
Abstract
Background and Objective
Sacituzumab govitecan (SG) is an antibody–drug conjugate composed of an antibody with affinity for Trop-2 coupled to SN-38 via hydrolyzable linker. SG is approved for patients with metastatic triple-negative breast cancer (mTNBC) who have received two or more prior chemotherapies (at least one in a metastatic setting) and for patients with pretreated hormone receptor positive (HR+)/human epidermal growth factor receptor 2 negative (HER2–) metastatic breast cancer.
Methods
In these analyses, the pharmacokinetics of SG, free SN-38, and total antibody (tAB) were characterized using data from 529 patients with mTNBC or other solid tumors across two large clinical trials (NCT01631552; ASCENT, NCT02574455). Three population pharmacokinetic models were constructed using non-linear mixed-effects modeling; clinically relevant covariates were evaluated to assess their impact on exposure. Models for SG and tAB were developed independently whereas free SN-38 was sequentially generated via a first-order release process from SG.
Results
Pharmacokinetics of the three analytes were each described by a two-compartment model with estimated body weight-based scaling exponents for clearance and volume. Typical parameter estimates for clearance and steady-state volume of distribution were 0.133 L/h and 3.68 L for SG and 0.0164 L/h and 4.26 L for tAB, respectively. Mild-to-moderate renal impairment, mild hepatic impairment, age, sex, baseline albumin level, tumor type, UGT1A1 genotype, or Trop-2 expression did not have a clinically relevant impact on exposure for any of the three analytes.
Conclusions
These analyses support the approved SG dosing regimen of 10 mg/kg as intravenous infusion on days 1 and 8 of 21-day cycles and did not identify a need for dose adjustment based on evaluated covariates or disease characteristics.
Similar content being viewed by others

Sacituzumab govitecan (SG) is a Trop-2-directed antibody-drug conjugate of free SN-38 approved for treating patients with metastatic triple-negative breast cancer (mTNBC) who received two or more prior chemotherapies, with at least one of the chemotherapies for metastatic cancer. |
Using data from 529 patients with mTNBC or other solid tumors, three population pharmacokinetic models for SG, free SN-38, and total antibody were developed and characterized the pharmacokinetics of SG and its components. |
The presented analyses demonstrate that age, sex, race, tumor type, renal impairment, hepatic impairment, UGT1A1 genotype, use of UGT1A1 inducers/inhibitors, and Trop-2-expression had no clinically relevant impact on exposure of SG or its components; these findings support that no dose adjustment is needed based on these evaluated covariates or disease characteristics. |
1 Introduction
Triple-negative breast cancer (TNBC), defined as a lack of tumor-cell expression of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 (HER2) [1], accounts for approximately 15% of invasive breast cancers [2,3,4] and is associated with aggressive tumor biology and a poor prognosis. At initial diagnosis, the majority of TNBC tumors are highly proliferative with stage II or III disease. TNBC is associated with earlier disease recurrence in comparison with other subtypes of breast cancer [1]. Upon disease relapse and progression to metastatic TNBC (mTNBC), patients generally receive sequential systemic chemotherapy [1]; however, a clear global standard of care does not exist. In recent years, targeted therapies such as polyadenosine diphosphate-ribose polymerase inhibitors and programmed cell-death protein 1 immune checkpoint inhibitors have improved progression-free survival (PFS) and have expanded treatment options for mTNBC [1]. Despite these advancements, the mTNBC 5-year survival rate is still 12%, highlighting the need for further clinical development [5].
Recently, two antibody–drug conjugate (ADC) therapies have been approved for the treatment of metastatic breast cancer (mBC)—sacituzumab govitecan (SG) and trastuzumab deruxtecan. Trastuzumab deruxtecan, approved for patients with pretreated unresectable or metastatic HER2-low breast cancer (BC), is a HER2-directed antibody and topoisomerase inhibitor conjugate [6, 7]. SG is a first-in-class ADC composed of a humanized monoclonal antibody with a nanomolar binding affinity for trophoblast cell-surface antigen-2 (Trop-2), coupled to SN-38, the active metabolite of the topoisomerase 1 inhibitor irinotecan, via a proprietary, hydrolyzable linker [8]. Because of its hydrolyzable linker, SG releases its SN-38 payload both intra- and extracellularly in the tumor microenvironment [9, 10] and delivers significantly greater amounts of SN-38 to a Trop-2–expressing tumor than conventional irinotecan chemotherapy [11]. This extracellular release of SN-38 from SG allows for localized bystander killing within the tumor environment of neighboring Trop-2 low/negative tumor cells [12, 13]. Thus, SG can deliver cytotoxic chemotherapy to tumors, including adjacent cancer cells, in concentrations higher than standard chemotherapy and may reduce toxic effects in normal tissues, where Trop-2 expression is lower.
SG is approved in multiple countries for the treatment of mTNBC following two or more systemic therapies (at least one of them for metastasis) and HR+/HER2– mBC following endocrine therapy and at least two additional systemic therapies in the metastatic setting. SG also has accelerated approval in the United States for the treatment of metastatic urothelial cancer following chemotherapy and immunotherapy [14]. Safety and efficacy of SG in mTNBC were evaluated in a phase I/II study, IMMU-132-01 (ClinicalTrials.gov identifier: NCT01631552), in patients with advanced epithelial cancers [15], and a phase III registration trial, ASCENT (IMMU-132-05, NCT02574455), which compared SG with single-agent treatment of physician’s choice (TPC) in patients with relapsed or refractory mTNBC who had received two or more prior chemotherapies (at least one in the metastatic setting) [16]. SG clinical benefit was observed for PFS (median 5.6 vs 1.7 months; hazard ratio 0.41; p < 0.001) and overall survival (median 12.1 vs 6.7 months; hazard ratio 0.48; p < 0.001); objective response rate for SG versus TPC was 35% versus 5%.
