
Abstract
Sphingosine-1-phosphate (S1P) is a bioactive lipid metabolite that exerts its actions by engaging 5 G-protein-coupled receptors (S1PR1-S1PR5). S1P receptors are involved in several cellular and physiological events, including lymphocyte/hematopoietic cell trafficking. An S1P gradient (low in tissues, high in blood), maintained by synthetic and degradative enzymes, regulates lymphocyte trafficking. Because lymphocytes live long (which is critical for adaptive immunity) and recirculate thousands of times, the S1P-S1PR pathway is involved in the pathogenesis of immune-mediated diseases. The S1PR1 modulators lead to receptor internalization, subsequent ubiquitination, and proteasome degradation, which renders lymphocytes incapable of following the S1P gradient and prevents their access to inflammation sites. These drugs might also block lymphocyte egress from lymph nodes by inhibiting transendothelial migration. Targeting S1PRs as a therapeutic strategy was first employed for multiple sclerosis (MS), and four S1P modulators (fingolimod, siponimod, ozanimod, and ponesimod) are currently approved for its treatment. New S1PR modulators are under clinical development for MS, and their uses are being evaluated to treat other immune-mediated diseases, including inflammatory bowel disease (IBD), rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and psoriasis. A clinical trial in patients with COVID-19 treated with ozanimod is ongoing. Ozanimod and etrasimod have shown promising results in IBD; while in phase 2 clinical trials, ponesimod has shown improvement in 77% of the patients with psoriasis. Cenerimod and amiselimod have been tested in SLE patients. Fingolimod, etrasimod, and IMMH001 have shown efficacy in RA preclinical studies. Concerns relating to S1PR modulators are leukopenia, anemia, transaminase elevation, macular edema, teratogenicity, pulmonary disorders, infections, and cardiovascular events. Furthermore, S1PR modulators exhibit different pharmacokinetics; a well-established first-dose event associated with S1PR modulators can be mitigated by gradual up-titration. In conclusion, S1P modulators represent a novel and promising therapeutic strategy for immune-mediated diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
S1PR modulators are immunomodulatory drugs that modulate lymphocyte egress from lymphoid organs, and four S1PR modulators are now approved to treat multiple sclerosis. |
S1P and its receptors are involved in multiple immune functions, and therapies targeting S1P signaling have been used and tested to treat immune-mediated diseases other than MS, including IBD, RA, SLE, and psoriasis, with promising results. |
More selective S1PR modulators with potentially superior drug efficacy and better side-effect profiles are in the development pipeline. Ozanimod and etrasimod have shown positive results in IBD, whereas cenerimod might provide therapeutic benefits for SLE. |
1 Introduction
Sphingosine-1-phosphate (S1P) is a multifunctional bioactive lipid involved in multiple physiological and pathophysiological processes. S1P signals through S1P receptors (S1PR1–S1PR5) [1, 2], and is relevant for the regulation of the immune, cardiovascular, and nervous systems [3]. Thus, it is not surprising that the S1P pathway has been implicated in the pathophysiology of immune-mediated diseases and cancer. S1PR agonists/modulators or targeting the S1P axis via inhibitors that target S1P generation, transport, and degradation may represent additional “druggable” approaches for the treatment of immune-mediated diseases [1]. Research on the S1P pathway has led to approval of safe and effective S1PR modulators to treat MS, such as ponesimod, ozanimod, siponimod, and fingolimod [4]. Several other agents are in clinical development for MS, including amiselimod, cerafilimod, etrasimod, and GSK2018682 [4]. Beyond MS, therapies targeting S1P pathways have expanded to treat other inflammatory conditions. Currently various clinical trials have been completed while others are still ongoing in settings such as renal transplantation, chronic inflammatory demyelinating polyradiculoneuropathy, stroke, amyotrophic lateral sclerosis, schizophrenia, Rett syndrome, chronic plaque psoriasis, IBD, SLE, primary biliary cholangitis, pyoderma gangrenosum, transplant rejection, macular degeneration, interstitial cystitis, endometriosis, asthma, pneumonia, and glioblastoma [5,6,7]. Trials evaluating the effects of S1PR modulators on uveitis, acute demyelinating optic neuritis, polymyositis, dermatomyositis, graft versus host disease and renal cell carcinoma were, however, terminated before completion [6]. Here, we review strategies that target S1P-S1PRs signaling under clinical development as therapeutics for immune-mediated diseases, with an emphasis on IBD.
2 Sphingosine-1-phosphate (S1P) Signaling in Health and Disease
S1P is a signaling molecule involved in physiological processes such as cellular chemotaxis, migration, growth, and proliferation. It also participates in neurogenesis, angiogenesis, heart chronotropism, and inotropism, endothelial permeability, and blood vessel integrity and tone [8]. S1P enhances the intestinal epithelial barrier by increasing vascular endothelial (VE)-cadherin [9] and has a role in pathological conditions such as inflammation and cancer, where immune surveillance plays a role [8]. S1P is secreted by erythrocytes, vascular, and lymphatic endothelial cells [10, 11]. Platelets and mast cells secrete and produce S1P when stimulated, but do not contribute to basal S1P levels [10, 11].
S1P is a metabolite of ceramide (the basic unit of sphingolipids) and consists of a long-chain fatty acyl group attached to sphingosine via its amino group [12]. Sphingosine and ceramide are associated with apoptosis and cellular growth arrest, whereas S1P is associated with apoptosis suppression and cellular survival [12]. Several steps are required to maintain homeostatic S1P levels such as phosphorylation, transport, and degradation [9]. Ceramide is broken down by ceramidases into sphingosine, which is phosphorylated by two sphingosine-kinases (SphK), SphK1 and SphK2, generating S1P. S1P could be reversibly dephosphorylated by S1P-phosphatase (SPP1 and SPP2), or irreversibly degraded by the S1P-lyase to hexadecanal and phosphoethanolamine (Fig. 1a, [1, 13,14,15,16]). This enzymatic pathway is responsible for the regulation of S1P levels, maintaining a lower concentration at tissues, lymphoid organs, and thymus, and a higher concentration in blood, where it can be bound to albumin or apolipoprotein M (ApoM) [17]. Free S1P or albumin-bound S1P is more susceptible to degradation than S1P bound to lipoproteins [10].

S1P synthesis, degradation, export, and intracellular signaling [1, 13,14,15,16]. (a) Ceramide is broken down by ceramidases into sphingosine, which is phosphorylated by two sphingosine kinases (SphK1, SphK2), generating S1P within lysosomes and the endoplasmic reticulum (ER). SphK1 is mainly cytosolic and is translocated to the plasma membrane upon activation. SPHK2 is located within the mitochondria, nucleus, and ER. At the ER, S1P might be dephosphorylated by S1P phosphatase (SPP1,2) to sphingosine, which can be used for ceramide synthesis or irreversibly degraded by S1P lyases. S1P is also produced in the mitochondria and nucleus by SPHK2 with intracellular targets such as prohibitin 2 (PHB2) and histone deacetylases (HDACs) [1]. PHB2 stabilizes cytochrome c oxidases (COX), whereas HDACs remove acetyl groups from histones, altering gene transcription. S1P is transported out of the cell by specific ATP-binding cassette transporters like ABCA1, ABCC1, ABCG2, and Spinter Homolog 2 (SPNS2) protein [1]. Extracellular S1P can act in a paracrine or autocrine fashion binding to S1PRs and initiating downstream signaling. (b) S1P generated by SPHK1 in response to TNF binds to the TNF-receptor-associated factor 2 (TRAF2), an E3 ubiquitin ligase, enhancing its activity and leading to lysine-63-linked polyubiquitination of the receptor-interacting protein 1 (RIP1) and activating the nuclear factor “kappa-light-chain-enhancer” within the NF-κB pathway, favoring transcription of proinflammatory cytokines [14, 21]. TRAF2 may alternatively bind to TRAF-interacting protein (TRIP), blocking S1P binding to TRAF2 and suppressing downstream signaling. (c) In response to IL-1, SPHK1 and the cellular inhibitor of apoptosis 2 (cIAP2) form a complex with interferon-regulatory factor 1 (IRF1) leading to its polyubiquitination and activation. Consequently, IRF1 enhances the expression of the chemokine CXCL10 and CCL5, which are important for mononuclear cell recruitment to sites of inflammation [15, 16]
2.1 Intracellular Targets of S1P
S1P also acts directly on several intracellular targets without engagement of S1PRs [1]. It directly binds to and inhibits histone deacetylases (HDAC) 1 and 2, preventing the removal of acetyl groups from lysine residues within histone tails, and influencing the contextual chromatin states that impact gene transcription [18, 19]. The consequences of HDAC 1 and 2 inhibition by S1P are incompletely understood but its effects might be associated with SphK2, which significantly increases S1P nuclear levels inducing p21 expression [18]. P21, a potent cyclin-dependent kinase inhibitor, regulates the cell cycle progression, and probably influences the balance between cytostasis and apoptosis of malignant cells (Fig. 1a) [18]. Other intracellular S1P targets include TNF receptor associated factor 2 (TRAF-2) and the apoptosis inhibitor cIAP2 [20]. TRAF2 is an E3 ubiquitin ligase that is a key component of the nuclear factor-κB (NF-κB) activation triggered by TNF. Engagement of the TNF receptor results from the assembly of receptor-associated signaling complexes by adaptors (TRAF-2, TNFR1-associated death domain (TRADD), and the receptor-interacting protein 1 (RIP1)). TRAF-2 and S1P produced by SphK1 are needed for the lysine-63-linked polyubiquitination RIP1 that works as a scaffold for the recruitment and stimulation of IkB kinase complex. The complex includes two homologous subunits IKKα and IKKß and a regulatory subunit IKKγ. The IKKα phosphorylation by IKK complex leads to its lysine 48 polyubiquitination and subsequent proteasomal degradation, liberating the NFκB dimer (containing the p50 and p65 subunits) that enters the nucleus and regulates proinflammatory cytokine gene transcription [14]. On the other hand, binding of the TRAF-interacting protein (TRIP) to TRAF-2 blocks S1P binding, inhibiting TNF-induced NF-κB activation and the transcription of TNFα, interleukin-6 (IL-6), and IL-1, along with antiapoptotic proteins (Fig. 1b) [14, 21]. Intracellular interaction between the apoptosis inhibitor cIAP2 and S1P is crucial for K63-linked polyubiquitination of the interferon regulatory factor 1 (IRF-1), leading to IL-1 induced expression of chemokines CCL5 and CXCL10, which recruit mononuclear cells, essential for sterile inflammation [15, 16]. Upon IL-1R activation, IL-1R recruits the interleukin-1 receptor-associated kinase-4 (IRAK-4), IRAK-1, mitogen-activated protein kinase kinase kinase 3 (MEKK-3), TRAF-6, and the MyD88 adapter, producing the recruitment of signaling molecules and activation of mitogen-activated protein kinases (MAPKs), followed by nuclear translocation of NF-kB to induce the expression of genes encoding IRF-1, cIAP2, and a variety of cytokines. In response to IL-1, SphK1 generates S1P, mediating the recruitment and polyubiquitination of cIAP2 to TRAF-6 and the K63 polyubiquitination of IRF1 [15, 16]. Then, IRF-1 translocates to the nucleus activating the expression of IRF1 as CXCL10 and CCL5 (Fig. 1c) [15, 16].
