
Abstract
Circadian rhythms are a collection of endogenously driven biochemical, physiological, and behavioral processes that oscillate in a 24-h cycle and can be entrained by external cues. Circadian clock molecules are responsible for the expression of regulatory components that modulate, among others, the cell’s metabolism and energy consumption. In clinical practice, the regulation of clock mechanisms is relevant to biotransformation of therapeutics. Accordingly, xenobiotic metabolism and detoxification, the two processes that directly influence drug effectiveness and toxicity, are direct manifestations of the daily oscillations of the cellular and biochemical processes taking place within the gastrointestinal, hepatic/biliary, and renal/urologic systems. Consequently, the impact of circadian timing should be factored in when developing therapeutic regimens aimed at achieving maximum efficacy, minimum toxicity, and decreased adverse effects in a patient. However, and despite a strong mechanistic foundation, only 0.16 % of ongoing clinical trials worldwide exploit the concept of ‘time-of-day’ administration to develop safer and more effective therapies. In this article, we (1) emphasize points of control at which circadian biology intersects critical processes governing treatment interventions; (2) explore the extent to which chronotherapeutics are incorporated into clinical trials; (3) recognize roadblocks; and (4) recommend approaches to precipitate the integration of chronobiological concepts into clinical practice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Circadian factors influence rhythmic oscillations in biochemical activity that modulate the body’s daily changes in physiology and behavior. |
Circadian timing is relevant to mechanisms involved in biotransformation of therapeutics influencing drug effectiveness and toxicity; however, its clinical application is poorly exploited. |
In this article, emphasis is placed on the current state of knowledge about the use of chronobiological concepts in clinical practice, including strategic approaches to facilitate their integration. |
1 Introduction
Circadian rhythms are daily cycles of physiological and behavioral processes endogenously generated by an organism that can be externally modulated by cues such as light, temperature, and food intake (for review see Dibner et al. [1]). Their relevance to human diseases has become evident as perturbation of circadian rhythms is associated with altered insomnia and sleep syndromes, jet-lag, allergies and asthma, cardiovascular diseases, stroke, hypertension, metabolic diseases, neurological and psychiatric disorders, and hormone-dependent cancers (see previous publications [2–9] and references within). As a result, there has been increased interest in deciphering the molecular regulatory systems to which all circadian components connect and, more importantly, in understanding how circadian-triggered output signals maintain the body’s physiology in harmony with its evolving environment. These findings have yielded far-reaching advances in many areas of science, health, and medicine, including a much-needed molecular foundation to the ancient field of chronotherapy, in which treatments for diseases are administered at times of the day most likely to yield the greatest efficacy.
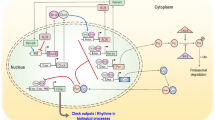
2 Clock Core Regulation of Output Signals
Core circadian clock genes are defined as genes whose protein products are necessary components for the generation and regulation of circadian rhythms. As such, clock proteins are interlocked through transcriptional/translational feedback loops that control circadian timing (for review, see Lowrey and Takahashi [10]). The chief mediator of the mammalian circadian clock is the heterodimer complex formed by the brain-muscle-aryl hydrocarbon receptor nuclear translocator-like protein 1 (BMAL1) and the circadian locomotor output cycles kaput protein (CLOCK; or its paralog neuronal PAS domain protein 2 [NPAS2]) (Fig. 1). Briefly, the positive arm of the feedback loop involves binding of CLOCK/BMAL1 to E-box response elements located in the regulatory region of target genes, of which PERIOD (PER1, PER2, and PER3) and CRYPTOCHROME (CRY1 and CRY2) genes are essential components [11–13]. The PER and CRY proteins accumulate in the cytosol, heterodimerize (PER/CRY), and translocate to the nucleus where they auto-repress their transcription by binding to and inhibiting CLOCK/BMAL1 [14]. Later, the PER/CRY repressor complex is degraded and CLOCK/BMAL1 is released and reassumes a new transcriptional cycle that generates the 24-h timekeeping system. Additionally, a mechanism that constitutes the negative feedback loop provides robustness to the cycle by controlling BMAL1 and, to a much lesser degree, CLOCK transcription [15]. In this scenario, CLOCK/BMAL1 transcriptionally activates the retinoic acid receptor-related orphan receptor (RORα/β/γ) and REV-ERB genes, which in turn bind to the retinoic acid-related orphan receptor elements (ROREs) located in the promoter region of BMAL1 and either activate or repress its expression, respectively (Fig. 1). The epigenetic regulation of circadian clock gene expression aligns well with diurnal variations in histone modifications, including phosphorylation, acetylation, and methylation, which are usually coupled to changes in cellular metabolism, as these modifications use varying metabolites and are influenced by environmental cues [16]. Selective chromatin remodeling of clock-controlled genes (ccgs) is maintained by balancing the intrinsic histone acetyltransferace (HAT) activity of CLOCK and the histone deacetylase activity of sirtuin 1 (SIRT1), an oxidized nicotinamide adenine dinucleotide (NAD+)-dependent enzyme whose activity exhibits robust circadian rhythmicity [17–19]. Interestingly, CLOCK, BMAL1, and SIRT1 transcriptionally regulate the expression of the rate-limiting enzyme in NAD+ biosynthesis, the nicotinamide phosphoribosyl-transferase (NAMPT), establishing a feedback loop in which SIRT1 regulates the levels of its own co-factor (Fig. 1) [20, 21].