Serum samples for pharmacokinetic analyses of SG and its components were collected from a subset of patients from the IMMU-132-01 study and all patients from the ASCENT study. The objective of these analyses was to characterize the population pharmacokinetics (PopPK) of SG (ADC), free SN-38 (payload), and total antibody (tAB) following administration of SG and assess the impact of clinically relevant covariates on their exposure in patients with mTNBC and other solid tumors [17].
2 Methods
2.1 Design of Clinical Studies Included in the Analyses and Pharmacokinetic Assessment
IMMU-132-01 was a phase I/II, open-label, single-arm, basket study evaluating the safety, efficacy, and pharmacokinetics of SG in patients with advanced solid tumors who had received at least one previous therapy for metastatic disease. In phase I, dose escalation was performed using a standard 3+3 design based on planned initial doses of 8, 12, and 18 mg/kg. A dose of 12 mg/kg was identified as the maximum tolerated dose but was associated with frequent dose delays and reductions. In phase II, 8 and 10 mg/kg doses were further evaluated in patients with mTNBC, HR+/HER2‒ mBC, and metastatic urothelial cancer (mUC). The regimen of 10 mg/kg on days 1 and 8 of the 21-day cycle was associated with a positive benefit/risk profile and was selected for further evaluation [18].
ASCENT was a phase III, randomized, open-label study of the efficacy and safety of SG in patients with locally advanced TNBC or mTNBC who were refractory or had relapsed after two prior chemotherapy regimens (at least one in the metastatic setting). Patients were randomized 1:1 to SG (10 mg/kg on days 1 and 8 of a 21-day cycle) or TPC (eribulin, capecitabine, gemcitabine, or vinorelbine). The primary endpoint was PFS, assessed by an independent review committee.
Concentration–time data from IMMU-132-01 and ASCENT patients who received at least one dose of SG and had at least one measurable concentration of SG or its components were included. Pharmacokinetic and anti-drug antibodies (ADA) serum sampling schemes for the two studies are in Table S1 (see electronic supplementary material [ESM]). Neither study had specific eligibility criteria regarding UGTIA1 genotype.
2.2 Bioanalytical Method
Separate validated liquid chromatography-tandem mass spectrometry methods were developed to measure concentrations of total SN-38 (CTotal SN-38) and free SN-38 (CFree SN-38). A validated sandwich electrochemiluminescence immunoassay was used to quantify tAB (hRS7 unconjugated or conjugated with SN-38) concentrations (ESM, section 1).
Concentrations of ADC (CSG) were calculated assuming a drug-to-antibody ratio (DAR) of 8 (ESM, section 2) using this equation:
For the detection of serum ADA and neutralizing ADAs, separate validated electrochemiluminescence immunoassays were developed (ESM, section 3).
2.3 Base Model Development
Three PopPK models were developed for SG, free SN-38, and tAB. Models for SG and tAB were developed independently using total dose of SG. The free SN-38 model was developed in a sequential manner. Formation of free SN-38 was driven by total amount of SG following first-order kinetics (KREL). Individual estimated concentrations of SG over time were the driver for release of free SN-38 following KREL. For the tAB model, the true clearance (CL) and volume parameters were calculated as model estimates multiplied by 0.92 (ratio of molecular weight of naked antibody to molecular weight of SG). For the free SN-38 model, due to lack of identifiability, the fraction metabolized could not be estimated; apparent clearance (CLSN38/F, QSN38/F) were estimated instead. Apparent volume of distribution terms for free SN-38 (V1SN38/F and V2SN38/F) were fixed based on literature-reported values [19].
The M6 method was used for handling the below limit of quantitation samples for SG and tAB whereas the M3 method was used for free SN-38 [20] (ESM, section 4). The first-order conditional estimation method of NONMEM (ICON plc, Dublin, Ireland) with interaction was used during all stages of the SG and tAB model development process. The LAPLACE estimation method was required for the development of the free SN-38 model based on the M3 method for handling the below limit of quantification data [20]. Untransformed tAB concentrations and log-transformed SG and free SN-38 concentrations were modeled. Base structural models (one and two compartment models with zero-order infusion rate) were evaluated based on standard goodness-of-fit (GOF) diagnostics. Interindividual variability (IIV) was modeled as follows:
where θi is the parameter for the ith participant, θT is the population typical value of the parameter, and ηi is a random interindividual effect with mean 0 and variance (ω2). ω2 was the diagonal element of variance-covariance matrix of the random effects (Ω). Models with only diagonal and those including nondiagonal Ω matrix were evaluated to quantify the correlation between η.
2.4 Covariate Assessments
Given the well-established correlation of body weight with CL and volume of distribution of monoclonal antibodies, the effects of baseline body weight were incorporated in the structural base model development. Following selection of the base model, including effects of body weight, other covariates were evaluated in a univariate covariate screening followed by stepwise covariate modeling. During univariate screening, covariates were added to the base model one at a time. Only covariates statistically significant in the univariate screening at a significance level of α = 0.01 based on the likelihood-ratio test were considered for stepwise forward addition.
Stepwise forward addition (α = 0.01) and backward elimination (α = 0.001) were used to select statistically significant covariates.
2.5 Model Evaluation
Final models were further evaluated using GOF plots and prediction-corrected visual predictive check (pcVPC) plots with 1000 simulations of concentration–time profiles based on the original analysis dataset. Model parameter distribution and uncertainty of the final pharmacokinetic parameters were evaluated by bootstrapping the analysis dataset (resampling with replacement) and re-estimating the pharmacokinetic model parameters.