2.2 S1P Receptors
S1P acts predominantly through activation of different subtypes of G protein-coupled receptors (S1PR). The physiological roles of S1PRs are shown in Table 1 [3, 6, 22]. S1PRs are differentially expressed in the immune system [10]. S1PR1 and S1PR4 are expressed by T cells, while B cells express S1PR1, S1PR2, S1PR3, and S1PR4. S1PR1 and S1PR2 are additionally found on macrophages and mast cells, whereas S1PR1 and S1PR5 are expressed by dendritic (DC) and natural killer (NK) cells [10, 13]. Interestingly, immune cells do not necessarily express all the S1PRs simultaneously, as there is differential expression during diverse stages of cell activation and maturation [17].
2.2.1 S1PR1
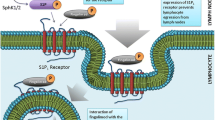
S1P signaling participates in immune cell traffic to lymphoid organs and exits to lymph and blood [10]. S1PR1 plays an essential role in the control of lymphocyte egress from the thymus, secondary lymphoid organs, and bone marrow [23, 24]. S1P-S1PR1-dependent chemotaxis requires an S1P concentration gradient, maintained by the degradative action of tissue S1P-lyase, which is absent in peripheral blood. This creates a gradient from tissues to blood/lymph, where S1P concentrations are highest [23]. The high S1P levels in the blood induce S1PR1 internalization (desensitization) in naïve T cells, whereas in lymph node and tissues, S1PR1 is re-expressed for several hours as CD69 forms a complex with S1PR1. Once S1PR1 is re-expressed, T cells then egress from the lymph node or tissue by sensing the higher S1P levels in blood [23, 24].
During inflammation, S1P-S1PR1 may play a role in lymphocyte retention within inflamed tissue [12]. S1PR1 and S1PR4 expression on T cells is induced by CXCL9 and CXCL11 chemokines, present on the endothelium, which engage CXCR3 on T cells [12]. Interaction of integrins with their ligands (lymphocyte function-associated antigen 1 (LFA-1)/intercellular adhesion molecule (ICAM-1), very late antigen-4 (VLA-4)/vascular cell adhesion molecule-1 (VCAM-1) enables T cells to arrest and extravasate into inflamed tissue. Finally, the heterotrimeric guanine-nucleotide-binding-coupled receptor-kinase2 (GRK2) downregulates S1PR1, retaining T cells within inflamed tissue [12]. S1PR1 participates in other immune regulatory functions such as inhibiting Foxp3+Treg differentiation, while promoting Th1 development and potentially Th17 polarization [25, 26].
The S1PR1/S1P axis participates in naïve B cell trafficking from bone marrow to blood, egress from secondary lymphoid organs [12], and marginal B cell localization in spleen [27]. S1PR1 also has a role in DC migration [28]. Furthermore, S1PR1 mediates suppression of sprouting angiogenesis and enhances cell-to-cell adhesion by controlling VE-cadherin at endothelial junctions, regulating lymphatic and vascular permeability. S1PR1 also participates in astrocyte proliferation, neural protection, heart rate, endothelial integrity, and ischemic reperfusion injury [28].
2.2.2 S1PR2 and S1PR3
S1PR2 receptors might have opposite functions to S1PR1 and a pro-inflammatory role has been described [29]. S1PR2 and S1PR3 mediate vascular, intestinal, bronchial, and bladder smooth muscle vasoconstriction [9]. Indeed, hypertension and ischemia-reperfusion renal injury have been reported with the use of non-selective S1PRs, likely due to S1PR2-S1PR3 modulation. S1PR3 agonism is linked to heart-rate effects and the risk for type I and type II atrioventricular block [4]. However, despite the lower affinity of new S1P modulators, there are still first-dose cardiac effects, suggesting that S1PR1 plays a role [4]. In addition, S1PR2 and S1PR3 have been implicated in pathophysiological processes including inflammation, fibrosis, cancer cell growth, and angiogenesis [9]. S1PR2 contributes to activated B-cell localization in follicles, promoting their local confinement within germinal centers [30, 31].
2.2.3 S1PR4
S1PR4 plays a role in immune cell migration and on the differentiation of certain immune cells. Immunosuppressive effects are mediated by inhibition of proliferation and secretion of effector cytokines, while enhancing IL-10 [12, 32]. S1PR4 expression on DC is involved in Th17 regulation and IL-27 production, which allows T-reg to suppress cytotoxic CD8+T cells [1, 13]. S1PR4 also enhances neutrophil trafficking from inflamed tissues to the draining lymph node [13].
2.2.4 S1PR5
NK cells employ S1PR5 [33] for trafficking from bone marrow and lymph nodes into tissues. It also controls brain endothelial barrier function, tight junctions, and permeability [34], and decreases NFκB activation on brain endothelial cells by lowering expression of adhesion molecules, inflammatory chemokines, and cytokines [34]. Oligodendrocytes, the myelin-forming cells of the CNS, express S1PR5, suggesting a role in myelination [35].
3 S1PR Agonists/Modulators in Immunological Disorders
Over the past few years, the S1P/S1PRs axis has been identified as a potential therapeutic target for immune-mediated diseases, and the safety and efficacy of several S1PRs modulators have been explored in MS, RA, SLE, psoriasis, atopic dermatitis (AD), ulcerative colitis (UC), and Crohn’s disease (CD) [23, 36]. S1PR1 agonists render lymphocytes incapable of sensing the S1P gradient and exiting lymph nodes, preventing their access to circulation and sites of inflammation. More than one mode of action has been proposed, acting as functional antagonists (binding internalizes receptors, desensitizing cells with a net effect of depressing S1PR-mediated signaling) and as traditional agonists (increasing activity through these pathways) [2, 4, 23]. Many S1PR modulators are prodrugs (e.g., fingolimod, amiselimod), which require conversion into their bioactive form by phosphorylation via sphingosine kinases (SphKs), whereas others are direct acting (e.g., ponesimod, siponimod, ozanimod) [37, 38]. The S1PR1 modulator/agonist-receptor complex is internalized, ubiquitinated, and degraded by the proteasome, leading to sustained desensitization, lymphocyte retention at lymph nodes, and peripheral lymphopenia [2, 23]. By contrast, native S1P induces S1PR internalization followed by recycling back to the cell surface, producing a transitory lymphopenia [2, 23]. These drugs also might block lymphocyte egress from lymph nodes by tightening the lymphatic endothelial barrier and inhibiting transendothelial passage from lymph nodes to blood [23]. S1PR1 participates in immune cell recruitment during both acute and chronic inflammation and a significant number of preclinical and clinical studies support their use in treating immune-mediated conditions [39]. Fingolimod/FTY720 was initially tested for the prevention of acute allograft rejection [40]. However, absence of efficacy in clinical trials led to its testing for other indications [41], eventually leading to its approval as the first oral non-selective S1PRs modulator for the treatment of relapsing-remitting MS [42, 43]. Fingolimod is a prodrug that requires phosphorylation by SphK2 to form the active metabolite fingolimod-P, a non-selective agonist of all S1PRs excluding S1PR2 [6]. Fingolimod-P regulates chemotactic responses and lymphocyte migration by inducing S1PR internalization/degradation, suppressing lymphocyte egress from thymus and secondary lymphoid organs (SLO). Its action on S1PR3, S1PR4 and S1PR5 may account for some adverse effects (hypertension, bradycardia, macular edema, reduced pulmonary function, hepatic adverse effects, and neoplasm), of which cardiovascular effects are the most common [44, 45]. Selective modulators continued to be developed to improve their safety profile [44, 45].
3.1 Inflammatory Bowel Diseases
IBD is a chronic immune-mediated inflammatory disease of the gastrointestinal tract with heterogeneous behavior and prognosis [46, 47]. Our understanding of leukocyte traffic orchestrating gut inflammation has led to the use of novel biologic agents directed against intestinal T-cell traffic, such as vedolizumab, an anti-integrin monoclonal antibody. However, all available agents in use today require parenteral administration. Small molecules that could be orally administered such as S1PR1 agonist/modulators could be a valuable addition. Fingolimod has not been tested in IBD despite its efficacy in colitic animal models, with more selective S1PR modulators tested instead [48,49,50,51,52,53].
3.1.1 Mocravimod
Mocravimod/KRP-203 is a prodrug agonist of S1PR1, S1PR4, and S1PR5 and partial agonist of S1PR3. Despite attenuating colitis in mouse models [2, 54], it was minimally effective in patients with UC (NCT01375179) [55]. A multicenter, double-blind, placebo-controlled, parallel-group study evaluated the efficacy, safety, and tolerability of KRP203 in moderately active UC refractory to 5-aminosalicylates over 8 weeks [55]. The primary endpoint was clinical remission defined as a partial Mayo Clinic Score (MCS) [56] of 0–1 and modified Baron Score [57] of 0–1 with a rectal bleeding subscore of 0 [55]. Clinical remission was achieved in 2/14 patients (14%) receiving KRP203 compared with none in the placebo group. This study did not meet the minimum clinically relevant threshold for efficacy [55] and was terminated (NCT01375179); however, its small sample size was a major limitation. Overall, KRP203 was well tolerated and safe, and no adverse cardiac events were reported [55].
