
Abstract
Background and Objective
More than half of all drugs are still prescribed off-label to children. Pharmacokinetic (PK) data are needed to support off-label dosing, however for many drugs such data are either sparse or not representative. Physiologically-based pharmacokinetic (PBPK) models are increasingly used to study PK and guide dosing decisions. Building compound models to study PK requires expertise and is time-consuming. Therefore, in this paper, we studied the feasibility of predicting pediatric exposure by pragmatically combining existing compound models, developed e.g. for studies in adults, with a pediatric and preterm physiology model.
Methods
Seven drugs, with various PK characteristics, were selected (meropenem, ceftazidime, azithromycin, propofol, midazolam, lorazepam, and caffeine) as a proof of concept. Simcyp® v20 was used to predict exposure in adults, children, and (pre)term neonates, by combining an existing compound model with relevant virtual physiology models. Predictive performance was evaluated by calculating the ratios of predicted-to-observed PK parameter values (0.5- to 2-fold acceptance range) and by visual predictive checks with prediction error values.
Results
Overall, model predicted PK in infants, children and adolescents capture clinical data. Confidence in PBPK model performance was therefore considered high. Predictive performance tends to decrease when predicting PK in the (pre)term neonatal population.
Conclusion
Pragmatic PBPK modeling in pediatrics, based on compound models verified with adult data, is feasible. A thorough understanding of the model assumptions and limitations is required, before model-informed doses can be recommended for clinical use.
Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Pediatric physiologically-based pharmacokinetic (PBPK) physiology models are scientifically well-founded with increasing information regarding developmental changes, while most published PBPK compound models have been created and verified to predict pharmacokinetics (PK) in adults. |
We describe a pragmatic modeling approach to predict PK in infants, children, and adolescents, based on combining existing compound models with pediatric physiology models in Simcyp®. |
Model-predicted PK captured clinical data for several drugs with diverse disposition pathways for most of the pediatric age groups. Predictive performance tends to decrease when predicting PK in the (pre)term neonatal population. |
1 Introduction
Children are often excluded from clinical trials due to ethical and logistical reasons, resulting in little to no dosing information for pediatrics after market authorization. Many initiatives, such as the Best Pharmaceuticals for Children Act (BPCA), the Pediatric Research Equity Act (PREA), and the European Paediatric Regulation have resulted in expanded drug labeling information on pediatric dosing through collection of pharmacokinetic (PK), pharmacodynamic (PD), and safety data in pediatric patients [1, 2]. Despite these efforts, so called ‘off-label’ drug use remains common practice in pediatrics, especially in very young children, for drugs whose pediatric absorption, distribution, metabolism, or excretion (ADME) data is lacking or non-representative for a broader pediatric population [3, 4]. Lack of PK and PK/PD knowledge puts children at increased risks of adverse events and therapeutic failure [5,6,7].
When clinical PK or PK/PD data are limited, modeling and simulation, especially physiologically-based pharmacokinetic (PBPK) modeling, may be helpful to support decision making with respect to dosing both in pediatric drug development as well as for dosing recommendations in clinical care. PBPK models are multi-compartmental mathematical models that incorporate systems data (e.g. population demographic information, anatomical and physiological parameters) as well as drug-specific data (e.g. physicochemical properties) [8]. Their mechanistic nature provides the possibility to conduct mechanism-based modeling. When simulating PK in pediatrics, age-related developmental changes such as organ maturation and the ontogeny profile of drug metabolizing enzymes can be included. Currently, pediatric PBPK models are scientifically well-founded and incorporate an increasing amount of information regarding these developmental changes [9,10,11,12,13,14]. Modeling results have been used in regulatory applications and by academic investigators to, for example, determine effects of non-maturational factors (e.g. disease) [10, 15].
Building PBPK models from scratch requires many different types of detailed data and knowledge and can be very time consuming. However, several PBPK software developers have constructed PBPK platforms incorporating physiological models representing a variety of populations of interest, including those describing pediatric physiology. Meanwhile, many PBPK compound models that have been used in conjunction with adult physiological parameters to predict PK in mature subjects have been published and are available in databases [16]. Due to the mechanistic nature of PBPK modeling, it is possible to combine any drug model with any defined virtual physiology model, and therefore also models that describe pediatric physiology. An interesting case example that used this pragmatic approach to support clinical implementation of an off-label drug dose is on chloroquine, which was indicated for pediatric COVID-19 at the beginning of the pandemic in the Netherlands [17]. The model-informed ‘off-label’ dosing was included in national guidelines until the drug was found to be ineffective in late 2020 [18].
Confidence in model performance increases with increasing knowledge of drug PK properties and age-dependent developmental changes [19]. However, using pediatric PBPK models in cases where PK data are limited to verify the model, requires strong confidence in the predictive performance of the pediatric and preterm physiology models. The objective of the present work is therefore to evaluate the feasibility of such a pragmatic PBPK modeling approach in which existing compound models are combined with existing pediatric physiology models, in order to predict PK in pediatric subjects and ultimately inform dosing. We do so by investigating model performance for predicting PK across the pediatric age range, for intravenous and oral administration routes, at several dose levels for a selected set of drugs that represent different elimination pathways.
2 Methods
In general, in this study, proof-of-concept drugs were identified by checking for relevance in pediatrics, the presence of existing compound models, and the availability of pediatric PK data in scientific literature. Following the pragmatic approach, the existing compound models were verified with published PK data against the adult, pediatric, and preterm neonatal physiology models in Simcyp®, consecutively. In this process, the compound model remained unchanged regardless of previous model performance in one of the age categories. This way, we were able to evaluate if our proposed pragmatic approach is feasible. A more detailed outline of the method is discussed in the following sections.