2.6 Model Predictions
The impact of covariates (included in final PopPK models) on maximum (peak) serum drug concentration (Cmax) and area under the serum concentration–time curve (AUC) in the first 21-day treatment cycle relative to the typical participant (based on the analysis dataset for each model) was visualized in tornado plots. The simulations accounted for parameter uncertainty by sampling 1000 sets of parameter estimates from the variance-covariance matrix of respective models and showed the effect of categorical covariates versus a reference category and continuous covariate values at the 5th and 95th percentiles of the covariate values in the analysis population with corresponding 95% confidence intervals (CIs).
To confirm lack of any missed clinically relevant covariate impact, individual estimated pharmacokinetic parameters from the final PopPK models were used to predict individual Cmax and AUC for the first treatment cycle of the 10-mg/kg SG dose. Exposures for participants were predicted under consideration of participants’ respective covariate values (including body weight). The predicted covariate correlations were visualized in forest plots (an additional set of plots for all covariates of interest, not only covariates included in the final models) showing reference covariate values and values used in the prediction of relative exposures.
2.7 Software
R (Version 4.4; The R Foundation for Statistical Computing, Vienna, Austria) was utilized for data analysis, model predictions, and simulations. NONMEM® (Versions 7.3.0 and 7.4.3; ICON Plc, Dublin, Ireland) and Perl-speaks NONMEM (Uppsala University, Sweden) were used for PopPK modeling.
3 Results
3.1 PopPK Analysis Datasets
A total of 529 patients were included. Among these, 276 were from the IMMU-132-01 study (mTNBC, n = 24; mUC, n = 36; HR+/HER2– mBC, n = 32) and 253 were from the ASCENT study (all mTNBC). The summary of demographics and clinically relevant baseline characteristics tested as covariates including sex, age, race, body weight, prior treatment, number of prior lines of chemotherapy, Eastern Cooperative Oncology Group (ECOG) status, tumor type, uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) genotype, liver function, renal function, and Trop-2 expression is provided in Table S2 (see ESM). Most patients were female (78%) with a median age of 58 years (range 27–88) and median body weight of 70 kg (range 37–140). In IMMU-132-01, Trop-2 expression was characterized as weak, moderate, or strong staining by immunohistochemistry [15], whereas in ASCENT, Trop-2 expression was characterized as a numerical histochemical score (range 0–300) [16].
3.2 Final PopPK Model for SG
The pharmacokinetics of SG were best described by a two-compartment model with first-order linear elimination with estimated body weight-based allometric scaling exponents on clearance (CLSG), intercompartment clearance (QSG), central volume of distribution (V1SG), peripheral volume of distribution (V2SG) (Fig. 1a), and IIV on CLSG. Typical parameter estimates based on the final SG PopPK model and median and 95% CIs of pharmacokinetic parameters based on bootstrap analysis were in good agreement (Table 1). The typical parameter estimates for CLSG, V1SG, and V2SG were 0.133 L/h, 2.77 L, and 0.908 L, respectively; allometric exponents for effect of body weight on clearance and volume parameters were estimated to be 0.508 and 0.532, respectively. All parameters of the final SG model were estimated with good precision with percentage of relative standard error (%RSE) <20% and acceptable shrinkage for η on CLSG (26%).

Final model schematic for SG, free SN-38, and tAB. CLSG clearance of SG, CLSN38/F apparent clearance of free SN-38, CLtAB clearance of tAB, IV intravenous, KREL first-order release rate constant for free SN-38, QSG intercompartmental clearance of SG, QSN38/F apparent intercompartmental clearance of free SN-38, QtAB intercompartmental clearance of tAB, SG sacituzumab govitecan, tAB total antibody, V1SG central volume of distribution of SG, V2SG peripheral volume of distribution of SG, V1SN38/F apparent central volume of distribution of free SN-38, V2SN38/F apparent peripheral volume of distribution of free SN-38, V1tAB central volume of distribution of tAB, V2tAB peripheral volume of distribution of tAB
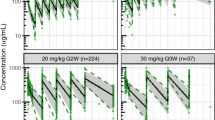
The effect of body weight-based scaling resulted in an approximately 5% increase in CLSG, QSG, V1SG, and V2SG with a 10% increase in body weight. Baseline albumin was a statistically significant covariate on CLSG with a 10% increase in baseline albumin corresponding to a 3% decrease in CLSG. The model included the effect of time after last dose on the residual unexplained variability (RUV). None of the other evaluated covariates (including markers of renal and hepatic function) had a statistically significant effect. Standard GOF plots for the final SG model (Fig. S1a, see ESM) showed weighted residuals evenly distributed around 0 with no trends over time or by concentration, indicating no meaningful bias in the model. The pcVPC plot (Fig. 2a) showed lack of model misspecification with good concordance between observed and simulated percentiles.

Prediction-corrected visual predictive check showing observed and simulated data versus time after last dose for the final PopPK models of SG (a), free SN-38 (b), and tAB (c). CI confidence interval, PopPK population pharmacokinetics, SG sacituzumab govitecan, tAB total antibody
3.3 Final PopPK Model for Free SN-38
The free SN-38 PopPK model was developed as a sequential model with free SN-38 being generated via a first-order release process from SG driven by individual model-predicted SG concentrations. The pharmacokinetics of free SN-38 were adequately described by a two-compartment model with estimated body weight-based allometric scaling exponent for apparent clearance (CLSN38/F) and intercompartmental clearance (QSN38/F), and apparent central and peripheral volumes of distribution (V1SN38/F and V2SN38/F, respectively) fixed to values reported in the literature (49 L and 2177 L, respectively) [19] to ensure identifiability of the model (Fig. 1a). IIV was included on first-order SG release rate (KREL) and CLSN38/F. A parameter estimating the covariance between individual KREL and CLSN38/F was included in the model. Typical parameter estimates based on the final free SN-38 model and median and 95% CI of pharmacokinetic parameters based on bootstrap analysis were in good agreement (Table 2). Parameters of the final free SN-38 model were estimated with good precision with %RSE <20%.
