:380–6.
Schuurman AG, van den Akker M, Ensinck KTJL, Metsemakers JFM, Knottnerus JA, Leentjens AFG, et al. Increased risk of Parkinson’s disease after depression: a retrospective cohort study. Neurology. 2002;58:1501–4.
Noyce AJ, Bestwick JP, Silveira-Moriyama L, Hawkes CH, Giovannoni G, Lees AJ, et al. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann Neurol. 2012;72:893–901.
Schrag A, Horsfall L, Walters K, Noyce A, Petersen I. Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. Lancet Neurol. 2015;14:57–64.
Walter U, Heilmann R, Kaulitz L, Just T, Krause BJ, Benecke R, et al. Prediction of Parkinson’s disease subsequent to severe depression: a ten-year follow-up study. J Neural Transm. 2015;122:789–97.
Piredda R, Desmarais P, Masellis M, Gasca-Salas C. Cognitive and psychiatric symptoms in genetically determined Parkinson’s disease: a systematic review. Eur J Neurol. 2020;27:229–34.
Lohmann E, Thobois S, Lesage S, Broussolle E, du Montcel ST, Ribeiro M-J, et al. A multidisciplinary study of patients with early-onset PD with and without parkin mutations. Neurology. 2009;72:110–6.
Srivastava A, Tang M-X, Mejia-Santana H, Rosado L, Louis ED, Caccappolo E, et al. The relation between depression and parkin genotype: The CORE-PD study. Parkinsonism Relat Disord. 2011;17:740–4.
Pankratz N, Marder KS, Halter CA, Rudolph A, Shults CW, Nichols WC, et al. Clinical correlates of depressive symptoms in familial Parkinson’s disease. Mov Disord. 2008;23:2216–23.
Simuni T, Uribe L, Cho HR, Caspell-Garcia C, Coffey CS, Siderowf A, et al. Clinical and dopamine transporter imaging characteristics of non-manifest LRRK2 and GBA mutation carriers in the Parkinson’s Progression Markers Initiative (PPMI): a cross-sectional study. Lancet Neurol. 2020;19:71–80.
Gaig C, Vilas D, Infante J, Sierra M, García-Gorostiaga I, Buongiorno M, et al. Nonmotor symptoms in LRRK2 G2019S associated Parkinson’s disease. PLoS ONE. 2014;9: e108982.
Kasten M, Kertelge L, Brüggemann N, van der Vegt J, Schmidt A, Tadic V, et al. Nonmotor symptoms in genetic Parkinson disease. Arch Neurol. 2010;67:670–6.
D’Souza T, Rajkumar AP. Systematic review of genetic variants associated with cognitive impairment and depressive symptoms in Parkinson’s disease. Acta Neuropsychiatr. 2020;32:10–22.
Srivatsal S, Cholerton B, Leverenz JB, Wszolek ZK, Uitti RJ, Dickson DW, et al. Cognitive profile of LRRK2-related Parkinson’s disease. Mov Disord. 2015;30:728–33.
Beavan M, McNeill A, Proukakis C, Hughes DA, Mehta A, Schapira AHV. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. 2015;72:201–8.
Burn DJ, Tiangyou W, Allcock LM, Davison J, Chinnery PF. Allelic variation of a functional polymorphism in the serotonin transporter gene and depression in Parkinson’s disease. Parkinsonism Relat Disord. 2006;12:139–41.
Dissanayaka NNW, Silburn PA, O’Sullivan JD, Mellick GD. Serotonin and dopamine transporter genes do not influence depression in Parkinson’s disease. Mov Disord. 2009;24:111–5.
Menza MA, Palermo B, DiPaola R, Sage JI, Ricketts MH. Depression and anxiety in Parkinson’s disease: possible effect of genetic variation in the serotonin transporter. J Geriatr Psychiatry Neurol. 1999;12:49–52.
Mössner R, Henneberg A, Schmitt A, Syagailo YV, Grässle M, Hennig T, et al. Allelic variation of serotonin transporter expression is associated with depression in Parkinson’s disease. Mol Psychiatry. 2001;6:350–2.
Barrero FJ, Ampuero I, Morales B, Vives F, de Dios Luna del Castillo J, Hoenicka J, et al. Depression in Parkinson’s disease is related to a genetic polymorphism of the cannabinoid receptor gene (CNR1). Pharmacogenomics J. 2005;5:135–41.
Haj-Dahmane S, Shen R-Y. Modulation of the serotonin system by endocannabinoid signaling. Neuropharmacology. 2011;61:414–20.
Berg D, Borghammer P, Fereshtehnejad S-M, Heinzel S, Horsager J, Schaeffer E, et al. Prodromal Parkinson disease subtypes — key to understanding heterogeneity. Nat Rev Neurol. 2021;17:349–61.
Sauerbier A, Jenner P, Todorova A, Chaudhuri KR. Non motor subtypes and Parkinson’s disease. Parkinsonism Relat Disord. 2016;22(Supplement 1):S41–6.