3.1.2 Ozanimod
Ozanimod/RPC1063/Zeposia is an oral S1PR modulator that selectively binds to S1PR1 and S1PR5 [58, 59], reducing concerns around S1PR3 engagement. Ozanimod induces S1PR1 internalization and degradation. Its effects are highest on lymphocytes that express the chemokine receptor 7 (CCR7+), (naïve and central memory lymph node-homing cells) with less or no effect on effector memory cells, thus maintaining some circulating protective immunity [58, 59]. Ozanimod is metabolized to two major metabolites (CC-112273 and CC1084037) and other minor active metabolites (RP101988 and RP101075) [58, 59]. Ozanimod decreases inflammation in animal models of immune-mediated diseases such as 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis, CD4+CD45RBhi T-cell adoptive transfer colitis, and experimental autoimmune encephalitis (EAE) [60]. These findings served as the foundation for clinical trials leading to its approval for the treatment of relapsing MS in the USA, based on the results of the RADIANCE (NCT02047734) and SUNBEAM (NCT02294058) trials [58, 61]. The TOUCHSTONE trial (NCT01647516) evaluated ozanimod’s efficacy and safety in moderate-to-severe UC [62]. 197 patients were randomized to receive ozanimod 0.5 mg or 1 mg or placebo orally for 32 weeks. The primary outcome defined as clinical remission at week 8 (MCS ≤ 2 with no subscore > 1) was achieved by the 1 mg dose versus placebo (16% vs. 6%; p = 0.048) [62]. The secondary outcomes at week 8, such as clinical response (decrease in MCS of ≥ 3 points and ≥ 30% decrease in MCS with ≥ 1-point decrease in rectal bleeding subscore or a subscore ≤ 1) were reached in 57%, 54%, and 37% for the 1 mg, 0.5 mg, and placebo groups, respectively. Mucosal improvement/healing was achieved in nearly 30% of the treated patients in comparison with 12% in the placebo group [62]. On follow-up (week 32), clinical remission rates were achieved in 21%, 26%, and 6% and clinical response rates of 51%, 35%, and 20% for 1 mg, 0.5 mg, and placebo, respectively. The most frequent adverse events were anemia and headache [62]. The open-label extension (OLE) of the TOUCHSTONE trial (n = 170) demonstrated that 1 mg ozanimod has a rapid onset, durable efficacy, and is well tolerated through 104 weeks [63]. The results from True North, a phase 3 placebo-controlled trial evaluating ozanimod as an induction and maintenance therapy in moderate to severe UC, were presented at the American Gastroenterology Association (ACG) 2020 [64]. Patients were randomized to receive ozanimod 1 mg (n = 429) or placebo (n = 216) in the induction period [64]. After induction, patients with clinical response were re-randomized to double-blind maintenance treatment with ozanimod or placebo through 52 weeks [64]. The primary aim for both periods was the proportion of patients in clinical remission per three-component MCS. This study met both primary endpoints, showing clinically and statistically significant results compared with placebo; clinical remission at induction 18.4% versus 6% (p < 0.0001) [64]. In the maintenance period 457 patients were re-randomized to ozanimod (n = 230) or placebo (n = 227), and obtained 37% versus 18.5% (p < 0.0001) clinical remission at week 52. Interestingly, in patients with prior anti-TNF exposure, the proportion with clinical remission was higher, but not statistically superior versus placebo at week 10 [64]. However, at maintenance, clinical remission and response showed a significant improvement regardless of anti-TNF exposure. Ozanimod demonstrated significant benefits on clinical, endoscopic, histologic, and mucosal healing endpoints for up to 52 weeks in moderate to severe UC [64]. The most common adverse events were anemia, nasopharyngitis, headache, and increased serum alanine aminotransferase or gamma-glutamyl transferase [64]. These results suggest that ozanimod could be appropriate for patients who fail anti-TNF, but the onset of action is slower. The full report of the True North trial is awaited. Currently, there are three active clinical trials for moderate-severe UC: NCT02531126, NCT01647516, NCT03915769, three of which are in phase 3. STEPSTONE, a phase 2, uncontrolled multicenter trial, recruited 69 patients with moderate to severe Crohn’s disease (CD), to examine endoscopic and clinical outcomes following treatment with ozanimod for 12 weeks [65]. All patients started with a 7-day dose-escalation treatment (days 1–4 on ozanimod 0.25 mg daily, followed by 3 days at 0.5 mg daily). Then, for the additional 11 weeks, patients received ozanimod 1 mg. The primary endpoint was the change in Simple Endoscopic Score for Crohn’s Disease (SES-CD) [66] from baseline to week 12, and the proportion of patients with an endoscopic response (≥ 50% decrease in SES-CD) and endoscopic remission (SES-CD ≤ 4 points and SES-CD decrease ≥ 2 points with no SES-CD subscore > 1 point) were also assessed at week 12. Secondary endpoints were changes in Crohn’s Disease Activity Index (CDAI) [67] from baseline to week 12, and the proportion of patients with clinical remission (CDAI score of < 150) and clinical response (CDAI reduction from baseline of ≥ 100 points) were also assessed at week 12. Patient-reported outcomes (PRO2) [68] were evaluated as the change from baseline to week 12 and changes in the histology activity using Geboes Histology Activity Score (GHAS) [69] and Robart’s Histopathology Index (RHI) [70]. The mean change from baseline at week 12 in SES-CD was − 2.2 (standard deviation (SD) = 6); 16/69 (23.2%) obtained endoscopic response whereas 7/69 (10.1%) achieved endoscopic remission. A reduction from baseline CDAI also was observed (mean change − 130.4, SD = 103.9). Clinical response was achieved in 39/69 (56.5%), and clinical remission in 27/69 (39.1%). The mean change from baseline in PRO2 score was − 66.1 (SD = 65.4). The mean change from baseline in GHAS was − 5.9 (SD = 11) and in RHI − 10.6 (SD = 25.1). CD flare was the most frequent adverse event 18/69 (26%), followed by abdominal pain 10/69 (15%), lymphopenia 9/69(13%), arthralgia 9/10 (13%), and nausea 8/69 (12%). Although in seven patients liver enzymes increased three or more times, none discontinued the study drug. No clinically significant changes in heart rate were observed following the first dose of ozanimod. Treatment-emergent adverse events reported were two cases of herpes zoster infection (mild severity) and one case of severe sepsis. In summary, clinical, endoscopic, and histological improvement were observed in this trial [65]. Currently four phase 3 ozanimod clinical trials in CD are ongoing: NCT03467958, NCT03440385, NCT03440372, and NCT03464097.
3.1.3 Etrasimod
Etrasimod/APD334, an oral selective modulator of S1PR1, S1PR4, and S1PR5, has shown efficacy in the CD4+CD45RBhi T-cell transfer mouse model of colitis [71]. Recently, the results from a phase 2, double-blind, parallel-group study in patients with moderate to severe UC (n = 156) have been published (NCT02447302) [72]. Patients were randomly assigned to receive once-daily etrasimod 1 mg (n = 52), etrasimod 2 mg (n = 50), or placebo (n = 54) groups for 12 weeks. The primary endpoint was mean improvement in modified MCS at week 12, whereas the secondary endpoints considered the proportion of patients with endoscopic improvement at week 12 [72]. The modified MCS, which ranged from 0 to 9 (instead of from 0 to 12), includes subscores for stool frequency, rectal bleeding, and endoscopic findings, where increasing numbers indicate higher disease activity. Both endpoints were met in the 2 mg group. A significant improvement in the modified MCS was observed for the etrasimod 2 mg group compared with placebo with a least-square mean difference of 0.99 (CI 0.30–1.68; p = 0.009) at week 12. Further, a significantly higher proportion, 41.8% versus 17%, in the group receiving etrasimod 2 mg versus placebo (CI 9.8–39.0%; p = 0.003) achieved endoscopic improvement. Histologic improvement was reported in 32% of patients receiving 2 mg versus 10% in those receiving placebo (p = 0.006). Histologic remission occurred in 20% of those treated with 2 mg compared with 6% of placebo recipients (p = 0.03). Adverse events were mild to moderate. Three patients had a transient, asymptomatic, low-grade atrioventricular (AV) block (second-degree AV type 1 in one patient, and first-degree AV in two patients) [72]. However, these patients had experienced AV block before etrasimod was initiated [72]. AV block represents a disturbance or delay in the electrical conduction from the atria to ventricles. This disruption could be transitory or permanent. There are three types of AV nodal block: first degree, second degree (type 1 or 2), and third degree. The first degree and second degree type 1 have a benign course with a better prognosis than second degree type 2 or third degree [73].
Currently, Arena Pharma is evaluating etrasimod’s use in other trials such as a phase 3 extension study for the treatment of moderate to severely active UC (ELEVATE OLE UC; NCT03950232) and a phase 2b trial evaluating etrasimod in moderately to severely active CD (CULTIVATE; NCT04173273). Etrasimod has also been evaluated in other conditions such as Primary Biliary Cholangitis (PBC) (NCT03155932) and Pyoderma gangrenosum (NCT03072953).