The final model of free SN-38 included body weight-based allometric scaling exponents for CLSN38/F and QSN38/F, and effects of time after last dose and study on RUV. No statistically significant effect of other covariates on CLSN38/F was identified. As the M3 method was used for the final free SN-38 model, in addition to the standard GOF plots, diagnostic plots based on normalized prediction distribution error were also generated (Fig. S1b, see ESM). Normalized prediction distribution errors were evenly distributed around 0 and had no trends over time or by concentration, indicating no meaningful bias in the model. The pcVPC plot (Fig. 2b) showed that the final pharmacokinetic model for free SN-38 slightly underpredicted the 5th percentile of the observed free SN-38 concentrations during the distribution phase.
3.4 Final PopPK Model for tAB
The pharmacokinetics of tAB were best described by a two-compartment model with first-order time-dependent elimination (Fig. 1b). The model included estimated body weight-based allometric scaling for CLtAB and QtAB and for V1tAB and V2tAB. IIV was estimated on CLtAB and V1tAB and a combined additive and proportional residual error model was utilized. Correlation of IIV on CLtAB and V1tAB as a covariance term was also included in the model. Typical parameter estimates based on the final tAB PopPK model were in good agreement with the median pharmacokinetic parameters based on bootstrap analysis (Table 3). Typical parameter estimates for CLtAB, V1tAB, and V2tAB were 0.016 L/h, 3.06 L, and 1.2 L, respectively. The model included a statistically significant effect of time on systemic CLtAB parametrized as a relative exponential onset model predicting a maximum reduction of the systemic CLtAB of 17% with a time to half-maximal reduction of approximately 48 days.
Based on the allometric scaling exponents for body weight, a 10% increase in body weight corresponded to a 3.6% increase in CLtAB and QtAB and 4.3% increase in V1tAB and V2tAB. Baseline serum albumin had a statistically significant impact on CLtAB with a 10% increase in baseline serum albumin leading to a 6.8% decrease in CLtAB. Other significant covariates included in the model were an effect for cancer type on CLtAB (13% lower CLtAB in patients with other tumor types including epithelial cancers, e.g., small-cell lung cancer, non-small-cell lung cancer, colorectal cancer, esophageal cancer, pancreatic ductal adenocarcinoma, etc. compared with those with mTNBC, mUC, and HR+/HER2– mBC), sex on V1tAB (12% higher V1tAB in males), and study on RUV (25% lower standard deviation of RUV in study 01 vs study 05). Standard GOF plots (Fig. S1c, see ESM) and pcVPC plots (Fig. 2c) showed no model misspecification and demonstrated that the final tAB PopPK model adequately characterized the observed concentrations.
3.5 Impact of Statistically Significant Covariates
Sensitivity analyses were conducted to assess the impact of statistically significant covariates identified in the final models on predicted first-cycle exposure (Cmax, AUC) of SG, free SN-38, and tAB, and these results were visualized using tornado plots. For SG, both body weight and serum albumin had a limited impact on AUC and Cmax, with predicted relative exposures within the 80–125% range (Fig. 3a,b). Body weights of 49 kg (5th percentile) and 110 kg (95th percentile) are estimated to result in 16% lower and 25% higher AUC for SG, respectively, than the typical exposure for a body weight of 70 kg. Similarly for albumin, values of 29 g/L (5th percentile) and 45 g/L (95th percentile) are estimated to result in 9% lower and 6% higher AUC of SG, respectively, than the typical exposure for albumin of 38 g/L. For free SN-38, body weight was the only statistically significant covariate and had a limited impact on AUC and Cmax (Fig. S2a, b, see ESM). Body weights of 49 kg and 110 kg are estimated to result in 18% lower and 25% higher AUC for free SN-38, respectively, than the typical exposure for a body weight of 70 kg. For tAB, the combined effect of body weight on CL and the body weight-based SG dosing regimen resulted in slightly higher exposure in patients with higher body weight, with a body weight of 110 kg (95th percentile) associated with an approximately 30% higher AUC and Cmax than a typical patient with a body weight of 70 kg (Fig. S2c, d, see ESM). Other statistically significant covariates including baseline serum albumin, sex, tumor type, and time after first dose had a very limited impact on tAB AUC and Cmax, with predicted relative exposures within the 80–125% range.

Impact of statistically significant covariates identified in the final model on predicted SG AUC (a) and Cmax (b). AUC and Cmax at the given covariate values were compared with those predicted for a typical patient with a body weight of 70 kg and baseline albumin of 38 g/L at the beginning of the study. Dots and error bars show the predicted relative exposure with 95% CI. ALB albumin, AUC area under the serum concentration-time curve, CI confidence interval, Cmax maximum (peak) serum drug concentration, SG sacituzumab govitecan
From the pharmacokinetic analysis dataset, no patients from IMMU-132-01 and four out 253 patients from ASCENT had treatment-emergent ADAs to SG. The reported ADA titer observed was low and varied from 10 to 30. Three patients with treatment-emergent ADAs were positive for neutralizing ADAs to SG.
3.6 Correlation of all Tested Covariates and Exposure
As an additional way to evaluate and visualize the covariate effects, the correlation between AUC or Cmax over the first treatment cycle and all covariates evaluated in the covariate assessment was visualized using forest plots for all three analytes with the respective final PopPK models. The covariates of mild or moderate renal impairment, mild hepatic impairment, age, sex, baseline albumin level, race, ECOG status, tumor type, UGT1A1 genotype, use of UGT1A1 inducers or inhibitors, or Trop-2 expression also did not have a clinically relevant correlation with SG AUC or Cmax (Fig. 4a, b). Analysis of the correlation between covariates and model-predicted free SN-38 and tAB exposure relative to a typical patient showed similar results (Figs. S3 and S4, see ESM).