Barber TR, Griffanti L, Muhammed K, Drew DS, Bradley KM, McGowan DR, et al. Apathy in rapid eye movement sleep behaviour disorder is associated with serotonin depletion in the dorsal raphe nucleus. Brain. 2018;141:2848–54.
Knudsen K, Fedorova TD, Hansen AK, Sommerauer M, Otto M, Svendsen KB, et al. In-vivo staging of pathology in REM sleep behaviour disorder: a multimodality imaging case-control study. Lancet Neurol. 2018;17:618–28.
Wilson H, Dervenoulas G, Pagano G, Koros C, Yousaf T, Picillo M, et al. Serotonergic pathology and disease burden in the premotor and motor phase of A53T α-synuclein parkinsonism: a cross-sectional study. Lancet Neurol. 2019;18:748–59.
Maillet A, Krack P, Lhommée E, Météreau E, Klinger H, Favre E, et al. The prominent role of serotonergic degeneration in apathy, anxiety and depression in de novo Parkinson’s disease. Brain. 2016;139:2486–502.
Braak H, Tredici KD, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211.
Adler CH, Beach TG, Zhang N, Shill HA, Driver-Dunckley E, Caviness JN, et al. Unified Staging system for lewy body disorders: clinicopathologic correlations and comparison to braak staging. J Neuropathol Exp Neurol. 2019;78:891–9.
Burke RE, Dauer WT, Vonsattel JPG. A critical evaluation of the Braak staging scheme for Parkinson’s disease. Ann Neurol. 2008;64:485–91.
Halliday G, McCann H, Shepherd C. Evaluation of the Braak hypothesis: how far can it explain the pathogenesis of Parkinson’s disease? Expert Rev Neurother. 2012;12:673–86.
Jellinger KA. Is Braak staging valid for all types of Parkinson’s disease? J Neural Transm. 2019;126:423–31.
Schrag A, Jahanshahi M, Quinn NP. What contributes to depression in Parkinson’s disease? Psychol Med. 2001;31:65–73.
Starkstein SE, Petracca G, Chemerinski E, Tesón A, Sabe L, Merello M, et al. Depression in classic versus akinetic-rigid Parkinson’s disease. Mov Disord. 1998;13:29–33.
Remy P, Doder M, Lees A, Turjanski N, Brooks D. Depression in Parkinson’s disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain J Neurol. 2005;128:1314–22.
Weintraub D, Newberg AB, Cary MS, Siderowf AD, Moberg PJ, Kleiner-Fisman G, et al. Striatal dopamine transporter imaging correlates with anxiety and depression symptoms in Parkinson’s disease. J Nucl Med. 2005;46:227–32.
Rektorova I, Srovnalova H, Kubikova R, Prasek J. Striatal dopamine transporter imaging correlates with depressive symptoms and tower of London task performance in Parkinson’s disease. Mov Disord. 2008;23:1580–7.
Hesse S, Meyer PM, Strecker K, Barthel H, Wegner F, Oehlwein C, et al. Monoamine transporter availability in Parkinson’s disease patients with or without depression. Eur J Nucl Med Mol Imaging. 2009;36:428–35.
Oh Y-S, Kim JH, Yoo S-W, Hwang E-J, Lyoo CH, Lee K-S, et al. Neuropsychiatric symptoms and striatal monoamine availability in early Parkinson’s disease without dementia. Neurol Sci. 2021;42:711–8.
Vriend C, Raijmakers P, Veltman DJ, van Dijk KD, van der Werf YD, Foncke EMJ, et al. Depressive symptoms in Parkinson’s disease are related to reduced [123I]FP-CIT binding in the caudate nucleus. J Neurol Neurosurg Psychiatry. 2014;85:159–64.
Frosini D, Unti E, Guidoccio F, Del Gamba C, Puccini G, Volterrani D, et al. Mesolimbic dopaminergic dysfunction in Parkinson’s disease depression: evidence from a 123I-FP-CIT SPECT investigation. J Neural Transm. 2015;122:1143–7.
Di Giuda D, Camardese G, Bentivoglio AR, Cocciolillo F, Guidubaldi A, Pucci L, et al. Dopaminergic dysfunction and psychiatric symptoms in movement disorders: a 123I-FP-CIT SPECT study. Eur J Nucl Med Mol Imaging. 2012;39:1937–48.
Koerts J, Leenders KL, Koning M, Portman AT, Van Beilen M. Striatal dopaminergic activity (FDOPA-PET) associated with cognitive items of a depression scale (MADRS) in Parkinson’s disease. Eur J Neurosci. 2007;25:3132–6.
Santangelo G, Vitale C, Picillo M, Cuoco S, Moccia M, Pezzella D, et al. Apathy and striatal dopamine transporter levels in de-novo, untreated Parkinson’s disease patients. Parkinsonism Relat Disord. 2015;21:489–93.
Meyer JH, Krüger S, Wilson AA, Christensen BK, Goulding VS, Schaffer A, et al. Lower dopamine transporter binding potential in striatum during depression. NeuroReport. 2001;12:4121–5.
Ceravolo R, Frosini D, Poletti M, Kiferle L, Pagni C, Mazzucchi S, et al. Mild affective symptoms in de novo Parkinson’s disease patients: relationship with dopaminergic dysfunction. Eur J Neurol. 2013;20:480–5.
