
Abstract
Despite the uncertainty of the pathogenesis of systemic lupus erythematosus, novel small molecules targeting specific intracellular mechanisms of immune cells are being developed to reverse the pathophysiological processes. These targeted molecules have the advantages of convenient administration, lower production costs, and the lack of immunogenicity. The Janus kinases, Bruton’s tyrosine kinases, and spleen tyrosine kinases are important enzymes for activating downstream signals from various receptors on immune cells that include cytokines, growth factor, hormones, Fc, CD40, and B-cell receptors. Suppression of these kinases impairs cellular activation, differentiation, and survival, leading to diminished cytokine actions and autoantibody secretion. Intracellular protein degradation by immunoproteasomes, levered by the cereblon E3 ubiquitin ligase complex, is an essential process for the regulation of cellular functions and survival. Modulation of the immunoproteasomes and cereblon leads to depletion of long-lived plasma cells, reduced plasmablast differentiation, and production of autoantibodies and interferon-α. The sphingosine 1-phosphate/sphingosine 1-phosphate receptor-1 pathway is responsible for lymphocyte trafficking, regulatory T-cell/Th17 cell homeostasis, and vascular permeability. Sphingosine 1-phosphate receptor-1 modulators limit the trafficking of autoreactive lymphocytes across the blood–brain barrier, increase regulatory T-cell function, and decrease production of autoantibodies and type I interferons. This article summarizes the development of these targeted small molecules in the treatment of systemic lupus erythematosus, and the future prospect for precision medicine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Small molecules that target the Janus kinases, Bruton’s tyrosine kinases, spleen tyrosine kinases, immunoproteasomes, cereblon, and sphingosine 1-phosphate receptor-1 are developed for the treatment of malignant and autoimmune disorders, including systemic lupus erythematosus. |
These targeted small molecules have the advantages of convenient administration, lower production costs, and the lack of immunogenicity. |
Some of these compounds, such as deucravacitinib, orelabrutinib, and iberdomide, have shown promise in phase II trials. Other small molecules, zetomipzomib and cenerimod, are undergoing phase II/III trials in systemic lupus erythematosus and lupus nephritis. |
Genetic and molecular profiling may help stratify patients to choose the most appropriate targeted therapies in future. |
With more treatment modalities available for systemic lupus erythematosus, the treat-to-target approach is increasingly feasible in clinical practice. |
1 Introduction
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease characterized by an unpredictable clinical course with periods of exacerbation and remission. The pathogenesis of SLE remains elusive, and multiple genetic, epigenetic, environmental, and hormonal factors contribute to loss of self-tolerance and aberration of the adaptive and innate immune systems [1, 2]. Clearance of apoptotic materials, nuclear antigens, nucleosomes, and immune complexes by macrophages and complements is defective in SLE [3]. In addition, dysregulated neutrophil apoptosis and inefficient degradation of the neutrophil extracellular traps that contain DNA, histones, cytoplasmic granules, and other mediators in SLE increase the burden of nuclear autoantigens to the immune system [3,4,5,6]. Interaction of excessive apoptotic materials and immune complexes with the toll-like receptors (TLRs) 7/9 leads to the activation of the plasmacytoid dendritic cells (pDCs) and release of type I interferons (IFNs) and interleukin (IL)-6, which in turn enhance monocyte maturation, impair apoptosis of T cells, and activate B-cell proliferation and autoantibody production [7,8,9]. Increased maturation of myeloid dendritic cells in SLE promotes IL-17 production by T cells [10] and the defective functions of the regulatory T cells (Tregs) and B cells also contributes to hyperactivity of the immune cells [11, 12].
Cytokines are secreted by cells of the immune systems for mutual communication and orchestration of the immune response [13], and may exhibit proinflammatory or anti-inflammatory properties, or both, depending on the microenvironment [14]. Production of cytokines is dysregulated in SLE, which may either be the primary pathogenetic process or secondary to the imbalance of immune pathways, such as the Th1/Th2 and Th17/Treg [15]. Patients with SLE have abnormal expression or levels of serum cytokines, such as the IFNs (IFNα, IFNγ), ILs (IL-2, IL-6, IL-10, IL-12, IL-15, IL-17, IL-21, IL-23), and B-cell activation factor (BAFF) [14]. The peripheral blood BAFF and IFN gene signatures of patients with SLE correlate with disease activity, particularly musculoskeletal and dermatological disease [16,17,18]. The dysregulation of the cytokine network contributes to inhibition of Treg activity but promotion of MHC expression, Th17 differentiation, T/B-cell activation and survival, and autoantibody production [15].
The increased knowledge of the intracellular mechanisms has led to the development of novel agents for the treatment of autoimmune diseases, including SLE. Inhibition of the receptor-associated Janus kinases (JAKs) provides a novel approach in suppressing the downstream signals of multiple cytokines and growth factors [1]. Suppression of the intracellular Bruton’s tyrosine kinase (BTK) and spleen tyrosine kinase (SYK), which are cytosolic non-receptor proteins essential for B-cell receptor signaling, leads to impaired B-cell activation, differentiation, and survival, as well as expression of the costimulatory molecules, and production of antibodies and cytokines [19, 20]. This BTK/SYK inhibition also affects the functions of other immune cell types such as the macrophages, neutrophils, mast cells, and basophils. Modulation of the immunoproteasomes and cereblon E3 ligase, which play an important role in intracellular protein degradation, results in depletion of long-lived plasma cells, reduction of B-cell differentiation to plasmablasts, and the production of autoantibodies and IFNα [21, 22]. The sphingosine 1-phosphate (S1P)/S1P receptor-1 (S1PR1) pathway influences lymphocyte trafficking, Treg/Th17 cell homeostasis, and vascular permeability [23]. The S1PR1 modulators diminish trafficking of autoreactive lymphocytes across the blood–brain barrier, increase Treg function, and decrease the production of autoantibodies and type I IFNs [24].
Small molecules that target JAKs, BTKs, SYKs, immunoproteasomes, cereblon, and S1PR1 are being developed for the treatment of malignant and autoimmune disorders. In contrast to the biologic disease-modifying anti-rheumatic drugs, which are large molecules that require parenteral administration, small molecules are orally available and enter the cellular cytoplasm to exert their effects directly. Targeted small molecules have the advantages of convenient administration, lower production costs, and the absence of immunogenicity. This article summarizes the current evidence of these small molecules in the treatment of SLE, and the prospect for precision medicine. Table 1 lists the different mechanisms of action of targeted small molecules that are being evaluated in SLE.
2 Janus Kinase (JAK) Inhibitors
The JAK-signal transducer and activator of transcription (STAT) is one of the most important intracellular signaling pathways that mediates proliferation, maturation, differentiation, activation, migration, and survival of almost all cell types [25]. The JAKs transduce signals from multiple cytokines of the IL and IFN families, hormones, and hemopoietic growth factors [26]. The type I cytokine receptors comprise common γ chain (IL-2, IL-4, IL-7, IL-9, IL-15, IL-21), gp130 family (IL-6), p40 subunit (IL-12, IL-23), and common β chain receptors (erythropoietin, thrombopoietin, and granulocyte-macrophage colony-stimulating factor), whereas the type II cytokine receptors are mainly associated with IL-10 and the type I/II IFNs [27]. Binding of cytokines and other soluble factors to their receptors results in activation of receptor-associated JAKs through cross-phosphorylation of each other [1]. The activation of the JAKs requires two JAK isoforms, either as homodimers or heterodimers, which in turn recruit and activate the STAT family of proteins that undergo phosphorylation of the tyrosine or serine residue. Upon activation, the STATs undergo a conformation change to form active homodimers, heterodimers, or tetramers that migrate to the nucleus and activate gene transcription [28]. There are four JAK enzymes, namely JAK1, JAK2, JAK3, and the non-receptor tyrosine protein kinase TYK2 and seven mammalian STATs, namely STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6 [29, 30].
The JAK inhibitors are small molecules that selectively block the adenosine triphosphate-binding site in the JH1 (catalytic kinase) domain of the JAK kinases [31]. With different selectivity to the JAKs and their corresponding STATs, the JAK inhibitors inhibit the actions of different combinations of cytokines and growth factors that are relevant to the pathophysiology of a number of immune and non-immune disorders such as SLE, inflammatory joint, skin, and bowel diseases, myeloproliferative disorders, and the cytokine storm related to severe COVID-19 infection [32].
2.1 JAK Inhibitors in Murine and Experimental Lupus
In murine models of SLE, JAK-2 inhibition reduced anti-dsDNA, serum cytokines, proteinuria, and IgG/C3 deposition in the glomeruli, leading to improvements of renal function, histology, and survival [33, 34]. Moreover, JAK2 inhibition suppressed the renal expression of monocyte chemotactic protein-1, IFNγ, and class II MHC and reduced infiltration by T cells and macrophages [33, 34]. In a study of NZB/WF1 mice, targeting JAK3 was effective in slowing down the course of experimental lupus nephritis (LN) [35]. Tofacitinib, a JAK1/3 inhibitor, has been shown to mitigate renal and skin disease in murine lupus by a reduction in IFN gene expression, anti-dsDNA levels, and CD4+ T-cell activation through upregulation of TGFβ type I receptor expression [36,37,38,39]. JAK1/2 inhibition by baricitinib has been shown to attenuate autoimmune features and renal inflammation in murine lupus by suppressing aberrant B-cell activation and podocyte abnormalities [40].
2.2 JAK Inhibitors in Human SLE
T lymphocytes from patients with active SLE were shown by in vitro studies to have an over-expression of the IFN regulatory factor-related genes, IFI35 and IFITM1, two JAK genes (JAK1/JAK2), and two STAT signaling genes (STAT1/STAT2) [41]. Pathway network analyses suggested that the IFN regulatory factor-related genes were regulated through the JAK-STAT pathway. Levels of STAT1 protein were significantly increased in SLE CD4+ T cells compared with patients with rheumatoid arthritis (RA) and healthy controls, and were associated with lower activated Treg counts and more SLE flares on the follow-up, suggesting enhanced STAT1 signaling may be involved in the dysregulation of Treg homeostasis [42]. Moreover, activation of the STAT protein upon stimulation by cytokines and IFNs appear to be related to genetic factors. The STAT4 risk allele rs7574865[T] in patients with SLE was associated with an increased phosphorylation response of the STAT4 protein to IL-12 and IFN-α stimulation [43]. Conversely, selective inhibition of TYK2 and JAK2 effectively blocked in-vitro IL-12 and IFN-γ-induced activation of the peripheral blood mononuclear cells (PBMCs) from patients with the STAT4-risk allele. In addition, autoantibody and IgG production from SLE B cells was abrogated by the addition of ruxolitinib (a JAK1/2 inhibitor) and stattic (a STAT3 inhibitor) to the culture system, indicating the JAK/STAT3 pathway is involved in the control of autoantibody production in SLE [44].
