
Abstract
Mavacamten (Camzyos™) is an oral small-molecule cardiac myosin inhibitor developed by MyoKardia, Inc., a wholly owned subsidiary of Bristol Myers Squibb, for the treatment of hypertrophic cardiomyopathy (HCM) and diseases of diastolic dysfunction. In April 2022, mavacamten was approved for use in the USA in the treatment of adults with symptomatic New York Heart Association (NYHA) class II-III obstructive HCM to improve functional capacity and symptoms. This article summarizes the milestones in the development of mavacamten leading to this first approval for the treatment of adults with symptomatic NYHA class II-III obstructive HCM.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Digital Features for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.20057537 |
A cardiac myosin inhibitor developed by MyoKardia, Inc., a wholly owned subsidiary of Bristol Myers Squibb, for the treatment of HCM and diseases of diastolic dysfunction |
Received its first approval on 28 April 2022 in the USA |
Approved for use in the treatment of adults with symptomatic NYHA class II-III obstructive HCM to improve functional capacity and symptoms |
1 Introduction
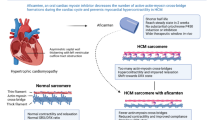
Hypertrophic cardiomyopathy (HCM) is one of the most common genetic cardiac disorders, affecting between 0.16% (1 in 625) and 0.29% (1 in 344) of the general adult population [1, 2]. It is characterized by primary left ventricular (LV) cardiac hypertrophy (with the greatest hypertrophy affecting the basal interventricular septum), decreased compliance and myocardial fibrosis. LV outflow obstruction at rest or on provocation is seen in approximately two-thirds of patients with HCM [1,2,3,4,5,6]. Dynamic LV outflow tract (LVOT) obstruction resulting from systolic anterior motion of the mitral valve is also common [2]. Although LV diastolic dysfunction often occurs, LV ejection fraction (LVEF) is preserved or increased [2]. HCM is recognised as a disease of the cardiac sarcomere [7]. Histological features of HCM include myocyte hypertrophy and disarray and interstitial fibrosis [2, 7]; at a molecular level, excess myosin actin cross-bridge formation and dysregulation of the super-relaxed state are evident [7, 8]. While HCM has a relatively benign course in most affected individuals, it is associated with chronic, progressive heart failure symptoms. It is also associated with an increased risk of atrial fibrillation and stroke and/or with sudden cardiac death, especially in adolescents and younger adults [2]. Current treatment for obstructive HCM focuses on pharmacological management of symptoms (e.g., β-blockers, non-dihydropyridine calcium channel blockers, disopyramide) and nonpharmacological options such as implantable cardiac defibrillators or septal reduction therapy (SRT) [6, 9, 10].

Key milestones in the development of mavacamten in the treatment of hypertrophic cardiomyopathy and diseases of diastolic dysfunction. EMA European Medicines Agency, MAA Marketing Authorization Application, NDA New Drug Application
While some patients benefit from pharmacological treatment, these agents often do not provide good control of LVOT gradients and symptoms, may not be well tolerated and do not target the underlying mechanisms and pathophysiology of HCM [4, 6].
Mavacamten (Camzyos™) is a small-molecule allosteric and reversible inhibitor of cardiac myosin ATPase [7, 8, 11]. It targets the sarcomere hypercontractility that is one of the characteristics of HCM [8, 12] and inhibits excessive myosin actin cross-bridge formation, shifting the overall myosin population towards an energy-sparing, recruitable, super-relaxed state [8]. Mavacamten received its first approval on 28 April 2022 in the USA for the treatment of adults with symptomatic New York Heart Association (NYHA) class II-III obstructive HCM to improve functional capacity and symptoms [8] and is under evaluation in the EU for the treatment of obstructive HCM [13]. The recommended starting dose of mavacamten is 5 mg once daily without regard to food; allowable subsequent doses with titration are 2.5, 5, 10, or 15 mg once daily. Regular LVEF and Valsalva (provoked) LVOT gradient assessment is required for careful dose titration to achieve an appropriate target Valsalva LVOT gradient, while maintaining LVEF ≥ 50% and avoiding heart failure symptoms. Daily dosing takes weeks to reach steady-state drug levels and therapeutic effects, and genetic variation in metabolism and drug interactions can cause large differences in exposure. Algorithms for initiation and maintenance dosing, patient monitoring schedules, and guidance for treatment interruption or discontinuation are provided in the prescribing information and should be followed [8].
The US prescribing information for mavacamten contains a boxed warning regarding heart failure. Mavacamten reduces LVEF and can cause heart failure due to systolic dysfunction [8]. Consequently, mavacamten is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the Camzyos REMS PROGRAM. Echocardiogram assessments of LVEF are required prior to and during treatment with mavacamten. Initiation of mavacamten treatment in patients with LVEF < 55% is not recommended; if LVEF is < 50% at any visit or if the patient experiences heart failure symptoms or worsening clinical status, mavacamten treatment should be interrupted. Coadministration of mavacamten with disopyramide in combination with verapamil or diltiazem should be avoided because such use has been associated with left ventricular systolic dysfunction and heart failure symptoms in patients with obstructive HCM [8].