2.1 Drug Selection
We selected proof-of-concept drugs as follows. First, the Model List of Essential Medicines for Children of the World Health Organization was screened [20]. Next, drugs were only selected if they were listed in the Dutch Pediatric Formulary [21], if a Simcyp® PBPK compound model was available, and if PK data in children was available. Then, drugs were selected which reflect different excretion pathways, such as renal clearance, cytochrome P450 (CYP)- and/or UDP-glucuronosyltransferase (UGT)-mediated metabolism, and biliary clearance.
2.2 PK Verification Data
To verify model predictions, for each drug PK data were retrieved from PubMed. Two standardized search queries were used, one to search for adult PK data and one for pediatric PK data (see section 1 of the electronic supplementary material [ESM]). Titles and abstracts of all search results were screened to check if actual PK data were provided in the publication. To extend our search strategy, the ‘Similar articles’ overview and references were checked to identify relevant articles that were missed with the initial search queries.
2.3 Model Verification
We used Simcyp® as it is one of the most frequently used PBPK modeling platforms with an easy-to-use user interface [16]. All simulations were conducted in Simcyp® v20 (Certara UK Limited, Simcyp Division, Sheffield, UK). Compound models were obtained from the Simcyp® software, the Simcyp® repository, or were described in scientific literature. Compound input parameters (e.g. Log PO:W and B/P) were checked prior to PBPK modeling but were left unchanged. Default physiology models of Simcyp® were used, namely ‘Sim-healthy volunteers’, ‘Sim-paediatric’ [22], and ‘Sim-preterm’ [23].
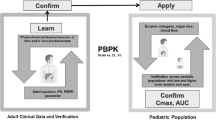
Our workflow for PBPK model validation is shown in Fig. 1. First, drug PBPK models were verified in the adult population to demonstrate adequate predictive performance in adults. This step was taken to avoid that compound models with poor predictive value in adults would impact the subsequent predictions in pediatric populations [24]. For simulations of adult PK, PK data upon single- and multidose intravenous (IV) administration were compared with published PK data. If the drug is administered orally in the clinical setting, and if PK data upon oral administration is available for this population, this step was followed by simulations of adult PK upon oral administration. After acceptance of adult model performance, simulations of pediatric single- and multidose IV and oral administration were conducted for age ranges and administration routes for which pediatric PK data were available.

Workflow of pragmatic pediatric PBPK modeling. Drug-specific physicochemical properties are defined in a compound model and age-dependent physiological parameters in distinct physiology models. The simulation trial design includes, amongst others, a description of the dosing schedule and the age of virtual subjects. The trial design is matched as closely as possible with the study design of reported PK studies (indicated with the dashed borders)
Predictive performance of PBPK models was evaluated by 1) calculating the ratio of predicted-to-observed PK parameters, and 2) a visual predictive check. Ratios within 0.5- to 2-fold range were considered acceptable as it is the most applied criterion [25]. For a visual check, observed plasma concentration–time profiles were extracted from literature, digitalized with WebPlotDigitizer v4.5, and compared with predicted plasma concentration–time curves. For each drug, visual predictive checks that represented the overall simulation results best, for each administration route and each age group, are shown in the manuscript. All other visual predictive checks are provided in the ESM (Fig. S1–8). To give an indication of overall model performance (i.e. multiple verification simulations were conducted per drug and per age group), prediction errors (PE) were calculated as described previously [26, 27], following the equation:
where Yobs,i is the ith observed plasma concentration in the clinical studies at a specific point in time and Ypred,i is the ith mean predicted plasma concentration for the same point in time. Variability between subjects and studies was expected to cancel out in the analysis; therefore, the median PE ideally equals zero. A median PE of ± 0.22, ± 0.40, ± 0.67, and ± 1.00 refers to 1.25-, 1.5-, 2-, and 3-fold median differences between observed and predicted values, respectively.
3 Results
3.1 Drug Selection and Collection of PK Studies
To assess the feasibility of the pragmatic PBPK modeling approach, seven drugs were selected: meropenem, ceftazidime, azithromycin, propofol, midazolam, lorazepam, and caffeine. The selected drugs with their elimination pathways and the source of compound models are shown in the ESM (section 3), as well as the adult and pediatric PK studies used for model verification and drug-dependent model input parameters (ESM sections 4 and 5, respectively).
3.2 PBPK Modeling
For every drug, simulations were conducted to match available clinical datasets for the different age categories: adults, adolescents, children, infants, and (pre)term neonates. Age ranges are defined in the ESM, section 2. Model performance, assessed by predicted-to-observed PK parameter ratios and visual checks, is shown in Figs. 2 and 3 and ESM section 6. Box–whisker plots for the PEs across all seven drugs for three age categories (adults, combined pediatric age groups, and [pre]term neonates) and per dosing route are provided in Fig. 4. Here, for the sake of concise presentation, PE values are shown irrespective of time, as also described earlier by Yamamoto et al. [27]. Full plots of PE values over time can be found in Fig. S9 of the ESM.