Engaging the mammalian clock regulatory network in signal transduction. Interlocked systems are represented by independent panels (i.e., core clock, cell division, nicotinamide adenine dinucleotide [NAD] metabolism, clock-controlled genes, and output metabolic processes) where major players and regulatory relationships are connected by arrows (dashed arrows indicate that other intermediaries or processes might exist). As summarized in the center-left panel, the core components of the mammalian clock consist of casein kinase 1ε/δ (CK1); chryptochrome proteins (CRY); period proteins (PER); circadian locomotor output cycles kaput protein (CLOCK); and brain-muscle-aryl hydrocarbon receptor nuclear translocator-like 1 protein (BMAL1). Briefly, CLOCK/BMAL1 heterodimers selectively bind to E-box enhancers and drive the expression of PER, CRY, and REV-ERBα genes. REV-ERBα proteins then repress BMAL1 transcription through Rev-Erbα/retinoic acid-related orphan receptor (ROR) elements (RORE) in its promoter. Thus, BMAL1 RNA falls while PER and CRY RNA levels peak. As the day progresses, PER proteins accumulate in the cytoplasm, become phosphorylated by CK1, ubiquitinated by E3-ligases (e.g., F-box/LRR-repeat protein 3 [Fbxl3] and F-box/WD repeat-containing protein 1 [Fbw1]), and targeted for degradation by the proteasome system. Later in the day, CRY accumulates, associates with PER/CK1, and the trimeric complex translocates to the nucleus where CRY disrupts the CLOCK/BMAL1-associated transcriptional complex, resulting in CRY, PER, and REV-ERBα transcriptional inhibition and de-repression of BMAL1 transcription. Thus, both transcriptional feedback loops are co-regulated by CLOCK/BMAL1. In addition, core clock components modulate the expression of clock-controlled genes (ccgs) that encode for intermediaries in processes that relate to cell growth, division, death, and maintenance, cell communication and metabolite transport, redox state, detoxification and stress response, carbohydrate, nucleobase, and amino acid metabolisms, extracellular adhesion and communication, protein turnover, hormone synthesis and secretion, and lipid synthesis and accumulation, among other clock-controlled responses (lower panels [34]). Points of intersection between circadian components and the cell division machinery exist and are relevant to the timely progression of the cell cycle (for review see Hunt and Sassone-Corsi [35] and Antoch and Kondratov [95]). For example, PER/CRY modulates CCND1, c-MYC, and the cyclin-dependent inhibitor p21 expression, therefore influencing G1 initiation and progression. Furthermore, CK1 activity is implicated in progression through the S and G2 phases by targeting cyclin A/Cdk complexes and expression of WEE1, a dual-specificity kinase that phosphorylates cyclin B/Cdc2 complex for inhibition and G2 arrest. CRY (through its interaction with Hausp) and PER (by stabilizing p53) influence checkpoint activation in response to genotoxic stress [74, 75]. An additional intersecting loop worth mentioning in the context of clock functioning refers to the role of the adenosine monophosphate-activated protein kinase (AMPK) as a metabolic sensor [76]. When activated, AMPK signals back to the clock core by phosphorylating PER and CRY and promoting their degradation, creating a reciprocal loop between the clock and metabolism. AMPK AMP-activated protein kinase, ATM ataxia-telangectasia mutated, ATR ataxia telangiectasia and Rad3-related protein, ATRIP ATR interacting protein, ccg clock-controlled gene, Cdc25A cell division cycle 25A phosphatase, Cdk2 cyclin-dependent kinase 2, Chk1 checkpoint kinase 1, Chk2 checkpoint kinase 2, cycA, B cyclins A and B, cycD cyclin D, cycE cyclin E, DBP D site of albumin promoter (albumin D-box) binding protein, GI gastrointestinal, G2 gap 2 phase, GSK3 glycogen synthase kinase-3, Hausp herpesvirus-associated ubiquitin-specific protease, M mitosis phase, NMNAT mononucleotide adenylyltransferase 1, NAMPT nicotinamide phosphoribosyl-transferase, P phosphate, PPARs peroxisome proliferator-activated receptors, REV-ERB (NR1D1) nuclear receptor subfamily 1, group D, member 1, S DNA synthesis phase, SIRT1 NAD-dependent deacetylase sirtuin-1, TIM timeless, WEE1 Mitosis inhibitor protein kinase
An additional level of regulation relates to the role of post-translational modifications in sustaining circadian periodicity by controlling protein stability. Accordingly, cytosolic phosphorylation of PERs and CRYs is mediated by casein kinase I ε and δ (CKIε/δ [22–25]), an event that triggers their binding to the E3-ligases F-box/WD repeat-containing protein 1 (Fbw1, also named β-TrCP1) and F-box/LRR-repeat protein 3 (Fbxl3), respectively, followed by ubiquitin-mediated proteasomal degradation (Fig. 1) [26–28]. It is not surprising to find that the extent of the regulatory kinases involved in controlling the clock mechanism is growing. For example, the α and β isoforms of glycogen synthase kinase-3 (GSK3) are now known to target PER, REV-ERBα, and CRY1/2 proteins and, conversely, GSK3 activity status is itself circadian-regulated in the suprachiasmatic nuclei of the brain [29, 30].
In addition to the aforementioned events, the CLOCK/BMAL1 heterodimer is responsible for coordinating the rhythmic transcriptional expression of ccgs in multiple tissues [31–33]. Among rhythmic transcripts are those encoding protein products involved in maintaining tissue homeostasis, cellular metabolism, and the regulation of cell division and death processes [32, 33]. Examples of ccgs include (1) the proto-oncogene c-MYC, which has a central role in G1 cell growth control, differentiation, and apoptosis; (2) the tumor-suppressor genes TP53 and GADD45α, whose protein products are responsible for most of the antiproliferative responses triggered in response to various forms of stress (endogenous and genotoxic); (3) the E3 ubiquitin–protein ligase mouse double minute 2 homolog (MDM2), a negative regulator of p53; and (4) caspases, G2/M kinases (e.g., WEE1), G1/S cyclins, cyclin-dependent kinase inhibitors, transcription factors, and ubiquitin-associated factors that are involved in regulating cell division and death (Fig. 1; Zhang et al. [34] and references within). An additional level of regulation results from unexpected findings that showed that clock components directly interact with key cell cycle regulators (Hunt and Sassone-Corsi [35] and references within). Specifically, PER1 interacts with the ataxia-telangectasia mutated (ATM) and checkpoint 2 (CHK2) proteins directly involved in the cellular response to ionizing radiation and double-strand DNA breaks. Another clock component, the human timeless homolog TIM, associates with the ATM-related kinase ataxia telangiectasia and Rad3-related protein (ATR) and its substrate ATR-interacting protein (ATRIP). More recent findings show that PER2 directly binds to p53, an association that influences p53 stability in unstressed conditions and activity in response to genotoxic stress (Fig. 1) [36, 37]. Despite emerging findings, we still lack a clear understanding of how molecules from the circadian and cell cycle circuitries are interlocked and operate to make cell proliferation or death decisions.
A multi-level analysis of the whole transcriptome in various clock tissues unveiled that only a small fraction of all clock-controlled transcripts consistently oscillate in all tissues, suggesting the existence of oscillatory patterns of expression that are tissue specific [31–33]. Furthermore, genome-wide studies have shown that only a fraction of cycling messenger RNAs exhibit a robust oscillation and that, more likely, post-transcriptional modifications (i.e., circadian control of poly(A) tail length) is responsible for the rhythmic behavior of most cell messengers [38–44]. Therefore, there is a direct impact in the time-dependent expression level and stability of key regulatory molecules and, consequently, in the various biochemical pathways that maintain tissue function and body’s homeostasis. Today, a comprehensive picture of the events that trigger, entrain (by means of Zeitgeber [‘time giver’] signals such as light and temperature [45]), and sustain circadian rhythms has been established and is supported by strong molecular data; the challenge lies in how to incorporate and implement this newly gained knowledge into the clinic to make treatment options as effective and safe as possible.
3 Coupling Metabolic Rhythms to the Body’s Daily Physiology
In-depth studies spanning decades of prolific work using experimental models, in vitro methods, and human clinical data cemented what once seemed a provocative hypothesis [46]: that the endogenous circadian and metabolic processes were intimately interconnected (for review see Bass and Takahashi [47], Asher and Schibler [48], and Sahar and Sassone-Corsi [49]). At present, examples of their crosstalk are numerous and varied and include a direct role for core clock components in the rhythmic biosynthesis of fatty acids, sterols, bile acids, gluconeogenesis, energy homeostasis, and lipid and glycogen catabolism (Fig. 1; see Bass and Takahashi [47] and references within). Accordingly, homozygous clock mutant mice exhibit hepatic steatosis, attenuated energy balance, hyperlipidemia, hyperleptinemia, hypertension, hyperglycemia, and insufficient compensatory insulin production; all indicators of a metabolic syndrome phenotype that, in this model, is also associated with obesity and the development of type 2 diabetes mellitus [50].
As direct molecular outputs of the clock, the rhythmic regulation of metabolic processes (e.g., gluconeogenesis, lipogenesis, oxidative metabolism, and respiration) influence, among others, daily changes in cerebral activity, vascular tone, blood pressure, heart rate, hormone levels (e.g., leptin, growth hormone, and insulin), cytokine secretion, body temperature, and renal activity (Fig. 1) [51]. Thus, not surprisingly, circadian desynchronization in humans (e.g., by shiftwork, high-fat diet intake, prolonged wakefulness) have direct implications in the temporal coordination of metabolic processes and, consequently, in health and disease [52, 53]. Epidemiological, clinical, and population-based statistics show that circadian–metabolism misalignment is emerging as the most common cause of cardiovascular disease, diabetes, inflammation, thrombosis, and even some forms of cancer in western societies [53]. The aforementioned connections stress the concept that the body is more susceptible to challenges (e.g., infections, toxins, injuries) at specific phases of the circadian cycle than at others and that circadian dysregulation is likely to favor a pathological condition. Most of all, it hints to ‘times-of-day’ at which a given tissue might be metabolically more sensitive to targeted treatment modalities in the clinic. Furthermore, the multiple oscillations in the expression and activity of rate-limiting regulatory enzymes in critical metabolic pathways establish times at which targeted treatments could yield the greatest results.