Chemical structure of mavacamten
Concomitant use of mavacamten with moderate to strong CYP2C19 inhibitors or strong CYP3A4 inhibitors, or with moderate to strong CYP2C19 inducers or moderate to strong CYP3A4 inducers is contraindicated. Mavacamten dosage adjustments may be required when concomitant administration of weak CYP2C19 inhibitors (e.g., omeprazole) or moderate CYP3A4 inhibitors (e.g., verapamil, diltiazem) is required. Avoid initiating concomitant weak CYP2C19 and moderate CYP3A4 inhibitors in patients who are on stable treatment with mavacamten 2.5 mg once daily, because a lower once-daily dose is not available. Mavacamten may cause fetal toxicity when administered to a pregnant female, based on findings in animal studies and effective contraception is advised during treatment and for 4 months after the last dose [8].
1.1 Company Agreements
In November 2020, Bristol Myers Squibb announced that they had closed the acquisition of MyoKardia, Inc. and that MyoKardia had become a wholly owned subsidiary of Bristol Myers Squibb [14]. In October 2020, Bristol Myers Squibb had announced a definitive merger agreement under which it would acquire MyoKardia [15].
In August 2020, MyoKardia and LianBio entered into a licensing agreement to develop and commercialize mavacamten in China, Hong Kong, Macau, Taiwan, Thailand and Singapore [16, 17]. The partnership will initially pursue a registration strategy for mavacamten in China for obstructive HCM (including a registrational phase 3 trial [16]), with plans for additional indications to follow consistent with MyoKardia’s development strategy [17].
In July 2019, MyoKardia re-acquired the US royalty rights to mavacamten from Sanofi [18]. In January 2019, MyoKardia regained worldwide rights to all programs (including those for mavacamten) covered under its license and collaboration agreement with Sanofi. The collaboration was not extended beyond the initial research term (which had commenced in August 2014 [19] and ended on December 2018 [20]) and was fully concluded on 1 April 2019 [20].
2 Scientific Summary
2.1 Pharmacodynamics
Mavacamten exerts its effect through the myosin-S1 motor and modulates multiple steps of the myosin chemomechanical cycle [12]. Contractility of cardiac myofibrils is modulated by ATPase activity; in vitro, mavacamten reduces steady-state ATPase activity by inhibiting the rate of phosphate release from β-cardiac myosin-S1 (the rate-limiting step of the myosin chemomechanical cycle) [12, 21]. Mavacamten also decreases the number of myosin heads that can enter “on actin” (power-generating) states, and reduces the probability of force-producing (systolic) and residual (diastolic) cross-bridge formation [8, 12, 22, 23]. In in vitro studies of isolated cells, muscle fibre preparations and engineered human heart tissue models, mavacamten stabilized cardiac myosin, inhibited cardiac muscle hypercontractility and improved active lusitropic function of cardiac muscles [12, 21, 24,25,26,27,28,29,30].
In a feline model of HCM, mavacamten reduced contractility, eliminated systolic anterior motion of the mitral valve and relieved LVOT pressure gradients in a dose-dependent manner [31]. Administration of mavacamten early in the course of the disease in a genetic mouse model of HCM (harbouring heterozygous human mutations in the myosin heavy chain) suppressed the development of ventricular hypertrophy, cardiomyocyte disarray and myocardial fibrosis and attenuated hypertrophic and profibrotic gene expression [21].
In the phase 2, placebo-controlled, dose-ranging MAVERICK-HCM (NCT03442764) safety and tolerability trial (n = 59), geometric mean N-terminal pro-B-type natriuretic peptide (NT-proBNP; a biomarker of cardiac wall stress) levels were reduced from baseline to a greater extent with mavacamten than placebo (−435 vs −6 pg/mL; p = 0.0005) in adults with symptomatic NYHA class II/III non-obstructive HCM [32]. Reductions from baseline in geometric mean cardiac troponin 1 levels were seen with mavacamten, but not with placebo (−0.008 vs +0.001 ng/mL; p = 0.009) [32]. Patients who completed MAVERICK-HCM were eligible to enrol in the MAVERICK-LTE cohort of the 5-year MAVA-LTE (NCT03723655) trial. Reductions in median NT-proBNP with mavacamten treatment at weeks 24 and 48 of treatment in the MAVERICK-LTE cohort were comparable with those seen in MAVERICK-HCM [33].
Treatment with mavacamten reduced LVOT obstruction, LV mass, left atrial (LA) volume and NT-proBNP in patients with symptomatic obstructive HCM in the 30-week, phase 3, randomized, placebo-controlled EXPLORER-HCM trial (n = 251; NCT05174416) [4, 34, 35]. Greater reductions from baseline in mean resting and Valsalva LVOT gradients were seen in mavacamten than placebo recipients; at 30 weeks, resting LVOT gradient was 14.1 vs 45.9 mmHg, respectively (baseline values 51.7 and 51.1 mmHg) and Valsalva LVOT gradients were 24.8 vs 62.7 mmHg (baseline values 72.4 and 73.9 mmHg) [4]. Mavacamten relieved LVOT obstruction (post-exercise gradient < 30 mmHg) in 57% of patients (vs 7% of patients in the placebo group) and reduced the gradient to below the standard threshold for invasive septal reduction therapy (< 50 mmHg) in 74% of patients (vs 21% in the placebo group) [4]. These improvements with mavacamten were seen from week 4 of treatment onwards. A complete response (a reduction in all LVOT gradients to < 30 mm Hg and reaching NYHA class I) was achieved in 27% mavacamten recipients compared with 1% of placebo recipients [4]. Mean resting LVEF at baseline was 74.1% and 74.2% in the mavacamten and placebo arms, respectively [4]. Decreases in LVEF accompanied reductions in Valsalva LVOT gradient in the mavacamten arm, but these were generally within the normal range (mean absolute change from baseline in LVEF over the 30-week period of −3.9%); the mean LVEF reduction in the placebo group was -0.01%. At week 38, following the 8-week treatment interruption, mean LVEF in both treatment arms was similar to baseline [4, 8].