Felicio AC, Moriyama TS, Godeiro-Junior C, Shih MC, Hoexter MQ, Borges V, et al. Higher dopamine transporter density in Parkinson’s disease patients with depression. Psychopharmacology. 2010;211:27–31.
Park SB, Kwon K-Y, Lee J-Y, Im K, Sunwoo J-S, Lee KB, et al. Lack of association between dopamine transporter loss and non-motor symptoms in patients with Parkinson’s disease: a detailed PET analysis of 12 striatal subregions. Neurol Sci. 2019;40:311–7.
Boileau I, Guttman M, Rusjan P, Adams JR, Houle S, Tong J, et al. Decreased binding of the D3 dopamine receptor-preferring ligand [11C]-(+)-PHNO in drug-naive Parkinson’s disease. Brain J Neurol. 2009;132:1366–75.
Murai T, Müller U, Werheid K, Sorger D, Reuter M, Becker T, et al. In vivo evidence for differential association of striatal dopamine and midbrain serotonin systems with neuropsychiatric symptoms in Parkinson’s disease. J Neuropsychiatry Clin Neurosci. 2001;13:222–8.
Qamhawi Z, Towey D, Shah B, Pagano G, Seibyl J, Marek K, et al. Clinical correlates of raphe serotonergic dysfunction in early Parkinson’s disease. Brain. 2015;138:2964–73.
Politis M, Wu K, Loane C, Turkheimer FE, Molloy S, Brooks DJ, et al. Depressive symptoms in PD correlate with higher 5-HTT binding in raphe and limbic structures. Neurology. 2010;75:1920–7.
Boileau I, Warsh JJ, Guttman M, Saint-Cyr JA, McCluskey T, Rusjan P, et al. Elevated serotonin transporter binding in depressed patients with Parkinson’s disease: a preliminary PET study with [11C]DASB. Mov Disord. 2008;23:1776–80.
Maillet A, Météreau E, Tremblay L, Favre E, Klinger H, Lhommée E, et al. Serotonergic and dopaminergic lesions underlying parkinsonian neuropsychiatric signs. Mov Disord. 2021;36:2888–900.
Ballanger B, Klinger H, Eche J, Lerond J, Vallet A-E, Le Bars D, et al. Role of serotonergic 1A receptor dysfunction in depression associated with Parkinson’s disease. Mov Disord. 2012;27:84–9.
Becker T, Becker G, Seufert J, Hofmann E, Lange KW, Naumann M, et al. Parkinson’s disease and depression: evidence for an alteration of the basal limbic system detected by transcranial sonography. J Neurol Neurosurg Psychiatry. 1997;63:590–5.
Berg D, Supprian T, Hofmann E, Zeiler B, Jäger A, Lange KW, et al. Depression in Parkinson’s disease: brainstem midline alteration on transcranial sonography and magnetic resonance imaging. J Neurol. 1999;246:1186–93.
Walter U, Prudente-Morrissey L, Herpertz SC, Benecke R, Hoeppner J. Relationship of brainstem raphe echogenicity and clinical findings in depressive states. Psychiatry Res. 2007;155:67–73.
Sommerauer M, Fedorova TD, Hansen AK, Knudsen K, Otto M, Jeppesen J, et al. Evaluation of the noradrenergic system in Parkinson’s disease: an 11C-MeNER PET and neuromelanin MRI study. Brain. 2018;141:496–504.
Bohnen NI, Albin RL. The cholinergic system and Parkinson disease. Behav Brain Res. 2011;221:564–73.
Bohnen NI, Kanel P, Zhou Z, Koeppe RA, Frey KA, Dauer WT, et al. Cholinergic system changes of falls and freezing of gait in Parkinson’s disease. Ann Neurol. 2019;85:538–49.
Kotagal V, Albin RL, Müller MLTM, Koeppe RA, Chervin RD, Frey KA, et al. Symptoms of rapid eye movement sleep behavior disorder are associated with cholinergic denervation in Parkinson disease. Ann Neurol. 2012;71:560–8.
Bohnen NI, Kaufer DI, Hendrickson R, Constantine GM, Mathis CA, Moore RY. Cortical cholinergic denervation is associated with depressive symptoms in Parkinson’s disease and parkinsonian dementia. J Neurol Neurosurg Psychiatry. 2007;78:641–3.
Meyer PM, Strecker K, Kendziorra K, Becker G, Hesse S, Woelpl D, et al. Reduced α4β2*–nicotinic acetylcholine receptor binding and its relationship to mild cognitive and depressive symptoms in Parkinson disease. Arch Gen Psychiatry. 2009;66:866–77.
Bohnen N, Muller M, Albin R, Scott P, Frey K, Koeppe R. Progression of regional cortical cholinergic denervation in Parkinson disease: a longitudinal acetycholinesterase 11C-PMP PET study. J Nucl Med. 2018;59:630–630.
Feldmann A, Illes Z, Kosztolanyi P, Illes E, Mike A, Kover F, et al. Morphometric changes of gray matter in Parkinson’s disease with depression: a voxel-based morphometry study. Mov Disord. 2008;23:42–6.