4 ‘Time-of-Day’ Medicine
Personalized medicine, a milestone that was unthinkable in the pre-human genome project era, is rapidly becoming a reality. Today, this innovative approach to disease prevention arises from the possibility of more precisely predicting a person’s risk of disease onset based on each person’s unique genetic makeup and clinical information [54]. Adding to this is the possibility of tailoring treatments based on the patient’s unique genetic background, lifestyle factors, and environmental exposure—an area of study usually referred to as personalized therapy or, more recently, precision medicine [55]. As a result, customizing an individual patient’s treatment weighs the entire spectrum of healthcare and creates important changes in clinical practice.
As precision medicine is formulated to achieve the goal of maximizing benefit and efficacy, lowering toxicity and side effects, and favoring the greatest possible quality of life for a patient, it is necessary to recognize that “effectiveness”, as broadly considered here, depends on multi-factorial elements (genetics included) that influence the distribution, uptake/efflux, and breakdown of a given therapy, and the elimination of any byproducts. What makes these biological processes worth re-examination in the context of seeking to achieve the greatest personalized therapeutic outcome is that all of them are periodic and most rate-limiting steps are clock-controlled [56]. A clear example comes from studies carried out using human and animal subjects in which the temporal abundance and distribution of plasma proteins, especially albumin and α-1-glycoprotein, was found to be relevant to drug binding and tissue absorption (for review see Musiek and Fitzgerald [57] and references within). In fact, early work showed that peak concentrations of most plasma proteins occurs in the early afternoon, albeit some variation in time exists between young adults and elderly subjects. Furthermore, their oscillations are independent of the proportion of red blood cells in the plasma and more likely reflect the daily variation in protein synthesis that results from liver metabolism [57]. As protein carrier levels oscillate, it is imperative to factor ‘time of administration’ into the treatment equation to optimize precision medicine and develop safer and more effective treatments.
Chronobiology is a broadly defined field of biology that is concerned with biological phenomena that exhibit cyclical behavior. It is divided into sub-disciplines that place specific emphasis on dynamic oscillatory signatures that either remain normal (chronophysiology) or are altered (chronopathology). Accordingly, chronophysiology focuses on the timely organization of biochemical and behavioral processes that, although tissue specific, need to be globally synchronized to ensure the body’s homeostasis. Conversely, chronopathology recognizes alterations in parameters over time that deviate from the mean value and, thus, serve as prognostic factors by helping to recognize a pre-pathological condition. These concepts are the foundation for a more specific field of study known as chronotherapy, in which ‘time’ becomes a critical variable when it comes to studying the effects of ‘when’ a given treatment modality needs to be administered. As chronotherapy represents the therapeutic application of chronopharmacology, it embraces several specific principles that relate to the type of disease being treated, the patient’s physiology, and the efficacy and tolerability that the patient exhibits to a given formulation considering biological timing and endogenous periodicities. Accordingly, the goal of chronotherapy is to ensure that, in vivo, the availability of a drug is timed to correspond with the rhythms of the disease and, thus, optimizing the therapeutic outcomes while minimizing adverse effects (for review see Kaur et al. [58]). As expected, many aspects of pharmacology are time-of-day-dependent, including (1) the abundance and activity of molecules involved in drug absorption and metabolism (considered under chronokinetics); (2) the physiological system targeted by the therapeutic (considered under chronodynamics); (3) the controlled-release formulations (considered under chronomodulated delivery and chronoformulation); and (4) the effectiveness of a therapeutic (considered under chronoeffectiveness) [56]. Lastly, as toxicological information is critical to the goals of chronotherapy, chronotoxicology considers the toxic responses and adverse effects of active substances and, thus, helps define optimal times at which medical regimens should be administered to enhance both effectiveness and tolerance [58].
Over the past several decades, circadian biologists have faced the challenge of identifying molecular players, uncovering elusive mechanistic details, and deciphering regulatory networks to explain how ‘time’ relates to processes relevant to normal physiology, disease onset and progression, and treatment outcome. Acquiring this knowledge helped expose the fundamental difference dividing the practice of clinical medicine versus the application of chronotherapy as most health professionals see the body’s physiology as a relatively invariant, rather than dynamic, homeostatic unit. The principles of chronotherapy do not disregard any valid or conventionally accepted approach or method to diagnose and treat diseases, nor do they disregard any new trends in modern medicine; instead, this discipline points to optimizing each aspect of the treatment process to achieve the greatest personalized therapeutic outcome.
5 Pharmacodynamics and the Molecular ‘Best Time’ to Administer a Therapy
There is a need to consider the many organizational levels, from molecular and cellular to tissue and organ, at which drugs can act when assessing their biochemical and physiological effects on the body. This concept, initially embraced within the basic principles of pharmacodynamics, now considers that different cells from different organs have different periodicities in the activity of their biochemical processes—from transcription/translation to metabolism—and in the temporal organization of their signal transduction cascades. Consequently, the notion of chronopharmacodynamics seems suited to weigh in on how circadian oscillation in a physiological system influences therapeutic outcomes.
