
Abstract
Background
Factor Xa inhibitors (FXaIs) are increasingly used without having sufficient drug–drug interaction data. Using a microdosed cocktail methodology could support filling the knowledge gap quickly.
Methods
In a randomised crossover trial, we investigated the drug–drug interactions between six oral azole antifungals and a microdosed FXaI cocktail containing 25 µg rivaroxaban, 25 µg apixaban, and 50 µg edoxaban. Additionally, different enzyme activities were also monitored using a microdosed cocktail approach. The six different azole antifungals were administered in therapeutic doses over a 24 h period, while the microdosed cocktails were administered 1 h after administration of the azole antifungals.
Results
Ketoconazole and posaconazole were the strongest perpetrators, showing similar increases as apixaban (area under the concentration–time curve ratio [AUCR] 1.64 and 1.62, respectively) and edoxaban (AUCR 2.08 and 2.1, respectively), whereas ketoconazole increased rivaroxaban 2.32-fold but only increased posaconazole 1.37-fold. All other azole antifungals showed less perpetrator effects on the FXaIs. Cytochrome P450 (CYP) 3A inhibition was confirmed using microdosed midazolam, with ketoconazole also the most potent perpetrator (8.42-fold).
Conclusion
Drug–drug interactions for three victim drugs of the same drug class (FXaIs) with different clearance mechanisms can be studied using a microdosed cocktail approach. Using members of the azole antifungal drug class as perpetrators, multiple interactions can be studied in one trial, and a more detailed insight into the underlying interaction mechanisms is possible.
Clinical Trial Registration
EudraCT number: 2017-004453-16.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
This is the first study that demonstrates the perpetrator characteristics of all available oral azole antifungal drugs on three microdosed oral factor Xa inhibitors (FXaIs). |
Of the azoles, ketoconazole showed the strongest inhibitory effects on all three oral FXaIs; however, the largest clearance reduction (46%) was observed with edoxaban following coadministration with posaconazole. |
These drug–drug interactions will be clinically manageable, with a dose reduction by half, at the most, although some combinations are not recommended by the manufacturers. |
1 Introduction
Factor Xa inhibitors (FXaIs) are commonly prescribed anticoagulants. They are usually taken over a long period of time and in combination with various other medications due to comorbidities. Accordingly, knowledge of the possible influence of comedication and its consequences for FXaI dosing is of importance.
Although three FXaIs share the same mechanism of action (rivaroxaban, apixaban and edoxaban), their elimination is quite different, with a greater or lesser extent of cytochrome P450 (CYP)-mediated metabolism and involvement of drug transporters such as P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). For example, edoxaban is mostly excreted unchanged, with only a minor role of CYP-mediated metabolism [1]. Apixaban metabolism involves mostly CYP3A, with only a minor contribution from CYP1A2, CYP2J2, CYP2C8, CYP2C9, and CYP2C19 [2], while rivaroxaban is metabolised by CYP3A and CYP2J2 [3, 4]. Therefore, drug–drug interactions (DDIs) of FXaIs in clinical practice are not a class phenomenon; rather, members of this drug class must be considered individually. Increased plasma concentrations are associated with an increased risk for bleeding, and decreased plasma concentrations might show reduced efficacy [5, 6]. Therefore, depending on the comedication, the best option would be to choose the specific FXaI that least interacts with the coadministered perpetrator; however, to date, only limited information from comparative interaction studies is available [7]. Coadministration of the strong CYP3A and P-gp inhibitor ketoconazole increased the area under the concentration–time curve (AUC) of apixaban 2.0-fold, 1.9-fold for edoxaban, and 2.6-fold for rivaroxaban [8,9,10]. Based on these data, the use of the entire class of azole antifungals with rivaroxaban and apixaban is not recommended [11, 12]. Furthermore, in the case of edoxaban, there is no recommendation for azoles except for coadministration with ketoconazole [13].