Predicted-to-observed ratios of the maximum concentration (Cmax), area under the curve (AUC), volume of distribution (Vd), clearance (CL), and half-life (t½), per drug, per age group (age ranges are defined in the electronic supplementary material, section 2). Single symbols represent a predicted-to-observed ratio of a single pharmacokinetic (PK) study. However, in case the age range of the simulated PK study covered more than one age group, the ratio is presented for each age group. ‘Neonates’ can refer to preterm and/or term neonates. The black lines represent the 2-fold range, the dashed lines the 1.5-fold range, the gray shaded area represents the 1.25-fold range and the gray line represents the unity line. Six ratios (i.e. PK parameter ratios of six simulated PK studies) fall outside the 0.2- to 5-fold range and are indicated with an asterisk


Prediction of drug plasma concentration-time profiles in the adult, pediatric and neonatal population. Meropenem (adults: Harrison et al., pediatrics: Blumer et al., and neonates: van den Anker et al.), ceftazidime (adults: Paradis et al., pediatrics: Bradley et al., and neonates: Mulhall & De Louvois), azithromycin (adults IV: Rodvold et al., adults PO: Barve et al., pediatrics IV: Jacobs et al., pediatrics PO: Stevens et al., neonates IV: Hassan et al.), propofol (adults: Knibbe et al. 1999, pediatrics: Jones et al., and neonates: Allegaert et al.), midazolam (adults IV: Smith et al., adults PO: Greenblatt et al., pediatrics IV: Reed et al., pediatrics PO: Reed et al., and neonates: Lee et al.), lorazepam (adults IV: Greenblatt et al. 1979a, adults PO: Friedman et al., and pediatrics IV: Chamberlain et al.), and caffeine (adults IV: Kennedy et al., adults PO: Thai et al., pediatrics PO: Akinyinka et al., neonates IV: Aranda et al., and neonates PO: Aranda et al.). References are provided in the electronic supplementary material. The solid line is the predicted mean and the shaded area represents the 5th and 95th percentile interval of the predicted plasma concentrations in the virtual population. Circles are the observed data. Insets depict log-transformed plasma concentration–time data

Box–whisker plots for the prediction errors (PEs) describing observed vs predicted plasma concentrations. Data are shown per age group and per dosing route. PE values for all observations (i.e. seven drugs, all studies, all time points) are presented as gray dots, irrespectively of time in the whisker plots. The box whiskers indicate the quartiles with the median and the minimum and maximum PE value. Note that positive values indicate an underprediction and negative values an overprediction by the model. Full plots of PE values over time can be found in the electronic supplementary material, section 6
3.2.1 Meropenem
In adults, children, infants and (pre)term neonates, the maximum concentration (Cmax) and area under the curve (AUC) predicted-to-observed ratios fall within 2-fold (Fig. 2). Also, the volume of distribution (Vd), clearance (CL), and half-life (t½) ratios fall within the 2-fold range for adults, children, and infants (Figs. 2, 3). Only in (pre)term neonates there is a tendency for Vd and t½ to be underpredicted and CL to be overpredicted (Figs. 2, 3). Evaluation of median PE (Fig. 4) and distribution of PEs over time (Fig. S9, see ESM) support this.
3.2.2 Ceftazidime
All predicted PK values fall within a 2-fold range of that observed for adults, adolescents, children, and infants, except for Vd which is underpredicted in the pediatric age groups (Fig. 2). In (pre)term neonates, Cmax is overpredicted and t½ is underpredicted when compared with several clinical studies (Fig. 2). These inaccuracies are reflected in the observed and predicted plasma concentration–time profile in (pre)term neonates (Fig. 3). Note that the median PE (Fig. 4) is close to 0 in all age groups. However, closer inspection of the PE values over time in Fig. S9 (see ESM) shows that this is the result of an initial overprediction of Cmax and a subsequent underprediction of t½ in children and (pre)term neonates. Inspecting the predictions for Vd and CL that drive the PBPK estimations of AUC and t½, it can be seen that predicted Vd and CL in (pre)term neonates generally falls within the 2-fold range, though a tendency to under- and overpredict can be observed, respectively (Fig. 2). Across the other pediatric age groups, clinical studies only sparsely reported Vd. For studies which reported Vd values, the predicted-to-observed ratios are bordering the 0.5-fold threshold (Fig. 2).
3.2.3 Azithromycin
Predicted Cmax for adults, adolescents, children, and infants generally fall within the 2-fold range (Fig. 2). This is also the case for prediction of the AUC, with the exception of two studies (one in adults and one in a combined population including both infants and children). When looking more closely at the predicted-to-observed ratios, it seems that the model has difficulty in capturing the clinical variability in exposure, as ratios spread widely within the acceptance interval for Cmax and beyond for AUC. This is further illustrated when evaluating predictions for Vd, CL and t½, where the acceptance interval thresholds are also violated in several cases (Fig. 2). Diving deeper into the differences between administration routes, predicted plasma concentration–time profiles after IV administration correspond well with observed profiles in adults, adolescents, children, and infants (Fig. 3 and ESM Fig. S9). In contrast, after oral administration, exposure is generally overpredicted in children, particularly in the absorption phase (Fig. 3 and ESM Fig. S9). This occurs in conjunction with predicted tmax values (time to maximum plasma concentration; Fig. S3, see ESM), which are generally lower than those clinically observed.
Note that exposure of azithromycin (IV) is highly underpredicted in preterm neonates. This cannot be taken from Fig. 2, since no clinical PK parameter values were available for comparison. Fig. 3 (as well as Fig. S3R and S3W in the ESM), does show that the reported plasma concentration–time curves were not captured by the model, which is further indicated in Fig. 4, where the median PE clearly deviates from 0.