Circadian variations in parameters associated with ligand (e.g., drug, hormone, neurotransmitter)–receptor interaction and signaling were among the earliest biological processes to be pharmacodynamically characterized. Findings determined the existence of diurnal variations in potency (related to receptor occupancy), efficacy (related to intracellular signaling system and tissue response), sensitivity (related to receptor binding) [59], and the level and activity of downstream signaling components triggered by ligand–receptor binding including enzymes, metabolites, and even secondary messengers (Musiek and Fitzgerald [57] and references within). Today, knowledge of the chronopharmacodynamic action of cardiovascular drugs, chemotherapeutic agents, analgesic and non-steroidal anti-inflammatory compounds, antidepressant drugs, anxiolytics, and antipsychotic compounds has conferred numerous benefits to patients with various clinical conditions. For example, cardiovascular disorders (e.g., hypertension, arrhythmias, coronary heart disease) are treated with different chemistries (lipophilic vs. hydrophilic) of β-receptor blocking drugs (e.g., atenolol, propranolol, acebutolol) following chronopharmacodynamic and kinetic rationales. This takes into account decades of research that established the rhythm organization of the cardiovascular system and is a reflection of circadian variations in hemodynamic parameters, ventricular stroke volume, heart rate, plasma concentration of hormones (e.g., noradrenaline, renin, angiotensin, aldosterone), and cyclic adenosine monophosphate [cAMP], as well as blood-associated parameters such as viscosity, platelet aggregability, and fibrinolytic activity (for review see Paschos et al. [60] and the classic work summarized in Lemmer et al. [61] and references within). Accordingly, the onset of cardiovascular diseases and early symptomatology have strong temporal dependencies. For example, morbid and mortal events in myocardial infarction and ischemic events peak between 6 a.m. and 12 p.m.; thus, the use of β-blockers such as oral doses of propranolol is recommended early in the morning. In fact, the therapeutic value of propranolol mostly comes from its dose–response relationship, which is dependent on the circadian phase at which it is administered, and less from its chronopharmacokinetics. Thus, ingestion of oral propranolol hours before wakening blocks β-adrenoreceptors in the sympathetic tissue, altering its tone, hemodynamic conditions, and heart rate at hours of early activity in individuals [61]. Compounds such as nitroglycerin and isosorbide mononitrate/dinitrate (ISMN/ISDN) act as vasodilators and are used to treat angina pectoris symptoms to relieve chest pain. The therapeutic action of nitrate derivatives results from activation of the vascular nitric oxide (NO)/cyclic guanosine monophosphate (cGMP) pathway, which, in turn, elicits venodilation and aortic and large elastic artery distensibility, promoting hemodynamic changes [62]. Interestingly, whereas ISDN exhibits only slight pharmacokinetic changes in relation to the time of drug administration, its therapeutic action is more pronounced when orally administered early in the morning (~2 a.m.), with most angina attacks taking place between 2 and 4 a.m. [61]. These are just a few examples in which time of administration is critical to restoring or eliciting aspects of vascular physiology that are either directly regulated by the master clock or indirectly modulated through slave oscillators. In fact, the release of glucocorticoids, angiotensin II, and catecholamines, as well as the activity of endothelial NO synthase is subject to diurnal variations, affecting vasoactive response and blood pressure. Similarly, processes such as vascular angiogenesis and thrombogenesis rely on signaling events that are clock controlled; thus, targeted therapies need to consider the time-of-day expression/activity of target molecules when developing protocols for administration. More recently, improvements in the therapeutic treatment of hypercholesterolemia with statins, a group of competitive inhibitors of hydroxymethylglutaryl (HMG)-coenzyme A reductase, have benefited from knowledge of the rhythmic activity of the enzyme. Accordingly, the optimal dose administration time for statins such as cerivastatin and simvastatin is in the evening, whereas fluvastatin benefits from a bedtime ingestion, lovastatin should be ingested after meals in the evening, and atorvastatin does not exhibit an optimal time-of-day dosing benefit [63]. As examples accumulate, and chronopharmacodynamic-based applications have been reviewed extensively elsewhere, we direct the reader to excellent disease-specific articles that summarize in great detail the current state of knowledge on time-dependent administration of therapeutics for various diseases and disorders of different type and pathogenesis [63–65].
6 Coordinating Drug Pharmacokinetics with Circadian Physiology
The rhythmic physiology of a specific group of organ systems—intestine, hepatobiliary, and renal—determines the effectiveness of drug absorption, distribution, metabolism, and excretion and, thus, its blood concentrations and bioavailability [56] (Fig. 2). Compelling molecular and biochemical data support the existence of a gut clock that modulates time-dependent gut physiology and, therefore, influences the absorption and distribution of drugs (for review see Dallmann et al. [56] and Musiek and Fitzgerald [57]). Accordingly, genetically modified mouse models carrying specific disruptions in circadian genes, for instance clock mutant mice [66], show a severe deregulation in the absorption of nutrients that is accompanied by disruptions in intestinal circadian activity.

Circadian timing, a new dimension in the pharmacological landscape of drug therapies: schematic representation of the disposition process of pharmaceutical therapeutics within the human body (absorption, distribution, metabolism, and excretion) and possible routes of drug administration (processes are indicated by blue text). Therapy administration strategies focus on attaining a therapeutic drug concentration in plasma within a window of values between the minimal effective concentration and the concentration that causes toxicity. Effective absorption results from a combination of biological and physicochemical factors, with the former being greatly influenced by circadian timing. Drug distribution is accomplished rapidly via circulation and is influenced by local changes in blood flow. Central compartments refer to major organs and tissues such as liver, lung, heart, brain, and kidney; peripheral compartments, on the other hand, refer to tissues where drugs are slowly metabolized and redistributed, such as adipose tissue and skeletal muscle. As many drugs bind to plasma proteins (e.g., albumin), ‘protein binding’ indicates an additional mode of distribution that also serves the purpose of facilitating elimination when the drug is secreted by renal glomerular filtration. Biotransformation takes place at various levels in the body, although the liver is the primary organ of drug metabolism and the gastrointestinal tract is the most important extrahepatic site. Other secondary organ sites, as indicated in the figure, carry out drug-metabolizing reactions. Cellular metabolism refers to the processes that occur mainly in the smooth endoplasmic reticulum of the cell and that chiefly involve oxidation reactions. The biochemical processes are divided into two phases in which drugs are chemically modified (Phase I) and conjugated (Phase II), if necessary, to facilitate their elimination. Lastly, excretion of drugs and byproducts is largely accomplished by the kidney, although compounds can also be eliminated via bile, sweat, saliva, exhaled air, or milk. The excretion process may involve renal glomerular filtration, renal tubular secretion, renal tubular reabsorption, and biliary excretion; thus, the nature of the mechanism impacts on the rate of elimination and can prolong the effect of the drug in the system. Yellow circles with an inclusive wave symbol indicate clock-controlled biochemical and physiological processes
Absorption of orally administered therapeutic agents in the gastrointestinal tract depends on key variables, including (1) the chemistry of the drug (i.e., hydrophobicity and degree of ionization as pH changes); (2) the rhythmic physiology of the gastrointestinal tract (i.e., circadian changes in gastric pH [67], gastrointestinal blood flow [68], gastric emptying time and motility [69, 70]); and (3) the expression of unique transporters on epithelial cells of the gut (Fig. 2) [71, 72]. Circadian patterns of absorption for lipophilic drugs and water-soluble compounds are markedly different, as documented in various animal models and human clinical studies in which a myriad of pharmacokinetic parameters were evaluated (e.g., maximum [peak] drug concentration [C max], time to reach C max following drug administration [t max], and area under the plasma concentration–time curve [AUC]) [57, 73]. Accordingly, most lipophilic compounds exhibit more rapid absorption in the morning (active period) than in the evening hours in humans as gut perfusion, gastric pH, and intestinal blood flow are simultaneously favored, a result that translates to higher C max and shorter t max values during morning administration [68, 73]. In contrast, absorption of water-soluble compounds does not exhibit circadian variation and, thus, assimilation remains steady throughout the day [73, 74]. To date, drugs spanning different chemistries and uptake mechanisms have confirmed diurnal variation in absorption, which argues for consideration of this when developing oral therapeutic administration regimens [57, 64]. Studies in healthy volunteers have examined the relevance of the method for delivering lipophilic compounds (e.g., immediate vs. sustained release, intravenous) and administration time in the pharmacokinetics of absorption. As the concept of bioavailability, defined as the extent and rate at which a substance becomes available at the site of action, relates to absorption, formulations of lipophilic drugs with immediate release have shown a circadian phase dependency on their action that has not been observed when using sustained-release formulations or after intravenous inoculation. Consequently, absorption of immediate-release preparations of, for example, nifedipine [75], cyclosporine [76], and non-steroidal anti-inflammatory drugs [77] peak in the early morning (e.g., 8 a.m., 22.5 min t max for nifedipine [peroral administration]; 6 a.m. for acetaminophen [peroral administration]; beginning of the active period for cyclosporine [78], for which drug bioavailability is greater than at night).
Effective drug distribution is largely dependent on its lipophilicity, affinity to, and abundance of carrier proteins in the blood flow (Fig. 2). These considerations impact a drug’s efficacy and the magnitude of adverse effects as unbound drug does not distribute within the target site and accumulates in unwanted tissues. Daily alterations in blood flow—daytime increase and night-time decline—depends on the circadian time-dependent function of the autonomic nervous system [79]. Accordingly, blood flow contributes to the difference in drug distribution observed when dosing takes place at different times [80].