Azole antifungals are known to differentially inhibit CYPs and drug transporters such as P-gp and BCRP:
-
Ketoconazole: Strong CYP3A and P-gp/BCRP inhibitor [14, 15]; additionally, a potent inhibitor of CYP2J2 [16]
-
Posaconazole: Strong CYP3A and in vitro P-gp/BCRP inhibitor [17]
-
Voriconazole: Strong CYP3A [14], but not P-gp/BCRP, inhibitor [17, 18]
-
Fluconazole: Mainly moderate CYP3A [14], but not P-gp/BCRP, inhibitor [17,18,19]; additionally, a strong CYP2C19 inhibitor and moderate CYP2C9 inhibitor [14]
-
Isavuconazole: Weak to moderate CYP3A [20] and P-gp inhibitor, but not BCRP inhibitor [21]
We recently suggested the use of a microdosed FXaI cocktail containing edoxaban, apixaban, and rivaroxaban for simultaneous DDI screening [22]. Following this approach, in the present study the influence of six different azole antifungals, each with different perpetrator properties, was tested for their individual inhibitory effect on the three FXaIs. In order to reduce the safety risks of the azoles, the pre-exposure of the perpetrators (azoles) was limited to 1 h before the administration of the victim substances (FXaIs) and the recommended dosing scheme was used for 1 day only. In this way, screening for possible interactions between FXaIs and azoles was carried out using the same conditions to elucidate possible interaction mechanisms.
2 Methods
The trial protocol (EudraCT 2017-004453-16) was approved by the responsible Ethics Committee of the Medical Faculty of Heidelberg University and the competent authority (BfArM, Germany). The clinical trial was carried out in accordance with the standards of Good Clinical Practice (GCP) and the specific legal requirements of German law, the applicable version of the Declaration of Helsinki, and the International Conference on Harmonisation (ICH) recommendations on GCP. The trial was conducted at the Clinical Research Centre of the Department of Clinical Pharmacology and Pharmacoepidemiology, Heidelberg University Hospital, which is certified according to DIN EN ISO 9001:2015. All participants gave their written informed consent.
2.1 Trial Design
This phase I, single-centre, open-label, seven-part, three-sequence, randomised crossover clinical trial investigated the DDI between six oral azole antifungals and a microdosed FXaI cocktail containing 25 µg rivaroxaban (Xarelto®, Bayer), 25 µg apixaban (Eliquis®, Bristol-Myers Squibb), and 50 µg edoxaban (Lixiana®, Daiichi Sankyo Europe). Additionally, to monitor enzyme activities, a cocktail of 10 µg midazolam (Dormicum®; 5 mg/5 mL solutions for infusion; Roche), 50 µg yohimbine (Yohimbinum hydrochloricum D4®, Deutsche Homöopathie-Union-Arzneimittel), and 100 µg esomeprazole (Nexium®; gastroresistant granules; Astra Zeneca) was administered, together with the FXaI cocktail.
The following oral azole antifungals were used as perpetrators only, over a time period of 24 h:
-
200 mg itraconazole twice daily (SEMPERA® Kapseln, Jansen-Cilag GmbH)
-
400 mg voriconazole twice daily (Voriconazol Zentiva®, Zentiva Pharma GmbH)
-
400 mg fluconazole once daily (Fluconazol HEXAL®, Hexal AG)
-
400 mg ketoconazole once daily (Ketoconazole HRA®, Laboratoire HRA Pharma)
-
300 mg posaconazole twice daily (Noxafil®, Merck Sharp & Dohme Ltd)
-
200 mg isavuconazole three times daily (CRESEMBA®, Pfizer Pharma PFE GmbH)
To assess the baseline pharmacokinetic parameters, in one part of the trial the FXaI cocktail was administered alone. Participants were randomised into three groups (n = 4 participants in each group), each with a different sequence of trial parts. Isavuconazole was always administered last due to its long terminal elimination half-life (t½). The washout period between trial parts was always 6 days.
Oral stock solutions of edoxaban (30 µg/mL), rivaroxaban (25 µg/mL), and apixaban (25 µg/mL) were prepared in compliance with good manufacturing practice (GMP) by the Pharmacy of Heidelberg University Hospital, according to pharmaceutical development protocols approved by the competent authority (BfArM, Germany). Oral solutions were freshly prepared within 30 min prior to administration; 10 mL of oral stock solutions of rivaroxaban and apixaban, as well as 1.66 mL of edoxaban solution, were transferred into 70 mL of tap water in a plastic cup.