Relationships between continuous (a) and categorical (b) covariates to SG AUC or Cmax over the first treatment cycle relative to exposures in a typical patient with the reference covariate value. Gray band shows the 80–125% range. Dots and error bars show predicted exposure and 90% CI, respectively. Categories with less than five patients (three patients with ECOG score of 2, one patient with moderate hepatic impairment, three patients with severe renal impairment, and one patient with missing renal impairment level) were excluded from the plot. ALB albumin, ALP alkaline phosphatase, ALT alanine aminotransferase, AST aspartate aminotransferase, AUC area under the serum concentration-time curve, BILI total bilirubin, CI confidence interval, CLCr creatinine clearance, Cmax maximum (peak) serum drug concentration, CPI checkpoint inhibitor, ECOG Eastern Cooperative Oncology Group, HER2 human epidermal growth factor receptor 2, HR hormonal receptor, mBC metastatic breast cancer, mTNBC metastatic triple-negative breast cancer, SG sacituzumab govitecan, Trop-2 trophoblast cell-surface antigen-2, UC urothelial cancer, UGT1A1 uridine 5'-diphospho-glucuronosyltransferase 1A1, ULN upper limit of normal
4 Discussion
These are the first analyses to characterize the PopPK of SG and its components. A robust dataset from two large trials of SG in patients with mTNBC and other solid tumors was used; the phase I/II trial that supported the accelerated approval, and the phase III pivotal trial that supported the full approval of SG in patients with unresectable, locally advanced or mTNBC who received two or more prior systemic therapies (at least one in the metastatic setting). The pharmacokinetics of SG, free SN-38, and tAB were each well characterized by two-compartment models with estimated body weight-based scaling exponents on CL and volume parameters with key clinical covariates assessed in the models. The PopPK models of SG and tAB were developed independently. The model of free SN-38 was developed in a sequential manner where individually estimated pharmacokinetic parameters from the SG PopPK model were used for predicting individual SG concentration–time profiles in the PopPK model of free SN-38, with a first-order release of free SN-38 from SG. All parameters in the final models for the three analytes, including covariate effects, were estimated with good precision and overall, the models provided a good description of the data. No major bias or trends were observed in the GOF plots. The pcVPC of all final models showed good concordance between observed and simulated percentiles.
Estimated exponents for the effect of body weight on the clearance and volume parameters provided better fit to the observed data in comparison with fixing the exponents. Body weight was a statistically significant covariate for the pharmacokinetic parameters of SG, free SN-38, and tAB. Effect of body weight on exposure of these analytes is accounted for with a body weight-based dosing regimen of SG. The remaining impact of body weight on exposure with the linear dose adjustment with body weight (relative to the shallower estimated exponents in the current analyses) is not considered clinically relevant given the overall variability of exposure of the evaluated analytes. CLtAB was found to decrease slightly with time after initiating treatment, with a maximum 17% decrease by approximately 6 months after starting treatment. A small time-dependent decrease in CL has also been reported for immune checkpoint inhibitor monoclonal antibodies in oncology patients and may be linked to reduced tumor burden and reduced cachexia with treatment [21, 22].
The covariate evaluation suggested that no dose adjustment is required based on sex, race (White vs Black/African American vs Asian vs other) or age (elderly vs younger patients). The impact of baseline Trop-2 expression was assessed separately for IMMU-132-01 and ASCENT as a categorical and continuous covariate, respectively, but was not found to be statistically significant for SG, free SN-38, or tAB clearance. Similarly, mild-to-moderate renal impairment and mild hepatic impairment did not have a clinically relevant impact on exposure of all three analytes. Due to limitation of available data, the potential impact of severe renal impairment and moderate or severe hepatic impairment could not be characterized. Of note, a phase I study is currently ongoing to assess the pharmacokinetics of SG in patients with metastatic solid tumors and moderate hepatic impairment (NCT04617522).
The UGT1A1 enzyme plays a key role in detoxifying SN-38 through glucuronidation [23]. Approximately 10% of North Americans have a UGT1A1 *28/*28 genotype, resulting in reduced UGT1A1 activity and an increased risk of neutropenia in patients receiving irinotecan, the prodrug for SN-38 [24]. Based on the current PopPK analyses using data from the IMMU-132-01 and ASCENT studies, UGT1A1 genotype was not a statistically significant covariate of SG, tAB, or free SN-38 pharmacokinetic parameters. Consistently, model-predicted exposures for UGT1A1 *1/*1, *1/*28, and *28/*28 genotypes in our study showed overlapping CIs. A small number of participants received UGT1A1 inhibitors (n = 14) or inducers (n = 5) during SG treatment. Because of such small numbers, the impact of these covariates could not be formally tested in these PopPK analyses. However, based on the limited data available, predicted individual SG or free SN-38 exposures in participants who received UGT1A1 inhibitors/inducers were comparable and within the range of exposures for participants who did not receive inhibitors/inducers, suggesting minimal impact of UGT1A1 inhibitors/inducers on exposure. The impact of UGT1A1 inhibitors and inducers on SG and free SN-38 exposure will be further assessed when additional data from ongoing clinical trials become available.
Tumor type was found to be a significant covariate for CLtAB, which was predicted to be 13% lower in participants with ‘other’ cancer types (including urothelial, small-cell and non-small-cell lung, colorectal, esophageal, endometrial, etc.) compared with mTNBC, mUC, and HR+/HER2‒ mBC. With this small, estimated difference in CLtAB, the predicted tAB exposure among all tumor types was comparable and within the range of observed IIV. These covariate evaluation results support the prescribing information of SG. Phase III studies evaluating the safety and efficacy of SG in TNBC, HR+/HER2‒ mBC, lung cancer, mUC, and different solid tumors in combination with other therapies are currently underway [25,26,27,28,29,30,31,32]. Furthermore, three among these studies are assessing the efficacy of SG for first-line treatment of mTNBC and early-stage TNBC [26, 27, 32]. These studies will further characterize the pharmacokinetics of SG in monotherapy and combination settings in other tumor types.