Genome-wide association studies have identified a number of genes that confer susceptibility to SLE, including the STAT genes [45,46,47,48]. The STAT4 risk allele was associated with the presence of the anti-dsDNA antibody [49] and more severe manifestations such as renal disease [50] in patients with SLE. While the exact functional significance of the STAT4 polymorphism remains to be elucidated, the STAT4 risk allele was postulated to confer an increased sensitivity of immune cells to IFNα signaling in SLE [43, 51], leading to enhanced cellular activation, B-cell differentiation, and autoantibody production [52].
2.2.1 Tofacitinib
Tofacitinib, a pan-JAK inhibitor with more potent action on JAK1/3, has been licensed for the treatment of RA and ulcerative colitis [53, 54]. In a phase Ib/IIa, double-blind, randomized controlled trial (RCT), 30 patients with SLE were randomized in a 2:1 ratio to receive tofacitinib (5 mg twice daily) or placebo (PBO) and the primary outcome was safety at day 84 [55]. Tofacitinib was well tolerated in patients studied without unexpected serious adverse events (SAEs), disease worsening, or thromboembolism (Table 2). Lipid profile, arterial stiffness, type I IFN gene signature, and circulating neutrophil extracellular traps improved with tofacitinib treatment. The improvement of laboratory parameters was more robust in patients with the STAT4 risk allele, which was associated with a more severe clinical phenotype of SLE. Two other phase I/II studies on cutaneous lupus were registered (NCT03159936; NCT03288324).
2.2.2 Solcitinib and Filgotinib
Solcitinib is a selective JAK1 inhibitor. A phase II RCT of solcitinib in non-renal SLE (NCT01777256) was prematurely terminated after recruiting 50 patients for a lack of efficacy (no improvement in IFN transcriptional biomarker expression) on an interim analysis, along with two cases (4%) of drug reaction with eosinophilia and systemic symptoms and four (8%) other cases of reversible liver derangement [56, 57] (Table 2). Filgotinib is another selective JAK1 inhibitor that has been approved for ulcerative colitis [58] and RA (in Europe) [59]. Phase II trials are being arranged for its efficacy in cutaneous lupus (NCT03134222) and membranous lupus nephropathy (NCT03285711) but there are no registered trials in non-renal SLE.
2.2.3 Baricitinib
Baricitinib is a JAK1/2 inhibitor approved for RA [60], alopecia areata [61], atopic dermatitis [62], and severe COVID-19 infection [63, 64]. In a phase II PBO-controlled RCT, 314 adult patients with autoantibody-positive SLE who had active joint and/or skin disease, and a clinical SLEDAI score ≥ 4 were randomly assigned to receive two doses of baricitinib (2 mg/day and 4 mg/day) or PBO in addition to standard-of-care (SOC) therapy that included glucocorticoids (GCs), antimalarials, non-steroidal anti-inflammatory drugs, or a single immunosuppressive agent (methotrexate, azathioprine, or mycophenolate mofetil) [65]. The primary efficacy endpoint was resolution of skin disease or arthritis at week 24, which was achieved in 67%, 58%, and 53%, respectively, of patients receiving baricitinib 4 mg, 2 mg, and PBO (baricitinib 4 mg vs PBO; p = 0.04). Secondary outcomes, namely the achievement of the SLE Responder Index-4 (SRI-4) response (64% vs 48%; p = 0.02) and the lupus low disease activity state (LLDAS) [38% vs 26%; p = 0.04], were also more frequent in baricitinib 4-mg than PBO-treated patients.
While the extent and severity of skin lesions (assessed by the Cutaneous Lupus Erythematosus Disease Area and Severity Index [CLASI]) did not improve any better with baricitinib, tender joint counts showed a greater reduction in the baricitinib 4-mg group than the PBO group of patients (− 6.9 vs − 5.6 joints; p = 0.04). Improvement in the number of swollen joints, however, was not greater with baricitinib. Moreover, baricitinib 4-mg treatment did not lead to greater changes in levels of anti-dsDNA and complement C3 compared to PBO, and no correlation could be observed between the drop in anti-dsDNA/IgG with the SRI-4 response [66]. Nevertheless, elevated levels of IL-12 p40, IL-6, messenger RNA expression of STAT1-target, STAT2-target, and STAT4-target and multiple IFN responsive genes were reduced with baricitinib treatment [67].
The occurrence of adverse events (AEs) was similar among the three groups of patients. Serious AEs were numerically more common in the baricitinib than PBO groups. The rate of serious infection was higher in the baricitinib 4-mg group (6%) than the baricitinib 2-mg group (2%) or PBO group (1%). Only one patient who tested positive for the antiphospholipid antibodies developed a deep vein thrombosis after baricitinib treatment (4 mg/day).
The favorable result of this phase II RCT led to two subsequent phase III RCTs of baricitinib in non-renal SLE (SLE-BRAVE I; NCT03616912 and SLE-BRAVE II; NCT03616964) [68, 69] (Table 2). Participants were patients with autoantibody-positive SLE with ≥ 1 BILAG A or ≥ 2 BILAG B scores, total SLEDAI ≥ 6, and clinical SLEDAI ≥ 4 and were receiving background therapy with stable doses of GCs, a single antimalarial, or other immunosuppressive drug. Similar to the phase II study [65], these patients were randomized to baricitinib 4 mg/day, 2 mg/day, or PBO. The primary efficacy endpoint was SRI-4 response at week 52, which was met in SLE-BRAVE-I (57% vs 46%; p = 0.02) but not SLE-BRAVE-II (47.4% vs 45.6%) despite an identical study design. Secondary endpoints that included SRI-4 response at week 24, time to first severe flare, GC sparing, and achievement of LLDAS were not met in either study. Although the musculoskeletal and mucocutaneous domains on SLEDAI and BILAG improved significantly with baricitinib 4 mg versus PBO in the SLE-BRAVE-I study, this was not observed in the SLE-BRAVE-II study. The safety of baricitinib was consistent with the known profile of the drug, with an increased risk of serious infections but not venous thromboembolism. The inconsistent results of these two RCTs render the efficacy of baricitinib in SLE inconclusive. A long-term extension study of baricitinib in SLE is in progress (SLE-BRAVE-X).
2.2.4 Deucravacitinib
Deucravacitinib is a selective Tyk-2 inhibitor that blocks the downstream signaling of IL-12, IL-23, IL-10, and the type I IFNs. In a phase II RCT (PAISLEY), 363 patients with autoantibody-positive SLE with active disease (≥ 1 BILAG A or ≥ 2 BILAG B scores, clinical SLEDAI-2K ≥ 6 with skin or joint involvement) were randomly assigned to receive three dosage regimens of deucravacitinib (3 mg twice daily, 6 mg twice daily, 12 mg once daily) or PBO in addition to background medications [70]. Subjects with severe or life-threatening organ manifestations of SLE were excluded. A protocol-based GC taper was required from week 8 to 20, whereas a further GC taper from week 32 to 40 was optional.
The primary endpoint of this RCT was the SRI-4 response at week 32, which was met by the deucravacitinib 3-mg and 6-mg twice-daily groups compared to PBO (58.2% vs 34.4%; p < 0.001 and 49.5% vs 34.4%; p = 0.02, respectively) [Table 2]. Secondary endpoints, such as the SRI-4 response, achievement of LLDAS, and the BICLA response at week 48 were also significantly higher in the deucravacitinib 3-mg twice-daily group versus PBO. Moreover, significantly more patients in the deucravacitinib 3-mg twice-daily group achieved a ≥ 50% reduction in the CLASI score and combined swollen/tender joint counts. Deucravacitinib treatment resulted in a greater improvement in anti-dsDNA titer and complement levels. Moreover, all dosages of deucravacitinib, but not PBO, were associated with a reduction in the IFN signature through 44 weeks of treatment.
Deucravacitinib was well tolerated, with no increase in AEs, SAEs, or infective complications including herpes zoster infection observed. The most common AEs (≥ 10%) reported in deucravacitinib-treated patients were upper respiratory tract infection, nasopharyngitis, headache, and urinary tract infection. Cancer incidence was similar between deucravacitinib and PBO and there were no deaths, thrombotic events, opportunistic infections, or tuberculosis. A phase III RCT (POETYK SLE-1) has just been registered (NCT05617677).
2.2.5 Beprocitinib
Beprocitinib is a selective JAK1/Tyk2 inhibitor with a promising mechanism in the treatment of SLE through inhibition of the downstream signal of IL-10, IL-12, IL-23, and the type I IFNs. A phase II RCT in non-renal SLE (NCT03845517) has just completed recruitment.
3 Bruton’s Tyrosine Kinase Inhibitors (BTKis)
The BTK is a cytoplasmatic tyrosine kinase belonging to the family of tyrosine kinase expressed in hepatocellular carcinoma [71] and expressed in most hematopoietic cells, including the B cells and terminally differentiated plasma cells, monocytes/macrophages, dendritic cells, natural killer cells, mast cells, and platelets [20]. The BTK mediates the signaling of several immune receptors, including the B-cell receptor and Fc receptor [72]. In B cells, BTK plays an essential role in the downstream signal pathways through the B-cell receptor [73, 74] and enhances the sensitivity of the B cells to the Toll-like receptor signaling event such as germinal center formation, CD80 expression, IL-1, IL-6, IFNγ, and anti-nuclear autoantibody production [75]. In addition, BTK is involved in differentiation, phagocytosis, production of cytokines, and other inflammatory mediators of other innate myeloid immune cells [76]. In mast cells, BTK plays an important role in mediating Fcε receptor signaling for the chemotactic response [77]. Finally, BTK also activates platelets via the glycoprotein VI receptor [78] and osteoclast differentiation [79].
The engagement of the B-cell receptor initiates intracellular signaling that involves phosphorylation of SYK, leading to partial activation of BTK, which in turn autophosphorylates to full activation and orchestrates consequent phosphorylation of its immediate downstream effector, phospholipase Cγ2, ultimately leading to calcium influx and activation of multiple downstream signaling pathways and transcription factors, including nuclear factor of activated T cells, extracellular signal-regulated kinase, and nuclear factor-κB [20]. Bruton’s tyrosine kinase and SYK are also involved in the signaling of the BAFF and CD40 receptors that activate the non-canonical nuclear factor-κB pathway [72]. These signals are crucial for the regulation of cellular differentiation, proliferation, survival, and activation that leads to costimulatory molecule expression and the production of antibodies and cytokines.