Circadian variation in the level of plasma proteins reflects the great extent of the fluctuations in metabolic activity in the liver. Examples include albumin, globulins, and α-glycoproteins, whose concentrations are elevated during the daytime, peak at noon, and fall to a minimum at night [81]. As a result, it is expected that drug–protein binding varies in a diurnal fashion depending on the concentration of the carrier molecule in the plasma (Fig. 2) [78, 82]. While this premise has been demonstrated to work in clinical settings for various antiepileptic agents, chemotherapeutics, and anti-inflammatory drugs (for review see Erkekoglu and Baydar [83]), other drugs with more specific carriers display peak binding at night (e.g., carbamazepine [peak at ~8 p.m.], as evaluated in patients with epilepsy not responding to conventional dosing schedules [84]) or early afternoon (e.g., 5-fluorouracil [optimal timing varies according to sex and genetic background], as evaluated in patients with metastatic colorectal cancer receiving standard dosage [85]). Unlike the case of hydrophobic molecules, distribution of water-soluble drugs in tissues requires the expression of transporters and channels that allow for transit to occur. In this scenario, drug distribution depends on the circadian expression of specific transporters [71, 72]. Beyond distribution, there is a point at which the dose becomes toxic to consider when the therapeutic treatment takes place at a constant rate over a 24-h period.
Xenobiotic detoxification takes place primarily in the liver, although other extrahepatic detoxification systems exist (e.g., brain, kidney), in a two-phase process in which drugs are chemically modified (Phase I) and then conjugated to facilitate their excretion into bile, feces, sweat, saliva, and urine (Phase II; Fig. 2). Remarkably, genome-wide studies of the liver transcriptome have confirmed early observations that most genes encoding members of the microsomal cytochrome P450 family of enzymes driving Phase I, as well as members of epoxide hydrolases, oxidoreductases and sulfo-, glutathione-S-, UDP-glucuronosyl-, methyl-, and acetyl-transferases relevant to Phase II, show a circadian oscillatory pattern of expression [32, 33, 86] and activity that is reflected in fluctuations in drug metabolites eliminated in urine (for review see previous publications [47, 48, 87]).
Lastly, the process of excretion of metabolized drugs via bile flow and urine depends on the functional capacity of the kidney to reabsorb or secrete drugs across the epithelium (Fig. 2). The rate of elimination and its efficiency depends on two critical renal physiological variables: (1) the volume of blood delivered to the kidney per unit of time—renal blood flow; and (2) the rate of filtered fluid through the kidney per unit of time—the glomerular filtration rate. The renal blood flow is largely responsible for the daily elimination of most ionized drugs into urine with a peak of secretion during the active phase [88]. Unlike renal blood flow, the glomerular filtration rate seems to be a steady process that persists even during the resting phase [88]. The role of clock components in regulating the rhythmic elimination of metabolites by the renal system became evident in CRY1/CRY2 knockout animals. Accordingly, CRY knockout mice exhibit an abnormal renal blood flow that most likely results from a disrupted renin-angiotensin-aldosterone system and, partially, by oscillations in arterial blood pressure and cardiac output [89]. Moreover, the expression of the sodium-proton exchanger 3 system, relevant for the daily variations in urine pH (from 4.5 to 8.0), is abrogated in the proximal tubule of CRY knockout animals, indicating the clock’s control of drug elimination via urine acidification. Not surprisingly, reabsorption/secretion of water-soluble drugs and small organic ions accompany a robust diurnal expression of membrane transporters (e.g., ABC and Slc) in the proximal tubule of the kidney [90].
In summary, it is evident that clock components act at multiple regulatory levels influencing several aspects of the body’s physiology and, consequently, a drug’s pharmacokinetics and therapeutic/toxicological outcomes.
7 Circadian Biology in Today’s Clinical Trials
The application of circadian timing to patient treatment is an emerging practice in clinical trials that is still in its infancy and is mostly tested with regards to neurological disorders. Today, the US National Institutes of Health provides, through the ClinicalTrials.gov website (https://clinicaltrials.gov), the most compelling global registry of publicly and privately supported clinical trials involving any category of circadian intervention applicable to human subjects. As of June 2016, a total of 217,258 registered studies are ongoing in the USA and 193 countries around the world. Of these, 37 % (81,111 studies) are located only in the USA, 46 % (100,114 studies) are located in non-US countries, and 6 % (12,469 studies) are international multi-site clinical trials (Fig. 3a). Registered studies are of various types, comprising interventional (~80 % of cases), observational (~19 %, including drug treatments, behavioral studies, and surgical procedures), and other US Food and Drug Administration (FDA)-regulated studies that meet the criteria of ‘expanded access’ (http://www.fda.gov/). Therefore, this database provides a comprehensive overview of the current status of chronotherapy being considered as part of the design of clinical trials.

The impact of chronotherapy in the clinical trial world. a Pie chart showing the distribution of currently listed clinical trials based on their geographic locations. b Pie chart illustrating clinical trials in which chronotherapy is considered part of the treatment strategy by geographic location. c Pie chart divided into sectors proportionate to the distribution of clinical trials involving a form of chronotherapy treatment categorized by disease condition. d Pie chart showing the pharmacological categories of anticancer drugs whose pharmacokinetics, pharmacodynamics, tolerability, and/or efficacy are influenced by circadian timing [55]. In all cases, categories are given as a percentage of the whole pie. Cox-2 cyclo-oxygenase-2, VEGF vascular endothelial growth factor
Presently, 348 registered clinical studies (~0.16 % of existing clinical trials) worldwide include a form of circadian intervention, of which the majority are in Europe (122 studies), North America (171 studies), and Asia (24 studies) (Fig. 3b). Of note, chronotherapy may not be a variable under consideration when it comes to medical studies in which human volunteers are administered slow and sustained-release pharmacological formulations; however, other chrono-interventions are possible either earlier, to ameliorate the symptoms of the disease, or later, as part of the recovery phase.
Of the 348 chronotherapy studies, 45 % are categorized as related to ‘behavior and mental disorders’ and ‘diseases of the nervous system’ (Fig. 3c) and span studies in which aggression, alcohol consumption and related disorders, Alzheimer’s disease, anxiety, Asperger’s syndrome, attention–deficit disorder with hyperactivity, attention–deficit and disruptive behavior disorders, autism spectrum disorder, various behavioral symptoms, bipolar disorder, bulimia nervosa, child development disorders, compulsive behavior, ataxia, arthralgia, aphasia, amnesia, acromegaly, acute pain, and cognition and communication disorders are among the most noticeable. The remaining ~55 % of trials are divided into various categories, with each representing a proportion from 0.2 % (viral diseases) to 4.6 % (nutritional and metabolic diseases) (Fig. 3c).