SG concentrations were calculated from bound SN-38 concentrations which were derived from measured total and free SN-38 concentrations in serum. The calculation for the ADC assumed a constant DAR of 8, while the payload is gradually released from the ADC over time in vivo. An approximation of a fixed DAR is typically utilized in ADC concentration determination due to the inability of the utilized bioanalytical assays to robustly resolve the different DARs over time.
Given the small number of patients who were ADA positive, it was not appropriate to formally test a possible relationship between immunogenicity and exposure. However, no relationship was apparent based on limited available data (data not shown). The data utilized in the present analyses were primarily from patients with mTNBC, and smaller subsets of patients had other tumor types. Additional analyses including pharmacokinetic data from ongoing phase III trials in other tumor types are warranted.
Pharmacokinetics of SG, free SN-38, and tAB following SG administration were well described with the developed population models. The effect of statistically significant covariates identified in the analyses was not considered clinically meaningful. Demographics and baseline characteristics of age, sex, race, body weight, or number of prior lines of chemotherapy, and key clinical covariates of renal impairment, hepatic impairment, tumor type, UGT1A1 genotype, use of UGT1A1-inducers or inhibitors, or Trop-2 expression did not have a clinically relevant impact on exposure. The analyses support the labeling recommendations for the approved 10-mg/kg clinical regimen. Additionally, body weight-based dosing of SG is appropriate in the target population and there is no need for further dose adjustment. The developed PopPK models were used to estimate exposures in the exposure–response analyses of SG efficacy and safety in patients with mTNBC. Detailed reports of these analyses are forthcoming.
Abbreviations
- %RSE:
-
Percentage of relative standard error
- ADA:
-
Anti-drug antibodies
- ADC:
-
Antibody-drug conjugate
- ALB:
-
Albumin
- ALP:
-
Alkaline phosphatase
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- AUC:
-
Area under the serum concentration-time curve
- BC:
-
Breast cancer
- BILI:
-
Total bilirubin
- CI:
-
Confidence interval
- CL:
-
Clearance
- CLCr:
-
Creatinine clearance
- CLSG :
-
Clearance of SG
- CLSN38/F:
-
Apparent clearance of free SN-38
- CLtAB :
-
Clearance of total antibody
- CFree SN38 :
-
Serum concentration of free SN-38
- CTotal SN38 :
-
Serum concentration of total SN-38
- Cmax :
-
Maximum (peak) serum drug concentration
- CPI:
-
Checkpoint inhibitor
- DAR:
-
Drug to antibody ratio
- ECOG:
-
Eastern Cooperative Oncology Group
- GOF:
-
Goodness-of-fit
- HER2:
-
Human epidermal growth factor receptor 2
- HER2–:
-
Human epidermal growth factor receptor 2-negative
- HR+:
-
Hormone receptor-positive
- IIV:
-
Interindividual variability
- KREL :
-
First-order release rate constant for free SN-38
- mBC:
-
Metastatic breast cancer
- mTNBC:
-
Metastatic triple-negative breast cancer
- mUC:
-
Metastatic urothelial cancer
- pcVPC:
-
Prediction-corrected visual predictive check
- PFS:
-
Progression-free survival
- PopPK:
-
Population pharmacokinetics
- QSG :
-
Intercompartmental clearance of SG
- QSN38/F:
-
Apparent intercompartmental clearance of free SN-38
- QtAB :
-
Intercompartmental clearance of total antibody
- RUV:
-
Residual unexplained variability
- SD:
-
Standard deviation
- SG:
-
Sacituzumab govitecan
- tAB:
-
Total antibody
- TNBC:
-
Triple-negative breast cancer
- TPC:
-
Treatment of physician’s choice
- TROP-2:
-
Trophoblast cell-surface antigen-2
- UGT1A1:
-
Uridine diphosphate glucuronosyltransferase 1A1
- V1SG :
-
Central volume of distribution of SG
- V2SG :
-
Peripheral volume of distribution of SG
- V1SN38/F:
-
Apparent central volume of distribution of free SN-38
- V2SN38/F:
-
Apparent peripheral volume of distribution of free SN-38
- V1tAB :
-
Central volume of distribution of total antibody
- V2tAB :
-
Peripheral volume of distribution of total antibody
References
Anders CK, Zagar TM, Carey LA. The management of early-stage and metastatic triple-negative breast cancer: a review. Hematol Oncol Clin North Am. 2013;27(4):737–49. https://doi.org/10.1016/j.hoc.2013.05.003. (viii).
DeSantis CE, Fedewa SA, Goding Sauer A, Kramer JL, Smith RA, Jemal A. Breast cancer statistics, 2015: convergence of incidence rates between black and white women. CA Cancer J Clin. 2016;66(1):31–42. https://doi.org/10.3322/caac.21320.
Kohler BA, Sherman RL, Howlader N, Jemal A, Ryerson AB, Henry KA, et al. Annual Report to the Nation on the Status of Cancer, 1975–2011, featuring incidence of breast cancer subtypes by race/ethnicity, poverty, and state. J Natl Cancer Inst. 2015;107(6): djv048. https://doi.org/10.1093/jnci/djv048.
Plasilova ML, Hayse B, Killelea BK, Horowitz NR, Chagpar AB, Lannin DR. Features of triple-negative breast cancer: analysis of 38,813 cases from the national cancer database. Medicine (Baltimore). 2016;95(35): e4614. https://doi.org/10.1097/MD.0000000000004614.