2.2 Trial Population
Participants were included if they had a good state of health (physically and mentally), a known CYP2C19 genotype, and no clinically relevant findings in medical history, physical examination, electrocardiogram, or routine laboratory analyses, including renal and liver function. However, participants were excluded if they were pregnant or lactating or had any known intolerance or contraindication to any component of the trial medication. No participants had participated in an interventional clinical trial within 30 days prior to inclusion in the study. During the entire study, other medications or substances (except oral contraceptives), including herbal drugs (especially St. John’s wort), smoking, nicotine, grapefruit and other citrus fruits, were not allowed. The same applied to the use of alcohol on trial days, and not more than 20 g of alcohol/day was allowed during the entire trial period. Furthermore, blood donations were not permitted 30 days (males) or 60 days (females) before commencement of the trial, during the trial, and for 2 months thereafter. All participants used two independent contraceptive methods (Pearl index <1%) during the trial and for a further 3-week period.
2.3 Trial Conduct
In each trial part, participants arrived fasting (for at least 8 h) and stayed fasting for approximately an additional 5 h. In each trial part, the first dose of the perpetrator was administered 1 h before administration of the FXaI cocktail, and blood samples (4.9 mL lithium heparin tubes) were taken before and 0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 2.5, 3, 4, 6, 8, 11, 23, and 24 h after administration of the FXaI cocktail. Urine was collected for 24 h. Blood samples were centrifuged for 10 min at 4 °C and 3600 rpm within 30 min after sampling. Urine samples and separated plasma aliquots were stored at −20 °C until analyses.
2.4 Analytical Quantification of Factor Xa Inhibitors (FXaIs), Midazolam, Yohimbine and Azole Antifungals
The FXaI plasma and urine concentrations were quantified by ultra-performance liquid chromatography coupled with tandem mass spectrometry (UPLC–MS/MS) according to published assays [23]. The method of FXaIs was modified to an ultrasensitive tandem mass spectrometer (Waters Xevo TQ-S, Waters, Germany) to increase sensitivity, as previously described [24]. Urine samples were diluted 1:10 with blank human plasma for analyses. Accuracy and precision values were less than or equal to ± 15%. The lower limits of quantification (LLOQ) were 2.5 pg/mL for all FXaIs.
Midazolam plasma concentrations were quantified by UPLC–MS/MS according to a published assay with an LLOQ of 0.37 pg/mL [25], while yohimbine plasma concentrations were determined using an UPLC–MS/MS assay with an LLOQ of 5 pg/mL [26]. An esomeprazole assay was established, however it was not applied to the plasma samples after we checked the accuracy of the 100 µg dose with the enteric-coated granules, which was not reproducible and inaccurate for unknown reasons.
Plasma azole concentrations were quantified by a UPLC–MS/MS (Waters TQD, Waters, Germany) assay developed earlier for therapeutic drug monitoring (TDM) purposes, and which was validated according to guidelines on bioanalytical method validation. Samples were spiked with stable isotope-labelled internal standards and purified by liquid–liquid extraction. Chromatographic separation was performed on an Acquity UPLC BEH C18 column (Waters, Germany) using a gradient program with UPLC eluents consisting of acetonitrile, 0.1% formic acid, and water. A linear calibration range was established from 0.1 to 25 µg/mL (50 µg/mL for fluconazole and isavuconazole). The LLOQ of all azoles was 0.1 µg/mL. Accuracy and precision values were less than or equal to ± 15%.
All assays fulfilled the pertinent guidelines on bioanalytical method validations from the European Medicines Agency and the US FDA [27, 28].