Consideration of circadian biology has been given in the treatment of diseases of the immune system, heart and blood, nutrition and metabolism, and cancer and neoplasms in clinical trials; however, the number of ongoing studies in each category is insignificant in the context of all ongoing studies in those specific areas. For example, only 3.4 % of clinical trials involving circadian interventions apply to any form of cancer study (Fig. 3c; 0.15 % when considering all existing cancer trials) despite mounting evidence of the relevance of timing in xenobiotic metabolism and detoxification of drugs and in processes that are critical for tumor initiation and progression [64, 91]. Presently, breast neoplasms of various origins, and to a lesser extent others from bone, brain, and colorectal tissues, are the largest group of proliferative diseases targeted for circadian interventions. However, the majority of those studies are of an observational type and aim to investigate, for example, whether disorders in sleep architecture (1) follow breast cancer surgery (ClinicalTrials.gov identifier NCT01171508); (2) are associated with chemotherapeutic treatment in patients with specific clock polymorphisms (e.g., NCT01887925); (3) act as a prognostic factor in the progression of metastatic breast cancer (e.g., NCT00519168); or (4) can be corrected using behavioral (e.g., NCT02002533) or bright light (e.g., NCT02658708) therapies in patients undergoing chemotherapy. In addition, a few interventional clinical trials seek to (1) determine the optimal circadian time for administering chemotherapeutic combinations in women with metastatic breast cancer (e.g., NCT00003730); and (2) monitor circadian biometrics using wearable devices as an attempt to develop supplemental screening technologies to mammography and ultrasound for the purpose of diagnosis (e.g., NCT02511301). Lastly, a limited number of interventional studies evaluate approaches for chemoprevention of breast and prostate cancer using dietary supplements (e.g., NCT01611038) and behavioral strategies (e.g., NCT00342407 and NCT02609373) in individuals experiencing shift-work schedules. Overall, the gold standard for treatment of cancer patients is the conventional 24-h schedule, with constant-rate infusion, of chemotherapeutics; a protocol that leaves no room for chronotherapeutic administration. Remarkably, this conventional approach has remained largely invariable despite the fact that 40 or more anticancer drugs have already been tested in animal models for their tolerability, pharmacokinetics, and/or tumor efficacy and have been shown to be influenced by circadian timing (Fig. 3d; for review see Levi et al. [64]).
Another example that exposes the limited application of circadian biology in clinical trials is its occurrence in studies involving nutritional and metabolic diseases (Fig. 3c; 4.6 % of all studies involving circadian interventions). When evaluated in this category, circadian interventions are repeatedly represented in studies of diabetes, glucose metabolism disorders, insulin resistance, metabolic diseases, and obesity. Accordingly, most clinical trials are directed at evaluating the effect of circadian misalignment on insulin sensitivity (e.g., NCT02580513), risk of diabetes (e.g., NCT01786564 and NCT00989534), daily oscillations in glucose values (e.g., NCT00848822), and variations in skeletal muscle energy metabolism (e.g., NCT02261168). In addition, studies explore alterations in the function of the circadian clockwork in subjects who are healthy, obese, and have type 2 diabetes (e.g., NCT02384148), the use of chronotherapeutic interventions for prevention of cardiovascular events (e.g., NCT00725127), the time-dependent safety and tolerability of drugs for the treatment of diabetes (e.g., NCT00377676), the relevance of time-restricted feeding for the control of blood sugar (e.g., NCT01895179), and whether circadian phase adjustments can improve metabolic conditions in night-shift workers (e.g., NCT02108353). A small number of additional trials tangentially explore a relationship between circadian biology and disorders in iron metabolism (e.g., NCT01810965), dyslipidemias (e.g., NCT00770445), and calcium metabolism (e.g., NCT00004358). Thus, whereas data from molecular, biochemical, and engineered animal models have bolstered our appreciation of the oscillatory behavior of metabolic processes in health and disease, the translation of this fundamental knowledge to the clinic has been limited. Yet most circadian studies remain in the domain of diseases and disorders of the nervous system (Fig. 3c, 44.1 % of all registered clinical studies using a form of chronotherapy), a proportion that becomes insignificant (0.07 %) in the context of the total number of ongoing clinical trials.
8 Filling the Research Translational Gap One Step at a Time
Circadian biology long ago moved from defining the genetic components and anatomical structures responsible for the generation of rhythms to, more recently, molecularly identifying clock-driven processes governing the body’s physiology and behavior (for review see Golombek and Rosenstein [45]). Thus, it seems reasonable to assume that concepts and processes associated with circadian biology would have been translated broadly into more effective treatment interventions. However, the initial success that followed the use of chronotherapy to alleviate symptoms associated with bronchial asthma, hypertension, rheumatoid arthritis, and gastric ulcers, and improved tolerability and efficacy of chemotherapeutic treatment regimens (e.g., cyclophosphamide, cisplatin, 5-fluorouracil [92, 93]) have not been enough of a catalyst to integrate chronotherapy widely in clinical trials. Among the top-100 best-selling branded drugs on the market (IMS Health, based on 2015 US data) and another 119 components named on the World Health Organization essential medicines list were drugs that directly target products of genes with rhythmic expression (e.g., Nexium® for esophagitis/gastritis [AstraZeneca], Advair Diskus® for asthma [GlaxoSmithKline], Ritalin® for attention–deficit hyperactivity disorder [Novartis], Tamiflu® for influenza [Genentech], Viagra® for erectile dysfunction [Pfizer] [34]). Furthermore, the majority of these drugs have a half-life shorter than 6 h; thus, patients would greatly benefit from a timed dosage. Consequently, there is a need for a multi-level strategy to be launched that would facilitate the translational transition and incorporation of circadian timing into practice. To accomplish this goal, we (1) urge health professionals to consider including newly gained knowledge in circadian biology, its relationship to health and disease, and its relevance to the clinical practice, longitudinally in the medical school curricula (as outlined by Selfridge et al. [94]); (2) recommend that clinical pharmacists and pharmacy practitioners emphasize, whenever possible (e.g., via use of standard of care sheets), the importance of ‘time of administration’ as a variable to augment treatment efficacy and avoid variations associated with drug pharmacokinetics; (3) emphasize the need for circadian biologists to weigh the relevance of other multi-components such as sex, age, ethnicity, body mass index, activity–rest schedule, and eating times (and the meal’s composition) in their studies to better address the likely impact of those variables on treatment strategies and to understand inter-individual differences; and, more importantly, (4) support an inclusive ‘team science’ approach, in which the summation of each individual’s expertise provides a more holistic and comprehensive understanding of a biomedical challenge, which would include circadian biologists to help accelerate scientific transitions and progress. Today, the numerous advances in molecular circadian biology, chronopharmacology, and the evolving landscape in translational research that has resulted from various strategic initiatives sponsored by the National Institutes of Health favor a scenario where all these recommendations can find fertile ground.
9 Conclusions
As in many aspects of life, there is a great deal of truth in the proclamation that ‘timing is everything’; thus, it should not come as a surprise that this saying also applies to the administration of medical treatments. Largely disregarded in the standard medical practice because it lacked a strong mechanistic foundation, circadian biology has waited decades to emerge as a complementary and inclusive scientific discipline capable of bridging molecular genetics, biochemistry, physiology, pharmacology, pathology, and clinical research. Given the possibilities that this field of research opens to clinicians today, it could impact the quality of life of patients and the effectiveness of disease therapies. The clinical translation of chronotherapy to diseases such as hypertension, arthritis, asthma, and peptic ulcers resulted in such significant benefits for patients that it is now standard practice [73]. Therefore, health professionals and regulatory agencies have a sense of urgency regarding the implementation of chronotherapy when designing clinical studies and evaluating trial results.
References
Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol. 2010;72:517–49.
Huang W, Ramsey KM, Marcheva B, Bass J. Circadian rhythms, sleep, and metabolism. J Clin Invest. 2011;121(6):2133–41.
Waterhouse J, Reilly T, Atkinson G, Edwards B. Jet lag: trends and coping strategies. Lancet. 2007;369(9567):1117–29.
Portaluppi F, Tiseo R, Smolensky MH, Hermida RC, Ayala DE, Fabbian F. Circadian rhythms and cardiovascular health. Sleep Med Rev. 2012;16(2):151–66.