3.2.4 Propofol
We could not assess the prediction accuracy of Cmax and AUC values, as these parameters were not reported in literature. However, predicted plasma concentration–time profiles in adults, adolescents, children, and infants were found to correspond well with observed profiles (Fig. 3 and Fig. S4 in the ESM). This is also reflected by the median PE (Fig. 4). Interestingly, Vd ratios vary widely from 0.53 to 6.12 in adults (and in children from 0.55 to 4.56; Fig. 2), due to a large variability in observed Vd values (Fig. S5, see ESM). Almost all CL and t½ ratios are within 2-fold range, the CL ratio in (pre)term neonates is 0.07 (not shown in Fig. 2). In addition, a large interindividual variability in observed plasma concentrations is seen compared to the predicted 5th and 95th percentile range in (pre)term neonates (Fig. 3).
3.2.5 Midazolam
Calculated Cmax and AUC ratios are almost all within the 2-fold range for all age groups (Fig. 2), with the exception of those after oral administration in adolescents, children, and infants (Fig. 2). The plasma concentration–time curves also indicate that after oral administration exposure is overpredicted in the pediatric age groups (Fig. 3 and Fig. S9 in the ESM). Furthermore, Vd tends to be underpredicted in adolescents, children, and infants, though not in (pre)term neonates, with an underprediction of CL in children and infants, while t½ tends to be overpredicted in all age groups except for adolescents (Fig. 2). Nevertheless, the observed plasma concentration–time profiles are captured adequately (Figs. 3, 4).
3.2.6 Lorazepam
In adults, almost all ratios for the different PK parameters are within 2-fold (Fig. 2), predicted plasma concentration–time curves correspond well with observed profiles, and PE values are acceptable (Figs. 3, 4). In contrast, in children, infants, and term neonates, AUC and Cmax tend to be overpredicted, while Vd and CL tend to be underpredicted (also in adolescents, Fig. 2). Nevertheless, the majority of the predicted-to-observed ratios remain within 2-fold. A visual check of predicted and observed plasma concentration–time profiles and corresponding median PE values (Figs. 3, 4) of adolescents, children, and infants supports adequate model performance. It is of note that the observed bodyweight-based CL (L/h/kg) decreases from infants to adolescents, while predicted CL values (L/h/kg) increase with increasing age.
3.2.7 Caffeine
In general, for all age groups Cmax, AUC, and t½ ratios fall within the 2-fold range (Fig. 2), which is in line with the visual checks (Fig. 3). Only for preterm neonates, exposure after oral administration is overpredicted. This statement is based on the degree to which the plasma concentration–time curves were captured by the model, as exposure estimates were not reported in the clinical paper (Figs. 3, 4). In preterm neonates, CL is predicted accurately, as all ratios are within 2-fold (Fig. 2). Vd tends to be underpredicted in preterm neonates; most ratios in Fig. 2 are bordering the 0.5-fold threshold. Looking more closely, especially with a multidose strategy, the Vd was underpredicted in preterm neonates (Fig. S8P, Q and R, see ESM). This was also the case for multidose simulations in adults (Fig. S8G in the ESM).
4 Discussion
We have shown the feasibility of using existing compound models and pediatric models to simulate pediatric PK, thus negating the need to build compound and physiology models from scratch. Generally, model predictions in adolescents, children, and infants were able to capture clinical observed PK data according to pre-defined criteria (Figs. 2, 4), and confidence in PBPK model performance was therefore considered high. In (pre)term neonates, predictive performance was generally lower. During our study, we identified underlying reasons for suboptimal model performance. A good understanding of these issues is imperative to improve performance and facilitates implementation of this pragmatic PBPK modeling approach.
4.1 Model Verification with Dissimilar Populations
While ceftazidime CL is predicted well for adults and children, Vd is generally underpredicted in children and (pre)term neonates. One explanation for these suboptimal predictions may be the impact of critical illness. For all adult PBPK model simulations, only PK studies including healthy volunteers are used for verification. In case of pediatrics, however, only PK data from pediatric patients are available for verification, while PK is predicted for healthy pediatric virtual subjects. Vd of many cephalosporins, including ceftazidime, is often increased in critical illness [28, 29], which is not considered in the physiology models used for the predictions. Ideally, a critically ill pediatric physiology model should be developed by adapting physiological parameters in the model which are influenced by severe infection. An adult critically ill septic population has been developed previously that included changes related to protein binding and body composition [30]. A similar pediatric model has not yet been developed, as the required pediatric-specific information is still unknown. Until sufficient physiological data are available to adjust the model mechanistically, adjusting the model in a non-mechanistic manner might be the best strategy, for instance, by optimizing the Vd by adjusting tissue-to-plasma partition coefficients or by using a higher fixed (user-defined) input parameter value for Vd.