The BTK inhibitors (BTKis) are small molecules that inhibit the activity of BTK and have been developed for the treatment of various B-cell malignancies for their anti-proliferation effects [80]. Ibrutinib is the first-in-class BTKi approved for B-cell proliferative disorders. However, AEs such as cardiotoxicity (atrial and ventricular arrythmia, cardiomyopathy, hypertension) and bleeding (platelet dysfunction) may result in treatment interruption or discontinuation [81]. Newer generation BTKis are now available to overcome treatment resistance to first-generation agents and minimize off-target kinase activity for better safety profiles [82].
3.1 BTKis and Murine Lupus
In two classical models of spontaneous murine lupus (NZB/W and MRL/lpr), BTK inhibition reduced the number of splenic B cells and anti-dsDNA titers, delayed the onset of proteinuria, and ameliorated kidney inflammation [83,84,85,86]. Cutaneous and neuropsychiatric lesions in the MRL/lpr mice were also attenuated by administration of a BTKi [87]. In the lupus-prone B6.Sle1 and B6.Sle1.Sle3 mice, BTK inhibition was effective in dampening humoral and cellular immunity, and glomerulonephritis [88]. Moreover, inhibiting BTK has been shown to reduce autoantibodies and suppress arthritis and nephritis in TLR7- and IFN-driven murine lupus models [89]. Finally, in the NZB/W F1 mouse model, evobrutinib, a newer BTKi, suppressed B-cell activation, reduced autoantibody production and plasma cell numbers, and normalized B- and T-cell subsets, leading to reduced kidney damage [90].
3.2 BTKis in Human SLE
Bruton’s tyrosine kinase expression was shown to be higher in PBMCs from patients with SLE than healthy controls, and correlated with the disease activity score, anti-dsDNA, complement levels, and proteinuria [91]. However, in patients with LN, no relationship between BTK expression and histologic activity index was observed. Bruton’s tyrosine kinase is an attractive target for SLE treatment because its modulation does not cause B-cell depletion. Moreover, BTK is expressed in multiple immune cell types and its inhibition may enhance therapeutic effect beyond B-cell modulation. A couple of clinical trials of BTKis have been performed in SLE.
Evobrutinib, a highly selective and central nervous system-penetrating oral BTKi, was tested in a phase II RCT of autoantibody-positive SLE [92]. In this study, 469 patients with SLE with a SLEDAI-2K score ≥ 6 (including a clinical SLEDAI-2K score ≥ 4) despite SOC treatment were randomly assigned to oral evobrutinib 25 mg once daily, 75 mg once daily, 50 mg twice daily, or PBO. Primary efficacy endpoints were SRI-4 at week 52 and SRI-6 at week 52 in the high disease activity subpopulation (Table 2). At week 52, none of the evobrutinib groups achieved a significantly higher SRI-4 rate than the PBO group. The SRI-6 response rates were also similar across all the four arms in the high disease activity subgroup. No clinically meaningful differences with evobrutinib versus PBO in the changes in organ-specific disease activity, lupus serology, immunoglobulin levels, annualized flare rate, quality of life, or GC usage were observed at week 52. All doses of evobrutinib were well tolerated, with no dose effect observed for treatment-emergent adverse events (TEAEs).
Fenebrutinib is a second-generation, non-covalent, highly selective, reversible oral BTKi that has shown efficacy in phase I/II RCTs of RA [93] and refractory B-cell malignancies [94]. A large phase II dose-ranging study was conducted in 260 patients with moderately and severely active autoantibody-positive SLE who were receiving SOC (SLEDAI-2K ≥ 8; Physician Global Assessment score ≥ 1) [95]. Patients with proliferative LN, nephrotic range proteinuria, neuropsychiatric disease, and the antiphospholipid syndrome were excluded. Participants were randomized to receive fenebrutinib 200 mg twice daily, 150 mg once daily, or PBO. A GC taper was recommended from weeks 0 to 12 and from weeks 24 to 36. The primary endpoint was SRI-4 response at week 48. Although fenebrutinib reduced the BTK-dependent plasmablast RNA signature, anti-dsDNA, and IgG/IgM levels but increased C4 levels relative to PBO, the proportion of patients who achieved the SRI-4 response was not significantly higher in the treatment groups than the PBO group at week 48 (52%/51% vs 44%) [Table 2]. Similarly, the BICLA response rate at week 48 was not significantly different between the fenebrutinib- and PBO-treated patients (42%/53% vs 41%). Adverse events, however, were not significantly more common with fenebrutinib compared with PBO but the frequency of SAEs was numerically higher.
Orelabrutinib is an oral, highly selective, irreversible inhibitor of BTK. A phase Ib/II RCT was conducted in China in 60 patients with seropositive SLE with active disease (SLEDAI score ≥ 5) [96]. Patients were randomized to receive three doses of orelabrutinib or PBO for 12 weeks in addition to the SOC. The primary outcome, SRI-4 response rate, was higher in any dose of orelabrutinib than PBO (Table 2). Overall, AEs were mild to moderate and the majority of TEAEs were not severe. The reasons for the discrepancies of results in these three RCTs are not immediately apparent and further phase III RCTs of the BTKis in SLE are of interest.
4 Spleen Tyrosine Kinase Inhibitors (SYKis)
Spleen tyrosine kinase is a non-receptor tyrosine kinase that belongs to the Zeta-associated protein kinase of the 70-kDa (ZAP70) family [97]. Spleen tyrosine kinase is primarily expressed in hemopoietic cells, including B cells, immature T cells, macrophages, neutrophils, and mast cells that are involved in both adaptive and innate immune responses. In immune cells, SYK signals through multiple receptors such as B cells, pre-T cells, Fc, and TLRs [98]. It catalyzes the phosphorylation of a receptor-associated protein complex known as an immunoreceptor tyrosine based-activation motif that further activates the SYK itself. Spleen tyrosine kinase autophosphorylates and activates adapter proteins that are involved in several intracellular signal transduction pathways, including PI3K/Akt, Ras/ERK, PLCγ/NFAT, Vav-1/Rac, and IKK/NKκB [99]. Via these actions, SYK regulates the proliferation, survival, differentiation, activation, degranulation, and cytokine production of the immune cells.
4.1 SYKi and Murine Lupus
Spleen tyrosine kinase expression is abnormally increased in the skin lesions of lupus prone mice [100]. The SYK inhibitor (SYKi), fostamatinib, has been shown to ameliorate kidney and skin disease in female MRL/lpr or BAK/BAX lupus-prone mice [100]. Administration of fostamatinib before or after disease onset was effective in delaying the onset of proteinuria and renal failure, reducing kidney infiltrates, and improving the survival of the NZB/NZW mice without suppressing autoantibody titers [101]. Lanraplenib, a selective SYKi, was also shown to retard the progression of LN-like disease in NZB/W mice and reduce glomerular IgG deposition and serum proinflammatory cytokines [102]. Other newer generation SYKis have also been shown to retard the progression of LN in the lupus mouse models [103, 104].
4.2 SYKi in Human Immune-Mediated Diseases
Fostamatinib has been tested in various immune-mediated diseases such as RA, chronic immune thrombocytopenia, autoimmune hemolytic anemia, and IgA nephropathy [105]. It is the first SYKi approved for the treatment of chronic immune thrombocytopenia by blocking signal transduction through Fcγ receptors involved in the antibody-mediated destruction of platelets by immune cells [106]. Fostamatinib was studied in patients with RA who had an inadequate response to methotrexate or the tumor necrosis factor inhibitors [107, 108]. In both phase III RCTs, although the efficacy of fostamatinib was observed, AEs of concern such as hypertension, diarrhea, neutropenia, headache, and elevation of liver parenchymal enzymes occurred not infrequently in a dose-dependent manner. A higher dose of fostamatinib would lead to better efficacy but is limited by its toxicities. Another phase II trial of a selective SYKi, GS-9876, in RA did not report efficacy, although the drug was well tolerated [109]. As a result, the drug is not further developed for RA.
Hyperexpression of SYK in lupus T cells influences the expression of a number of cytokines, enzymes, and receptors that are involved in the pathogenesis in SLE [110, 111]. Increased frequency of a SYK bright CD27- memory-like B-cell population, which might be a source of increased plasma cells, was demonstrated in patients with SLE compared with healthy controls [112]. Lanraplenib was tested in 19 patients with moderate-to-severe cutaneous lupus in a phase II proof-of-concept RCT [113]. Although the drug was well tolerated, the primary efficacy endpoint of the change in CLASI score was not met. Another phase II study of filgotinib and lanraplenib in nine patients with lupus membranous nephropathy reported some efficacy of the former. However, because of the limited number of participants receiving lanraplenib and the high drop-out rate, no conclusion could be drawn [114]. There are no other registered studies of the SYKis in SLE or LN.
5 Proteasome Inhibitors
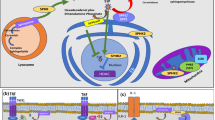
The ubiquitin-proteasome system (UPS) is the key mechanism for selective degradation of the majority of intracellular proteins that is critical for the maintenance of cellular homeostasis and regulation of cellular functions such as survival and proliferation [21]. Alterations in the UPS are linked to oncogenesis [115]. The 26S constitutive proteasome is expressed ubiquitously in body tissues, including the heart, kidney, and liver, whereas the variant proteasome, known as the immunoproteasome, which has a high homology in the catalytic activity subunits to the constitutive proteasome, is expressed in immune cells, such as lymphocytes and monocytes [116]. Inhibition of both types of proteasomes leads to increased cellular apoptosis and reduced proliferation, whereas selective inhibition of the immunoproteasomes results in cytokine suppression and anti-inflammatory activities in ex-vivo models [117]. Bortezomib, carfilzomib, and ixazomib are non-selective proteasome inhibitors that simultaneously suppress both the constitutive and immunoproteasomes. They are developed for their anti-tumor effects to treat multiple myeloma and mantle cell lymphoma.
Long-lived plasma cells are capable of producing protective antibodies but also play an essential role in autoreactive immunologic memory that leads to the generation of autoantibodies in SLE [118]. In contrast to short-lived plasmablasts, long-lived plasma cells are resistant to conventional immunosuppressive and B-cell depletion therapies. Persistence of these cells is associated with refractory disease activity or flares in patients with SLE.
5.1 Proteasome Inhibitors in Murine Lupus
Bortezomib and delanzomib have been shown to deplete plasma cells, reduce anti-dsDNA titers, and alleviate renal disease in the NZB/W F1 and MRL/lpr mice [119,120,121]. Treatment of the lupus-prone mice with carfilzomib, bortezomib, or the immunoproteasome-specific inhibitors such as ONX0914 prevented disease progression and abrogated glomerulonephritis, along with a reduction in autoantibody levels and production of IFNα by TLR-activated pDCs in vitro and in vivo [122]. Administration of zetomipzomib (KZR-616), a selective immunoproteasome inhibitor, in lupus mice led to a durable improvement of renal disease, a reduction in anti-dsDNA antibodies, and renal IgG deposition without affecting normal T-cell-dependent responses [123].