3.1.4 Amiselimod
Amiselimod/MT-1303 is an oral prodrug that is converted to its active metabolite (MT-1303-P) by sphingosine kinases (SphKs); it has been evaluated for various immune-mediated diseases such as MS, SLE, IBD, and psoriasis [37, 74,75,76,77]. Amiselimod has a higher affinity for S1PR1 than for S1PR2–5, obtaining a more favorable cardiac safety profile than non-selective S1PRs modulators [78]. In a prospective, randomized, placebo-controlled clinical trial in active CD patients with elevated inflammatory markers (n = 180), the primary endpoint (clinical response defined as a drop in the CDAI of 100 points at week 12) was not achieved (48.7% for amiselimod vs. 54.1% on placebo) [78]. The high placebo response rate and weak peripheral lymphocyte reduction observed might explain the unmet endpoints [78]. Its clinical development was discontinued by Biogen in 2016, and subsequently its rights were returned to Mitsubishi Tanabe Pharma [79]. Amiselimod shows a favorable safety profile in CD and might be promising for the treatment of other autoimmune/immune-mediated diseases, including UC [80]. In Japan, a phase 2 clinical trial in CD is ongoing [81]. Results from a randomized, double-blind, multiple-dose, placebo-controlled phase 1 study (n = 190) were recently presented at the 2021 Crohn's and Colitis Congress [82]. Healthy individuals were randomized 2:1:1 to receive a single placebo dose followed by oral amiselimod in upwardly titrated doses to achieve 0.4 mg and 0.8 mg steady-state doses, moxifloxacin 400 mg followed by placebo, or placebo followed by moxifloxacin 400 mg single dose. The study aimed to evaluate the amiselimod safety profile. The discontinuation rate was 8% (n = 8) in the amiselimod group versus 4% (n=4) in the moxifloxacin group. Three patients discontinued amiselimod because of low lymphocyte counts, and one patient had atrial fibrillation requiring hospitalization and cardioversion, which led to discontinuation. All other adverse events were mild to moderate in severity. The authors conclude that upwardly titrated amiselimod was well tolerated in healthy individuals [82].
3.2 Psoriasis
Psoriasis is a chronic immune-mediated skin condition characterized by abnormal keratinocyte proliferation and lymphocyte infiltration of the dermis and epidermis [83]. Thus, drugs that induce lymphopenia and prevent lymphocyte migration might be effective.
3.2.1 Ponesimod
Ponesimod/ACT-128800, a selective modulator of S1PR1, S1PR4, and S1PR5 was evaluated in a phase 2 study (NST0128090), leading to a 75% reduction in area of involvement and severity index in 77% of patients [84, 85]. The most frequent adverse events were dyspnea, elevated liver enzymes, headache, nasopharyngitis, dizziness, bradycardia, pruritus, and cough [85]. Interestingly, liver enzyme abnormalities were significantly higher in the drug group, and these frequencies were higher than those seen in the phase 2 MS study [86, 87]. Regarding heart rate and conduction events, in this study the effect of the first-dose effect of ponesimod on AV conduction and heart rate in cardiomyocytes might be due to vagomimetic S1PR1 activity [85]. Cardiomyocytes rapidly lose sensitivity to ponesimod, which results in resolution of effects on AV conduction and heart rate [85]. In this study all heart rate and AV conduction adverse events were dose dependent and limited by up-titration [85]. A gradual up-titration with ponesimod markedly mitigated the initial cardiodynamic effects [88]. Information available in ClinicalTrials.gov states that ponesimod has not progressed to a phase 3 clinical trials in patients with psoriasis.
3.2.2 Other S1P modulators
Amiselimod: The results of a phase 2 dose-finding study of amiselimod in chronic plaque psoriasis are not yet published (NCT01987843).
IMMH002: IMMH002, a novel orally active S1PR1 modulator, showed skin improvement and decreased T-lymphocyte infiltration at the preclinical level in a mouse model, without adverse events such as bradycardia [89].
3.3 Atopic Dermatitis
Atopic dermatitis (AD) is a chronic allergic relapsing inflammatory skin disease characterized by an impairment of epidermal barrier function, and an immune response skewed towards T-helper 2 [90]. Currently, the safety and efficacy of etrasimod in AD are being evaluated in a phase 2 placebo-controlled, dose-finding trial (ADVISE; NCT04162769).
3.4 Rheumatoid Arthritis
RA is a chronic immune-mediated joint disease characterized by joint infiltration by inflammatory cells and increased cytokine and chemokine secretion leading to joint destruction and systemic inflammatory burden [91]. S1P demonstrated a role in RA by increasing TNF production, which in turn upregulates S1PRs and increases inflammatory infiltration [92, 93]. Fingolimod decreased IL-6 and TNF in synovium in mouse models of RA. Etrasimod has also shown efficacy in a collagen-induced model of arthritis [94, 95]. Recently, IMMH001 (SYL930), a S1PR1 and S1PR4 modulator, inhibited arthritis progression in Sprague-Dawley rats, improving hind paw swelling and arthritis index, diminishing pro-inflammatory cytokines and chemokines in damaged joints. Despite these encouraging results, there are no ongoing clinical studies in RA [96].
3.5 Systemic Lupus Erythematosus
SLE is an autoimmune disease characterized by aberrant B- and T-cell activation, leading to increased production of IL-1, IL-6, and autoantibodies against multiple cytoplasmic and nuclear antigens in various organ systems [97]. In preclinical studies in murine models, fingolimod, ozanimod, mocravimod, and amiselimod have shown efficacy [37, 92, 98]. Of these, mocravimod was tested in cutaneous lupus erythematosus (CLE) (NCT01294774), but the results of the trial are not yet published. No additional trials for CLE are ongoing [99].
3.5.1 Cenerimod
Cenerimod/ACT-334441, an oral S1PR1 modulator, ameliorated organ-specific and systemic autoimmunity and preserved organ function in models of SLE and Sjögren’s syndrome [100]. A phase 2 (NCT02472795) clinical trial that assessed the effects of cenerimod on SLE activity and circulating lymphocytes in patients with mild to moderate SLE showed efficacy and an acceptable safety profile [101]. These findings led to an ongoing phase 2 study in patients with moderate to severe SLE (NCT03742037).
Cenerimod was tested for efficacy in a translational study with an MRL/lpr mouse SLE model and clinically in patients with SLE. The MRL/lpr mouse is an SLE model that reflects the autoimmunity and organ damage associated with SLE and was used to evaluate preclinical efficacy. Seven-week-old MRL/lpr females were treated with vehicle or cenerimod food admix. At week 11, 30% in the vehicle group died, whereas all cerenimod-treated mice survived. A significant B and T blood lymphocyte reduction was observed, along with decreased organ damage and a pronounced suppression of the inflammatory environment. Inflammatory biomarkers and lymphocyte subsets were characterized in a 12-week phase 2 clinical study (NCT02472795) in which patients were treated with multiple doses of a placebo or cenerimod. In SLE patients, cenerimod treatment reduced T and B blood lymphocytes, reduced interferon-associated biomarkers, and normalized blood antibody-secreting cell numbers [102].
3.5.2 Amiselimod
Recently, the results of a phase 1b multicenter, open-label amiselimod trial in SLE were published [103]. The primary objective was to evaluate the safety of amiselimod in SLE patients. Seventeen patients received 0.2 or 0.4 mg for 24 weeks. It was well tolerated, without serious adverse events or infections. The total SLE disease index 2000 score [104] decreased ≥ 4 points at 24 weeks in 7/17 patients. These data suggest the safety and efficacy of amiselimod in SLE [103].
Apart from the agents described above, there are multiple S1PR agonists/modulators that are being investigated for use in autoimmune/immune-mediated disorders (Table 1). These include ceralifimod (S1PR1,5; RRMS. NCT01081782, NCT01226745), CS-077 (S1PR1; MS) [105], sonepcizumab (S1PR1; macular degeneration, NCT00767949), LC51-0255 (S1PR1; UC. NCT04096573, NCT04360343), OPL-002 (S1PR1; UC/healthy subjects, NCT04451811), GSK2018682 [106] (S1PR1; healthy subjects, GSK2018682, NCT01387217, NCT01431937), and ASP4058 (S1PR1, S1PR5; healthy subjects, NCT01998646) [6].
4 Safety and Adverse Events
4.1 Infections
Since S1PRs agonist/modulators have limited effects on effector memory T-cell recirculation, patients appear to maintain immune surveillance [23]. However, upper respiratory infections and urinary tract infections have been reported. Serious infections such as herpes and disseminated varicella zoster (VZV) are rare but have been reported with fingolimod [107]. VZV serology and administration of the recombinant zoster vaccine (Shingrix, GlaxoSmithKline) might decrease the risk of infection and associated complications [23]. Cases of progressive multifocal leukoencephalopathy (PML) have been reported with fingolimod, but the risk is 1:10,000 patients treated. A few cases of cryptococcal meningitis and disseminated cryptococcal infections have been reported with the use of fingolimod [23, 108].
4.2 Cardiovascular Events
First-degree AV block, sinus bradycardia, orthostatic hypotension, and hypertension have been reported [23, 58]. The first two aforementioned cardiac events usually occur within hours of the first dose, and are attributed to S1PR1, S1PR2, and S1PR3 agonism [72]. Initially, heart rate effects were attributed to S1PR3 agonism, because fingolimod resulted in bradycardia in wild-type but not in S1PR3 knockout mice [109]. However, subsequent research shows that S1PR1 also mediates this effect [109]. The S1PR1 activation on cardiomyocytes produces activation of G protein-coupled inwardly rectifying potassium channel, which plays a role in heart rate and cardiac conduction [72]. Then, temporary bradycardia might be explained due to myocyte hyperpolarization and transitory reduction in excitability induced by S1P receptor activation of G protein-coupled inwardly rectifying potassium channels, before S1P receptor internalization or S1P antagonism [72]. An up-titration regimen can mitigate the heart rate reduction produced by S1PR modulators exhibiting short half-lives (e.g., ponesimod, siponimod) [110]. Conversely, up-titration is not required for compounds with long half-lives (fingolimod, cerenimod) due to the less pronounced first-dose-related negative chronotropic effects, presumably due to “built-in up-titration” [4, 110]. Interestingly, an inadvertent first-dose effect is possible after a brief treatment interruption of S1PR modulators with short half-lives but without long half-life active metabolites (siponimod and ponesimod). In that situation, drug reinitiation requires a dose titration [4]. An electrocardiogram is recommended prior to initiating S1P modulators [58, 110,111,112]. Patients with a history of unstable angina, heart attack, stroke, transient ischemic attack, decompensated heart failure, class III or IV heart failure, Mobitz type II second-degree or third-degree AV block, sino-atrial block, or sick sinus syndrome (except patients with a pacemaker) should avoid their use [58, 110,111,112].