4.2 Large Clinical Variability in PK
Predictions of propofol PK for adults and the distinct pediatric groups are accurate. However, Vd in adults exhibits a large variability (from 77.6 to 922.4 L), while predicted Vd values are much less variable (from 426.3 to 504.27 L; Fig. S5 in the ESM). This results in a large range of predicted-to-observed Vd ratios (Fig. 2). PK studies for model verification included healthy volunteers, but also ASA PS (American Society of Anesthesiologist Physical Status) classification grade 1 or 2 patients undergoing surgery (Table S5, see ESM). Grade 2 indicates a patient with mild systemic disease including, for example, current smokers or well-controlled diabetes mellitus, but it also includes obesity (30 < BMI < 40) [31]. There is, however, no clear correlation between the type of population studied (healthy volunteer, ASA 1 or 2-graded patient population) and size of the Vd. Also, no BMI range or individual age, weight, and height characteristics were provided in the studies, therefore, it is impossible to check if a large subgroup of patients with obesity might explain observed variability. The large clinical variability thus remains inexplicable. Still, the predicted values do fall within the observed range. In general, it should be noted that in case there are only a few PK studies available that report Vd, the chance of falsely claiming an over- or underprediction of Vd is high. This complicates the model verification process.
4.3 Missing or Incorrect Relevant ADME Processes
Azithromycin plasma exposure is predicted accurately in adults and pediatrics after IV administration, however, simulations of pediatric PK after oral administration show a discrepancy between observed and predicted plasma concentrations in the absorption phase (overpredicted Cmax and a shorter tmax). According to the compound summary document, available from the Simcyp® repository, the fraction available from dosage form (fa) and the first-order absorption rate constant (ka) are fitted to observed values. This is reasonable considering that the model is developed to be a fit-for-purpose model (i.e. as a CYP3A4 inhibitor model compound). Nonetheless, it is not specified which clinical study was used for parameter fitting. This is important as the input parameters for the compound model are dependent on the clinical study used. Hence, in case of high interindividual variability in the population, deriving input data from a small number of subjects can result in input parameter values that are biased. This may be the case for azithromycin, as substantial variability in pediatrics is observed in the clinic regarding both absorption and clearance [32].
A similar phenomenon may also be applicable to meropenem CL. Surprisingly, when we verified model performance in adults (the population for which the model was developed), this parameter tended to be underpredicted. This may be explained by the fact that we verified the model against a much wider array of published PK studies compared with the number of PK studies initially used for model development [33]. This may indicate that model parameterization of clearance may have been biased. In such cases, optimizing model parameterization to better reflect adult PK first could be considered in order to provide a better basis for extrapolation to pediatrics.
In contrast to the tendency to underpredict meropenem CL in adults and children, CL tends to be overpredicted in the (pre)term neonatal population. This overprediction is presumably caused by inaccurate mechanistic parameterization of the model. Meropenem is mainly cleared renally via glomerular filtration and to a large extent via tubular secretion by organic anion transporters 1 and 3 (OAT1 and OAT3) [34, 35]. However, in the current model, elimination is defined as total renal clearance with an additional systemic clearance component (Table S9, see ESM) and total renal clearance is based on maturation of glomerular filtration rate, solely. OAT1 and OAT3 protein abundance are significantly lower in term neonates and infants compared with older age groups [36]. A more recently published PBPK model of Ganguly and colleagues, which more accurately predicted PK in (pre)term neonates compared with our predictions, included OAT3 in a mechanistic manner, by including OAT3 ontogeny as well as Michaelis constant (Km) and maximum velocity (Vmax) values [37]. Moreover, renal meropenem metabolism by dehydropeptidase-I (DHP-1) is included in their model instead of using a general, non-specific, additional systemic clearance component. However, DHP-1 ontogeny is unknown and the authors therefore incorporated a theoretical function of age-based maturation of kidney weight to reflect DHP-1 ontogeny. No substantial difference is expected when including an additional systemic clearance compared with including DHP-1 metabolism, as additional systemic clearance is allometrically scaled (i.e. allometric scaling and kidney weight maturation functions are alike). Better performance of the model of Ganguly and colleagues can therefore primarily be assigned to the addition of OAT3-mediated tubular secretion, in a mechanistic manner.
Finally, midazolam PBPK modeling illustrates an example in which missing mechanistic ADME data impacts simulation accuracy. The model tends to underpredict Vd in adolescents, children, and infants (Fig. 3). Looking closer, it appeared that this finding is not surprising, since the midazolam model in Simcyp® consists of a minimal distribution model with a fixed value for Vd (i.e. 0.88 L/kg). This means that the Vd is fixed for all pediatric age groups and hence age-dependent changes in body composition are not taken into consideration. However, from clinical measurements of the Vd of midazolam, it is known that Vd is higher in children (i.e. 1.7 L/kg) [38] and in preterm neonates (i.e. 1.1 L/kg) [39] compared with adults. It is therefore logical that the simulated Vd values do not correspond to the clinical data. Still, the impact of Vd parameterization on the predicted exposure profiles is limited.
4.4 Inadequate Ontogeny Patterns
Propofol CL is predicted accurately in adults and children, but not in (pre)term neonates. A closer look at the compound model and Simcyp® settings revealed that while both CYP2B6 and UGT1A9 intrinsic clearance values were included in the model, the ontogeny profile of CYP2B6 was not. Simcyp’s default expression level of CYP2B6 in preterm neonates is set to zero while Upreti and Wahlstrom reported that CYP2B6 activity in this age group is comparable to adult activity [40]. Absent CYP2B6 activity in this age group naturally results in the underpredicted CL by our model, underlining the importance of incorporating appropriate ontogeny profiles. Aside from CYP2B6, the UGT1A9 ontogeny profile in Simcyp® corresponds with literature, indicating no activity at birth [41, 42]. Propofol is to a lesser extent also hydroxylated by CYP2C9 (Table S1, see ESM), but this was also not included in the tested compound model. Adding this would presumably contribute to better predictions as Michelet et al. showed that another propofol compound model, incorporating CYP2C9 and CYP2B6 activity, did result in accurate predictions in (pre)term neonates [43]. Of note, Michelet et al. calculated activity adjustment factors for CYP2B6 and CYP2C9 (with data from in vitro experiments with probe substrates), while intrinsic clearance values in the models used in this study were empirically scaled based on propofol data only [44] in order to correct for observed differences between in vitro and in vivo activity.