5.2 Proteasome Inhibitors in Human SLE
Small open case series have shown the efficacy of bortezomib in refractory human SLE manifestations, including LN [124,125,126,127,128]. A significant depletion of both short- and long-lived plasma cells in peripheral blood and bone marrow, leading to a reduction in autoantibody and serum immunoglobulin levels, was observed after bortezomib administration [129]. In a series of 12 patients with refractory SLE, bortezomib treatment led to a sustained improvement in disease activity for 6 months [125]. Serum antibody levels significantly declined, with a greater effect on anti-dsDNA than vaccine-induced protective antibody titers. However, 11 (92%) patients experienced AEs and four (33%) experienced SAEs. The commonly reported AEs were infections (16%), nausea (16%), headache (16%), polyneuropathy (11%), fever (11%), and allergic skin reactions (11%). Although most AEs were mild/moderate in severity and resolved completely, bortezomib was discontinued in seven (58%) patients. Another series of 12 female patients with refractory LN also reported persistent hypogammaglobulinemia (16.6%) and sensory neuropathy (16.6%), which led to bortezomib withdrawal [127]. In a small RCT conducted in Japan, 14 patients with SLE with refractory disease were treated with either bortezomib or PBO [130] (Table 2). Efficacy was not demonstrated at week 24. Four (50%) patients treated with bortezomib withdrew from the protocol and three others (38%) did not complete the minimal protocol requirement because of SAEs. The action of bortezomib is short-lived and continuous B-cell inhibition may be needed to achieve sustained plasma cell depletion and renal efficacy [131]. In fact, sequential administration of bortezomib and belimumab has been used successfully in two patients with SLE with refractory renal disease and/or pulmonary hemorrhage [132].
The narrow therapeutic index of the current non-selective proteasome inhibitors limits their clinical use in SLE. Highly selective immunoproteasome inhibitors are developed to enhance tolerability. Zetomipzomib is the first-in-class irreversible, tripeptide epoxyketone-based, selective immunoproteasome inhibitor that specifically targets the inflammatory cells [133]. The drug was shown to reduce the production of proinflammatory cytokines from human PBMCs, block T-cell production of IFN-γ, tumor necrosis factor-α, and granulocyte-macrophage colony-stimulating factor, and the differentiation of B cells to plasmablasts [123]. Interim results from a phase Ib open-labeled study (NCT03393013) showed the efficacy of zetomipzomib in SLE and LN with an acceptable AE profile [134]. Further RCTs are expected.
6 Cereblon E3 Ligase Modulators
As previously mentioned, degradation of intracellular protein by the UPS is a major mechanism for cellular hemostasis and survival. The protein cereblon (CRBN) is a substrate receptor of the cullin-ring ligase-4 (CRL4CRBN) E3 ubiquitin ligase complex that allows tagging of polyubiquitin chains to promote degradation of target proteins that are traditionally difficult to modulate by direct pharmacological means (e.g., transcription factors and oncoprotein) [135]. Cereblon E3 ligase modulators (CELMoDs) are synthetic agents that leverage the UPS to enhance the selective degradation of disease-promoting proteins. Drugs such as lenalidomide and pomalidomide were developed for treating myeloma before the actual mechanisms are known. In fact, they are CELMoDs that induce degradation of Ikaros and Aiolos, which are transcriptional factors with a zinc finger-based structure that regulate multiple genes involved in lymphocyte function and differentiation [22]. Highly potent CELMODs such as iberdomide (CC-220) and mezigdomide are now undergoing clinical trials in myeloma and autoimmune diseases [135, 136].
Ikaros and Aiolos are encoded by the IKZF1 and IKZF3 genes, respectively [137]. Polymorphisms in these two genes are associated with SLE susceptibility [138, 139]. Ikaros is widely expressed in hemopoietic precursor cells and plays a role in the development of B cells and pDCs, which are the main source of type I IFN in patients with SLE. In contrast, Aiolos has a more restricted expression in pre-B/mature B cells and is required for the differentiation into long-lived plasma cells [137]. The CRBN, IKZF1, and IKZF3 genes were over-expressed in PBMCs of patients with SLE compared with healthy controls and in-vitro iberdomide administration resulted in a reduced production of anti-dsDNA and antiphospholipid antibodies from cultured PBMCs [140]. Moreover, peripheral blood CD19+ B cells isolated from patients with SLE showed a significant reduction in TLR7 and IFNα-mediated production of immunoglobulins, reduced differentiation into plasmablasts and antibody production, as well as IKZF1 and IKZF3 gene expression (naive B cells and plasmablasts) upon iberdomide administration in vitro [141].
6.1 Cereblon Modulators in Murine Models
In female NZB/WF1 mice, treatment with thalidomide (10 mg/kg) showed a significant reduction in proteinuria, immune complex accumulation, and glomerular and tubular damage, which was coupled with a decrease in serum anti-dsDNA, IgG2a/2b, and nuclear translocation of NF-κB in kidney tissues [142]. In vitro treatment with thalidomide has also been shown to reduce proliferation and co-stimulatory molecule expression of splenic CD4+ T cells isolated from C57BL/6 mice [143].
6.2 Cereblon Modulators in Human SLE
Thalidomide and lenalidomide are effective treatments for refractory cutaneous lupus [144]. However, most studies were small, retrospective, and lacked a control group. Peripheral polyneuropathy was reported in 15–80% of thalidomide-treated patients without a clear relationship with the duration of use, although reversibility was observed in 70% of cases upon drug withdrawal. Lenalidomide appeared to be less neurotoxic, but relapses occurred in 25–75% cases upon drug cessation. Teratogenicity, cardiovascular toxic effects, and thromboembolism, especially in older and high-risk patients [145], limit their use in SLE.
Iberdomide is a high-affinity CELMoD that targets the hemopoietic transcription factors Ikaros and Aiolos for proteasomal degradation [146]. In a 12-week, proof-of-concept, phase IIa, PBO-controlled dose-escalating RCT, 42 patients (33 patients completed the protocol) with active SLE were assigned to receive four doses of iberdomide or PBO [146]. The most common TEAEs were nausea, diarrhea, and upper respiratory tract infections that were not severe. There was a dose-dependent reduction in peripheral blood total B cells and pDCs, coupled with an improvement of the Physician Global Assessment and the CLASI scores.
A subsequent phase II RCT of 288 patients with seropositive SLE with moderate-to-severe disease activity (SLEDAI score ≥ 6 and clinical SLEDAI ≥ 4) was performed. Subjects with severe neuropsychiatric renal disease or the antiphospholipid syndrome were excluded. Patients were randomly assigned to three doses of iberdomide (0.15 mg, 0.30 mg, 0.45 mg/day) or PBO [147] (Table 2). The primary endpoint was the SRI-4 response at week 24, which was achieved at a significantly higher rate in the iberdomide 0.45-mg group than PBO (54% vs 35%; p = 0.01). The SRI-4 rates were higher in those with a SLEDAI score ≥ 10, high IKZF3 (Aiolos) expression, and a high IFN signature at baseline [147, 148]. Iberdomide treatment resulted in a SRI-4 response in all patients (100%) with an extremely high IFN signature and reduced anti-dsDNA in those with elevated levels at baseline. In patients with a CLASI score ≥ 10, numerically more patients had a CLASI-50 improvement in the iberdomide 0.45-mg group than the PBO group. Changes in joint counts and other secondary outcomes were not significantly different between the treatment and PBO groups. Moreover, neutropenia, upper respiratory tract, and urinary tract infection were more common in iberdomide-treated patients. Regarding laboratory parameters, iberdomide treatment reduced peripheral B cells (including those expressing the BLyS receptor gene and in switched memory B cells) and pDCs, but increased Tregs and IL-2 at week 24 in a dose-dependent manner [148]. This suggests that iberdomide is capable of reversing the immunological abnormalities in SLE. Further clinical trials of iberdomide in SLE are warranted.
7 Sphingosine 1-Phosphate Receptor (S1PR) Modulators
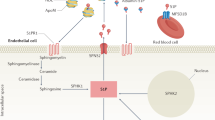
Sphingosine 1-phosphate (S1P) is a bioactive lipid molecule that binds to G protein-coupled S1P receptors (S1PRs) and affects cell proliferation, survival, and migration. Among the S1PR isoforms, S1PR1, expressed on leukocytes and endothelial cells, is an important mediator of lymphocyte trafficking, Treg/Th17 cell homeostasis, and vascular permeability [149]. Four S1PR1 modulators have been approved for the treatment of multiple sclerosis and ulcerative colitis [23]. Fingolimod binds to multiple S1PRs, halts lymphocyte egress from secondary lymphoid tissues, and reduces inflammation in the central nervous system. Newer generation compounds such as ozanimod, siponimod, and ponesimod have greater specificity for S1PR1, which may contribute to fewer AEs.
The S1PR1 modulators could ameliorate disease activity of SLE by reducing the trafficking of autoreactive lymphocytes and differentiation of the Th17 cells, enhancing the number and function of the Tregs, and decreasing autoantibody production [150]. In addition, these modulators increase the endothelial cell barrier function and blood–brain barrier function, and reduce expression of the adhesion molecules for leukocyte transmigration and type I IFN production by pDCs in response to viral or oligonucleotide stimulation [24]. These mechanisms are potentially beneficial for renal, neuropsychiatric disease and atherosclerotic injury in SLE.
7.1 S1PR1 Modulators in Murine Lupus
Modulators of the S1PR1, such as ozanimod, fingolimod, amiselimod, cenerimod, and KRP-203, have been shown to attenuate renal disease and improve survival in multiple murine lupus models [151,152,153,154,155,156]. Fingolimod reduced the number of T cells and B cells in the thymus, indicating increased lymphocyte apoptosis is a major mechanism of the drug [154, 157]. Fingolimod has also been shown to improve certain neuropsychiatric features of the MRL/lpr mice, such as depression-like behavior, memory deficits, and leukocyte infiltration of the choroid plexus [158, 159]. The protective effects of fingolimod on the central nervous system are likely contributed to by the direct action on the microvascular endothelial cells and strengthening of the blood–brain barrier. Cenerimod or amiselimod was shown to reduce peripheral blood CD19+ B cells, CD4+ and CD8+ T cells, plasma cells, anti-dsDNA antibodies splenomegaly, lymphadenopathy, plasma and tissue levels of IFNα, as well as tumor necrosis factor, IL-6, BAFF, and IL-10 in the lupus mice [151, 155]. Other S1PR1 modulators have also been shown to induce peripheral lymphopenia and reduce lymphocyte infiltration and IgG/C3 deposition to the kidneys of these mice [152, 153, 155, 156].