Goto M, Kamagata K, Hatano T, Hattori N, Abe O, Aoki S, et al. Depressive symptoms in Parkinson’s disease are related to decreased left hippocampal volume: correlation with the 15-item shortened version of the Geriatric Depression Scale. Acta Radiol. 2018;59:341–5.
Kostić VS, Filippi M. Neuroanatomical correlates of depression and apathy in Parkinson’s disease: magnetic resonance imaging studies. J Neurol Sci. 2011;310:61–3.
Mayberg HS, Starkstein SE, Sadzot B, Preziosi T, Andrezejewski PL, Dannals RF, et al. Selective hypometabolism in the inferior frontal lobe in depressed patients with Parkinson’s disease. Ann Neurol. 1990;28:57–64.
Mentis MJ, McIntosh AR, Perrine K, Dhawan V, Berlin B, Feigin A, et al. Relationships among the metabolic patterns that correlate with mnemonic, visuospatial, and mood symptoms in Parkinson’s disease. Am J Psychiatry. 2002;159:746–54.
Ring HA, Bench CJ, Trimble MR, Brooks DJ, Frackowiak RS, Dolan RJ. Depression in Parkinson’s disease. A positron emission study. Br J Psychiatry. 1994;165:333–9.
Huang C, Ravdin LD, Nirenberg MJ, Piboolnurak P, Severt L, Maniscalco JS, et al. Neuroimaging markers of motor and nonmotor features of Parkinson’s disease: an 18f fluorodeoxyglucose positron emission computed tomography study. Dement Geriatr Cogn Disord. 2013;35:183–96.
Huang P, Xuan M, Gu Q, Yu X, Xu X, Luo W, et al. Abnormal amygdala function in Parkinson’s disease patients and its relationship to depression. J Affect Disord. 2015;183:263–8.
Matsui H, Nishinaka K, Oda M, Niikawa H, Komatsu K, Kubori T, et al. Depression in Parkinson’s disease. Diffusion tensor imaging study. J Neurol. 2007;254:1170–3.
Huang P, Xu X, Gu Q, Xuan M, Yu X, Luo W, et al. Disrupted white matter integrity in depressed versus non-depressed Parkinson’s disease patients: a tract-based spatial statistics study. J Neurol Sci. 2014;346:145–8.
Wu J-Y, Zhang Y, Wu W-B, Hu G, Xu Y. Impaired long contact white matter fibers integrity is related to depression in Parkinson’s disease. CNS Neurosci Ther. 2018;24:108–14.
Haghshomar M, Dolatshahi M, Ghazi Sherbaf F, Sanjari Moghaddam H, Shirin Shandiz M, Aarabi MH. Disruption of inferior longitudinal fasciculus microstructure in Parkinson’s disease: a systematic review of diffusion tensor imaging studies. Front Neurol. 2018;9:598.
Tinaz S, Kamel S, Aravala SS, Sezgin M, Elfil M, Sinha R. Distinct neural circuits are associated with subclinical neuropsychiatric symptoms in Parkinson’s disease. J Neurol Sci. 2021;423: 117365.
Sheng K, Fang W, Su M, Li R, Zou D, Han Y, et al. Altered spontaneous brain activity in patients with parkinson’s disease accompanied by depressive symptoms, as revealed by regional homogeneity and functional connectivity in the prefrontal-limbic system. PLoS ONE. 2014;9: e84705.
Lou Y, Huang P, Li D, Cen Z, Wang B, Gao J, et al. Altered brain network centrality in depressed Parkinson’s disease patients. Mov Disord. 2015;30:1777–84.
Lin H, Cai X, Zhang D, Liu J, Na P, Li W. Functional connectivity markers of depression in advanced Parkinson’s disease. NeuroImage Clin. 2020;25: 102130.
Drevets WC, Price JL, Simpson JR, Todd RD, Reich T, Vannier M, et al. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–7.
Bohnen NI, Albin RL. White matter lesions in Parkinson disease. Nat Rev Neurol. 2011;7:229–36.
Patterson L, Rushton SP, Attems J, Thomas AJ, Morris CM. Degeneration of dopaminergic circuitry influences depressive symptoms in Lewy body disorders. Brain Pathol. 2019;29:544–57.
Pessoa Rocha N, Reis HJ, Vanden Berghe P, Cirillo C. Depression and cognitive impairment in Parkinson’s disease: a role for inflammation and immunomodulation? NeuroImmunoModulation. 2014;21:88–94.
Lindqvist D, Kaufman E, Brundin L, Hall S, Surova Y, Hansson O. Non-motor symptoms in patients with Parkinson’s disease—correlations with inflammatory cytokines in serum. PLoS ONE. 2012;7: e47387.
Hassin-Baer S, Cohen OS, Vakil E, Molshazki N, Sela B-A, Nitsan Z, et al. Is C-reactive protein level a marker of advanced motor and neuropsychiatric complications in Parkinson’s disease? J Neural Transm. 2011;118:539–43.