Reductions in NT-proBNP in the mavacamten arm of EXPLORER-HCM were observed by week 4 and were maintained throughout treatment. At week 30, the reduction in NT-proBNP from baseline in the mavacamten group was 80% greater than in the placebo group and the reduction in high-sensitivity cardiac troponin 1 was 41% greater [4]. Interim, longer-term data from the EXPLORER-LTE cohort of MAVA-LTE extension study showed that clinically meaningful improvements in LVOT gradients and reductions in NT-proBNP consistent with those seen with mavacamten in the 30-week EXPLORER-HCM trial which were evident at 48 weeks (n = 206) and up to 84 weeks (n = 66) of mavacamten treatment. At week 84, 83.5% of patients had LVOT gradients < 30 mmHg and NT-proBNP had reduced from baseline by 63% [36, 37]. Reductions in NT-proBNP were significantly associated with improvements in several echocardiographic parameters of cardiac structure and function in mavacamten recipients, the strongest of which was the association with the reduction in resting LVOT gradient (p < 0.0001) [34].
In patients with mitral valve systolic anterior motion at baseline in the EXPLORER-HCM trial (94 mavacamten and 97 placebo recipients), significantly more mavacamten than placebo recipients showed complete resolution after 30 weeks of treatment (80.9% vs 34.0%; p < 0.0001) [34]. Mavacamten recipients also showed a mean reduction from baseline in LV mass index at 30 weeks, whereas placebo recipients showed a mean increase (−7.4 vs +8.9 g/m2; p < 0.0001). LA volume index (LAVI) was reduced to a greater extent with mavacamten than placebo (−7.5 vs −0.1 mL/m2; p < 0.0001), and this was associated with significantly improved peak O2 consumption (p = 0.04). Lateral E/e’ (−3.8 vs +0.04; p < 0.0001) and septal E/e’ (−3.5 vs −0.3; p < 0.0001) were also significantly improved with mavacamten. Significant improvements in LAVI, lateral E/e’ and septal E/e’ with mavacamten were evident from week 18 onwards [34]. In a cardiac magnetic resonance substudy of EXPLORER-HCM (n = 35 patients randomized; 17 to mavacamten, 18 to placebo), significantly greater reductions in mean LV mass index (primary endpoint) from baseline to week 30 were seen with mavacamten (−17.4 vs −1.6 g/m2; p < 0.0001). LV mass (between-group difference −30.0 g; p < 0.0001) and maximum LAVI (mean between group difference −10.3 mL/m2; p = 0.0004) were also reduced to a greater extent in the mavacamten arm [35].
A meta-analysis of the clinical studies of mavacamten in patients with HCM did not show clinically relevant increases in the QTc interval in the therapeutic exposure range [8]. After administration of multiple doses of mavacamten in healthy volunteers, a concentration-dependent increase in the QTc interval was seen at doses up to 25 mg; however, no acute changes in QTc interval were seen at similar exposures after a single dose of the drug. The mechanism of this QT prolongation is unknown [8]. Nonclinical data indicates that mavacamten is not torsadogenic [38].
2.2 Pharmacokinetics
The pharmacokinetics of once-daily oral mavacamten are generally dose proportional over a dose range of 1–15 mg [8]. In patients with HCM, exposures of mavacamten are 170% higher than those in healthy individuals receiving the same dose. The estimated oral bioavailability of mavacamten is ≥ 85% and Tmax is 1 h. Administration with food (a high fat meal) has no clinically significant effect on mavacamten pharmacokinetics. Mavacamten is highly protein bound in plasma (97–98%). The peak-to-trough plasma concentration ratio of mavacamten at steady state after once daily dosing is ≈ 1.5 [8].
Features and properties of mavacamten
Alternative names | Camzyos; HCM 1; MAVA-Bristol Myers Squibb/MyoKardia; MYK-461; SAR-439152 |
Class | Cardiovascular therapies; Ethylamines; Heart failure therapies; Pyrimidinones; Small molecules |
Mechanism of action | Cardiac myosin inhibitors |
Route of administration | Oral |
Pharmacodynamics | Stabilizes cardiac myosin; decreases the number of myosin heads that can enter “on actin” states and reduces the probability of systolic and diastolic cross-bridge formation. Reduces LVOT obstruction, LV mass, LA volume and NT-proBNP levels |
Pharmacokinetics | Median Tmax 1 h, 97–98% plasma protein bound. Extensively metabolized in the liver, primarily via CYP2C19 (74%), CYP3A4 (18%) and CYP2C9 (8%); elimination depends on polymorphic CYP2C19 status (mean AUC∞ ↑241%, mean Cmax ↑ 47%, t1/2 ↑ to 23 days in CYP2C19 poor metabolizers). Mainly excreted in urine |
Adverse events | |
Most frequent | Dizziness, syncope |
Occasional | Reversible ↓ in LVEF to < 50% |
ATC codes | |
WHO ATC code | C01E-B24 (Mavacamten) |
EphMRA ATC code | C1 (Cardiac Therapy) |
Chemical name | 3-(1-methylethyl)-6-[[(1S)-1-phenylethyl]amino]-2,4(1H,3H)-pyrimidinedione |
Mavacamten is extensively metabolized in the liver, primarily via CYP2C19 (74%), CYP3A4 (18%) and CYP2C9 (8%) [8]. Mavacamten elimination is variable and depends largely on polymorphic CYP2C19 status. Normal metabolizers carry two normal function alleles, whereas poor metabolizers carry two nonfunctional alleles. A small proportion of individuals with European (≈ 2%) or African (≈ 4%) ancestry are poor metabolizers; the prevalence of poor metabolizers is higher in Asian populations (e.g., ≈ 13% in East Asians). After a single 15 mg dose, mavacamten exposure is increased (AUC∞ by 241% and Cmax by 47%) in CYP2C19 poor metabolizers compared with normal metabolizers, and t1/2 is prolonged (23 vs 6–9 days, respectively). In CYP2C19 normal metabolizers, the accumulation ratio for Cmax is ≈ 2-fold and that for AUC is ≈ 7-fold. Accumulation in CYP2C19 poor metabolizers is considerably greater than in CYP2C19 normal metabolizers [8].