7.2 S1PR1 Modulators in Human SLE
An open-label phase Ib safety trial of amiselimod was conducted in 17 patients with SLE with mild/moderate activity [160]. Lymphopenia was observed in all patients after treatment but none developed serious infections, cardiotoxicity, or SAEs. A reduction in anti-dsDNA antibodies occurred in the majority of patients who had elevated levels before treatment (Table 2).
A proof-of-concept PBO-controlled RCT of oral cenerimod was conducted in 49 patients with seropositive SLE with active mucocutaneous or musculoskeletal disease [161]. Cenerimod treatment led to a significant dose-dependent reduction in the total lymphocyte count. At week 12, further improvement in the modified SLEDAI-2K score and anti-dsDNA titer was observed in the treatment than the PBO group (Table 2). No increase in TEAEs was reported with cenerimod but a small but non-clinically relevant drop in the heart rate was observed in the first 6 h of drug administration. A phase II RCT has just completed (NCT03742037) and a phase III RCT (OPUS-1) has started recruitment (NCT05648500).
8 Tailor-Made Therapy for SLE: Are We There Yet?
Systemic lupus erythematosus is a clinically and serologically heterogeneous disease. There are considerable inter-ethnic differences in the tolerability of medications and the treatment responses to unified protocols in research settings [162,163,164]. In two pivotal RCTs of anifrolumab (a monoclonal antibody against type I IFN receptor) in non-renal SLE [165, 166], greater treatment responses relative to PBO were achieved in patients with high IFN signatures at baseline [167]. A phase Ib/IIa RCT of tofacitinib in SLE also revealed a stronger reduction in IFN signatures in patients with the STAT4 risk allele [55]. Finally, patients with higher IFN and IKZF3 expression were found to have a better clinical response to iberdomide [147, 148]. Collectively, these observations suggest the possibility of genetic profiling to determine the choice of targeted therapies in patients with SLE to achieve the best therapeutic effects. Urine proteomics and molecular profiling of renal tissues by transcriptomic analyses may help reflect intrarenal activity that correlates with treatment refractoriness to guide therapeutic approaches [168,169,170]. However, until these genomic and proteomic biomarkers are adequately validated in different ethnic groups, the choice of treatment modalities in SLE still depends on clinical judgment based on ethnicity, anticipated treatment adherence and tolerability, organ function, and the presence of medical comorbidities. It is hoped that patient stratification by comprehensive molecular techniques is possible in the future to help patients choose the most appropriate and cost-effective individualized therapies.
9 Conclusions
The development of novel therapeutics in SLE is fraught with difficulty and disappointment. Many novel agents have halted progression for the negative results from pivotal RCTs. With the improvement in patient stratification, adjustment of background immunosuppression, and assessment of study endpoints, we are now having more approved drugs in SLE [171]. A number of targeted small molecules are undergoing clinical trials in patients with SLE. Tyk2 inhibition appears to be most promising [70] and phase II/III results are eagerly awaited. However, the recent concern of thromboembolism and cancer risk in post-marketing studies of RA, particularly in older patients with a cardiovascular risk [172], has led to caution of the use of JAK inhibitors in patients with SLE, who are also prone to thrombosis and malignancies. Two BTKis did not show benefits in SLE [92, 95] but a third showed promising results [96]. Although the SYKis showed efficacy in RA [107, 108], toxicities limited their further development in RA and SLE. The narrow therapeutic index of the conventional non-selective proteasome inhibitors such as bortezomib has limited their clinical use. However, the selective immunoproteasome inhibitor, zetomipzomib, has an improved safety profile [134] and is undergoing further trials in SLE. The cereblon modulator, iberdomide, presented encouraging results in SLE from a recent phase II RCT [147]. Finally, a selective modulator of the S1PR1 receptor, such as cenerimod, has started phase II/III studies in SLE. A new era of SLE therapies is expected in the next couple of years when the results of these trials are ready, and the treat-to-target approach in SLE is increasing feasible.
References
Mok CC. The Jakinibs in systemic lupus erythematosus: progress and prospects. Expert Opin Investig Drugs. 2019;28:85–92.
Tsokos GC. Systemic lupus erythematosus. N Engl J Med. 2011;365:2110–21.
Mahajan A, Herrmann M, Muñoz LE. Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol. 2016;7:35.
Fresneda Alarcon M, McLaren Z, Wright HL. Neutrophils in the pathogenesis of rheumatoid arthritis and systemic lupus erythematosus: Same foe different M.O. Front Immunol. 2021;12: 649693.
Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. 2012;188:3522–31.
Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. 2010;107:9813–8.
Sakata K, Nakayamada S, Miyazaki Y, Kubo S, Ishii A, Nakano K, et al. Up-regulation of TLR7-mediated IFN-α production by plasmacytoid dendritic cells in patients with systemic lupus erythematosus. Front Immunol. 2018;9:1957.
Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Investig. 2005;115:407–17.
Eloranta ML, Alm GV, Rönnblom L. Disease mechanisms in rheumatology–tools and pathways: plasmacytoid dendritic cells and their role in autoimmune rheumatic diseases. Arthritis Rheumatol. 2013;65:853–63.
Fransen JH, van der Vlag J, Ruben J, Adema GJ, Berden JH, Hilbrands LB. The role of dendritic cells in the pathogenesis of systemic lupus erythematosus. Arthritis Res Ther. 2010;12:207.
Scheinecker C, Bonelli M, Smolen JS. Pathogenetic aspects of systemic lupus erythematosus with an emphasis on regulatory T cells. J Autoimmun. 2010;35:269–75.
Fujio K, Okamura T, Sumitomo S, Yamamoto K. Regulatory cell subsets in the control of autoantibody production related to systemic autoimmunity. Ann Rheum Dis. 2013;72(Suppl 2):ii85–9.
Apostolidis SA, Lieberman LA, Kis-Toth K, Crispín JC, Tsokos GC. The dysregulation of cytokine networks in systemic lupus erythematosus. J Interferon Cytokine Res. 2011;31:769–79.
Su DL, Lu ZM, Shen MN, Li X, Sun LY. Roles of pro- and anti-inflammatory cytokines in the pathogenesis of SLE. J Biomed Biotechnol. 2012;2012: 347141.
Jacob N, Stohl W. Cytokine disturbances in systemic lupus erythematosus. Arthritis Res Ther. 2011;13:228.
Petri M, Stohl W, Chatham W, McCune WJ, Chevrier M, Ryel J, et al. Association of plasma B lymphocyte stimulator levels and disease activity in systemic lupus erythematosus. Arthritis Rheumatol. 2008;58:2453–9.
Mai L, Asaduzzaman A, Noamani B, Fortin PR, Gladman DD, Touma Z, et al. The baseline interferon signature predicts disease severity over the subsequent 5 years in systemic lupus erythematosus. Arthritis Res Ther. 2021;23:29.
Petri M, Fu W, Ranger A, Allaire N, Cullen P, Magder LS, et al. Association between changes in gene signatures expression and disease activity among patients with systemic lupus erythematosus. BMC Med Genom. 2019;12:4.
Tang S, Yu Q, Ding C. Investigational spleen tyrosine kinase (SYK) inhibitors for the treatment of autoimmune diseases. Expert Opin Investig Drugs. 2022;31:291–303.
Neys SFH, Rip J, Hendriks RW, Corneth OBJ. Bruton’s tyrosine kinase inhibition as an emerging therapy in systemic autoimmune disease. Drugs. 2021;81:1605–26.
Xi J, Zhuang R, Kong L, He R, Zhu H, Zhang J. Immunoproteasome-selective inhibitors: an overview of recent developments as potential drugs for hematologic malignancies and autoimmune diseases. Eur J Med Chem. 2019;182: 111646.
Fuchs O. Targeting cereblon in hematologic malignancies. Blood Rev. 2023;57: 100994.
Tsai HC, Han MH. Sphingosine-1-phosphate (S1P) and S1P signaling pathway: therapeutic targets in autoimmunity and inflammation. Drugs. 2016;76:1067–79.
Burg N, Salmon JE, Hla T. Sphingosine 1-phosphate receptor-targeted therapeutics in rheumatic diseases. Nat Rev Rheumatol. 2022;18:335–51.
Stark GR, Darnell JE. The JAK-STAT pathway at twenty. Immunity. 2012;36:503–14.
Tanaka Y, Luo Y, O’Shea JJ, Nakayamada S. Janus kinase-targeting therapies in rheumatology: a mechanisms-based approach. Nat Rev Rheumatol. 2022;18:133–45.
Choy EH. Clinical significance of Janus Kinase inhibitor selectivity. Rheumatology (Oxford). 2019;58:953–62.
O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013;368:161–70.
Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;17:78.
Gadina M, Johnson C, Schwartz D, Bonelli M, Hasni S, Kanno Y, et al. Translational and clinical advances in JAK-STAT biology: the present and future of jakinibs. J Leukoc Biol. 2018;104:499–514.
Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. 2017;77:521–46.
Luo Y, Alexander M, Gadina M, O’Shea JJ, Meylan F, Schwartz DM. JAK-STAT signaling in human disease: from genetic syndromes to clinical inhibition. J Allergy Clin Immunol. 2021;148:911–25.
Lu LD, Stump KL, Wallace NH, Dobrzanski P, Serdikoff C, Gingrich DE, et al. Depletion of autoreactive plasma cells and treatment of lupus nephritis in mice using CEP-33779, a novel, orally active, selective inhibitor of JAK2. J Immunol. 2011;187:3840–53.
Wang S, Yang N, Zhang L, Huang B, Tan H, Liang Y, et al. Jak/STAT signaling is involved in the inflammatory infiltration of the kidneys in MRL/lpr mice. Lupus. 2010;19:1171–80.
Ripoll È, de Ramon L, DraibeBordignon J, Merino A, Bolaños N, Goma M, et al. JAK3-STAT pathway blocking benefits in experimental lupus nephritis. Arthritis Res Ther. 2016;18:134.
Ikeda K, Hayakawa K, Fujishiro M, Kawasaki M, Hirai T, Tsushima H, et al. JAK inhibitor has the amelioration effect in lupus-prone mice: the involvement of IFN signature gene downregulation. BMC Immunol. 2017;18:41.
Lin J, Zhang Y, Wang M, Zhang Y, Li P, Cao Y, et al. Therapeutic effects of tofacitinib on pristane-induced murine lupus. Arch Rheumatol. 2022;37:195–204.
Furumoto Y, Smith CK, Blanco L, Zhao W, Brooks SR, Thacker SG, et al. Tofacitinib ameliorates murine lupus and its associated vascular dysfunction. Arthritis Rheumatol. 2017;69:148–60.