Felger JC, Li Z, Haroon E, Woolwine BJ, Jung MY, Hu X, et al. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol Psychiatry. 2016;21:1358–65.
Santiago JA, Littlefield AM, Potashkin JA. Integrative transcriptomic meta-analysis of Parkinson’s disease and depression identifies NAMPT as a potential blood biomarker for de novo Parkinson’s disease. Sci Rep. 2016;6:34579.
Lubomski M, Tan AH, Lim S-Y, Holmes AJ, Davis RL, Sue CM. Parkinson’s disease and the gastrointestinal microbiome. J Neurol. 2020;267:2507–23.
Pachana NA, Egan SJ, Laidlaw K, Dissanayaka N, Byrne GJ, Brockman S, et al. Clinical issues in the treatment of anxiety and depression in older adults with Parkinson’s disease. Mov Disord. 2013;28:1930–4.
Skapinakis P, Bakola E, Salanti G, Lewis G, Kyritsis AP, Mavreas V. Efficacy and acceptability of selective serotonin reuptake inhibitors for the treatment of depression in Parkinson’s disease: a systematic review and meta-analysis of randomized controlled trials. BMC Neurol. 2010;10:49.
Troeung L, Egan SJ, Gasson N. A meta-analysis of randomised placebo-controlled treatment trials for depression and anxiety in Parkinson’s disease. PLoS ONE. 2013;8: e79510.
Weintraub D, Morales KH, Moberg PJ, Bilker WB, Balderston C, Duda JE, et al. Antidepressant studies in Parkinson’s disease: a review and meta-analysis. Mov Disord. 2005;20:1161–9.
Weintraub D, Moberg PJ, Duda JE, Katz IR, Stern MB. Recognition and treatment of depression in Parkinson’s disease. J Geriatr Psychiatry Neurol. 2003;16:178–83.
Leentjens AFG, Vreeling FW, Luijckx G-J, Verhey FRJ. SSRIs in the treatment of depression in Parkinson’s disease. Int J Geriatr Psychiatry. 2003;18:552–4.
Wermuth L, Sørensen PS, Timm S, Christensen B, Utzon NP, Boas J, et al. Depression in idiopathic Parkinson’s disease treated with citalopram: A placebo-controlled trial. Nord J Psychiatry. 1998;52:163–9.
Castrioto A, Thobois S, Anheim M, Quesada JL, Lhommée E, Klinger H, et al. A randomized controlled double-blind study of rotigotine on neuropsychiatric symptoms in de novo PD. NPJ Park Dis. 2020;6:1–6.
Barone P, Poewe W, Albrecht S, Debieuvre C, Massey D, Rascol O, et al. Pramipexole for the treatment of depressive symptoms in patients with Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9:573–80.
Pahwa R, Stacy MA, Factor SA, Lyons KE, Stocchi F, Hersh BP, et al. Ropinirole 24-hour prolonged release: Randomized, controlled study in advanced Parkinson disease. Neurology. 2007;68:1108–15.
Rektorova I, Balaz M, Svatova J, Zarubova K, Honig I, Dostal V, et al. Effects of ropinirole on nonmotor symptoms of Parkinson disease: a prospective multicenter study. Clin Neuropharmacol. 2008;31:261–6.
Wang H-T, Wang L, He Y, Yu G. Rotigotine transdermal patch for the treatment of neuropsychiatric symptoms in Parkinson’s disease: a meta-analysis of randomized placebo-controlled trials. J Neurol Sci. 2018;393:31–8.
Seppi K, Weintraub D, Coelho M, Perez-Lloret S, Fox SH, Katzenschlager R, et al. The movement disorder society evidence-based medicine review update: treatments for the non-motor symptoms of Parkinson’s disease. Mov Disord. 2011;26:S42-80.
Rektorová I, Rektor I, Bareš M, Dostál V, Ehler E, Fanfrdlová Z, et al. Pramipexole and pergolide in the treatment of depression in Parkinson’s disease: a national multicentre prospective randomized study. Eur J Neurol. 2003;10:399–406.
Barone P, Scarzella L, Marconi R, Antonini A, Morgante L, Bracco F, et al. Pramipexole versus sertraline in the treatment of depression in Parkinson’s disease. J Neurol. 2006;253:601–7.
Antonini A, Tesei S, Zecchinelli A, Barone P, De Gaspari D, Canesi M, et al. Randomized study of sertraline and low-dose amitriptyline in patients with Parkinson’s disease and depression: effect on quality of life. Mov Disord. 2006;21:1119–22.
Thobois S, Lhommée E, Klinger H, Ardouin C, Schmitt E, Bichon A, et al. Parkinsonian apathy responds to dopaminergic stimulation of D2/D3 receptors with piribedil. Brain. 2013;136:1568–77.
Barone P, Santangelo G, Morgante L, Onofrj M, Meco G, Abbruzzese G, et al. A randomized clinical trial to evaluate the effects of rasagiline on depressive symptoms in non-demented Parkinson’s disease patients. Eur J Neurol. 2015;22:1184–91.