While lorazepam model performance in adults is accurate, in pediatrics and term neonates CL is underpredicted. Lorazepam is, like propofol, mainly metabolized by glucuronidation. The UGT enzymes UGT2B4, UGT2B7, and UGT2B15 are involved in lorazepam metabolism (Table S1, see ESM). According to the compound model, total liver clearance has been held responsible for approximately 95% of the total excretion (human liver microsomal data) with the remaining 5% attributable to renal clearance. In the Simcyp® preterm physiology model, activity of UGT2B7 and UGT2B15 is set to zero. However, it has been reported that hepatic UGT2B7 and UGT2B15 protein abundance in neonates is 13 and 38.6% of adult values, respectively [42]. This suggests their contribution to clearance is not negligible. Omitting UGT2B7 and UGT2B15 ontogeny might explain the large underprediction of CL in term neonates. Furthermore, according to Bhatt et al. [42], 50% of liver protein abundance for UGT2B7 in adults was reached at the age of 2.6 years. In contrast, in the model used by us, UGT2B7 abundance at this age was only around 20%. This discrepancy may explain the underpredicted CL, also in infants.
4.5 Different Contribution of ADME Pathways in Preterm Neonates Compared with Adults
Beforehand, we recognized that the contribution of eliminating pathways to total predicted caffeine clearance in preterm neonates deviates from what has been reported in scientific literature. Determination of caffeine and its metabolites in urine and enzyme expression and activity data showed that caffeine metabolism in preterm neonates is negligible [13, 45]. However, in the virtual physiology model, CYP1A2 activity increases from 1.8 to 23.5% (fraction of adult activity) in preterm neonates with a postmenstrual age (PMA) of 25 and 41 weeks, respectively. When conducting simulations, CYP1A2-mediated metabolism was predicted to be responsible for 91.5 to 97.4% of total caffeine clearance in preterm neonates (PMA of 25 and 39 weeks, respectively). This contrasts with the clinical PK data and highlights that an adequate simulation of total body clearance does not necessarily imply a correct mechanistic parameterization of clearance.
4.6 General Limitations
Our paper comes with some limitations. First, we only focused on Simcyp® for the pragmatic approach and did not include other PBPK software programs such as PK-Sim or GastroPlus. Although the general principle of the approach outlined in the present work should be applicable to other platforms as well, thorough assessment of model performance in these platforms is required to be able to conclude that the proposed pragmatic modeling approach is indeed feasible across the different platforms. Several underlying reasons for suboptimal performance identified here may be relevant for other software platforms as well, such as the large clinical variability in PK. On the other hand, suboptimal model performance due to, for example, inadequate ontogeny patterns can be highly platform-dependent, depending to what extent physiological data has been included in the physiology models these platforms employ.
For the predicted-to-observed ratios in our analysis, we have mainly focused on the 2-fold acceptance range. However, it is under debate if this criterion is stringent enough [46], e.g. for drugs with a small therapeutic window. More stringent ranges for predicted-to-observed ratios could thus be considered as well (e.g. 1.5- or 1.25-fold ranges), which are indicated in Fig. 2. 85.4% of all ratios fall within the 2-fold range, while 62.7% and 41.4% fall within the 1.5-fold and 1.25-fold range, respectively.
Finally, it should be noted that no plasma concentration–time profiles were available for lorazepam and no PK parameter values were reported for azithromycin in (pre)term neonates. When such clinical PK data are missing from the literature, model verification is less thorough.
5 Conclusion
PK of various drugs with distinct characteristics in adult and pediatric age groups were modeled via a pragmatic approach by combining existing compound models with existing pediatric physiology models. In most cases, this approach was successful, supporting the off-the-shelf use of PBPK models for dose selections in the pediatric and preterm population when PK data is absent or sparse. PBPK modeling can thus be a quick, ethical, and relevant approach to generate digital pieces of evidence that can help guide drug dosing in this special patient population. Still, the identified underlying reasons resulting in suboptimal model performance need to be considered when applying the pragmatic approach and a modeling workflow that tackles these challenges is key to move forward. The method should be combined with a decision framework that considers all clinical benefits, risks, and uncertainties of the model-recommended dose, in order to implement the pragmatic PBPK modeling approach into clinical practice.
References
Rodriguez W, et al. Improving pediatric dosing through pediatric initiatives: what we have learned. Pediatrics. 2008;121(3):530–9. https://doi.org/10.1542/peds.2007-1529.
The 2017 State of paediatric medicines in the EU commission report: 10 years of the EU Paediatric Regulation. 182. 3 May 2022. Available from: https://ec.europa.eu/health/system/files/2017-11/2017_childrensmedicines_report_en_0.pdf. Accessed: 3 May 2022.
Allen HC, et al. Off-label medication use in children, more common than we think: a systematic review of the literature. J Okla State Med Assoc. 2018;111(8):776–83.
Balan S, Hassali MAA, Mak VSL. Two decades of off-label prescribing in children: a literature review. World J Pediatr. 2018;14(6):528–40. https://doi.org/10.1007/s12519-018-0186-y.