Yan Q, Chen W, Song H, Long X, Zhang Z, Tang X, et al. Tofacitinib ameliorates lupus through suppression of T cell activation mediated by TGF-beta type I receptor. Front Immunol. 2021;12: 675542.
Lee J, Park Y, Jang SG, Hong SM, Song YS, Kim MJ, et al. Baricitinib attenuates autoimmune phenotype and podocyte injury in a murine model of systemic lupus erythematosus. Front Immunol. 2021;12: 704526.
Kawasaki M, Fujishiro M, Yamaguchi A, Nozawa K, Kaneko H, Takasaki Y, et al. Possible role of the JAK/STAT pathways in the regulation of T cell-interferon related genes in systemic lupus erythematosus. Lupus. 2011;20:1231–9.
Goropevšek A, Gorenjak M, Gradišnik S, Dai K, Holc I, Hojs R, et al. Increased levels of STAT1 protein in blood CD4 T cells from systemic lupus erythematosus patients are associated with perturbed homeostasis of activated CD45RA-FOXP3hi regulatory subset and follow-up disease severity. J Interferon Cytokine Res. 2017;37:254–68.
Hagberg N, Joelsson M, Leonard D, Reid S, Eloranta ML, Mo J, et al. The STAT4 SLE risk allele rs7574865[T] is associated with increased IL-12-induced IFN-γ production in T cells from patients with SLE. Ann Rheum Dis. 2018;77:1070–7.
de la Varga MR, Rodríguez-Bayona B, Añez GA, Medina Varo F, Pérez Venegas JJ, Brieva JA, et al. Clinical relevance of circulating anti-ENA and anti-dsDNA secreting cells from SLE patients and their dependence on STAT-3 activation. Eur J Immunol. 2017;47:1211–9.
Wang JM, Xu WD, Huang AF. Association of STAT4 Gene Rs7574865, Rs10168266 polymorphisms and systemic lupus erythematosus susceptibility: a meta-analysis. Immunol Investig. 2021;50:282–94.
Tangtanatakul P, Thumarat C, Satproedprai N, Kunhapan P, Chaiyasung T, Klinchanhom S, et al. Meta-analysis of genome-wide association study identifies FBN2 as a novel locus associated with systemic lupus erythematosus in Thai population. Arthritis Res Ther. 2020;22:185.
Alarcón-Riquelme ME, Ziegler JT, Molineros J, Howard TD, Moreno-Estrada A, Sánchez-Rodríguez E, et al. Genome-wide association study in an Amerindian ancestry population reveals novel systemic lupus erythematosus risk loci and the role of European admixture. Arthritis Rheumatol. 2016;68:932–43.
Zheng J, Yin J, Huang R, Petersen F, Yu X. Meta-analysis reveals an association of STAT4 polymorphisms with systemic autoimmune disorders and anti-dsDNA antibody. Hum Immunol. 2013;74:986–92.
Chung SA, Taylor KE, Graham RR, Nititham J, Lee AT, Ortmann WA, et al. Differential genetic associations for systemic lupus erythematosus based on anti-dsDNA autoantibody production. PLoS Genet. 2011;7: e1001323.
Bolin K, Sandling JK, Zickert A, Jönsen A, Sjöwall C, Svenungsson E, et al. Association of STAT4 polymorphism with severe renal insufficiency in lupus nephritis. PLoS One. 2013;8: e84450.
Kariuki SN, Kirou KA, MacDermott EJ, Barillas-Arias L, Crow MK, Niewold TB. Cutting edge: autoimmune disease risk variant of STAT4 confers increased sensitivity to IFN-alpha in lupus patients in vivo. J Immunol. 2009;182:34–8.
Crow MK, Ronnblom L. Type I interferons in host defence and inflammatory diseases. Lupus Sci Med. 2019;6: e000336.
van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508–19.
Sandborn WJ, Su C, Sands BE, D’Haens GR, Vermeire S, Schreiber S, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2017;376:1723–36.
Hasni SA, Gupta S, Davis M, Poncio E, Temesgen-Oyelakin Y, Carlucci PM, et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat Commun. 2021;12:3391.
Kahl L, Patel J, Layton M, Binks M, Hicks K, Leon G, et al. Safety, tolerability, efficacy and pharmacodynamics of the selective JAK1 inhibitor GSK2586184 in patients with systemic lupus erythematosus. Lupus. 2016;25:1420–30.
van Vollenhoven RF, Layton M, Kahl L, Schifano L, Hachulla E, Machado D, et al. DRESS syndrome and reversible liver function abnormalities in patients with systemic lupus erythematosus treated with the highly selective JAK-1 inhibitor GSK2586184. Lupus. 2015;24:648–9.
Feagan BG, Danese S, Loftus EV, Vermeire S, Schreiber S, Ritter T, et al. Filgotinib as induction and maintenance therapy for ulcerative colitis (SELECTION): a phase 2b/3 double-blind, randomised, placebo-controlled trial. Lancet. 2021;397:2372–84.
Genovese MC, Kalunian K, Gottenberg JE, Mozaffarian N, Bartok B, Matzkies F, et al. Effect of filgotinib vs placebo on clinical response in patients with moderate to severe rheumatoid arthritis refractory to disease-modifying antirheumatic drug therapy: the FINCH 2 Randomized Clinical Trial. JAMA. 2019;322:315–25.
Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, Del Carmen ML, Reyes Gonzaga J, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. N Engl J Med. 2017;376:652–62.
King B, Ohyama M, Kwon O, Zlotogorski A, Ko J, Mesinkovska NA, et al. Two phase 3 trials of baricitinib for alopecia areata. N Engl J Med. 2022;386:1687–99.
Reich K, Kabashima K, Peris K, Silverberg JI, Eichenfield LF, Bieber T, et al. Efficacy and safety of baricitinib combined with topical corticosteroids for treatment of moderate to severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156:1333–43.
Kalil AC, Patterson TF, Mehta AK, Tomashek KM, Wolfe CR, Ghazaryan V, et al. Baricitinib plus remdesivir for hospitalized adults with Covid-19. N Engl J Med. 2021;384:795–807.
Ely EW, Ramanan AV, Kartman CE, de Bono S, Liao R, Piruzeli MLB, et al. Efficacy and safety of baricitinib plus standard of care for the treatment of critically ill hospitalised adults with COVID-19 on invasive mechanical ventilation or extracorporeal membrane oxygenation: an exploratory, randomised, placebo-controlled trial. Lancet Respir Med. 2022;10:327–36.
Wallace DJ, Furie RA, Tanaka Y, Kalunian KC, Mosca M, Petri MA, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2018;392:222–31.
Dörner T, van Vollenhoven RF, Doria A, Jia B, Ross Terres JA, Silk ME, et al. Baricitinib decreases anti-dsDNA in patients with systemic lupus erythematosus: results from a phase II double-blind, randomized, placebo-controlled trial. Arthritis Res Ther. 2022;24:112.
Dörner T, Tanaka Y, Petri MA, Smolen JS, Wallace DJ, Dow ER, et al. Baricitinib-associated changes in global gene expression during a 24-week phase II clinical systemic lupus erythematosus trial implicates a mechanism of action through multiple immune-related pathways. Lupus Sci Med. 2020;7: e000424.
Morand EF, Vital EM, Petri M, van Vollenhoven R, Wallace DJ, Mosca M, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 3 trial (SLE-BRAVE-I). Lancet. 2023. https://doi.org/10.1016/S0140-6736(22)02607-1.
Petri M, Bruce IN, Dörner T, Tanaka Y, Morand EF, Kalunian KC, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 3 trial (SLE-BRAVE-II). Lancet. 2023. https://doi.org/10.1016/S0140-6736(22)02546-6.
Morand E, Pike M, Merrill JT, van Vollenhoven R, Werth VP, Hobar C, et al. Deucravacitinib, a tyrosine kinase 2 inhibitor, in systemic lupus erythematosus: a phase II, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 2023;75:242–52.
CarneroContentti E, Correale J. Bruton’s tyrosine kinase inhibitors: a promising emerging treatment option for multiple sclerosis. Expert Opin Emerg Drugs. 2020;25:377–81.
Neys SFH, Hendriks RW, Corneth OBJ. Targeting Bruton’s tyrosine kinase in inflammatory and autoimmune pathologies. Front Cell Dev Biol. 2021;9: 668131.
Liang C, Tian D, Ren X, Ding S, Jia M, Xin M, et al. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: a mini-review. Eur J Med Chem. 2018;151:315–26.
Zhang D, Gong H, Meng F. Recent advances in BTK inhibitors for the treatment of inflammatory and autoimmune diseases. Molecules. 2021;26:4907.
Rip J, de Bruijn MJW, Appelman MK, Pal Singh S, Hendriks RW, Corneth OBJ. Toll-like receptor signaling drives Btk-mediated autoimmune disease. Front Immunol. 2019;10:95.
López-Herrera G, Vargas-Hernández A, González-Serrano ME, Berrón-Ruiz L, Rodríguez-Alba JC, Espinosa-Rosales F, et al. Bruton’s tyrosine kinase: an integral protein of B cell development that also has an essential role in the innate immune system. J Leukoc Biol. 2014;95:243–50.
Kuehn HS, Rådinger M, Brown JM, Ali K, Vanhaesebroeck B, Beaven MA, et al. Btk-dependent Rac activation and actin rearrangement following FcepsilonRI aggregation promotes enhanced chemotactic responses of mast cells. J Cell Sci. 2010;123:2576–85.
Quek LS, Bolen J, Watson SP. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8:1137–40.
Shinohara M, Koga T, Okamoto K, Sakaguchi S, Arai K, Yasuda H, et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008;132:794–806.
Liu J, Chen C, Wang D, Zhang J, Zhang T. Emerging small-molecule inhibitors of the Bruton’s tyrosine kinase (BTK): current development. Eur J Med Chem. 2021;217: 113329.
Christensen BW, Zaha VG, Awan FT. Cardiotoxicity of BTK inhibitors: ibrutinib and beyond. Expert Rev Hematol. 2022;15:321–31.
Thompson PA, Burger JA. Bruton’s tyrosine kinase inhibitors: first and second generation agents for patients with chronic lymphocytic leukemia (CLL). Expert Opin Investig Drugs. 2018;27:31–42.
Chalmers SA, Glynn E, Garcia SJ, Panzenbeck M, Pelletier J, Dimock J, et al. BTK inhibition ameliorates kidney disease in spontaneous lupus nephritis. Clin Immunol. 2018;197:205–18.
Kim YY, Park KT, Jang SY, Lee KH, Byun JY, Suh KH, et al. HM71224, a selective Bruton’s tyrosine kinase inhibitor, attenuates the development of murine lupus. Arthritis Res Ther. 2017;19:211.