National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) Program. Cancer stat facts: female breast cancer subtypes. https://seer.cancer.gov/statfacts/html/breast-subtypes.html. Accessed Aug 7, 2023.
ENHERTU® (fam-trastuzumab deruxtecan-nxki) [prescribing information] Daiichi Sankyo, Inc., Basking Ridge, NJ; Nov 2022.
Modi S, Jacot W, Yamashita T, Sohn J, Vidal M, Tokunaga E, et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N Engl J Med. 2022;387(1):9–20. https://doi.org/10.1056/NEJMoa2203690.
Goldenberg DM, Sharkey RM. Sacituzumab govitecan, a novel, third-generation, antibody-drug conjugate (ADC) for cancer therapy. Expert Opin Biol Ther. 2020;20(8):871–85. https://doi.org/10.1080/14712598.2020.1757067.
Goldenberg DM, Cardillo TM, Govindan SV, Rossi EA, Sharkey RM. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget. 2015;6(26):22496–512. https://doi.org/10.18632/oncotarget.4318.
Govindan SV, Cardillo TM, Sharkey RM, Tat F, Gold DV, Goldenberg DM. Milatuzumab-SN-38 conjugates for the treatment of CD74+ cancers. Mol Cancer Ther. 2013;12(6):968–78. https://doi.org/10.1158/1535-7163.MCT-12-1170.
Sharkey RM, McBride WJ, Cardillo TM, Govindan SV, Wang Y, Rossi EA, et al. Enhanced delivery of SN-38 to human tumor xenografts with an anti-Trop-2-SN-38 antibody conjugate (sacituzumab govitecan). Clin Cancer Res. 2015;21(22):5131–8. https://doi.org/10.1158/1078-0432.CCR-15-0670.
Perrone E, Lopez S, Zeybek B, Bellone S, Bonazzoli E, Pelligra S, et al. Preclinical activity of sacituzumab govitecan, an antibody-drug conjugate targeting trophoblast cell-surface antigen 2 (Trop-2) linked to the active metabolite of irinotecan (SN-38), in ovarian cancer. Front Oncol. 2020;10:118. https://doi.org/10.3389/fonc.2020.00118.
Zeybek B, Manzano A, Bianchi A, Bonazzoli E, Bellone S, Buza N, et al. Cervical carcinomas that overexpress human trophoblast cell-surface marker (Trop-2) are highly sensitive to the antibody-drug conjugate sacituzumab govitecan. Sci Rep. 2020;10(1):973. https://doi.org/10.1038/s41598-020-58009-3.
Trodelvy (sacituzumab govitecan-hziy) [prescribing information] Gilead Sciences, Inc., Foster City, CA. ; 2022.
Bardia A, Messersmith WA, Kio EA, Berlin JD, Vahdat L, Masters GA, et al. Sacituzumab govitecan, a Trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann Oncol. 2021;32(6):746–56. https://doi.org/10.1016/j.annonc.2021.03.005.
Bardia A, Hurvitz SA, Tolaney SM, Loirat D, Punie K, Oliveira M, et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N Engl J Med. 2021;384(16):1529–41. https://doi.org/10.1056/NEJMoa2028485.
Sathe AG, Singh I, Singh P, Diderichsen P, Wang X, Chang P, et al. 189P Pharmacokinetics (PK) of sacituzumab govitecan (SG) in patients (Pts) with metastatic triple-negative breast cancer (mTNBC) and other solid tumors. Ann Oncol. 2022;33:S214. https://doi.org/10.1016/j.annonc.2022.03.208.
Ocean AJ, Starodub AN, Bardia A, Vahdat LT, Isakoff SJ, Guarino M, et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: safety and pharmacokinetics. Cancer. 2017;123(19):3843–54. https://doi.org/10.1002/cncr.30789.
Klein CE, Gupta E, Reid JM, Atherton PJ, Sloan JA, Pitot HC, et al. Population pharmacokinetic model for irinotecan and two of its metabolites, SN-38 and SN-38 glucuronide. Clin Pharmacol Ther. 2002;72(6):638–47. https://doi.org/10.1067/mcp.2002.129502.
Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28(5):481–504. https://doi.org/10.1023/a:1012299115260.
Li H, Yu J, Liu C, Liu J, Subramaniam S, Zhao H, et al. Time dependent pharmacokinetics of pembrolizumab in patients with solid tumor and its correlation with best overall response. J Pharmacokinet Pharmacodyn. 2017;44(5):403–14. https://doi.org/10.1007/s10928-017-9528-y.
Marchand M, Zhang R, Chan P, Quarmby V, Ballinger M, Sternheim N, et al. Time-dependent population PK models of single-agent atezolizumab in patients with cancer. Cancer Chemother Pharmacol. 2021;88(2):211–21. https://doi.org/10.1007/s00280-021-04276-4.
Chabot GG. Clinical pharmacokinetics of irinotecan. Clin Pharmacokinet. 1997;33(4):245–59. https://doi.org/10.2165/00003088-199733040-00001.
Dean L. Irinotecan therapy and UGT1A1 genotype. https://www.ncbi.nlm.nih.gov/pubmed/28520360. Accessed Jun 15, 2023.
US National Library of Medicine. Study of sacituzumab govitecan-hziy (IMMU-132) versus treatment of physician's choice in participants with metastatic or locally advanced unresectable urothelial cancer (TROPiCS-04). ClinicalTrials.gov identifier: NCT04527991. Last update December 9, 2022 https://clinicaltrials.gov/ct2/show/NCT04527991. Accessed Jun 1, 2023.