2.5 Calculations and Statistical Analysis
Standard pharmacokinetic parameters of each FXaI were calculated using Kinetica 5.0 (ThermoFisher Scientific, Waltham, MA, USA) in a non-compartmental analysis, and described with descriptive statistics. Calculated parameters included maximum plasma concentration (Cmax), time to Cmax (tmax), area under the plasma concentration–time curve extrapolated to infinity (AUC), apparent volume of distribution based on the terminal phase (Vz/F), apparent oral clearance (CL/F [F = bioavailability]), t½, apparent volume of distribution, renal clearance (CLrenal; expressed as the amount excreted unchanged in urine divided by the AUC), and non-renal clearance (CLnonrenal; as the difference between CL/F and CLrenal). AUC was calculated using a mixed log-linear model. Data are reported as geometric mean along with their lower and upper 95% confidence interval (CI), while Tmax is reported as median and range. A repeated measure analysis of variance (ANOVA) of the logarithmic transformed data was performed to test for differences against each FXaI separately without any azole (baseline), with an α error of 5%. The ratio of AUC at baseline (without azole) and during the azole (AUCR) was calculated separately for each antifungal and reported as AUCRs. The same applies to other ratios. A possible relationship between azole exposure and the corresponding AUCR of the three FXaIs was explored by correlation analysis.
To determine the effect of the azoles on CYP3A activity, the previously established limited sampling method for midazolam was applied to determine midazolam partial metabolic clearance (CLmet) by using midazolam concentrations at 2, 2.5, 3, and 4 h after dosing [29,30,31]. These data have been previously published [32].
To evaluate the effect of the azole antifungals on other drug-metabolising enzymes, yohimbine (CYP2D6) and esomeprazole (CYP2C19) were coadministered as a microdosed cocktail. No data on esomeprazole are available (see Sect. 2.4). As a CYP2D6 activity measure, yohimbine clearance was determined and compared using repeated measure ANOVA of the logarithmic transformed clearance.
Graphical and statistical analyses were performed using Prism 8.1.1 (GraphPad Software Inc., San Diego, CA, USA). A p value < 0.05 was considered significant.
3 Results
Twelve healthy volunteers (five females) with a median age of 24.5 years (range 20–48) and a body mass index (BMI) of 24.5 kg/m2 (range 21.9–29.9) completed all study parts (median duration of 45 days [range 44–120]). None of the participants was a CYP2C19 poor metaboliser. All safety data have already been comprehensively reported [32], with voriconazole causing the highest number of adverse events.
Over the observation period of 24 h, no apixaban concentrations were below the LLOQ. Rivaroxaban concentrations below the LLOQ at the 23 and 24 h samples were observed in one volunteer in two study parts, and in another volunteer in six study parts. In five volunteers, edoxaban concentrations were below the LLOQ at 23 and 24 h in two study parts, and in one volunteer in four study parts.
3.1 Baseline Microdosed FXaIs
The detailed pharmacokinetics of the microdosed FXaIs are shown in Tables 1, 2, 3.
3.2 Apixaban and Azole Antifungals
Compared with baseline, AUC and Cmax were significantly increased under treatment with ketoconazole (AUC 1.64-fold; Cmax 1.5-fold), posaconazole (AUC 1.62-fold; Cmax 1.58-fold), itraconazole (AUC 1.42-fold; Cmax 1.45-fold) and isavuconazole (AUC 1.33-fold; Cmax 1.44-fold). Voriconazole significantly increased AUC (1.24-fold) but not Cmax (1.24-fold), while fluconazole did not significantly alter apixaban pharmacokinetics. None of the azoles significantly changed tmax or t½ (Fig. 1). With the exception of fluconazole, all other azoles resulted in a significant reduction in total and non-renal clearance (Table 1). The amount excreted in urine as unchanged apixaban (Ae) increased significantly when treated with posaconazole (1.7-fold), isavuconazole (1.48-fold), ketoconazole (1.39-fold), and itraconazole (1.38-fold); however, none of the azoles affected CLrenal.