Rankin AL, Seth N, Keegan S, Andreyeva T, Cook TA, Edmonds J, et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J Immunol. 2013;191:4540–50.
Mina-Osorio P, LaStant J, Keirstead N, Whittard T, Ayala J, Stefanova S, et al. Suppression of glomerulonephritis in lupus-prone NZB × NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheumatol. 2013;65:2380–91.
Chalmers SA, Wen J, Doerner J, Stock A, Cuda CM, Makinde HM, et al. Highly selective inhibition of Bruton’s tyrosine kinase attenuates skin and brain disease in murine lupus. Arthritis Res Ther. 2018;20:10.
Hutcheson J, Vanarsa K, Bashmakov A, Grewal S, Sajitharan D, Chang BY, et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res Ther. 2012;14:R243.
Bender AT, Pereira A, Fu K, Samy E, Wu Y, Liu-Bujalski L, et al. Btk inhibition treats TLR7/IFN driven murine lupus. Clin Immunol. 2016;164:65–77.
Haselmayer P, Camps M, Liu-Bujalski L, Nguyen N, Morandi F, Head J, et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor evobrutinib in autoimmune disease models. J Immunol. 2019;202:2888–906.
Kong W, Deng W, Sun Y, Huang S, Zhang Z, Shi B, et al. Increased expression of Bruton’s tyrosine kinase in peripheral blood is associated with lupus nephritis. Clin Rheumatol. 2018;37:43–9.
Wallace DJ, Dörner T, Pisetsky DS, Sanchez-Guerrero J, Patel AC, Parsons-Rich D, et al. Efficacy and safety of the Bruton’s tyrosine kinase inhibitor evobrutinib in systemic lupus erythematosus: results of a phase II, randomized, double-blind, placebo-controlled dose-ranging trial. ACR Open Rheumatol. 2023;5:38–48.
Cohen S, Tuckwell K, Katsumoto TR, Zhao R, Galanter J, Lee C, et al. Fenebrutinib versus placebo or adalimumab in rheumatoid arthritis: a randomized, double-blind, phase II trial (ANDES Study). Arthritis Rheumatol. 2020;72:1435–46.
Byrd JC, Smith S, Wagner-Johnston N, Sharman J, Chen AI, Advani R, et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget. 2018;9:13023–35.
Isenberg D, Furie R, Jones NS, Guibord P, Galanter J, Lee C, et al. Efficacy, safety, and pharmacodynamic effects of the Bruton’s tyrosine kinase inhibitor fenebrutinib (GDC-0853) in systemic lupus erythematosus: results of a phase II, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 2021;73:1835–46.
Li R, Zhu X, Liu S, Zhang X, Xie C, Fu Z, et al. Orelabrutinib, an irreversible inhibitor of Bruton’s tyrosine kinase (btk), for the treatment of systemic lupus erythematosus (SLE): results of a randomized, double-blind, placebo-controlled, phase Ib/IIa dose-finding study. Ann Rheum Dis. 2022;81(Suppl. 1):210.
Deng GM, Kyttaris VC, Tsokos GC. Targeting Syk in autoimmune rheumatic diseases. Front Immunol. 2016;7:78.
Aouar B, Kovarova D, Letard S, Font-Haro A, Florentin J, Weber J, et al. Dual role of the tyrosine kinase Syk in regulation of Toll-like receptor signaling in plasmacytoid dendritic cells. PLoS One. 2016;11: e0156063.
Kelly V, Genovese M. Novel small molecule therapeutics in rheumatoid arthritis. Rheumatology (Oxford). 2013;52:1155–62.
Deng GM, Liu L, Bahjat FR, Pine PR, Tsokos GC. Suppression of skin and kidney disease by inhibition of spleen tyrosine kinase in lupus-prone mice. Arthritis Rheumatol. 2010;62:2086–92.
Bahjat FR, Pine PR, Reitsma A, Cassafer G, Baluom M, Grillo S, et al. An orally bioavailable spleen tyrosine kinase inhibitor delays disease progression and prolongs survival in murine lupus. Arthritis Rheumatol. 2008;58:1433–44.
Pohlmeyer CW, Shang C, Han P, Cui ZH, Jones RM, Clarke AS, et al. Characterization of the mechanism of action of lanraplenib, a novel spleen tyrosine kinase inhibitor, in models of lupus nephritis. BMC Rheumatol. 2021;5:15.
Kitai M, Fukuda N, Ueno T, Endo M, Maruyama T, Abe M, et al. Effects of a spleen tyrosine kinase inhibitor on progression of the lupus nephritis in mice. J Pharmacol Sci. 2017;134:29–36.
Cho S, Jang E, Yoon T, Hwang H, Youn J. A novel selective spleen tyrosine kinase inhibitor SKI-O-703 (cevidoplenib) ameliorates lupus nephritis and serum-induced arthritis in murine models. Clin Exp Immunol. 2022;211:31–45.
Matsukane R, Suetsugu K, Hirota T, Ieiri I. Clinical pharmacokinetics and pharmacodynamics of fostamatinib and its active moiety R406. Clin Pharmacokinet. 2022;61:955–72.
Paik J. Fostamatinib: a review in chronic immune thrombocytopenia. Drugs. 2021;81:935–43.
Genovese MC, van der Heijde DM, Keystone EC, Spindler AJ, Benhamou C, Kavanaugh A, et al. A phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study of 2 dosing regimens of fostamatinib in patients with rheumatoid arthritis with an inadequate response to a tumor necrosis factor-α antagonist. J Rheumatol. 2014;41:2120–8.
Weinblatt ME, Genovese MC, Ho M, Hollis S, Rosiak-Jedrychowicz K, Kavanaugh A, et al. Effects of fostamatinib, an oral spleen tyrosine kinase inhibitor, in rheumatoid arthritis patients with an inadequate response to methotrexate: results from a phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheumatol. 2014;66:3255–64.
Kivitz AJ, Mehta DP, Matzkies F, Mozaffarian A, Kunder R, Di Paolo J, et al. GS-9876, a novel, highly selective, SYK inhibitor in patients with active rheumatoid arthritis: safety, tolerability and efficacy results of a phase 2 study [abstract 2518]. Arthritis Rheumatol. 2018;70(Suppl. 9).
Grammatikos AP, Ghosh D, Devlin A, Kyttaris VC, Tsokos GC. Spleen tyrosine kinase (Syk) regulates systemic lupus erythematosus (SLE) T cell signaling. PLoS One. 2013;8: e74550.
Krishnan S, Juang YT, Chowdhury B, Magilavy A, Fisher CU, Nguyen H, et al. Differential expression and molecular associations of Syk in systemic lupus erythematosus T cells. J Immunol. 2008;181:8145–52.
Fleischer SJ, Giesecke C, Mei HE, Lipsky PE, Daridon C, Dörner T. Increased frequency of a unique spleen tyrosine kinase bright memory B cell population in systemic lupus erythematosus. Arthritis Rheumatol. 2014;66:3424–35.
Werth VP, Fleischmann R, Robern M, Touma Z, Tiamiyu I, Gurtovaya O, et al. Filgotinib or lanraplenib in moderate to severe cutaneous lupus erythematosus: a phase 2, randomized, double-blind, placebo-controlled study. Rheumatology (Oxford). 2022;61:2413–23.
Baker M, Chaichian Y, Genovese M, Derebail V, Rao P, Chatham W, et al. Phase II, randomised, double-blind, multicentre study evaluating the safety and efficacy of filgotinib and lanraplenib in patients with lupus membranous nephropathy. RMD Open. 2020;6: e001490.
Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. 2017;14:417–33.
Wang J, Fang Y, Fan RA, Kirk CJ. Proteasome inhibitors and their pharmacokinetics, pharmacodynamics, and metabolism. Int J Mol Sci. 2021;22:11595.
Eskandari SK, Seelen MAJ, Lin G, Azzi JR. The immunoproteasome: an old player with a novel and emerging role in alloimmunity. Am J Transplant. 2017;17:3033–9.
Hiepe F, Radbruch A. Plasma cells as an innovative target in autoimmune disease with renal manifestations. Nat Rev Nephrol. 2016;12:232–40.
Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. 2008;14:748–55.
Seavey MM, Lu LD, Stump KL, Wallace NH, Ruggeri BA. Novel, orally active, proteasome inhibitor, delanzomib (CEP-18770), ameliorates disease symptoms and glomerulonephritis in two preclinical mouse models of SLE. Int Immunopharmacol. 2012;12:257–70.
Lee SW, Kim BS. Comparison of therapeutic efficacy between bortezomib and combination treatment of prednisolone and mycophenolate mofetil on nephritis in NZB/WF1 mice. Clin Exp Rheumatol. 2010;28:393–6.
Ichikawa HT, Conley T, Muchamuel T, Jiang J, Lee S, Owen T, et al. Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheumatol. 2012;64:493–503.
Muchamuel T, Anderl JL, Fan RA, Johnson HWB, Kirk CJ, Lowe ET. KZR-616, a selective inhibitor of the immunoproteasome, blocks the disease progression in multiple models of systemic lupus erythematosus (SLE). Ann Rheum Dis. 2018;77(Suppl):A685.
Walhelm T, Gunnarsson I, Heijke R, Leonard D, Trysberg E, Eriksson P, et al. Clinical experience of proteasome inhibitor bortezomib regarding efficacy and safety in severe systemic lupus erythematosus: a nationwide study. Front Immunol. 2021;12: 756941.
Alexander T, Sarfert R, Klotsche J, Kühl AA, Rubbert-Roth A, Lorenz HM, et al. The proteasome inhibitor bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann Rheum Dis. 2015;74:1474–8.
Quartuccio L, Rupolo M, Michieli M, De Vita S. Efficacy and tolerability of repeated cycles of a once-weekly regimen of bortezomib in lupus. Rheumatology (Oxford). 2014;53:381–2.
Segarra A, Arredondo KV, Jaramillo J, Jatem E, Salcedo MT, Agraz I, et al. Efficacy and safety of bortezomib in refractory lupus nephritis: a single-center experience. Lupus. 2020;29:118–25.
Zhang H, Liu Z, Huang L, Hou J, Zhou M, Huang X, et al. The short-term efficacy of bortezomib combined with glucocorticoids for the treatment of refractory lupus nephritis. Lupus. 2017;26:952–8.
Alexander T, Cheng Q, Klotsche J, Khodadadi L, Waka A, Biesen R, et al. Proteasome inhibition with bortezomib induces a therapeutically relevant depletion of plasma cells in SLE but does not target their precursors. Eur J Immunol. 2018;48:1573–9.
Ishii T, Tanaka Y, Kawakami A, Saito K, Ichinose K, Fujii H, et al. Multicenter double-blind randomized controlled trial to evaluate the effectiveness and safety of bortezomib as a treatment for refractory systemic lupus erythematosus. Mod Rheumatol. 2018;28:986–92.