Mavacamten is primarily excreted in urine. After administration of a single 25 mg dose of radiolabelled mavacamten, 85% of the dose was recovered in urine (3% as the unchanged drug) and 7% in faeces (1% as the unchanged drug) [8].
In patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment, mavacamten exposure (AUC) increased up to 220%; however, no additional dosage adjustment is required with the recommended dose titration algorithm and monitoring plan in this patient group. The effect of severe (Child-Pugh C) hepatic impairment on mavacamten pharmacokinetics is unknown. Mild to moderate renal impairment had no clinically significant effect on the pharmacokinetics of mavacamten; the effects of severe renal impairment and kidney failure on mavacamten pharmacokinetics is unknown [8].
In vitro, mavacamten does not inhibit CYP1A2, CYP2B6, or CYP2C8 and at clinically relevant drug concentrations does not inhibit CYP2D6, CYP2C9, CYP2C19, or CYP3A4; however, mavacamten induces CYP3A4, CYP2C9, CYP2C19 and CYP2B6. Mavacamten does not inhibit the transporters P-gp, BCRP, BSEP, MATE1, MATE2-K, OATPs, OCTs or OATs [5, 8].
Inducers and inhibitors of CYP2C19 and moderate to strong inhibitors or inducers of CYP3A4 may affect the mavacamten exposure [8]. Coadministration of mavacamten with moderate to strong CYP2C19 inhibitors or strong CYP3A4 inhibitors increases mavacamten exposure, while coadministration with moderate to strong CYP2C19 inducers or moderate to strong CYP3A4 inducers decreases mavacamten exposure, both of which may have a clinical impact on patient management. Concomitant use of mavacamten and drugs that are CYP3A4, CYP2C19, or CYP2C9 substrates may decrease plasma concentration of these drugs and close monitoring is required. As the hormonal contraceptive components progestin and ethinyl estradiol are CYP3A4 substrates, alternative contraceptive methods are required during concomitant use and for 4 months after the last mavacamten dose [8]. Consult mavacamten prescribing information for detailed information regarding guidance on the management of potential drug interactions [8].
Key clinical trials of mavacamten
Drug(s) | Indication | Phase | Status | Location | Sponsor(s) | Identifier |
|---|---|---|---|---|---|---|
Mavacamten, placebo | Symptomatic oHCM | 3 | Active | USA | MyoKardia, Inc.a | NCT04349072; VALOR-HCM |
Mavacamten, placebo | Symptomatic oHCM | 3 | Active | China | LianBio | NCT05174416; EXPLORER-CN; CTR20212890 |
Mavacamten, placebo | Symptomatic oHCM | 3 | Completed | Global | MyoKardia, Inc.a | NCT03470545; EXPLORER-HCM; EudraCT 2017-002530-23 |
Mavacamten | oHCM, nHCM | 2/3 | Active | Global | MyoKardia, Inc.a | NCT03723655; MAVA-LTE; EudraCT 2018-004039-64 |
Mavacamten, | Symptomatic oHCM | 2 | Active | USA | MyoKardia, Inc.a | NCT03496168; PIONEER-OLE |
Mavacamten | Symptomatic oHCM | 2 | Completed | USA | MyoKardia, Inc.a | NCT02842242; PIONEER-HCM |
Mavacamten, placebo | Symptomatic nHCM | 2 | Completed | USA | MyoKardia, Inc.a | NCT03442764; MAVERICK-HCM |
Mavacamten, | HF with preserved ejection fraction | 2 | Active | USA | MyoKardia, Inc.a | NCT04766892; EMBARK-HFpEF |
2.3 Therapeutic Trials
2.3.1 Phase 3 Trials
Treatment with mavacamten improved cardiac function in adult patients with symptomatic obstructive HCM in the phase 3 randomized, double-blind, placebo-controlled EXPLORER-HCM (NCT03470545) [4]. Significantly more mavacamten (n = 123) than placebo (n = 128) recipients achieved either improvement of peak oxygen consumption (pVO2) by ≥ 1.5 mL/kg/min plus improvement in NYHA class by at least 1 or improvement of pVO2 by ≥ 3.0 mL/kg/min plus no worsening in NYHA class at 30 weeks [37% vs 17%, respectively; p = 0.0005 (primary composite functional endpoint)]. More than twice as many mavacamten than placebo recipients had a change from baseline in pVO2 ≥ 1.5 mL/kg/min and a decreased NYHA class (33% vs 14%) or a change from baseline in pVO2 ≥ 3 mL/kg/min with no increase in NYHA class (23% vs 11%) [4, 8]. Treatment with mavacamten also significantly improved LVOT obstruction, functional capacity and health status compared with placebo. Significant changes versus placebo from baseline to week 30 were evident in post-exercise LVOT gradient (mean −47 vs −10 mmHg, respectively; p < 0.0001), pVO2 (mean +1.4 vs −0.1 mL/kg/min; p < 0.0006), and the number of patients with NYHA Class improved ≥ 1 (65% vs 31%; p < 0.0001). Measures of health-related quality of life [Kansas City Cardiomyopathy Questionnaire-23 Clinical Summary Score (KCCQ-23 CSS), KCCQ-23 overall score, Hypertrophic Cardiomyopathy Symptom Questionnaire Shortness of Breath (HCMSQ SoB) subscore [4, 8, 39, 40], EuroQoL 5-dimension 5-level (EQ-5D-5L) index score and EuroQoL visual analog scale (EQ-VAS) score] were all improved from baseline to a significantly greater extent with mavacamten than placebo (p < 0.05) [41]. Additional assessment using cardiopulmonary exercise testing showed that mavacamten significantly improved other parameters of exercise performance compared with placebo (p < 0.05 vs placebo for peak VE/VCO2, peak metabolism equivalents, peak circulatory power, peak exercise time, VE/VCO2 slope and ventilatory power) [42].