4.3 Malignancy
Cases of skin cancer (squamous cell carcinoma, melanoma, and basal cell carcinoma) have been related to fingolimod and ozanimod [23, 58, 113]. Therefore, patients should be periodically evaluated by a dermatologist.
Other concerns related to S1PR agonist/modulators are leukopenia (reversible with drug discontinuation), teratogenicity, pulmonary disorders, transaminase elevation, anemia, macular edema, headache, and human papillomavirus infections [12, 58, 113, 114].
The identification of new S1PR agonist/modulators with a shorter half-life is desirable yet challenging. Recovery of peripheral lymphocyte counts following fingolimod requires > 5 weeks, which could complicate treatment of opportunistic infections [95]. Teratogenicity has been observed in rodents treated with fingolimod, and the long half-life in humans (8 days) requires contraception for 2 months after stopping treatment, if pregnancy is planned [58, 95]. Ozanimod has a 17- to 21-h half-life while long-acting active metabolites have half-lives of approximately 10 days, thus it must be discontinued for 3 months before conception [58]. The promising results of NCT02447302 showed etrasimod’s rapid onset and offset, rapid depletion of circulating lymphocytes (approximately 53% depletion at day 3), and rapid recovery to 5% of baseline within a week of treatment cessation [72]. However, the clinical significance of receptor specificity between the drugs needs to be assessed by head-to-head comparisons in large trials [72]. Finally, enhanced selectivity and a short half-life are desirable traits for the new S1PR agonist/modulators under development as lymphocyte counts will rebound rapidly upon cessation in case of adverse events, particularly infections.
5 Future Directions for S1P Pathway Targeting
Dysregulation of the S1P pathway is observed in the intestine of IBD patients. Sphingosine kinases (SphK1) and phosphatases (SPP) are upregulated, whereas SP1-lyase is downregulated, resulting in higher S1P levels [54]. Thus, modulation of pathways involved in synthesis, degradation, and transport of S1P might represent novel druggable targets. In addition, SphK1 upregulation has been implicated in colitis-associated cancer and other cancers (diffused B-cell lymphoma, breast cancer, colorectal cancer, esophageal cancer, gastric cancer, and prostate cancer) [115,116,117].
5.1 Sphingosine Kinase Inhibitors
SphKs catalyze the phosphorylation of sphingosine to S1P [118]. SphKs may represent additional therapeutic targets, and preclinical studies have shown promising results [118]. SphK1 is ubiquitously expressed and has been reported to be involved in RA, asthma, and IBD [119]. Proinflammatory cytokines such as TNF activate SphK1, and this enzyme induces COX2 expression, increasing prostaglandin E2 production [119]. LCL351, a SphK1 inhibitor, induced a reduction in TNF, CXCL1, CXCL2 expression, and neutrophil infiltration in the DSS-induced model of colitis [119]. Other SphK1 inhibitors (ABC747080 and ABC294640) also modified TNF-mediated inflammation and colitis severity [117, 119]. Furthermore, in a mast cell-dependent mouse model of allergic asthma treatment with SphK1 inhibitor (SK1-I) attenuated mast cell-dependent allergic inflammation and airway hyper-responsiveness, supporting the potential use of SphK1 inhibitors for the treatment of allergic airway inflammation [120]. SphK2 inhibition ameliorated inflammation by blocking Th17 differentiation of CD4 naïve cells in a mouse model of psoriasis [121].
5.2 Sphingosine Phosphatases
The S1P phosphatases (SPP1 and SPP2) catalyze the dephosphorylation of S1P to sphingosine, participating in the regulation of S1P levels. SPP2 is found in the gastrointestinal tract, and its expression is elevated in patients with UC [122]. Moreover, in the DSS-induced colitis model, deficiency of SPP2 improved mucosal barrier integrity, suggesting that inhibition of SPP2 may be further explored in IBD [122].
5.3 S1P-lyase
This enzyme is abundant in tissue and degrades S1P irreversibly, maintaining an S1P gradient and promoting lymphocyte recirculation from tissues to blood. In a TNF-overproducing model of Crohn-like ileitis (TNF∆ARE), S1P-lyase inhibitors altered the S1P gradient and attenuated ileitis [24]. In addition, an S1P-lyase inhibitor ameliorated DSS-induced colitis [123]. However, other studies in mice show that a deficit of intestinal S1P-lyase worsened disease [124]. Additional studies are needed to clarify the potential role of S1P-lyase inhibition in IBD. In preclinical models of RA, LX3305 (an S1P-lyase inhibitor) induced a reduction in the inflammatory response. Safety, tolerability, and efficacy were evaluated in a phase 2 trial (NCT00903383) in patients with active RA and showed potential clinical benefit and a favorable safety profile [125].
5.4 Spinster Homolog 2 (SPNS2)
This carrier exports S1P out of the cell, and its deletion attenuates inflammation in mouse models, such as airway inflammation, hypersensitivity, DSS- and oxazolone-induced colitis, experimental autoimmune encephalitis, and collagen-induced arthritis [126].
5.5 S1P Agonists for Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)
Coronavirus disease-2019 (COVID-19) is currently a global pandemic [127]. Severe COVID-19 cases have been reported with the use of fingolimod in MS [128]. On the other hand, S1PR agonists (e.g., fingolimod) have been proposed as a strategy to dampen cytokine and chemokine releases in those patients displaying excessive immune responses. A phase 2 clinical trial is evaluating ozanimod efficacy on COVID-19 (NCT04405102), whereas a fingolimod trial (NCT04280588) was discontinued. Further studies are needed to evaluate their safety and efficacy in patients with COVID-19 [129].
6 Positioning S1P Modulators in IBD: Pros and Cons
New orally administered small molecules such as S1P modulators offer a viable alternative to the current therapeutic arsenal for many immune-mediated diseases, especially in IBD where to date most second-tier drugs require parenteral administration. In contrast to these biological therapies used in immune-mediated and autoimmune diseases, these drugs have the advantage of their lack of immunogenicity, oral administration, safety profile, low manufacturing cost, and shorter half-life [39]. S1P modulators are well tolerated but might have first-dose effects on heart rate and immune and pulmonary systems [88]. Importantly, S1P modulators have different pharmacokinetic properties despite their similar pharmacodynamic effects [39]. Those with short half-lives (ponesimod, siponimod) have a rapid elimination with rapid reversibility of their pharmacodynamic effects, but require up-titration initially [39]. In comparison, the S1P modulators with a long half-life (e.g., fingolimod) have a built-in up-titration [39]. Although ozanimod has a relatively short half-life, its metabolites lead to a sustained effect on cell count [39]. Short half-lives have the advantages of faster clearance in case of infections, pregnancy, or any other adverse events.
Selective S1P modulators such as ozanimod, siponimod, and ponesimod have demonstrated an improved safety profile in comparison with non-selective S1P modulators [130]. Adverse effects such as macular edema, dysregulated lung function and hypertension are attributed to non-specific S1PR1-5 activity [71]. S1PR2 and S1PR3 have been implicated in processes such as fibrosis and vasoconstriction, along with S1PR3 in cardiac conduction [71]. Regarding infections, research reveals that only specific subsets are affected, such as T (CD3) and B (CD20) lymphocytes [131, 132], whereas natural killer, NKT, granulocyte, and monocyte counts should be not affected, maintaining immune surveillance against cancer and viral infections [39, 134, 135]. Table 2 compares features of S1PR modulators mentioned in this review [4, 39, 58, 72, 74, 101, 130, 133,134,135,136,137,138].
Between 30 and 50% of patients with IBD show a primary non-response to biologics and many more lose response every year. In general, response to a second biologic is lower in patients previously treated with anti-TNF compared with biologic-naïve patients [139, 140]. S1P modulators might be promising in this group of IBD patients. Recent data from the ozanimod North Study showed efficacy in both anti-TNF naïve and anti-TNF non-naïve patients. Head-to-head trials that compare similar drugs such as etrasimod and ozanimod will be needed to address superiority. If S1P modulators are eventually approved for IBD, they could be particularly useful in anti-TNF refractory patients, those with conditions such as arthritis, psoriasis, multiple sclerosis, and in cases where the use of anti-TNF agents is contraindicated, such as demyelinating disease, psoriasis induced by anti-TNF, hypersensitivity reactions, and/or immunogenicity to anti-TNF agents.
JAK inhibitors could be seen as alternative orally administered small molecules as they have shown efficacy in patients refractory to anti-TNF. However, adverse events such as infections and thrombosis are important considerations. Drugs with better safety profiles and distinct mechanisms of action are needed. S1PR modulators might represent a viable alternative to widely used parenteral biologics.
7 Conclusions
S1P and its receptors are involved in many physiologic and pathologic processes, having a critical role in the immune response by regulating immune cell migration, driving the differentiation of immune cells, and changing their functional phenotype. S1P and SphKs may additionally participate in inflammatory cytokine signaling. Thus, modulation of S1P-S1PR signaling is a promising area for drug development. Encouraging preclinical data show the potential of targeting SphKs, SPPs, S1P-lyase, and transporters. The quest for novel efficacious oral S1PR agonist/modulators with improved selectivity, higher potency, and favorable pharmacokinetic properties and safety profile may expand the current therapeutic arsenal against IBD and other immune-mediated diseases.
Change history
30 July 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40265-021-01577-z
References
Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov. 2013;12(9):688–702.
Park SJ, Im DS. Sphingosine 1-phosphate receptor modulators and drug discovery. Biomol Ther (Seoul). 2017;25(1):80–90.
Peyrin-Biroulet L, Christopher R, Behan D, Lassen C. Modulation of sphingosine-1-phosphate in inflammatory bowel disease. Autoimmun Rev. 2017;16(5):495–503.
Chun J, Giovannoni G, Hunter SF. Sphingosine 1-phosphate receptor modulator therapy for multiple sclerosis: differential downstream receptor signalling and clinical profile effects. Drugs. 2021;81(2):207–31.