Poewe W, Hauser RA, Lang A, ADAGIO Investigators. Effects of rasagiline on the progression of nonmotor scores of the MDS-UPDRS. Mov Disord 2015;30:589–92.
Smith KM, Eyal E, Weintraub D, for the ADAGIO Investigators. Combined rasagiline and antidepressant use in Parkinson disease in the ADAGIO Study: effects on nonmotor symptoms and tolerability. JAMA Neurol. 2015;72:88–95.
Cattaneo C, Müller T, Bonizzoni E, Lazzeri G, Kottakis I, Keywood C. Long-term effects of safinamide on mood fluctuations in Parkinson’s disease. J Park Dis. 2017;7:629–34.
Valldeoriola F, Catalán MJ, Escamilla-Sevilla F, Freire E, Olivares J, Cubo E, et al. Patient and caregiver outcomes with levodopa-carbidopa intestinal gel in advanced Parkinson’s disease. NPJ Park Dis. 2021;7:1–9.
Ciurleo R, Corallo F, Bonanno L, Lo Buono V, Di Lorenzo G, Versaci R, et al. Assessment of Duodopa® effects on quality of life of patients with advanced Parkinson’s disease and their caregivers. J Neurol. 2018;265:2005–14.
Martínez-Fernández R, Schmitt E, Martinez-Martin P, Krack P. The hidden sister of motor fluctuations in Parkinson’s disease: a review on nonmotor fluctuations. Mov Disord. 2016;31:1080–94.
Zibetti M, Merola A, Ricchi V, Marchisio A, Artusi CA, Rizzi L, et al. Long-term duodenal levodopa infusion in Parkinson’s disease: a 3-year motor and cognitive follow-up study. J Neurol. 2013;260:105–14.
Merola A, Zibetti M, Angrisano S, Rizzi L, Lanotte M, Lopiano L. Comparison of subthalamic nucleus deep brain stimulation and Duodopa in the treatment of advanced Parkinson’s disease. Mov Disord. 2011;26:664–70.
Tsunemi T, Oyama G, Saiki S, Hatano T, Fukae J, Shimo Y, et al. Intrajejunal infusion of levodopa/carbidopa for advanced Parkinson’s disease: a systematic review. Mov Disord. 2021;36:1759–71.
Katzenschlager R, Poewe W, Rascol O, Trenkwalder C, Deuschl G, Chaudhuri KR, et al. Apomorphine subcutaneous infusion in patients with Parkinson’s disease with persistent motor fluctuations (TOLEDO): a multicentre, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2018;17:749–59.
Devos D, Dujardin K, Poirot I, Moreau C, Cottencin O, Thomas P, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord. 2008;23:850–7.
Menza M, Dobkin RD, Marin H, Mark MH, Gara M, Buyske S, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology. 2009;72:886–92.
Richard IH, McDermott MP, Kurlan R, Lyness JM, Como PG, Pearson N, et al. A randomized, double-blind, placebo-controlled trial of antidepressants in Parkinson disease. Neurology. 2012;78:1229–36.
Akca A, Mehmet A, Sutcugil L, Ozsahin A, Kutukcu Y. Comparison of sertraline and venlafaxine treatments for depression in parkinson’s disease. Arch Neuropsychiatry. 2011;48:201–7.
Andersen J, Aabro E, Gulmann N, Hjelmsted A, Pedersen HE. Anti-depressive treatment in Parkinson’s disease. A controlled trial of the effect of nortriptyline in patients with Parkinson’s disease treated with L-DOPA. Acta Neurol Scand. 1980;62:210–9.
Serrano-Dueñas M. A comparison between low doses of amitriptyline and low doses of fluoxetin used in the control of depression in patients suffering from Parkinson’s disease. Rev Neurol. 2002;35:1010–4.
Liu J, Dong J, Wang L, Su Y, Yan P, Sun S. Comparative Efficacy and acceptability of antidepressants in Parkinson’s disease: a network meta-analysis. PLoS ONE. 2013;8: e76651.
Lemke MR. Effect of reboxetine on depression in Parkinson’s disease patients. J Clin Psychiatry. 2002;63:300–4.
Weintraub D, Mavandadi S, Mamikonyan E, Siderowf AD, Duda JE, Hurtig HI, et al. Atomoxetine for depression and other neuropsychiatric symptoms in Parkinson disease. Neurology. 2010;75:448–55.
Takahashi M, Tabu H, Ozaki A, Hamano T, Takeshima T, REBORN Study Group. Antidepressants for depression, apathy, and gait instability in Parkinson’s disease: a multicenter randomized study. Intern Med Tokyo Jpn. 2019;58:361–8.
Meloni M, Puligheddu M, Carta M, Cannas A, Figorilli M, Defazio G. Efficacy and safety of 5-hydroxytryptophan on depression and apathy in Parkinson’s disease: a preliminary finding. Eur J Neurol. 2020;27:779–86.
Richard IH, Maughn A, Kurlan R. Do serotonin reuptake inhibitor antidepressants worsen Parkinson’s disease? A retrospective case series. Mov Disord. 1999;14:155–7.