Interim, longer-term data from the EXPLORER-LTE cohort of the 5-year MAVA-LTE (NCT03723655) extension study showed that clinically meaningful improvements in NYHA Class and shortness of breath with exercise consistent with those seen with mavacamten in EXPLORER-HCM were evident in mavacamten recipients at 48 weeks (n = 206) and up to 84 weeks (n = 66). 68% of patients improved by ≥ 1 NYHA class and the proportion of patients with NYHA class III disease decreased from 29% at baseline to 4.9% at 48 weeks. The proportion of patients with shortness of breath with exercise halved between baseline and 48 weeks (from 94% to 45%) [36, 37].
Eligible patients in EXPLORER-HCM had symptomatic NYHA class II and III obstructive HCM, LVEF ≥ 55%, at least one LVOT peak gradient ≥50 mmHg at rest or with provocation, and Valsalva LVOT peak gradient >30 mmHg and were randomized to receive either a starting dose of mavacamten 5 mg or placebo once daily for 30 weeks. The dose was periodically adjusted to optimize patient response (i.e., a decrease in Valsalva LVOT gradient) and maintain LVEF ≥ 50%. At baseline, ≈ 73% of patients were NYHA class II and ≈ 27% were NYHA class III, mean LVEF was 74% and mean Valsalva LVOT gradient was 73 mmHg. 92% of trial participants remained on background β-blockers or calcium-channel blockers. Exclusion criteria included dual β-blocker and calcium channel blocker treatment, monotherapy with disopyramide or ranolazine and known infiltrative or storage disorders [4, 6]. 231 of 244 patients from EXPLORER- HCM were enrolled in the EXPLORER-LTE cohort of MAVA-LTE, a long-term dose-blinded extension study that also enrolled patients from the phase 2 MAVERICK-HCM trial. Patients restarted/started treatment with mavacamten (5 mg once daily on study entry), with subsequent dose adjustments using echo-guided titration to achieve therapeutic targets [36, 37].
Preliminary results of the phase 3, randomized, double-blind, placebo-controlled VALOR-HCM trial (NCT04349072) indicate that treatment with mavacamten significantly reduced the need for SRT in patients with severely symptomatic drug-refractory obstructive HCM who met guideline criteria of eligibility for SRT [43]. After 16 weeks of treatment, the percentage of patients who proceeded with SRT prior to or at week 16 or remained eligible for SRT at week 16 (primary composite endpoint [44]) was significantly lower with mavacamten (10/56 patients) than with placebo (43/56) [(17.9% vs 76.8%; p < 0.0001]. Significantly more mavacamten than placebo recipients had improved ≥ 1 NYHA Class (62.5% vs 21.4%; p < 0.0001) and improvements in KCCQ-23 CSS average scores (10.4 ±16.1 vs 1.9 ±12.0; p < 0.0001) at week 16. Post-exercise LVOT peak gradient was also decreased from baseline to a greater extent in the mavacamten group (−39.1 vs −1.8 mmHg) at week 16 [43]. VALOR-HCM was a 138-week trial that consisted of a 2-week screening period followed by a 16-week placebo-controlled treatment period, a 16-week active blinded treatment period, a 96-week long-term extension and an 8-week post-treatment follow-up visit [44]. The study enrolled 112 patients with symptomatic obstructive HCM (NYHA Class III-IV or Class II with exertional syncope or near syncope) who met the 2011 ACC/AHA Guideline criteria for SRT (LVOT gradient of ≥ 50mm Hg and NYHA Class III-IV or Class II with syncope) and were referred for SRT. Trial participants were receiving maximally tolerated standard HCM treatment and remained on this throughout the study [43]. During the placebo-controlled dosing period, patients were randomized to receive mavacamten 5 mg or placebo once daily followed by echo-guided dose titration at weeks 4, 8, and 12. In the active-controlled period, mavacamten recipients continued on mavacamten and placebo recipients received mavacamten 5 mg, followed by echo-guided dose titration at weeks 20, 24 and 28. Patients continued mavacamten treatment during the long-term extension, from week 32 to week 128 [44].