Khodadadi L, Cheng Q, Alexander T, Sercan-Alp Ö, Klotsche J, Radbruch A, et al. Bortezomib plus continuous B cell depletion results in sustained plasma cell depletion and amelioration of lupus nephritis in NZB/W F1 mice. PLoS One. 2015;10: e0135081.
Sjöwall C, Hjorth M, Eriksson P. Successful treatment of refractory systemic lupus erythematosus using proteasome inhibitor bortezomib followed by belimumab: description of two cases. Lupus. 2017;26:1333–8.
Fang Y, Johnson H, Anderl JL, Muchamuel T, McMinn D, Morisseau C, et al. Role of epoxide hydrolases and cytochrome P450s on metabolism of KZR-616, a first-in-class selective inhibitor of the immunoproteasome. Drug Metab Dispos. 2021;49:810–21.
Furie R, Parikh S, Maiquez A, Khan A, Moreno O, Soneira M, et al. Treatment of SLE with the immunoproteasome inhibitor KZR-616: results from the first four cohorts of the MISSION study, an open-label phase 1b dose escalation trial. Ann Rheum Dis. 2020;79(Suppl. 1):1482.
Richardson PG, Mateos MV, Vangsted AJ, Ramasamy K, Abildgaard N, Ho PJ, et al. The role of E3 ubiquitin ligase in multiple myeloma: potential for cereblon E3 ligase modulators in the treatment of relapsed/refractory disease. Expert Rev Proteomics. 2022;19:235–46.
Matyskiela ME, Zhang W, Man HW, Muller G, Khambatta G, Baculi F, et al. A cereblon modulator (CC-220) with improved degradation of Ikaros and Aiolos. J Med Chem. 2018;61:535–42.
John LB, Ward AC. The Ikaros gene family: transcriptional regulators of hematopoiesis and immunity. Mol Immunol. 2011;48:1272–8.
Lessard CJ, Adrianto I, Ice JA, Wiley GB, Kelly JA, Glenn SB, et al. Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. Am J Hum Genet. 2012;90:648–60.
Wang C, Ahlford A, Järvinen TM, Nordmark G, Eloranta ML, Gunnarsson I, et al. Genes identified in Asian SLE GWASs are also associated with SLE in Caucasian populations. Eur J Hum Genet. 2013;21:994–9.
Schafer PH, Ye Y, Wu L, Kosek J, Ringheim G, Yang Z, et al. Cereblon modulator iberdomide induces degradation of the transcription factors Ikaros and Aiolos: immunomodulation in healthy volunteers and relevance to systemic lupus erythematosus. Ann Rheum Dis. 2018;77:1516–23.
Rivellese F, Manou-Stathopoulou S, Mauro D, Goldmann K, Pyne D, Rajakariar R, et al. Effects of targeting the transcription factors Ikaros and Aiolos on B cell activation and differentiation in systemic lupus erythematosus. Lupus Sci Med. 2021;8: e000445.
Lee SW, Park YB, Yang J, Park KH, Lee SK, Choi KH, et al. Attenuation of nephritis in lupus-prone mice by thalidomide. Rheumatology (Oxford). 2012;51:2131–40.
Kim EJ, Lee JG, Kim JY, Song SH, Joo DJ, Huh KH, et al. Enhanced immune-modulatory effects of thalidomide and dexamethasone co-treatment on T cell subsets. Immunology. 2017;152:628–37.
Yuki EFN, Silva CA, Aikawa NE, Romiti R, Heise CO, Bonfa E, et al. Thalidomide and lenalidomide for refractory systemic/cutaneous lupus erythematosus treatment: a narrative review of literature for clinical practice. J Clin Rheumatol. 2021;27:248–59.
Li W, Garcia D, Cornell RF, Gailani D, Laubach J, Maglio ME, et al. Cardiovascular and thrombotic complications of novel multiple myeloma therapies: a review. JAMA Oncol. 2017;3:980–8.
Furie RA, Hough DR, Gaudy A, Ye Y, Korish S, Delev N, et al. Iberdomide in patients with systemic lupus erythematosus: a randomised, double-blind, placebo-controlled, ascending-dose, phase 2a study. Lupus Sci Med. 2022;9: e000581.
Merrill JT, Werth VP, Furie R, van Vollenhoven R, Dörner T, Petronijevic M, et al. Phase 2 trial of iberdomide in systemic lupus erythematosus. N Engl J Med. 2022;386:1034–45.
Lipsky PE, van Vollenhoven R, Dörner T, Werth VP, Merrill JT, Furie R, et al. Biological impact of iberdomide in patients with active systemic lupus erythematosus. Ann Rheum Dis. 2022;81:1136–42.
Pérez-Jeldres T, Alvarez-Lobos M, Rivera-Nieves J. Targeting sphingosine-1-phosphate signaling in immune-mediated diseases: beyond multiple sclerosis. Drugs. 2021;81:985–1002.
Chen H, Wang J, Zhang C, Ding P, Tian S, Chen J, et al. Sphingosine 1-phosphate receptor, a new therapeutic direction in different diseases. Biomed Pharmacother. 2022;153: 113341.
Strasser DS, Froidevaux S, Sippel V, Gerossier E, Grieder U, Pierlot GM, et al. Preclinical to clinical translation of cenerimod, a novel S1P1 receptor modulator, in systemic lupus erythematosus. RMD Open. 2020;6: e001261.
Ando S, Amano H, Amano E, Minowa K, Watanabe T, Nakano S, et al. FTY720 exerts a survival advantage through the prevention of end-stage glomerular inflammation in lupus-prone BXSB mice. Biochem Biophys Res Commun. 2010;394:804–10.
Okazaki H, Hirata D, Kamimura T, Sato H, Iwamoto M, Yoshio T, et al. Effects of FTY720 in MRL-lpr/lpr mice: therapeutic potential in systemic lupus erythematosus. J Rheumatol. 2002;29:707–16.
Wenderfer SE, Stepkowski SM, Braun MC. Increased survival and reduced renal injury in MRL/lpr mice treated with a novel sphingosine-1-phosphate receptor agonist. Kidney Int. 2008;74:1319–26.
Sugahara K, Maeda Y, Shimano K, Murase M, Mochiduki S, Takemoto K, et al. Amiselimod (MT-1303), a novel sphingosine 1-phosphate receptor-1 modulator, potently inhibits the progression of lupus nephritis in two murine SLE models. J Immunol Res. 2019;2019: 5821589.
Taylor Meadows KR, Steinberg MW, Clemons B, Stokes ME, Opiteck GJ, Peach R, et al. Ozanimod (RPC1063), a selective S1PR1 and S1PR5 modulator, reduces chronic inflammation and alleviates kidney pathology in murine systemic lupus erythematosus. PLoS One. 2018;13: e0193236.
Suzuki S, Li XK, Shinomiya T, Enosawa S, Amemiya H, Amari M, et al. The in vivo induction of lymphocyte apoptosis in MRL-lpr/lpr mice treated with FTY720. Clin Exp Immunol. 1997;107:103–11.
Shi D, Tian T, Yao S, Cao K, Zhu X, Zhang M, et al. FTY720 attenuates behavioral deficits in a murine model of systemic lupus erythematosus. Brain Behav Immun. 2018;70:293–304.
Mike EV, Makinde HM, Der E, Stock A, Gulinello M, Gadhvi GT, et al. Neuropsychiatric systemic lupus erythematosus is dependent on sphingosine-1-phosphate signaling. Front Immunol. 2018;9: 2189.
Tanaka Y, Kondo K, Ichibori A, Yanai Y, Susuta Y, Inoue S, et al. Amiselimod, a sphingosine 1-phosphate receptor-1 modulator, for systemic lupus erythematosus: a multicenter, open-label exploratory study. Lupus. 2020;29:1902–13.
Hermann V, Batalov A, Smakotina S, Juif PE, Cornelisse P. First use of cenerimod, a selective S1P1 receptor modulator, for the treatment of SLE: a double-blind, randomised, placebo-controlled, proof-of-concept study. Lupus Sci Med. 2019;6: e000354.
Appel GB, Contreras G, Dooley MA, Ginzler EM, Isenberg D, Jayne D, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol. 2009;20:1103–12.
Ginzler EM, Dooley MA, Aranow C, Kim MY, Buyon J, Merrill JT, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med. 2005;353:2219–28.
Ginzler E, Guedes Barbosa LS, D’Cruz D, Furie R, Maksimowicz-McKinnon K, Oates J, et al. Phase III/IV, randomized, fifty-two-week study of the efficacy and safety of belimumab in patients of black African ancestry with systemic lupus erythematosus. Arthritis Rheumatol. 2022;74:112–23.
Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. 2020;382:211–21.
Furie RA, Morand EF, Bruce IN, Manzi S, Kalunian KC, Vital EM, et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): a randomised, controlled, phase 3 trial. Lancet Rheumatol. 2019;1:e208–19.
Vital EM, Merrill JT, Morand EF, Furie RA, Bruce IN, Tanaka Y, et al. Anifrolumab efficacy and safety by type I interferon gene signature and clinical subgroups in patients with SLE: post hoc analysis of pooled data from two phase III trials. Ann Rheum Dis. 2022;81:951–61.
Parikh SV, Malvar A, Song H, Shapiro J, Mejia-Vilet JM, Ayoub I, et al. Molecular profiling of kidney compartments from serial biopsies differentiate treatment responders from non-responders in lupus nephritis. Kidney Int. 2022;102(4):845–65.
Der E, Suryawanshi H, Morozov P, Kustagi M, Goilav B, Ranabothu S, et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat Immunol. 2019;20:915–27.
Mok CC, Mohan C. Urinary biomarkers in lupus nephritis: are we there yet? Arthritis Rheumatol. 2021;73:194–6.
Mok CC. The dawn of a new era of therapies in systemic lupus erythematosus. Rheumatol Immunol Res. 2020;1:31–7.
Ytterberg SR, Bhatt DL, Mikuls TR, Koch GG, Fleischmann R, Rivas JL, et al. Cardiovascular and cancer risk with tofacitinib in rheumatoid arthritis. N Engl J Med. 2022;386:316–26.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were received for the preparation of this article.
Conflict of Interest
Chi Chiu Mok has received speaker honoraria from GSK and Pfizer during the recent APLAR 2022 meeting.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Data Availability
Not applicable.
Code Availability
Not applicable.
Author Contributions
The sole author, Chi Chiu Mok, is responsible for the drafting of the manuscript and the literature search.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mok, C.C. Targeted Small Molecules for Systemic Lupus Erythematosus: Drugs in the Pipeline. Drugs 83, 479–496 (2023). https://doi.org/10.1007/s40265-023-01856-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-023-01856-x