Avila A, Cardona X, Martin-Baranera M, Maho P, Sastre F, Bello J. Does nefazodone improve both depression and Parkinson disease? A pilot randomized trial. J Clin Psychopharmacol. 2003;23:509–13.
DeKarske D, Alva G, Aldred JL, Coate B, Cantillon M, Jacobi L, et al. An open-label, 8-week study of safety and efficacy of pimavanserin treatment in adults with Parkinson’s disease and depression. J Parkinsons Dis. 2020;10:1751–61.
Thomas AJ, Burn DJ, Rowan EN, Littlewood E, Newby J, Cousins D, et al. A comparison of the efficacy of donepezil in Parkinson’s disease with dementia and dementia with lewy bodies. Int J Geriatr Psychiatry. 2005;20:938–44.
Athauda D, Maclagan K, Budnik N, Zampedri L, Hibbert S, Skene SS, et al. What effects might exenatide have on non-motor symptoms in Parkinson’s disease: a post hoc analysis. J Park Dis. 2018;8:247–58.
Funkiewiez A, Ardouin C, Caputo E, Krack P, Fraix V, Klinger H, et al. Long term effects of bilateral subthalamic nucleus stimulation on cognitive function, mood, and behaviour in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2004;75:834–9.
Lhommée E, Wojtecki L, Czernecki V, Witt K, Maier F, Tonder L, et al. Behavioural outcomes of subthalamic stimulation and medical therapy versus medical therapy alone for Parkinson’s disease with early motor complications (EARLYSTIM trial): secondary analysis of an open-label randomised trial. Lancet Neurol. 2018;17:223–31.
Tröster AI, Jankovic J, Tagliati M, Peichel D, Okun MS. Neuropsychological outcomes from constant current deep brain stimulation for Parkinson’s disease. Mov Disord. 2017;32:433–40.
Thobois S, Ardouin C, Lhommée E, Klinger H, Lagrange C, Xie J, et al. Non-motor dopamine withdrawal syndrome after surgery for Parkinson’s disease: predictors and underlying mesolimbic denervation. Brain. 2010;133:1111–27.
Czernecki V, Schüpbach M, Yaici S, Lévy R, Bardinet E, Yelnik J, et al. Apathy following subthalamic stimulation in Parkinson disease: a dopamine responsive symptom. Mov Disord. 2008;23:964–9.
Dafsari HS, Petry-Schmelzer JN, Ray-Chaudhuri K, Ashkan K, Weis L, Dembek TA, et al. Non-motor outcomes of subthalamic stimulation in Parkinson’s disease depend on location of active contacts. Brain Stimul. 2018;11:904–12.
Voon V, Krack P, Lang AE, Lozano AM, Dujardin K, Schüpbach M, et al. A multicentre study on suicide outcomes following subthalamic stimulation for Parkinson’s disease. Brain J Neurol. 2008;131:2720–8.
Giannini G, Francois M, Lhommée E, Polosan M, Schmitt E, Fraix V, et al. Suicide and suicide attempts after subthalamic nucleus stimulation in Parkinson disease. Neurology. 2019;93:e97-105.
Weintraub D, Duda JE, Carlson K, Luo P, Sagher O, Stern M, et al. Suicide ideation and behaviours after STN and GPi DBS surgery for Parkinson’s disease: results from a randomised, controlled trial. J Neurol Neurosurg Psychiatry. 2013;84:1113–8.
Chen J, He P, Zhang Y, Gao Y, Qiu Y, Li Y, et al. Non-pharmacological treatment for Parkinson disease patients with depression: a meta-analysis of repetitive transcranial magnetic stimulation and cognitive-behavioral treatment. Int J Neurosci. 2021;131:411–24.
Borisovskaya A, Bryson WC, Buchholz J, Samii A, Borson S. Electroconvulsive therapy for depression in Parkinson’s disease: systematic review of evidence and recommendations. Neurodegener Dis Manag. 2016;6:161–76.
Takamiya A, Seki M, Kudo S, Yoshizaki T, Nakahara J, Mimura M, et al. Electroconvulsive therapy for Parkinson’s disease: a systematic review and meta-analysis. Mov Disord. 2021;36:50–8.
Dobkin RD, Mann SL, Weintraub D, Rodriguez KM, Miller RB, St. Hill L, et al. Innovating Parkinson’s care: a randomized controlled trial of telemedicine depression treatment. Mov Disord. 2021;36:2549–58.
Dobkin RD, Mann SL, Gara MA, Interian A, Rodriguez KM, Menza M. Telephone-based cognitive behavioral therapy for depression in Parkinson disease. Neurology. 2020;94:e1764–73.
Wuthrich VM, Rapee RM. Telephone-delivered cognitive behavioural therapy for treating symptoms of anxiety and depression in Parkinson’s disease: a pilot trial. Clin Gerontol. 2019;42:444–53.
Dobkin RD, Menza M, Allen LA, Gara MA, Mark MH, Tiu J, et al. Cognitive behavior therapy for depression in Parkinson’s disease: a randomized controlled trial. Am J Psychiatry. 2011;168:1066–74.