Tsai HC, Han MH. Sphingosine-1-phosphate (S1P) and S1P signaling pathway: therapeutic targets in autoimmunity and inflammation. Drugs. 2016;76(11):1067–79.
Stepanovska B, Huwiler A. Targeting the S1P receptor signaling pathways as a promising approach for treatment of autoimmune and inflammatory diseases. Pharmacol Res. 2020;154:104170.
Cartier A, Hla T. Sphingosine 1-phosphate: lipid signaling in pathology and therapy. Science. 2019;366(6463):eaar5551.
Currò D, Pugliese D, Armuzzi A. Frontiers in drug research and development for inflammatory bowel disease. Front Pharmacol. 2017;8:400.
Argollo M, Furfaro F, Gilardi D, et al. Modulation of sphingosine-1-phosphate in ulcerative colitis. Expert Opin Biol Ther. 2020;20(4):413–20.
Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol. 2008;8(10):753–63.
Proia RL, Hla T. Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest. 2015;125(4):1379–87.
Aoki M, Aoki H, Ramanathan R, Hait NC, Takabe K. Sphingosine-1-phosphate signaling in immune cells and inflammation: roles and therapeutic potential. Mediators Inflamm. 2016;2016:8606878.
Olesch C, Ringel C, Brüne B, Weigert A. Beyond immune cell migration: the emerging role of the sphingosine-1-phosphate receptor S1PR4 as a modulator of innate immune cell activation. Mediat Inflamm. 2017;2017:6059203.
Park ES, Choi S, Shin B, et al. Tumor necrosis factor (TNF) receptor-associated factor (TRAF)-interacting protein (TRIP) negatively regulates the TRAF2 ubiquitin-dependent pathway by suppressing the TRAF2-sphingosine 1-phosphate (S1P) interaction. J Biol Chem. 2015;290(15):9660–73.
Wang T, Wang J. K63-linked polyubiquitination of IRF1: an essential step in the IL-1 signaling cascade. Cell Mol Immunol. 2014;11(5):407–9.
Harikumar KB, Yester JW, Surace MJ, et al. K63-linked polyubiquitination of transcription factor IRF1 is essential for IL-1-induced production of chemokines CXCL10 and CCL5. Nat Immunol. 2014;15(3):231–8.
Blaho VA, Hla T. An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res. 2014;55(8):1596–608.
Spiegel S, Milstien S, Grant S. Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene. 2012;31(5):537–51.
Hait NC, Allegood J, Maceyka M, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325(5945):1254–7.
Hait NC, Maiti A. The role of sphingosine-1-phosphate and ceramide-1-phosphate in inflammation and cancer. Mediat Inflamm. 2017;2017:4806541.
Alvarez SE, Harikumar KB, Hait NC, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465(7301):1084–8.
Nielsen OH, Li Y, Johansson-Lindbom B, Coskun M. Sphingosine-1-phosphate signaling in inflammatory bowel disease. Trends Mol Med. 2017;23(4):362–74.
Pérez-Jeldres T, Tyler CJ, Boyer JD, et al. Targeting cytokine signaling and lymphocyte traffic via small molecules in inflammatory bowel disease: JAK inhibitors and S1PR agonists. Front Pharmacol. 2019;10:212.
Karuppuchamy T, Tyler CJ, Lundborg LR, et al. Sphingosine-1-phosphate lyase inhibition alters the S1P gradient and ameliorates Crohn’s-like ileitis by suppressing thymocyte maturation. Inflamm Bowel Dis. 2020;26(2):216–28.
Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nat Immunol. 2010;11(11):1047–56.
Garris CS, Wu L, Acharya S, et al. Defective sphingosine 1-phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17-mediated autoimmune neuroinflammation. Nat Immunol. 2013;14(11):1166–72.
Cinamon G, Matloubian M, Lesneski MJ, et al. Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat Immunol. 2004;5(7):713–20.
Karuppuchamy T, Behrens EH, González-Cabrera P, et al. Sphingosine-1-phosphate receptor-1 (S1P1) is expressed by lymphocytes, dendritic cells, and endothelium and modulated during inflammatory bowel disease. Mucosal Immunol. 2017;10(1):162–71.
Blankenbach KV, Schwalm S, Pfeilschifter J, Heringdorf DMZ. Sphingosine-1-phosphate receptor-2 antagonists: therapeutic potential and potential risks. Front Pharmacol. 2016;7:167.
Green JA, Suzuki K, Cho B, et al. The sphingosine 1-phosphate receptor S1P2 maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat Immunol. 2011;12(7):672–80.
Muppidi JR, Schmitz R, Green JA, et al. Loss of signalling via Gα13 in germinal centre B-cell-derived lymphoma. Nature. 2014;516(7530):254–8.
Wang W, Graeler MH, Goetzl EJ. Type 4 sphingosine 1-phosphate G protein-coupled receptor (S1P4) transduces S1P effects on T cell proliferation and cytokine secretion without signaling migration. FASEB J. 2005;19(12):1731–3.
Jenne CN, Enders A, Rivera R, et al. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med. 2009;206(11):2469–81.
van Doorn R, Lopes Pinheiro MA, Kooij G, et al. Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J Neuroinflamm. 2012;9:133.
O’Sullivan C, Schubart A, Mir AK, Dev KK. The dual S1PR1/S1PR5 drug BAF312 (Siponimod) attenuates demyelination in organotypic slice cultures. J Neuroinflamm. 2016;13:31.
Sukocheva OA, Lukina E, McGowan E, Bishayee A. Sphingolipids as mediators of inflammation and novel therapeutic target in inflammatory bowel disease. Adv Protein Chem Struct Biol. 2020;120:123–58.
Sugahara K, Maeda Y, Shimano K, et al. Amiselimod, a novel sphingosine 1-phosphate receptor-1 modulator, has potent therapeutic efficacy for autoimmune diseases, with low bradycardia risk. Br J Pharmacol. 2017;174(1):15–27.
Piali L, Birker-Robaczewska M, Lescop C, et al. Cenerimod, a novel selective S1P1 receptor modulator with unique signaling properties. Pharmacol Res Perspect. 2017;5(6):e00370.
Juif PE, Kraehenbuehl S, Dingemanse J. Clinical pharmacology, efficacy, and safety aspects of sphingosine-1-phosphate receptor modulators. Expert Opin Drug Metab Toxicol. 2016;12(8):879–95.
Japtok L, Kleuser B. The role of sphingosine-1-phosphate receptor modulators in the prevention of transplant rejection and autoimmune diseases. Curr Opin Investig Drugs. 2009;10(11):1183–94.
Salvadori M, Budde K, Charpentier B, et al. FTY720 versus MMF with cyclosporine in de novo renal transplantation: a 1-year, randomized controlled trial in Europe and Australasia. Am J Transplant. 2006;6(12):2912–21.
Calabresi PA, Radue EW, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(6):545–56.
La Mantia L, Tramacere I, Firwana B, Pacchetti I, Palumbo R, Filippini G. Fingolimod for relapsing-remitting multiple sclerosis. Cochrane Database Syst Rev. 2016;4:CD009371.
Baker KF, Isaacs JD. Novel therapies for immune-mediated inflammatory diseases: What can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease and ulcerative colitis? Ann Rheum Dis. 2018;77(2):175–87.
Khatri BO, Pelletier J, Kappos L, et al. Effect of prior treatment status and reasons for discontinuation on the efficacy and safety of fingolimod vs. interferon β-1a intramuscular: Subgroup analyses of the Trial Assessing Injectable Interferon vs. Fingolimod Oral in Relapsing-Remitting Multiple Sclerosis (TRANSFORMS). Mult Scler Relat Disord. 2014;3(3):355–63.
Ramos GP, Papadakis KA. Mechanisms of disease: inflammatory bowel diseases. Mayo Clin Proc. 2019;94(1):155–65.
Uhlig HH, Powrie F. Translating immunology into therapeutic concepts for inflammatory bowel disease. Annu Rev Immunol. 2018;36:755–81.
Mizushima T, Ito T, Kishi D, et al. Therapeutic effects of a new lymphocyte homing reagent FTY720 in interleukin-10 gene-deficient mice with colitis. Inflamm Bowel Dis. 2004;10(3):182–92.
Huwiler A, Zangemeister-Wittke U. The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: Recent findings and new perspectives. Pharmacol Ther. 2018;185:34–49.
Deguchi Y, Andoh A, Yagi Y, et al. The S1P receptor modulator FTY720 prevents the development of experimental colitis in mice. Oncol Rep. 2006;16(4):699–703.
Daniel C, Sartory N, Zahn N, Geisslinger G, Radeke HH, Stein JM. FTY720 ameliorates Th1-mediated colitis in mice by directly affecting the functional activity of CD4+CD25+ regulatory T cells. J Immunol. 2007;178(4):2458–68.
Radi ZA, Heuvelman DM, Masferrer JL, Benson EL. Pharmacologic evaluation of sulfasalazine, FTY720, and anti-IL-12/23p40 in a TNBS-induced Crohn’s disease model. Dig Dis Sci. 2011;56(8):2283–91.
Montrose DC, Scherl EJ, Bosworth BP, et al. S1P1 localizes to the colonic vasculature in ulcerative colitis and maintains blood vessel integrity. J Lipid Res. 2013;54(3):843–51.
Gonzalez-Cabrera PJ, Brown S, Studer SM, Rosen H. S1P signaling: new therapies and opportunities. F1000Prime Rep. 2014;6:109.
Radeke H, Stein J, Van Assche G, Rogler G, Lakatos P, Muellershausen F, Moulin P, Jarvis P, Colin L, Gergely P, Kruis W. A multicentre, double-blind, placebo-controlled, parallel-group study to evaluate the efficacy, safety, and tolerability of the s1p receptor agonist KRP203 in patients with moderately active refractory ulcerative colitis. Inflamm Intest Dis. 2020;5(4):180–90.
Lewis JD, Chuai S, Nessel L, Lichtenstein GR, Aberra FN, Ellenberg JH. Use of the noninvasive components of the Mayo score to assess clinical response in ulcerative colitis. Inflamm Bowel Dis. 2008;14(12):1660–6.