Apixaban plasma concentration-time curves after administration of apixaban 25 µg alone (baseline; red points and dotted line) and during coadministration with different azole fungistatic drugs (itraconazole: yellow squares and line; voriconazole: light green upside triangles and line; fluconazole: dark green quadrates; ketoconazole: orange points; posaconazole: light blue hexagons; isavuconazole: purple downside triangles and line) in linear and logarithmic presentation. Error bars (standard deviation) are only presented for the baseline part for clarity reasons
3.3 Edoxaban and Azole Antifungals
Compared with baseline, AUC and Cmax were significantly increased under treatment with ketoconazole (AUC 2.08; Cmax 2.47-fold), posaconazole (AUC 2.1; Cmax 2.15-fold), itraconazole (AUC 1.52; Cmax 1.59-fold), voriconazole (AUC 1.27; Cmax 1.24-fold) and isavuconazole (AUC 1.52; Cmax 1.59-fold) [Table 2]. Fluconazole did not alter any of the pharmacokinetic parameters of edoxaban. All other azoles reduced total and non-renal apparent clearance, without changing CLrenal (Table 2). None of the azoles significantly influenced tmax and t½ (Fig. 2). Compared with baseline, the total clearance was 47.4% lower with ketoconazole, 44% lower with posaconazole, 42.7% lower with itraconazole, 30.6% lower with isavuconazole and 19.1% lower with voriconazole.

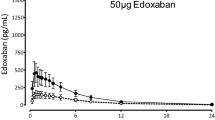
Edoxaban plasma concentration-time curves after administration of edoxaban 50 µg alone (baseline; red points and dotted line) and during coadministration with different azole fungistatic drugs (itraconazole: yellow squares and line; voriconazole: light green upside triangles and line; fluconazole: dark green quadrates; ketoconazole: orange points; posaconazole: light blue hexagons; isavuconazole: purple downside triangles and line) in linear and logarithmic presentation. Error bars (standard deviation) are only presented for the baseline part for clarity reasons
3.4 Rivaroxaban and Azole Antifungals
Rivaroxaban AUC and Cmax were significantly increased under treatment with ketoconazole (AUC 2.32-fold; Cmax 1.69-fold), itraconazole (AUC 1.47-fold; Cmax 1.33-fold) and posaconazole (AUC 1.37-fold; Cmax 1.39-fold) compared with baseline (Fig. 3). Voriconazole and fluconazole significantly increased AUC (1.16-fold and 1.27, respectively) but Cmax was not altered. No significant changes in the rivaroxaban pharmacokinetic parameters were observed during treatment with isavuconazole (Table 3). All other fungistatic drugs significantly decreased the total clearance of rivaroxaban, i.e. ketoconazole by 56%, itraconazole by 31%, posaconazole by 25%, fluconazole by 20%, and voriconazole by 13. While posaconazole (1.33-fold), itraconazole (1.29-fold) and ketoconazole (1.2-fold) significantly increased Ae, only ketoconazole led to a significant reduction in CLrenal (−48.2%, 95% CI 37.1–57.3%). No significant changes in tmax and t½ were observed during treatment with the azoles.

Rivaroxaban plasma concentration-time curves after administration of rivaroxaban 25 µg alone (baseline; red points and dotted line) and during coadministration with different azole fungistatic drugs (itraconazole: yellow squares and line; voriconazole: light green upside triangles and line; fluconazole: dark green quadrates; ketoconazole: orange points; posaconazole: light blue hexagons; isavuconazole: purple downside triangles and line) in linear and logarithmic presentation. Error bars (standard deviation) are only presented for the baseline part for clarity reasons
3.5 Comparison between the Three Different FXaIs
A single oral dose of any azole administered 1 h before microdosed midazolam resulted in potent CYP3A inhibition. The CL/F values of the FXaIs during treatment with different azoles (expressed as a percentage of baseline CL/F) are shown in Fig. 4, together with the concurrently observed midazolam clearances as previously reported [32].

Radar chart of the apparent oral clearances of apixaban (green circles), edoxaban (blue squares), rivaroxaban (red diamonds), and the metabolic clearance of midazolam (orange triangles) in relation to the different azole fungistatic drugs
No significant changes in CYP2D6 activity by the different azole antifungals were observed. The effect of different azole perpetrator drugs on yohimbine CL/F is demonstrated in Fig. 5. There were no CYP2D6 poor metabolisers in this study.