Roberts R, et al. Pediatric drug labeling: improving the safety and efficacy of pediatric therapies. JAMA. 2003;290(7):905–11. https://doi.org/10.1001/jama.290.7.905.
Choonara I. Unlicensed and off-label drug use in children: implications for safety. Expert Opin Drug Saf. 2004;3(2):81–3. https://doi.org/10.1517/eods.3.2.81.27342.
Rieder M. Adverse drug reactions in children: pediatric pharmacy and drug safety. J Pediatr Pharmacol Ther. 2019;24(1):4–9. https://doi.org/10.5863/1551-6776-24.1.4.
Jamei M, et al. The Simcyp population-based ADME simulator. Expert Opin Drug Metab Toxicol. 2009;5(2):211–23. https://doi.org/10.1517/17425250802691074.
Cheung KWK, et al. Incorporating ontogeny in physiologically based pharmacokinetic modeling to improve pediatric drug development: what we know about developmental changes in membrane transporters. J Clin Pharmacol. 2019;59(Suppl 1):S56–69. https://doi.org/10.1002/jcph.1489.
Verscheijden LFM, et al. Physiologically-based pharmacokinetic models for children: starting to reach maturation? Pharmacol Ther. 2020;211: 107541. https://doi.org/10.1016/j.pharmthera.2020.107541.
Wang K, et al. Physiologically based pharmacokinetic models are effective support for pediatric drug development. AAPS PharmSciTech. 2021;22(6):208. https://doi.org/10.1208/s12249-021-02076-w.
Ince I, et al. Predictive pediatric modeling and simulation using ontogeny information. J Clin Pharmacol. 2019;59(Suppl 1):S95–103. https://doi.org/10.1002/jcph.1497.
van Groen BD, et al. Ontogeny of hepatic transporters and drug-metabolizing enzymes in humans and in nonclinical species. Pharmacol Rev. 2021;73(2):597–678. https://doi.org/10.1124/pharmrev.120.000071.
Kiss M, et al. Ontogeny of small intestinal drug transporters and metabolizing enzymes based on targeted quantitative proteomics. Drug Metab Dispos. 2021;49(12):1038–46. https://doi.org/10.1124/dmd.121.000559.
Zhang X, et al. Application of PBPK modeling and simulation for regulatory decision making and its impact on US prescribing information: an update on the 2018–2019 submissions to the US FDA’s Office of Clinical Pharmacology. J Clin Pharmacol. 2020;60(Suppl 1):S160–78. https://doi.org/10.1002/jcph.1767.
El-Khateeb E, et al. Physiological-based pharmacokinetic modeling trends in pharmaceutical drug development over the last 20-years; in-depth analysis of applications, organizations, and platforms. Biopharm Drug Dispos. 2021;42(4):107–17. https://doi.org/10.1002/bdd.2257.
Verscheijden LFM, et al. Chloroquine dosing recommendations for pediatric COVID-19 supported by modeling and simulation. Clin Pharmacol Ther. 2020;108(2):248–52. https://doi.org/10.1002/cpt.1864.
Singh B, et al. Chloroquine or hydroxychloroquine for prevention and treatment of COVID-19. Cochrane Database Syst Rev. 2021;2(2):CD013587. https://doi.org/10.1002/14651858.CD013587.pub2.
Grimstein M, et al. Physiologically based pharmacokinetic modeling in regulatory science: an update from the US Food and Drug Administration’s Office of Clinical Pharmacology. J Pharm Sci. 2019;108(1):21–5. https://doi.org/10.1016/j.xphs.2018.10.033.
World Health Organization Model List of Essenital Medicines for Children, 7th List, 2019, in 7th List. 2019. Geneva: World Health Organization. p. Licence: CC BY-NC-SA 3.0 IGO.
van der Zanden TM, et al. Developing a paediatric drug formulary for the Netherlands. Arch Dis Child. 2017;102(4):357–61. https://doi.org/10.1136/archdischild-2016-311674.
Johnson TN, Rostami-Hodjegan A, Tucker GT. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin Pharmacokinet. 2006;45(9):931–56. https://doi.org/10.2165/00003088-200645090-00005.
Abduljalil K, et al. A preterm physiologically based pharmacokinetic model. Part I: physiological parameters and model building. Clin Pharmacokinet. 2020;59(4):485–500. https://doi.org/10.1007/s40262-019-00825-6.
Yellepeddi V, et al. State-of-the-art review on physiologically based pharmacokinetic modeling in pediatric drug development. Clin Pharmacokinet. 2019;58(1):1–13. https://doi.org/10.1007/s40262-018-0677-y.
Sager JE, et al. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab Dispos. 2015;43(11):1823–37. https://doi.org/10.1124/dmd.115.065920.
Verscheijden LFM, et al. Physiologically based pharmacokinetic/pharmacodynamic model for the prediction of morphine brain disposition and analgesia in adults and children. PLoS Comput Biol. 2021;17(3): e1008786. https://doi.org/10.1371/journal.pcbi.1008786.
Yamamoto Y, et al. Predicting drug concentration–time profiles in multiple CNS compartments using a comprehensive physiologically-based pharmacokinetic model. CPT Pharmacometrics Syst Pharmacol. 2017;6(11):765–77. https://doi.org/10.1002/psp4.12250.
Roberts JA, Lipman J. Pharmacokinetic issues for antibiotics in the critically ill patient. Crit Care Med. 2009;37(3):840–51. https://doi.org/10.1097/CCM.0b013e3181961bff (quiz 859).