Baron JH, Connell AM, Lennard-Jones JE. variation between observers in describing mucosal appearances in proctocolitis. Br Med J. 1964;1(5375):89–92.
Lamb YN. Ozanimod: first approval. Drugs. 2020;80(8):841–8.
Hemperly A, Sandborn WJ, Vande CN. Clinical pharmacology in adult and pediatric inflammatory bowel disease. Inflamm Bowel Dis. 2018;24(12):2527–42.
Scott FL, Clemons B, Brooks J, et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br J Pharmacol. 2016;173(11):1778–92.
Derfuss T, Mehling M, Papadopoulou A, Bar-Or A, Cohen JA, Kappos L. Advances in oral immunomodulating therapies in relapsing multiple sclerosis. Lancet Neurol. 2020;19(4):336–47.
Sandborn WJ, Feagan BG, Wolf DC, et al. Ozanimod induction and maintenance treatment for ulcerative colitis. N Engl J Med. 2016;374(18):1754–62.
Sandborn WJ, Feagan BG, Hanauer S, et al. Long-term efficacy and safety of ozanimod in moderate-to-severe ulcerative colitis: results from the open-label extension of the randomized, phase 2 touchstone study [published online ahead of print, 2021 Jan 13]. J Crohns Colitis. 2021;jjab012.
Sandborn, WJ, D’Haens G, Wolf D, Hanauer S, Jovanovic I, Ghosh S, et al. “LB5—(Late-Breaking Abstract) Ozanimod for Moderate-to-Severe Ulcerative Colitis: Efficacy, Safety, and Histology Results from the Induction and Maintenance Periods of the Phase 3 True North Study,” ACG 2020. https://www.eventscribe.com/2020/ACG/fsPopup.asp?efp=RElCSUFHUk02NDI2&PresentationID=783287&rnd=0.4752997&mode=presinfo. Accessed 24 Dec 2020.
Feagan BG, Sandborn WJ, Danese S, et al. Ozanimod induction therapy for patients with moderate to severe Crohn’s disease: a single-arm, phase 2, prospective observer-blinded endpoint study. Lancet Gastroenterol Hepatol. 2020;5(9):819–28.
Ferrante M, Colombel JF, Sandborn WJ, et al. Validation of endoscopic activity scores in patients with Crohn’s disease based on a post hoc analysis of data from SONIC. Gastroenterology. 2013;145(5):978-986.e5.
Best WR, Becktel JM, Singleton JW, Kern F Jr. Development of a Crohn’s disease activity index. National Cooperative Crohn’s Disease Study. Gastroenterology. 1976;70(3):439–44.
Khanna R, Zou G, D’Haens G, et al. A retrospective analysis: the development of patient reported outcome measures for the assessment of Crohn’s disease activity. Aliment Pharmacol Ther. 2015;41(1):77–86.
Geboes K, Riddell R, Ost A, Jensfelt B, Persson T, Löfberg R. A reproducible grading scale for histological assessment of inflammation in ulcerative colitis. Gut. 2000;47(3):404–9.
Pai RK, Khanna R, D’Haens GR, et al. Definitions of response and remission for the Robarts Histopathology Index. Gut. 2019;68(11):2101–2.
Al-Shamma H, Lehmann-Bruinsma K, Carroll C, et al. The selective sphingosine 1-phosphate receptor modulator etrasimod regulates lymphocyte trafficking and alleviates experimental colitis. J Pharmacol Exp Ther. 2019;369(3):311–7.
Sandborn WJ, Peyrin-Biroulet L, Zhang J, et al. Efficacy and safety of etrasimod in a phase 2 randomized trial of patients with ulcerative colitis. Gastroenterology. 2020;158(3):550–61.
Kashou AH, Goyal A, Nguyen T, Chhabra L. Atrioventricular block. In: StatPearls. Treasure Island: StatPearls Publishing; 2021.
Sugahara K, Maeda Y, Shimano K, et al. Amiselimod (MT-1303), a novel sphingosine 1-phosphate receptor-1 modulator, potently inhibits the progression of lupus nephritis in two murine SLE models. J Immunol Res. 2019;2019:5821589.
Shimano K, Maeda Y, Kataoka H, et al. Amiselimod (MT-1303), a novel sphingosine 1-phosphate receptor-1 functional antagonist, inhibits progress of chronic colitis induced by transfer of CD4+CD45RBhigh T cells. PLoS ONE. 2019;14(12):e0226154.
Kappos L, Arnold DL, Bar-Or A, et al. Two-year results from a phase 2 extension study of oral amiselimod in relapsing multiple sclerosis. Mult Scler. 2018;24(12):1605–16.
A clinical study to investigate the effects of MT-1303 on patients with moderate to severe Chronic Plaque Psoriasis focusing on how safe, tolerable and effective in treating the above condition MT-1303 is. https://www.cochranelibrary.com/es/central/doi/10.1002/central/CN-01799880/full. Accessed 12 Apr 2021.
D’Haens G, Danese S, Hibi T, Watanabe M, Davies M. 1005–a controlled trial of amiselimod, a selective S1P receptor modulator in Crohn’s disease. Gastroenterology. 2019;156:S-217. https://doi.org/10.1016/S0016-5085(19)37337-8.
Bausch Health picks up IBD drug amiselimod rejected by Biogen. https://www.thepharmaletter.com/article/bausch-health-picks-up-ibd-drug-amiselimod-rejected-by-biogen. Accessed 7 Feb 2021.
D’Haens, G. R. A. M., Slatkin, N, Israel, R, Heimanson Z. P097(Abstract).Favorable safety profile for amiselimod, a selective s1p receptor modulator, in Crohn’s Disease. Gastroenterology. 2020;150(3S):S2.
MT. Pharma. “6 State of New Product development”. https://www.mt-pharma.co.jp/e/develop/assets/pdf/e_pipeline2012.pdf. Accessed 7 Feb 2021.
Stephen H, O’Reilly T, Lester R, Slatkin N, Lee J, Franklin H, Bulawski A, Israel R. Safety of amiselimod in healthy subjects: results from a phase 1 randomized, double-blind, placebo-controlled study. Inflamm Bowel Dis (Abstract). 2021;27(Supplement_1):S1. https://doi.org/10.1093/ibd/izaa347.001
Greb JE, Goldminz AM, Elder JT, et al. Psoriasis. Nat Rev Dis Primers. 2016;2:16082.
Bell M, Foley D, Naylor C, et al. Discovery of super soft-drug modulators of sphingosine-1-phosphate receptor 1. Bioorg Med Chem Lett. 2018;28(19):3255–9.
Vaclavkova A, Chimenti S, Arenberger P, et al. Oral ponesimod in patients with chronic plaque psoriasis: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet. 2014;384(9959):2036–45.
Tokuyama M, Mabuchi T. New treatment addressing the pathogenesis of psoriasis. Int J Mol Sci. 2020;21(20):7488.
Ryan C, Menter A. Ponesimod—a future oral therapy for psoriasis? Lancet. 2014;384(9959):2006–8.
Juif PE, Hoch M, Vaclavkova A, Krause A, Bush J, Dingemanse J. Mitigation of initial cardiodynamic effects of the S1P1 receptor modulator ponesimod using a novel up-titration regimen. J Clin Pharmacol. 2017;57(3):401–10.
Jin J, Xue N, Liu Y, et al. A novel S1P1 modulator IMMH002 ameliorates psoriasis in multiple animal models. Acta Pharm Sin B. 2020;10(2):276–88.
Kusari A, Han AM, Schairer D, Eichenfield LF. Atopic dermatitis: new developments. Dermatol Clin. 2019;37(1):11–20.
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–38.
Mao-Draayer Y, Sarazin J, Fox D, Schiopu E. The sphingosine-1-phosphate receptor: a novel therapeutic target for multiple sclerosis and other autoimmune diseases. Clin Immunol. 2017;175:10–5.
Zhao C, Fernandes MJ, Turgeon M, et al. Specific and overlapping sphingosine-1-phosphate receptor functions in human synoviocytes: impact of TNF-alpha. J Lipid Res. 2008;49(11):2323–37.
Tsunemi S, Iwasaki T, Kitano S, Imado T, Miyazawa K, Sano H. Effects of the novel immunosuppressant FTY720 in a murine rheumatoid arthritis model. Clin Immunol. 2010;136(2):197–204.
Buzard DJ, Kim SH, Lopez L, et al. Discovery of APD334: design of a clinical stage functional antagonist of the sphingosine-1-phosphate-1 receptor. ACS Med Chem Lett. 2014;5(12):1313–7.
Jin J, Ji M, Fu R, et al. Sphingosine-1-phosphate receptor subtype 1 (S1P1) modulator IMMH001 regulates adjuvant- and collagen-induced arthritis. Front Pharmacol. 2019;10:1085.
Fava A, Petri M. Systemic lupus erythematosus: diagnosis and clinical management. J Autoimmun. 2019;96:1–13.
Taylor Meadows KR, Steinberg MW, Clemons B, et al. Ozanimod (RPC1063), a selective S1PR1 and S1PR5 modulator, reduces chronic inflammation and alleviates kidney pathology in murine systemic lupus erythematosus. PLoS ONE. 2018;13(4):e0193236.
Little AJ, Vesely MD. Cutaneous lupus erythematosus: current and future pathogenesis-directed therapies. Yale J Biol Med. 2020;93(1):81–95.
Gerossier E, Nayar S, Smith C, Froidevaux S, Barone F, Martinic M. Cenerimod, a potent and selective sphingosine-1-phosphate receptor 1 modulator, controls systemic autoimmunity and organ pathology in mouse models of systemic lupus erythematosus and Sjögren’s syndrome [abstract]. Arthritis Rheumatol. 2019;71(suppl 10). https://acrabstracts.org/abstract/cenerimod-a-potent-and-selective-sphingosine-1-phosphate-receptor-1-modulator-controls-systemic-autoimmunity-and-organ-pathology-in-mouse-models-of-systemic-lupus-erythematosus-and-sjogrens/. Accessed 25 Sept 2020.