Wu P-L, Lee M, Huang T-T. Effectiveness of physical activity on patients with depression and Parkinson’s disease: a systematic review. PLoS ONE. 2017;12: e0181515.
Dauwan M, Begemann MJH, Slot MIE, Lee EHM, Scheltens P, Sommer IEC. Physical exercise improves quality of life, depressive symptoms, and cognition across chronic brain disorders: a transdiagnostic systematic review and meta-analysis of randomized controlled trials. J Neurol. 2021;268:1222–46.
Sacheli MA, Neva JL, Lakhani B, Murray DK, Vafai N, Shahinfard E, et al. Exercise increases caudate dopamine release and ventral striatal activation in Parkinson’s disease. Mov Disord. 2019;34:1891–900.
Sagarwala R, Nasrallah HA. The effects of yoga on depression and motor function in patients with Parkinson’s disease: a review of controlled studies. Ann Clin Psychiatry. 2020;32:209–15.
Speelman AD, van de Warrenburg BP, van Nimwegen M, Petzinger GM, Munneke M, Bloem BR. How might physical activity benefit patients with Parkinson disease? Nat Rev Neurol. 2011;7:528–34.
Bega D, Palmentera P, Wagner A, Hovde M, Barish B, Kwasny MJ, et al. Laughter is the best medicine: the Second City® improvisation as an intervention for Parkinson’s disease. Parkinsonism Relat Disord. 2017;34:62–5.
Rutten S, Vriend C, Smit JH, Berendse HW, van Someren EJW, Hoogendoorn AW, et al. Bright light therapy for depression in Parkinson disease: a randomized controlled trial. Neurology. 2019;92:e1145–56.
Di Rocco A, Rogers JD, Brown R, Werner P, Bottiglieri T. S-Adenosyl-Methionine improves depression in patients with Parkinson’s disease in an open-label clinical trial. Mov Disord. 2000;15:1225–9.
Papakostas GI, Mischoulon D, Shyu I, Alpert JE, Fava M. S-adenosyl methionine (SAMe) augmentation of serotonin reuptake inhibitors for antidepressant nonresponders with major depressive disorder: a double-blind, randomized clinical trial. Am J Psychiatry. 2010;167:942–8.
Curcio M, Catto E, Stramentinoli G, Algeri S. Effect of S-adenosyl-L-methionine on serotonin metabolism in rat brain. Prog Neuropsychopharmacol. 1978;2:65–71.
Blier P, Ward HE, Tremblay P, Laberge L, Hébert C, Bergeron R. Combination of antidepressant medications from treatment initiation for major depressive disorder: a double-blind randomized study. Am J Psychiatry. 2010;167:281–8.
Goetz CG, Tanner CM, Klawans HL. Bupropion in Parkinson’s disease. Neurology. 1984;34:1092–4.
Leentjens AF, Verhey FR, Vreeling FW. Successful treatment of depression in a Parkinson disease patient with bupropion. Ned Tijdschr Geneeskd. 2000;144:2157–9.
Załuska M, Dyduch A. Bupropion in the treatment of depression in Parkinson’s disease. Int Psychogeriatr. 2011;23:325–7.
Hauser RA, Olanow CW, Kieburtz KD, Pourcher E, Docu-Axelerad A, Lew M, et al. Tozadenant (SYN115) in patients with Parkinson’s disease who have motor fluctuations on levodopa: a phase 2b, double-blind, randomised trial. Lancet Neurol. 2014;13:767–76.
Müller T. Experimental dopamine reuptake inhibitors in Parkinson’s disease: a review of the evidence. J Exp Pharmacol. 2021;13:397–408.
Rascol O. Tesofensine (NS 2330), a monoamine reuptake inhibitor, in patients with advanced parkinson disease and motor fluctuations: the ADVANS study. Arch Neurol. 2008;65:577.
Williams JR, Hirsch ES, Anderson K, Bush AL, Goldstein SR, Grill S, et al. A comparison of nine scales to detect depression in Parkinson disease: which scale to use? Neurology. 2012;78:998–1006.
Stephenson D, Hill D, Cedarbaum JM, Tome M, Vamvakas S, Romero K, et al. The qualification of an enrichment biomarker for clinical trials targeting early stages of Parkinson’s disease. J Park Dis. 2019;9:553–63.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL. Stéphane Prange was supported by a research fellowship from the Alexander von Humboldt foundation for this review.
Conflict of interest
Stéphane Prange, Hélène Klinger, Chloé Laurencin, Teodor Danaila, and Stéphane Thobois declare no competing interests.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Authors’ contributions
SP performed the literature search and analysis, created the figures, and wrote the first draft of the manuscript. HK, CL, TD and ST contributed to data interpretation and reviewed and edited the manuscript. All authors have read and approve the final submitted manuscript and agree to be accountable for the work.
Additional information
The Original online version of this article was revised: The Open Access funding information was missed and published in the original version.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Prange, S., Klinger, H., Laurencin, C. et al. Depression in Patients with Parkinson’s Disease: Current Understanding of its Neurobiology and Implications for Treatment. Drugs Aging 39, 417–439 (2022). https://doi.org/10.1007/s40266-022-00942-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40266-022-00942-1