2.3.2 Phase 2 Trial
Mavacamten reduced LVOT obstruction and improved exercise capacity and dyspnoea in patients with symptomatic obstructive HCM in the phase 2 open-label PIONEER-HCM trial (NCT02842242) [45]. Patients (n = 21) were divided into two treatment cohorts: cohort A (n = 11) received mavacamten 10–20 mg day with no background HCM treatment and cohort B (n = 10) received mavacamten 2 mg/day, increasing to 5 mg/day at the end of week 4 if the decrease in resting LVOT gradient was < 50% from baseline, while continuing prior β-blocker treatment. At 12 weeks, mean postexercise LVOT gradient was significantly reduced from baseline (primary endpoint) in both treatment cohorts [cohort A mean change −89.5 mm Hg (p = 0.008); cohort B mean change −25.0 mm (p = 0.02)]. In both cohorts, peak VO2 increased (mean change from baseline of 3.5 and 1.7 mL/kg/min, respectively) and dyspnoea scores improved (mean change from baseline of −3.1 and −3.0). In the 13 patients who re-initiated treatment with mavacamten in the PIONEER-OLE (NCT03496168) extension study, significant reductions from baseline in mean post-exercise LVOT gradient were noted at the 48-week assessment; the extent of improvements at this timepoint were comparable to those achieved in PIONEER-HCM [46]. Eligible patients in PIONEER-HCM had a diagnosis of HCM based on the presence of LV hypertrophy, LVOT obstruction and NYHA functional class II or III [45]. Patients who had completed PIONEER-HCM were eligible to enrol in the 3-year PIONEER-OLE (NCT03496168) [46].
2.4 Adverse Events
Mavacamten was generally well tolerated in clinical trials in patients with symptomatic obstructive HCM. Treatment-emergent adverse events (TEAEs) were reported in 88% of mavacamten 2.5–15 mg once daily recipients (n = 123) and 79% of placebo recipients (n = 128) in the phase 3 randomized, double-blind, placebo-controlled EXPLORER-HCM trial (NCT03470545) [4]. Dizziness (27% vs 18%) and syncope (6% vs 2%) were the most frequent adverse reactions occurring in > 5% of patients and at a higher incidence in the mavacamten arm [8]. Serious adverse events occurred in 8% of mavacamten recipients and 9% of placebo recipients; the most frequent serious adverse events were atrial fibrillation (2% vs 3%), syncope (2% vs 1%) and stress cardiomyopathy (2% vs 0%) [4]. The only adverse drug reaction leading to discontinuation of mavacamten treatment in EXPLORER-HCM was syncope (0.8%) [8]. Reversible reductions in LVEF to < 50% (median 48%) were evident in 7 (6%) mavacamten and 2 (2%) placebo recipients during treatment; reductions were asymptomatic in 3 mavacamten recipients and 1 placebo recipient. LVEF recovered after treatment interruption in all 7 mavacamten recipients (3 mavacamten recipients had a temporary interruption; 2 resumed treatment at the same dose and 1 had the dose reduced from 10 mg to 5 mg) [8]. The median duration of mavacamten exposure in EXPLORER-HCM was 30 weeks (range 2–40 weeks) [8]. In the phase 2 MAVERICK-HCM trial (n = 58), treatment-emergent adverse events were reported in 90% of mavacamten and 68% of placebo recipients; the most frequent adverse event occurring with a higher incidence in the mavacamten group was dizziness (17.9% vs 5.3%) [32].
Most TEAEs reported in the EXPLORER-LTE (n = 137) [47] and MAVERICK-LTE (n = 43) [33] cohorts of MAVA-LTE (NCT03723655) were mild or moderate in severity and were not related to mavacamten treatment. In the MAVERICK-LTE cohort [33], nine patients temporarily discontinued treatment because of LVEF < 50%; eight patients resumed treatment at a lower mavacamten dose after recovery of LVEF and one discontinued treatment permanently. In EXPLORER-LTE, 12 patients temporarily discontinued treatment because of LVEF < 50%; 7 patients resumed mavacamten treatment and 5 discontinued treatment permanently [48]. In the EXPLORER-LTE cohort (n = 231) [48], TEAEs of any severity were reported in 87.0% of patients; serious adverse events were reported in 14.7%. Drug-related TEAEs were reported in 17.3% of patients; drug-related cardiovascular TEAEs occurred in 8.2% [48].
In the phase 3, randomized, double-blind, placebo-controlled VALOR-HCM trial (NCT04349072) in patients with severely symptomatic drug-refractory obstructive HCM receiving mavacamten, no patients permanently discontinued therapy due to LVEF ≤30%, with none experiencing serious adverse events of congestive heart failure, syncope or sudden cardiac death, according to preliminary safety findings [43].
2.5 Ongoing Clinical Trials
Ongoing trials of mavacamten in HCM consist of the phase 3 VALOR-HCM (NCT04349072) study, MAVA-LTE (NCT03723655), a long-term open-label extension trial that has enrolled patients from MAVERICK-HCM and EXPLORER-HCM and the phase 2 open-label extension of PIONEER (PIONEER-OLE; NCT03496168). The phase 3 EXPLORER-CN (NCT05174416) study in Chinese patients with symptomatic obstructive HCM is recruiting. DISCOVER-HCM, a prospective registry study, will assess real-world safety and effectiveness of mavacamten in patients with symptomatic HCM in the USA. A phase 2 proof-of-concept trial of mavacamten in patients with heart failure and preserved ejection fraction (EMBARK-HFpEF; NCT04766892) is recruiting.