US National Library of Medicine. A randomized, open-label, phase 3 study of sacituzumab govitecan versus treatment of physician's choice in patients with previously untreated, locally advanced, inoperable or metastatic triple-negative breast cancer whose tumors do not express PD-L1 or in patients previously treated with anti-PD-(L)1 agents in the early setting whose tumors do express PD-L1. https://classic.clinicaltrials.gov/ct2/show/NCT05382299. Accessed Aug 28, 2023.
US National Library of Medicine. A randomized, open-label, phase 3 study of adjuvant sacituzumab govitecan and pembrolizumab versus treatment of physician's choice in patients with triple negative breast cancer who have residual invasive disease after surgery and neoadjuvant therapy. https://classic.clinicaltrials.gov/ct2/show/NCT05633654. Accessed Aug 28, 2023.
US National Library of Medicine. A randomized, open-label, phase 3 study of sacituzumab govitecan versus treatment of physician's choice in patients with hormone receptor-positive (HR+)/human epidermal growth factor receptor 2 negative (HER2-) (HER2 IHC0 or HER2-low [IHC 1+, IHC 2+/ISH-]) inoperable, locally advanced, or metastatic breast cancer and have received endocrine therapy. https://classic.clinicaltrials.gov/ct2/show/NCT05840211. Accessed Aug 28, 2023.
US National Library of Medicine. A phase 3 Asian study of sacituzumab govitecan (IMMU-132) versus treatment of physician's choice (TPC) in subjects with hormonal receptor-positive (HR+)/human epidermal growth factor receptor 2 negative (HER2-) metastatic breast cancer (MBC) who have failed at least 2 prior chemotherapy regimens. https://clinicaltrials.gov/study/NCT04639986?term=NCT04639986&rank=1. Accessed Aug 28, 2023.
US National Library of Medicine. Open-label, global, multicenter, randomized, phase 3 study of sacituzumab govitecan versus docetaxel in patients with advanced or metastatic non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy and anti-PD-1/PD-L1 immunotherapy. https://classic.clinicaltrials.gov/ct2/show/NCT05089734. Accessed Aug 28, 2023.
US National Library of Medicine. An open-label, multicenter, phase 3 randomized, active-comparator-controlled clinical study of pembrolizumab (MK-3475) in combination with sacituzumab govitecan versus MK-3475 monotherapy as first-line treatment in participants with PD L1 TPS greater than or equal to 50% metastatic non-small cell lung cancer (KEYNOTE D46/EVOKE-03). https://classic.clinicaltrials.gov/ct2/show/NCT05609968. Accessed Aug 28, 2023.
US National Library of Medicine. A randomized, open-label, phase 3 study of sacituzumab govitecan and pembrolizumab versus treatment of physician's choice and pembrolizumab in patients with previously untreated, locally advanced inoperable or metastatic triple-negative breast cancer, whose tumors express PD-L1. https://classic.clinicaltrials.gov/ct2/show/NCT05382286. Accessed Aug 29, 2023.
Acknowledgments
We want to thank the study participants and their caregivers for their participation and commitment to clinical research. Thanks also to the clinical study investigators and study teams. Medical writing and editorial assistance were provided by Gwendolyn F. Elphick, PhD, and Ben Labbe, PhD, of Parexel, and funded by Gilead Sciences, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding Information
This study was sponsored by Gilead Sciences, Inc.
Conflict of Interest
Abhishek G. Sathe is an employee of and holds stocks in Gilead Sciences, Inc. Indrajeet Singh was an employee at Gilead Sciences, Inc. Pratap Singh was an employee at Gilead Sciences, Inc. Paul M. Diderichsen is an employee of Certara and received support for the current manuscript and consulting fees from Gilead Sciences, Inc. Xiaohui Wang is an employee of Certara and received support for the current manuscript and consulting fees from Gilead Sciences, Inc. Peter Chang is an employee of and holds stock in Certara and received support for the current manuscript and consulting fees from Gilead Sciences, Inc. Atiya Taqui is an employee of and holds stocks in Gilead Sciences, Inc. See Phan is an employee of and holds stocks in Gilead Sciences, Inc. Sandhya Girish is an employee of and holds stocks in Gilead Sciences, Inc. and received grants and consulting fees from Certara. Ahmed A. Othman is an employee of and holds stocks in Gilead Science, Inc.
Availability of Data
Gilead Sciences, Inc. shares anonymized individual patient data upon request or as required by law or regulation with qualified external researchers based on submitted curriculum vitae and reflecting non-conflict of interest. The request proposal must also include a statistician. Approval of such requests is at the discretion of Gilead Sciences, Inc. and is dependent on the nature of the request, the merit of the research proposed, the availability of the data, and the intended use of the data. Data requests should be sent to datarequest@gilead.com.
Ethics Approval/Consent to Participate
The samples used were obtained from two clinical trials, IMMU-132-01 and [18] ASCENT [16]. These clinical trial study protocols were approved by the Institutional Review Boards at the participating study sites, conducted in compliance with the ethical principles originating in the Declaration of Helsinki, and conducted according to the International Conference on Harmonisation Guidelines for Good Clinical Practices.
Code Availability
Model code for SG, free SN-38, and total antibody final models are reported in the electronic supplementary material, Section 6.
Author Contributions
AGS, IS, and AAO wrote the manuscript, designed and performed research, and analyzed data. PS designed research. PMD designed and performed research and analyzed data. XW and PC performed research and analyzed data. AT performed research. SP and SG analyzed data. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Sathe, A.G., Singh, I., Singh, P. et al. Population Pharmacokinetics of Sacituzumab Govitecan in Patients with Metastatic Triple-Negative Breast Cancer and Other Solid Tumors. Clin Pharmacokinet (2024). https://doi.org/10.1007/s40262-024-01366-3
Accepted:
Published:
DOI: https://doi.org/10.1007/s40262-024-01366-3