Shea SA, Hilton MF, Hu K, Scheer FA. Existence of an endogenous circadian blood pressure rhythm in humans that peaks in the evening. Circ Res. 2011;108(8):980–4.
Masri S, Sassone-Corsi P. The circadian clock: a framework linking metabolism, epigenetics and neuronal function. Nat Rev Neurosci. 2013;14(1):69–75.
Kronfeld-Schor N, Einat H. Circadian rhythms and depression: human psychopathology and animal models. Neuropharmacology. 2012;62(1):101–14.
Markt SC, Valdimarsdottir UA, Shui IM, Sigurdardottir LG, Rider JR, Tamimi RM, et al. Circadian clock genes and risk of fatal prostate cancer. Cancer Causes Control. 2015;26(1):25–33.
Monsees GM, Kraft P, Hankinson SE, Hunter DJ, Schernhammer ES. Circadian genes and breast cancer susceptibility in rotating shift workers. Int J Cancer. 2012;131(11):2547–52.
Lowrey PL, Takahashi JS. Genetics of circadian rhythms in Mammalian model organisms. Adv Genet. 2011;74:175–230.
Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280(5369):1564–9.
Hogenesch JB, Gu YZ, Jain S, Bradfield CA. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc Natl Acad Sci USA. 1998;95(10):5474–9.
Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, et al. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell. 1999;98(2):193–205.
Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107(7):855–67.
Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, et al. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110(2):251–60.
Sahar S, Sassone-Corsi P. Metabolism and cancer: the circadian clock connection. Nat Rev Cancer. 2009;9(12):886–96.
Doi M, Hirayama J, Sassone-Corsi P. Circadian regulator CLOCK is a histone acetyltransferase. Cell. 2006;125(3):497–508.
Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, et al. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134(2):329–40.
Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403(6771):795–800.
Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324(5927):654–7.
Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, et al. Circadian clock feedback cycle through NAMPT-mediated NAD + biosynthesis. Science. 2009;324(5927):651–4.
Eide EJ, Woolf MF, Kang H, Woolf P, Hurst W, Camacho F, et al. Control of mammalian circadian rhythm by CKIepsilon-regulated proteasome-mediated PER2 degradation. Mol Cell Biol. 2005;25(7):2795–807.
Kurabayashi N, Hirota T, Harada Y, Sakai M, Fukada Y. Phosphorylation of mCRY2 at Ser557 in the hypothalamic suprachiasmatic nucleus of the mouse. Chronobiol Int. 2006;23(1–2):129–34.
Lee C, Weaver DR, Reppert SM. Direct association between mouse PERIOD and CKIepsilon is critical for a functioning circadian clock. Mol Cell Biol. 2004;24(2):584–94.
Yin L, Wang J, Klein PS, Lazar MA. Nuclear receptor Rev-erbalpha is a critical lithium-sensitive component of the circadian clock. Science. 2006;311(5763):1002–5.
Busino L, Bassermann F, Maiolica A, Lee C, Nolan PM, Godinho SI, et al. SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science. 2007;316(5826):900–4.
Ohsaki K, Oishi K, Kozono Y, Nakayama K, Nakayama KI, Ishida N. The role of {beta}-TrCP1 and {beta}-TrCP2 in circadian rhythm generation by mediating degradation of clock protein PER2. J Biochem. 2008;144(5):609–18.
Siepka SM, Yoo SH, Park J, Song W, Kumar V, Hu Y, et al. Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell. 2007;129(5):1011–23.
Gallego M, Virshup DM. Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Biol. 2007;8(2):139–48.
Besing RC, Paul JR, Hablitz LM, Rogers CO, Johnson RL, Young ME, et al. Circadian rhythmicity of active GSK3 isoforms modulates molecular clock gene rhythms in the suprachiasmatic nucleus. J Biol Rhythms. 2015;30(2):155–60.
Miller BH, McDearmon EL, Panda S, Hayes KR, Zhang J, Andrews JL, et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci USA. 2007;104(9):3342–7.
Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109(3):307–20.
Storch KF, Lipan O, Leykin I, Viswanathan N, Davis FC, Wong WH, et al. Extensive and divergent circadian gene expression in liver and heart. Nature. 2002;417(6884):78–83.
Zhang R, Lahens NF, Ballance HI, Hughes ME, Hogenesch JB. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci USA. 2014;111(45):16219–24.
Hunt T, Sassone-Corsi P. Riding tandem: circadian clocks and the cell cycle. Cell. 2007;129(3):461–4.
Gotoh T, Vila-Caballer M, Liu J, Schiffhauer S, Finkielstein CV. Association of the circadian factor Period 2 to p53 influences p53′s function in DNA-damage signaling. Mol Biol Cell. 2015;26(2):359–72.
Gotoh T, Vila-Caballer M, Santos CS, Liu J, Yang J, Finkielstein CV. The circadian factor Period 2 modulates p53 stability and transcriptional activity in unstressed cells. Mol Biol Cell. 2014;25(19):3081–93.
Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, et al. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. 2011;331(6022):1315–9.
Hatanaka F, Matsubara C, Myung J, Yoritaka T, Kamimura N, Tsutsumi S, et al. Genome-wide profiling of the core clock protein BMAL1 targets reveals a strict relationship with metabolism. Mol Cell Biol. 2010;30(24):5636–48.
Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, et al. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338(6105):349–54.
Kojima S, Sher-Chen EL, Green CB. Circadian control of mRNA polyadenylation dynamics regulates rhythmic protein expression. Genes Dev. 2012;26(24):2724–36.
Le Martelot G, Canella D, Symul L, Migliavacca E, Gilardi F, Liechti R, et al. Genome-wide RNA polymerase II profiles and RNA accumulation reveal kinetics of transcription and associated epigenetic changes during diurnal cycles. PLoS Biol. 2012;10(11):e1001442.
Menet JS, Rodriguez J, Abruzzi KC, Rosbash M. Nascent-Seq reveals novel features of mouse circadian transcriptional regulation. eLife. 2012;1:e00011.
Rey G, Cesbron F, Rougemont J, Reinke H, Brunner M, Naef F. Genome-wide and phase-specific DNA-binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biol. 2011;9(2):e1000595.
Golombek DA, Rosenstein RE. Physiology of circadian entrainment. Physiol Rev. 2010;90(3):1063–102.
Rutter J, Reick M, McKnight SL. Metabolism and the control of circadian rhythms. Annu Rev Biochem. 2002;71:307–31.
Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330(6009):1349–54.
Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011;13(2):125–37.
Sahar S, Sassone-Corsi P. Circadian rhythms and memory formation: regulation by chromatin remodeling. Front Mol Neurosci. 2012;5:37.
Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308(5724):1043–5.
Feng D, Lazar MA. Clocks, metabolism, and the epigenome. Mol Cell. 2012;47(2):158–67.
Dibner C, Schibler U. Circadian timing of metabolism in animal models and humans. J Intern Med. 2015;277(5):513–27.
Roenneberg T, Merrow M. The circadian clock and human health. Curr Biol. 2016;26(10):R432–43.
Goldberger JJ, Buxton AE. Personalized medicine vs guideline-based medicine. JAMA. 2013;309(24):2559–60.
Ashley EA. The precision medicine initiative: a new national effort. JAMA. 2015;313(21):2119–20.
Dallmann R, Brown SA, Gachon F. Chronopharmacology: new insights and therapeutic implications. Annu Rev Pharmacol Toxicol. 2014;54:339–61.
Musiek ES, Fitzgerald GA. Molecular clocks in pharmacology. Handb Exp Pharmacol. 2013;217:243–60.
Kaur G, Phillips C, Wong K, Saini B. Timing is important in medication administration: a timely review of chronotherapy research. Int J Clin Pharm. 2013;35(3):344–58.