Change history
03 August 2022
A Correction to this paper has been published: https://doi.org/10.1007/s40265-022-01758-4
References
Maron BJ, Gardin JM, Flack JM, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92(4):785–9.
Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. 2017;121(7):749–70.
Maron BJ. Clinical course and management of hypertrophic cardiomyopathy. N Engl J Med. 2018;379(20):1977.
Olivotto I, Oreziak A, Barriales-Villa R, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396(10253):759–69.
Grillo MP, Erve JCL, Dick R, et al. In vitro and in vivo pharmacokinetic characterization of mavacamten, a first-in-class small molecule allosteric modulator of beta cardiac myosin. Xenobiotica. 2019;49(6):718–33.
Ho CY, Olivotto I, Jacoby D, et al. Study design and rationale of EXPLORER-HCM: evaluation of mavacamten in adults with symptomatic obstructive hypertrophic cardiomyopathy. Circ Heart Fail. 2020;13(6):e006853.
Edelberg JM, Sehnert AJ, Mealiffe ME, et al. The impact of mavacamten on the pathophysiology of hypertrophic cardiomyopathy: a narrative review. Am J Cardiovasc Drugs. 2022. https://doi.org/10.1007/s40256-022-00532-x.
MyoKardia, Inc., a wholly owned subsidiary of Bristol Myers Squibb. CAMZYOSTM (mavacamten): US prescribing information. 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214998s000lbl.pdf. Accessed 18 May 2022
Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA Guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2011;58(25):e212–60.
Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733–79.
Papadakis M, Basu J, Sharma S. Mavacamten: treatment aspirations in hypertrophic cardiomyopathy. Lancet. 2020;396(10253):736–7.
Kawas RF, Anderson RL, Ingle SRB, et al. A small-molecule modulator of cardiac myosin acts on multiple stages of the myosin chemomechanical cycle. J Biol Chem. 2017;292(40):16571–7.
Bristol Myers Squibb. European Medicines Agency validates Bristol Myers Squibb's application for mavacamten for the treatment of obstructive hypertrophic cardiomyopathy [media release]. 1 Oct 2021. http://www.bms.com.
Bristol Myers Squibb, MyoKardia. Bristol Myers Squibb completes acquisition of MyoKardia, strengthening company's leading cardiovascular franchise [media release]. 17 Nov 2020. http://www.bms.com.
Bristol Myers Squibb. Bristol Myers Squibb to acquire MyoKardia for $13.1 billion in cash [media release]. 5 Oct 2020. http://www.bms.com.
LianBio. LianBio Announces first patient dosed in registrational phase 3 EXPLORER-CN trial of mavacamten in Chinese patients with obstructive hypertrophic cardiomyopathy [media release]. 10 Jan 2022. http://www.lianbio.com.
MyoKardia. MyoKardia and LianBio form strategic partnership to develop and commercialize mavacamten together in Greater China [media release]. 11 Aug 2020. http://www.myokardia.com.
MyoKardia. MyoKardia announces HCM program updates: accelerates timing for mavacamten topline phase 3 data; re-acquires U.S. royalty rights to HCM programs from Sanofi [media release]. 18 Jul 2019. http://www.myokardia.com.
MyoKardia. MyoKardia announces advancement to next phase of global collaboration with Sanofi [media release]. 3 Jan 2017. https://www.globenewswire.com/en/news-release/2017/01/03/902799/37418/en/MyoKardia-Announces-Advancement-to-Next-Phase-of-Global-Collaboration-with-Sanofi.html.
MyoKardia. MyoKardia regains global rights to mavacamten and MYK-491 programs from Sanofi [media release]. 2 Jan 2019. http://www.myokardia.com.
Green EM, Wakimoto H, Anderson RL, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351(6273):617–21.
Anderson RL, Trivedi DV, Sarkar SS, et al. Deciphering the super relaxed state of human β-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc Natl Acad Sci USA. 2018;115(35):E8143–52.
Rohde JA, Roopnarine O, Thomas DD, et al. Mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin. Proc Natl Acad Sci USA. 2018;115(32):E7486–94.
Scellini B, Piroddi N, Dente M, et al. Mavacamten has a differential impact on force generation in myofibrils from rabbit psoas and human cardiac muscle. J Gen Physiol. 2021;153(7):05.
Awinda PO, Bishaw Y, Watanabe M, et al. Effects of mavacamten on Ca2+ sensitivity of contraction as sarcomere length varied in human myocardium. Br J Pharmacol. 2020;177(24):5609–21.
Awinda PO, Watanabe M, Bishaw Y, et al. Mavacamten decreases maximal force and Ca2+ sensitivity in the N47K-myosin regulatory light chain mouse model of hypertrophic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2021;320(2):H881–90.
Sparrow AJ, Watkins H, Daniels MJ, et al. Mavacamten rescues increased myofilament calcium sensitivity and dysregulation of Ca2+ flux caused by thin filament hypertrophic cardiomyopathy mutations. Am J Physiol Heart Circ Physiol. 2020;318(3):H715–22.
Halder SS, Sewanan LR, Rynkiewicz MJ, et al. Mavacamten and danicamtiv reverse respective contractile abnormalities in engineered heart tissue models of hypertrophic and dilated cardiomyopathy [abstract no. P459]. Circ Res. 2021;129(Suppl 1).