Gomez CM, Cordingly JJ, Palazzo MG. Altered pharmacokinetics of ceftazidime in critically ill patients. Antimicrob Agents Chemother. 1999;43(7):1798–802. https://doi.org/10.1128/AAC.43.7.1798.
Radke C, et al. Development of a physiologically based pharmacokinetic modelling approach to predict the pharmacokinetics of vancomycin in critically ill septic patients. Clin Pharmacokinet. 2017;56(7):759–79. https://doi.org/10.1007/s40262-016-0475-3.
American Society for Anesthesiology; Committee on Economics. ASA Physical Status Classification System. 181. [cited 2022 06-06-2022]. Available from: https://www.asahq.org/standards-and-guidelines/asa-physical-status-classification-system. Accessed: 06-06-2022.
Stevens RC, et al. Pharmacokinetics of azithromycin after single- and multiple-doses in children. Pharmacotherapy. 1997;17(5):874–80.
Zhou W, et al. Predictive performance of physiologically based pharmacokinetic and population pharmacokinetic modeling of renally cleared drugs in children. CPT Pharmacometr Syst Pharmacol. 2016;5(9):475–83. https://doi.org/10.1002/psp4.12101.
Bax RP, et al. The pharmacokinetics of meropenem in volunteers. J Antimicrob Chemother. 1989;24(Suppl 1):311–20. https://doi.org/10.1093/jac/24.suppl_a.311.
Shibayama T, et al. Characterization of CS-023 (RO4908463), a novel parenteral carbapenem antibiotic, and meropenem as substrates of human renal transporters. Drug Metab Pharmacokinet. 2007;22(1):41–7. https://doi.org/10.2133/dmpk.22.41.
Cheung KWK, et al. A comprehensive analysis of ontogeny of renal drug transporters: mRNA analyses, quantitative proteomics, and localization. Clin Pharmacol Ther. 2019;106(5):1083–92. https://doi.org/10.1002/cpt.1516.
Ganguly S, et al. Physiologically based pharmacokinetic modeling of meropenem in preterm and term infants. Clin Pharmacokinet. 2021;60(12):1591–604. https://doi.org/10.1007/s40262-021-01046-6.
de Wildt SN, et al. Population pharmacokinetics and metabolism of midazolam in pediatric intensive care patients. Crit Care Med. 2003;31(7):1952–8. https://doi.org/10.1097/01.ccm.0000084806.15352.da.
de Wildt SN, et al. Pharmacokinetics and metabolism of intravenous midazolam in preterm infants. Clin Pharmacol Ther. 2001;70(6):525–31. https://doi.org/10.1067/mcp.2001.120683.
Upreti VV, Wahlstrom JL. Meta-analysis of hepatic cytochrome P450 ontogeny to underwrite the prediction of pediatric pharmacokinetics using physiologically based pharmacokinetic modeling. J Clin Pharmacol. 2016;56(3):266–83. https://doi.org/10.1002/jcph.585.
Badee J, et al. The ontogeny of UDP-glucuronosyltransferase enzymes, recommendations for future profiling studies and application through physiologically based pharmacokinetic modelling. Clin Pharmacokinet. 2019;58(2):189–211. https://doi.org/10.1007/s40262-018-0681-2.
Bhatt DK, et al. Age- and genotype-dependent variability in the protein abundance and activity of six major uridine diphosphate-glucuronosyltransferases in human liver. Clin Pharmacol Ther. 2019;105(1):131–41. https://doi.org/10.1002/cpt.1109.
Michelet R, et al. The use of PBPK modeling across the pediatric age range using propofol as a case. J Pharmacokinet Pharmacodyn. 2018;45(6):765–85. https://doi.org/10.1007/s10928-018-9607-8.
Gill KL, et al. Application of a physiologically based pharmacokinetic model to assess propofol hepatic and renal glucuronidation in isolation: utility of in vitro and in vivo data. Drug Metab Dispos. 2013;41(4):744–53. https://doi.org/10.1124/dmd.112.050294.
Aldridge A, Aranda JV, Neims AH. Caffeine metabolism in the newborn. Clin Pharmacol Ther. 1979;25(4):447–53. https://doi.org/10.1002/cpt1979254447.
Guest EJ, et al. Critique of the two-fold measure of prediction success for ratios: application for the assessment of drug–drug interactions. Drug Metab Dispos. 2011;39(2):170–3. https://doi.org/10.1124/dmd.110.036103.
Acknowledgements
We thank Bart Bolier for his assistance with the modeling and Dr. Ping Zhao for useful discussions that improved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Funding
This publication is based on research funded by the Bill & Melinda Gates Foundation (INV-001822). The findings and conclusions contained within are those of the authors and do not necessarily reflect positions or policies of the Bill & Melinda Gates Foundation.
Conflict of interest
All authors declare that they have no conflict of interest.
Author contributions
All authors wrote the manuscript. RG and SW designed the research. JH, JF, LB, SD, and AO performed the research. All authors analyzed the data.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
van der Heijden, J.E.M., Freriksen, J.J.M., de Hoop-Sommen, M.A. et al. Feasibility of a Pragmatic PBPK Modeling Approach: Towards Model-Informed Dosing in Pediatric Clinical Care. Clin Pharmacokinet 61, 1705–1717 (2022). https://doi.org/10.1007/s40262-022-01181-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-022-01181-8