Hermann V, Batalov A, Smakotina S, Juif PE, Cornelisse P. First use of cenerimod, a selective S1P1 receptor modulator, for the treatment of SLE: a double-blind, randomised, placebo-controlled, proof-of-concept study. Lupus Sci Med. 2019;6(1):e000354.
Strasser DS, Froidevaux S, Sippel V, et al. Preclinical to clinical translation of cenerimod, a novel S1P1 receptor modulator, in systemic lupus erythematosus. RMD Open. 2020;6(2):e001261.
Tanaka Y, Kondo K, Ichibori A, et al. Amiselimod, a sphingosine 1-phosphate receptor-1 modulator, for systemic lupus erythematosus: a multicenter, open-label exploratory study. Lupus. 2020;29(14):1902–13.
Gladman DD, Ibañez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol. 2002;29(2):288–91.
Moberly JB, Ford DM, Zahir H, et al. Pharmacological effects of CS-0777, a selective sphingosine 1-phosphate receptor-1 modulator: results from a 12-week, open-label pilot study in multiple sclerosis patients. J Neuroimmunol. 2012;246(1–2):100–7.
Xu J, Gray F, Henderson A, et al. Safety, pharmacokinetics, pharmacodynamics, and bioavailability of GSK2018682, a sphingosine-1-phosphate receptor modulator, in healthy volunteers. Clin Pharmacol Drug Dev. 2014;3(3):170–8.
Arvin AM, Wolinsky JS, Kappos L, et al. Varicella-zoster virus infections in patients treated with fingolimod: risk assessment and consensus recommendations for management. JAMA Neurol. 2015;72(1):31–9.
Chong I, Wang KY, Lincoln CM. Cryptococcal meningitis in a multiple sclerosis patient treated with fingolimod: a case report and review of imaging findings. Clin Imaging. 2019;54:53–6.
Urbano M, Guerrero M, Rosen H, Roberts E. Modulators of the sphingosine 1-phosphate receptor 1. Bioorg Med Chem Lett. 2013;23(23):6377–89.
Juif PE, Ufer M, Dingemanse J. Cardiodynamic interactions between two S1P1 receptor modulators in an experimental clinical setting: different pharmacokinetic properties as an opportunity to mitigate first-dose heart rate effects. Int J Mol Sci. 2019;20(13):3232.
Al-Salama ZT. Siponimod: first global approval. Drugs. 2019;79(9):1009–15.
Novartis Pharmaceuticals Corporation. MAYZENT® (siponimod): US prescribing information 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/209884s000lbl.pdf. Accessed 7 Feb 2021.
Cohen JA, Tenenbaum N, Bhatt A, Zhang Y, Kappos L. Extended treatment with fingolimod for relapsing multiple sclerosis: the 14-year LONGTERMS study results. Ther Adv Neurol Disord. 2019;12:1756286419878324.
Benedetti MD, Marangi A, Bozzetti S, et al. HPV-related papillary squamous cell carcinoma of the tonsil during treatment with fingolimod. Mult Scler Relat Disord. 2018;23:24–6.
Yuza K, Nagahashi M, Shimada Y, et al. Upregulation of phosphorylated sphingosine kinase 1 expression in colitis-associated cancer. J Surg Res. 2018;231:323–30.
Khoei SG, Sadeghi H, Samadi P, Najafi R, Saidijam M. Relationship between Sphk1/S1P and microRNAs in human cancers. Biotechnol Appl Biochem. 2020. https://doi.org/10.1002/bab.1922.
Sukocheva OA, Furuya H, Ng ML, et al. Sphingosine kinase and sphingosine-1-phosphate receptor signaling pathway in inflammatory gastrointestinal disease and cancers: A novel therapeutic target. Pharmacol Ther. 2020;207:107464.
Cao M, Ji C, Zhou Y, et al. Sphingosine kinase inhibitors: a patent review. Int J Mol Med. 2018;41(5):2450–60.
Pulkoski-Gross MJ, Uys JD, Orr-Gandy KA, et al. Novel sphingosine kinase-1 inhibitor, LCL351, reduces immune responses in murine DSS-induced colitis. Prostaglandins Other Lipid Mediat. 2017;130:47–56.
Price MM, Oskeritzian CA, Falanga YT, et al. A specific sphingosine kinase 1 inhibitor attenuates airway hyperresponsiveness and inflammation in a mast cell-dependent murine model of allergic asthma. J Allergy Clin Immunol. 2013;131(2):501-11.e1.
Shin SH, Cho KA, Hahn S, et al. Inhibiting sphingosine kinase 2 derived-sphingosine-1-phosphate ameliorates psoriasis-like skin disease via blocking Th17 differentiation of naïve CD4 T lymphocytes in mice. Acta Derm Venereol. 2019;99(6):594–601.
Huang WC, Liang J, Nagahashi M, et al. Sphingosine-1-phosphate phosphatase 2 promotes disruption of mucosal integrity, and contributes to ulcerative colitis in mice and humans. FASEB J. 2016;30(8):2945–58.
Shirakabe K, Higashiyama M, Furuhashi H, et al. Amelioration of colitis through blocking lymphocytes entry to Peyer’s patches by sphingosine-1-phosphate lyase inhibitor. J Gastroenterol Hepatol. 2018. https://doi.org/10.1111/jgh.14092.
Degagné E, Pandurangan A, Bandhuvula P, et al. Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J Clin Invest. 2014;124(12):5368–84.
Fleischmann RM, Poiley JE, Stoilov R, et al. The oral S1P lyase inhibitor LX3305 (LX2931) demonstrates favorable safety and potential clinical benefit at 12-weeks in a phase 2 proof-of-concept trial in patients with active rheumatoid arthritis on stable methotrexate. Presentation Number: 2593. 2011. https://acr.confex.com/acr/2011/webprogram/Paper19634.html. Accessed 25 Sept 2020
Donoviel MS, Hait NC, Ramachandran S, et al. Spinster 2, a sphingosine-1-phosphate transporter, plays a critical role in inflammatory and autoimmune diseases. FASEB J. 2015;29(12):5018–28.
Naz F, Arish M. Battling COVID-19 pandemic: sphingosine-1-phosphate analogs as an adjunctive therapy? Front Immunol. 2020;11:1102.
Foerch C, Friedauer L, Bauer B, Wolf T, Adam EH. Severe COVID-19 infection in a patient with multiple sclerosis treated with fingolimod. Mult Scler Relat Disord. 2020;42:102180.
Catanzaro M, Fagiani F, Racchi M, Corsini E, Govoni S, Lanni C. Immune response in COVID-19: addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct Target Ther. 2020;5(1):84.
Comi G, Hartung HP, Bakshi R, Williams IM, Wiendl H. Benefit-risk profile of sphingosine-1-phosphate receptor modulators in relapsing and secondary progressive multiple sclerosis. Drugs. 2017;77(16):1755–68.
Brinkmann V, Billich A, Baumruker T, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9(11):883–97.
Brossard P, Derendorf H, Xu J, Maatouk H, Halabi A, Dingemanse J. Pharmacokinetics and pharmacodynamics of ponesimod, a selective S1P1 receptor modulator, in the first-in-human study. Br J Clin Pharmacol. 2013;76(6):888–96.
David OJ, Kovarik JM, Schmouder RL. Clinical pharmacokinetics of fingolimod. Clin Pharmacokinet. 2012;51(1):15–28.
Dash RP, Rais R, Srinivas NR. Ponesimod, a selective sphingosine 1-phosphate (S1P1) receptor modulator for autoimmune diseases: review of clinical pharmacokinetics and drug disposition. Xenobiotica. 2018;48(5):442–51.
Boof ML, van Lier JJ, English S, Fischer H, Ufer M, Dingemanse J. Absorption, distribution, metabolism, and excretion of cenerimod, a selective S1P1 receptor modulator in healthy subjects. Xenobiotica. 2020;50(8):947–56.
Juif PE, Baldoni D, Reyes M, et al. Pharmacokinetics, pharmacodynamics, tolerability, and food effect of cenerimod, a selective S1P1 receptor modulator in healthy subjects. Int J Mol Sci. 2017;18(12):2636.
Gardin A, Gray C, Neelakantham S, et al. Siponimod pharmacokinetics, safety, and tolerability in combination with rifampin, a CYP2C9/3A4 inducer, in healthy subjects. Eur J Clin Pharmacol. 2018;74(12):1593–604.
Schreiber S, Morgan M, Christopher R, et al. Etrasimod (APD334), a Potent, Selective, Oral S1P Receptor Modulator With Preclinical Autoimmune Disease-modifying Activity Exhibits Favorable PK/PD Properties in Healthy Volunteers (Abstract). http://www.arenapharm.com/wp-content/uploads/2016/12/A23434E8.pdf. Accessed 7 Feb 2021.
Hazel K, O’Connor A. Emerging treatments for inflammatory bowel disease. Ther Adv Chronic Dis. 2020;11:2040622319899297.
Singh S, Murad MH, Fumery M, Dulai PS, Sandborn WJ. First- and second-line pharmacotherapies for patients with moderate to severely active ulcerative colitis: an updated network meta-analysis. Clin Gastroenterol Hepatol. 2020;18(10):2179-2191.e6.
PONVORYTM (ponesimod): US prescribing information 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213498s000lbl.pdf. Accessed 5 May 2021.
ZEPOSIA® (ozanimod): US prescribing information 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/209899s000lbl.pdf. Accessed 5 May 2021.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by grants from the National Institutes of Health (DK108670, DK118927, AI149636), VA Merit BLRD-I01 BX003436 to JRN and San Diego Digestive Diseases Research Center (P30 DK120515).
Conflict of interest
The authors have no relevant conflicts of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material (data transparency)
Not applicable.
Code availability
Not applicable.
Author contributions
TP-J, MA-L, and JR-N provided the concept and design, and drafted and revised the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Pérez-Jeldres, T., Alvarez-Lobos, M. & Rivera-Nieves, J. Targeting Sphingosine-1-Phosphate Signaling in Immune-Mediated Diseases: Beyond Multiple Sclerosis. Drugs 81, 985–1002 (2021). https://doi.org/10.1007/s40265-021-01528-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01528-8