Sewanan LR, Shen S, Campbell SG. Mavacamten preserves length-dependent contractility and improves diastolic function in human engineered heart tissue. Am J Physiol Heart Circ Physiol. 2021;320(3):H1112–23.
Gollapudi SK, Ma W, Chakravarthy S, et al. Two classes of myosin inhibitors, para-nitroblebbistatin and mavacamten, stabilize β-cardiac myosin in different structural and functional states. J Mol Biol. 2021;433(23):167295.
Stern JA, Markova S, Ueda Y, et al. A small molecule inhibitor of sarcomere contractility acutely relieves left ventricular outflow tract obstruction in feline hypertrophic cardiomyopathy. PLoS ONE. 2016;11(12):e0168407.
Ho CY, Mealiffe ME, Bach RG, et al. Evaluation of mavacamten in symptomatic patients with nonobstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2020;75(21):2649–60.
Owens A, Sherrid MV, Wong TC, et al. Long-term efficacy and safety of mavacamten in patients with non-obstructive hypertrophic cardiomyopathy: interim results from the MAVERICK-LTE cohort of the MAVA-LTE study [abstract no. 9685]. Circulation. 2021;144(Suppl 1).
Hegde SM, Lester SJ, Solomon SD, et al. Effect of mavacamten on echocardiographic features in symptomatic patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2021;78(25):2518–32.
Saberi S, Cardim N, Yamani M, et al. Mavacamten favorably impacts cardiac structure in obstructive hypertrophic cardiomyopathy: EXPLORER-HCM cardiac magnetic resonance substudy analysis. Circulation. 2021;143(6):606–8.
Bristol Myers Squibb. Bristol Myers Squibb announces data from EXPLORER-LTE demonstrating sustained improvements in clinically meaningful cardiovascular outcomes at weeks 48 and 84 in patients with symptomatic obstructive hypertrophic cardiomyopathy receiving mavacamten [media release]. 3 Apr 2022. http://www.bms.com.
American College of Cardiology. Efficacy, safety of mavacamten in treating obstructive HCM holds up in EXPLORER-LTE cohort of MAVA-LTE study. 2022. https://www.acc.org/Latest-in-Cardiology/Articles/2022/04/02/13/22/Sun-945am-Treatment-Mavacamten-acc-2022. Accessed 22 May 2022.
Data on file, Bristol Myers Squibb, 2022.
Spertus JA, Fine JT, Elliott P, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): health status analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2021;397(10293):2467–75.
Naidu NS, Saberi SS, Reaney MR, et al. Mavacamten improves symptoms of shortness of breath in patients with obstructive hypertrophic cardiomyopathy (HCM): analysis of the HCM Symptom Questionnaire in the phase 3 EXPLORER trial. Eur J Heart Fail. 2021;23(Supplement 2):252.
Xie J, Wang Y, Xu Y, et al. Assessing health-related quality-of-life in patients with symptomatic obstructive hypertrophic cardiomyopathy: EQ-5D-based utilities in the EXPLORER-HCM trial. J Med Econ. 2022;25(1):51–8.
Wheeler MT, Olivotto I, Elliott PM, et al. The effect of mavacamten on cardiopulmonary exercise testing performance of patients with obstructive hypertrophic cardiomyopathy in EXPLORER-HCM [abstract no. 1005-03]. J Am Coll Cardiol. 2022;79(9 Suppl A):237.
Bristol Myers Squibb. Mavacamten demonstrated significant reduction in need for septal reduction therapy in symptomatic obstructive HCM patients in phase 3 VALOR trial [media release]. 2 Apr 2022. http://www.bms.com.
Desai MY, Wolski K, Owens A, et al. Study design and rationale of VALOR-HCM: evaluation of mavacamten in adults with symptomatic obstructive hypertrophic cardiomyopathy who are eligible for septal reduction therapy. Am Heart J. 2021;239:80–9.
Heitner SB, Jacoby D, Lester SJ, et al. Mavacamten treatment for obstructive hypertrophic cardiomyopathy: a clinical trial. Ann Intern Med. 2019;170(11):741–8.
Sutton M, Heitner SB, Balaratnam G, et al. Targeted pharmacological treatment for obstructive hypertrophic cardiomyopathy with mavacamten: one-year results from PIONEER-OLE [abstract no. P015]. Heart Lung. 2020;49(5):670.
Rader F, Choudhury L, Saberi S, et al. Long-term safety of mavacamten in patients with obstructive hypertrophic cardiomyopathy:iInterim results of the MAVA-Long Term Extension (LTE) study [abstract no. 1033-03]. J Am Coll Cardiol. 2021;77(18 Suppl 1):532.
Rader F, Choudhury L, Saberi S, et al. Updated cumulative results of treatment with mavacamten from the EXPLORER-LTE cohort of the MAVA-LTE study in patients with obstructive hypertrophic cardiomyopathy [oral presentation]. In: American College of Cardiology Meeting. 2022.
U.S. Food & Drug Administration. FDA approves new drug to improve heart function in adults with rare heart condition [media release]. 29 April 2022. https://www.fda.gov/.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Susan J. Keam is a contracted employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
The original article has been revised due to retrospective open choice order.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Keam, S.J. Mavacamten: First Approval. Drugs 82, 1127–1135 (2022). https://doi.org/10.1007/s40265-022-01739-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-022-01739-7