Reinberg AE. Concepts in chronopharmacology. Annu Rev Pharmacol Toxicol. 1992;32:51–66.
Paschos GK, Baggs JE, Hogenesch JB, FitzGerald GA. The role of clock genes in pharmacology. Annu Rev Pharmacol Toxicol. 2010;50:187–214.
Lemmer B, Scheidel B, Behne S. Chronopharmacokinetics and chronopharmacodynamics of cardiovascular active drugs. Propranolol, organic nitrates, nifedipine. Ann N Y Acad Sci. 1991;618:166–81.
Bode-Boger SM, Kojda G. Organic nitrates in cardiovascular disease. Cell Mol Biol (Noisy-le-grand). 2005;51(3):307–20.
Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005;19(1):117–25.
Levi F, Okyar A, Dulong S, Innominato PF, Clairambault J. Circadian timing in cancer treatments. Annu Rev Pharmacol Toxicol. 2010;50:377–421.
Dallmann R, Okyar A, Levi F. Dosing-time makes the poison: circadian regulation and pharmacotherapy. Trends Mol Med. 2016;22(5):430–45.
Pan X, Hussain MM. Clock is important for food and circadian regulation of macronutrient absorption in mice. J Lipid Res. 2009;50(9):1800–13.
Bron R, Furness JB. Rhythm of digestion: keeping time in the gastrointestinal tract. Clin Exp Pharmacol Physiol. 2009;36(10):1041–8.
Lemmer B, Nold G. Circadian changes in estimated hepatic blood flow in healthy subjects. Br J Clin Pharmacol. 1991;32(5):627–9.
Goo RH, Moore JG, Greenberg E, Alazraki NP. Circadian variation in gastric emptying of meals in humans. Gastroenterology. 1987;93(3):515–8.
Kumar D, Wingate D, Ruckebusch Y. Circadian variation in the propagation velocity of the migrating motor complex. Gastroenterology. 1986;91(4):926–30.
Ando H, Yanagihara H, Sugimoto K, Hayashi Y, Tsuruoka S, Takamura T, et al. Daily rhythms of P-glycoprotein expression in mice. Chronobiol Int. 2005;22(4):655–65.
Stearns AT, Balakrishnan A, Rhoads DB, Ashley SW, Tavakkolizadeh A. Diurnal rhythmicity in the transcription of jejunal drug transporters. J Pharmacol Sci. 2008;108(1):144–8.
Baraldo M. The influence of circadian rhythms on the kinetics of drugs in humans. Expert Opin Drug Metab Toxicol. 2008;4(2):175–92.
Sukumaran S, Almon RR, DuBois DC, Jusko WJ. Circadian rhythms in gene expression: Relationship to physiology, disease, drug disposition and drug action. Adv Drug Deliv Rev. 2010;62(9–10):904–17.
Lemmer B, Nold G, Behne S, Kaiser R. Chronopharmacokinetics and cardiovascular effects of nifedipine. Chronobiol Int. 1991;8(6):485–94.
Baraldo M, Risaliti A, Bresadola F, Chiarandini P, Dalla Rocca G, Furlanut M. Circadian variations in cyclosporine C2 concentrations during the first 2 weeks after liver transplantation. Transplant Proc. 2003;35(4):1449–51.
Levi F, Schibler U. Circadian rhythms: mechanisms and therapeutic implications. Annu Rev Pharmacol Toxicol. 2007;47:593–628.
Ritschel WA, Forusz H. Chronopharmacology: a review of drugs studied. Methods Find Exp Clin Pharmacol. 1994;16(1):57–75.
Anderson NH, Devlin AM, Graham D, Morton JJ, Hamilton CA, Reid JL, et al. Telemetry for cardiovascular monitoring in a pharmacological study: new approaches to data analysis. Hypertension. 1999;33(1 Pt 2):248–55.
Innominato PF, Levi FA, Bjarnason GA. Chronotherapy and the molecular clock: clinical implications in oncology. Adv Drug Deliv Rev. 2010;62(9–10):979–1001.
Scheving LE, Pauly JE, Tsai TH. Circadian fluctuation in plasma proteins of the rat. Am J Physiol. 1968;215(5):1096–101.
Lemmer B, Bruguerolle B. Chronopharmacokinetics: are they clinically relevant? Clin Pharmacokinet. 1994;26(6):419–27.
Erkekoglu P, Baydar T. Chronopharmacokinetics of drugs in toxicological aspects: a short review for pharmacy practitioners. J Res Pharm Pract. 2012;1(1):3–9.
Yegnanarayan R, Mahesh SD, Sangle S. Chronotherapeutic dose schedule of phenytoin and carbamazepine in epileptic patients. Chronobiol Int. 2006;23(5):1035–46.
Ortiz-Tudela E, Innominato PF, Rol MA, Levi F, Madrid JA. Relevance of internal time and circadian robustness for cancer patients. BMC Cancer. 2016;16:285.
Akhtar RA, Reddy AB, Maywood ES, Clayton JD, King VM, Smith AG, et al. Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus. Curr Biol. 2002;12(7):540–50.
Bellet MM, Sassone-Corsi P. Mammalian circadian clock and metabolism—the epigenetic link. J Cell Sci. 2010;123(Pt 22):3837–48.
Koopman MG, Koomen GC, Krediet RT, de Moor EA, Hoek FJ, Arisz L. Circadian rhythm of glomerular filtration rate in normal individuals. Clin Sci (Lond). 1989;77(1):105–11.
Doi M, Takahashi Y, Komatsu R, Yamazaki F, Yamada H, Haraguchi S, et al. Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat Med. 2010;16(1):67–74.
Zuber AM, Centeno G, Pradervand S, Nikolaeva S, Maquelin L, Cardinaux L, et al. Molecular clock is involved in predictive circadian adjustment of renal function. Proc Natl Acad Sci USA. 2009;106(38):16523–8.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Gorbacheva VY, Kondratov RV, Zhang R, Cherukuri S, Gudkov AV, Takahashi JS, et al. Circadian sensitivity to the chemotherapeutic agent cyclophosphamide depends on the functional status of the CLOCK/BMAL1 transactivation complex. Proc Natl Acad Sci USA. 2005;102(9):3407–12.
Zhang X, Diasio RB. Regulation of human dihydropyrimidine dehydrogenase: implications in the pharmacogenetics of 5-FU-based chemotherapy. Pharmacogenomics. 2007;8(3):257–65.
Selfridge JM, Moyer K, Capelluto DG, Finkielstein CV. Opening the debate: how to fulfill the need for physicians’ training in circadian-related topics in a full medical school curriculum. J Circadian Rhythms. 2015;13:7.
Antoch MP, Kondratov RV. Circadian proteins and genotoxic stress response. Circ Res. 2010;106(1):68–78.
Acknowledgments
The authors would like to thank all members of the Finkielstein Laboratory for their help and discussions. We would also like to thank Dr J. Webster for comments and manuscript edition. JMS and CVF conceived the manuscript. CVF wrote the manuscript. The authors apologize to the many outstanding colleagues in the specific fields whose work has not been cited due to space limitations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared that no conflicting interest exists.
Funding
This work was supported by grants from the National Science Foundation (MCB-1517298) and the Fralin Life Science Institute to CVF.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Selfridge, J.M., Gotoh, T., Schiffhauer, S. et al. Chronotherapy: Intuitive, Sound, Founded…But Not Broadly Applied. Drugs 76, 1507–1521 (2016). https://doi.org/10.1007/s40265-016-0646-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-016-0646-4