Effect of different azole perpetrator drugs on yohimbine clearance (geometric mean and 95% confidence interval) in relation to baseline clearance; the dashed line depicts the baseline clearances for reason of comparison. CL/F apparent oral clearance
3.6 Pharmacokinetics of Azole Antifungals
The plasma concentration–time profiles of voriconazole, itraconazole, fluconazole, ketoconazole, posaconazole, and isavuconazole were determined. The washout period of 6 days was sufficient for all azole antifungals since no measurable concentrations from the previous part of the study were found in the predose samples, except for fluconazole, where, in 9 of 12 participants, concentrations close to the LOQ were measured in the predose samples of the following study part. Most of the itraconazole concentrations were below the LLOQ, with no AUC calculation performed. No correlation was found between the exposure (AUClast) of each azole and the corresponding AUCR of each FXaI, which is demonstrated representatively for ketoconazole (electronic supplementary Fig. 1).
4 Discussion
With this single trial, a large amount of data could be collected effectively and with reduced safety risk. We have recently shown that the pharmacokinetics of microdosed FXaIs are comparable with the pharmacokinetics after a therapeutic dose [22]. Also in this trial, FXaI pharmacokinetics are in good agreement with earlier findings [22, 24], with only minor deviations. The slightly lower Ae values of edoxaban and rivaroxaban are likely due to the shorter urine sampling period (24 vs. 48 h). The 30% higher average CL/F of edoxaban compared with published values from 18 study participants [24] seems relevant, but the observed individual CL/F of 600–2200 mL/min in this study is in a very similar range to the previous study (490–1460 mL/min) [24]. All results are interpreted recognising that the azole antifungals exhibit competitive or mixed-type inhibition [33, 34], although some in vitro data point towards time-dependent (mechanism-based) inhibition for itraconazole and posaconazole [35]. A recently developed physiologically based pharmacokinetic (PBPK) model for posaconazole to simulate drug interactions, with posaconazole as the perpetrator, has used competitive CYP3A inhibition [36].
4.1 Apixaban
The effect of ketoconazole on apixaban pharmacokinetics was confirmed in this study, with a slightly smaller increase in AUC and Cmax (1.64-fold and 1.5-fold, respectively) compared with the published data with similar perpetrator doses but longer pretreatment (1.99 and 1.6-fold, respectively) [8]. This can be explained by the difference in the study design because the exposure with ketoconazole in this study was only a single oral dose of 400 mg administered 1 h before the FXaI cocktail, whereas in the earlier trial. ketoconazole was started 24 h before administration of the victim drug [8]. In line with these findings is also the observation that t½ in the earlier trial was 2.5 h longer, whereas it was unchanged in our trial. The inhibition of CYP3A4 and intestinal P-gp/BCRP are considered to be the major mechanisms for this interaction [37]. In contrast, fluconazole and voriconazole are regarded as moderate and strong inhibitors of CYP3A without effects on P-gp/BCRP. Because fluconazole had no effect, and voriconazole had little effect, on apixaban pharmacokinetics, the contribution of CYP3A to the interaction is probably small and a more important role is played by P-gp/BCRP as a target structure. Isavuconazole, as only a weak to moderate inhibitor of CYP3A but with weak inhibitory effect on P-gp (but not BCRP) [21], resulted in a 1.33-fold increase in AUC and 1.44-fold increase in Cmax. This is in the range of the reported effect of clarithromycin (strong CYP3A and P-gp inhibitor), naproxen (P-gp inhibitor, CYP3A and BCRP are unclear) and diltiazem (moderate CYP3A and P-gp inhibitor), with increases in AUC and Cmax in the range of 1.4- to 1.6-fold and 1.3- to 1.5 fold, respectively [8, 38, 39].
Besides CYP3A, apixaban is metabolised by multiple other CYP enzymes [2], but with only a small (6%) contribution to the overall elimination [7], explaining the absence of fluconazole effects (strong CYP2C19, moderate CYP2C9 inhibitor) in the present trial.
In summary, these findings suggest that a relevant interaction with apixaban can only be achieved if multiple elimination pathways are modified, and if one of them is intestinal P-gp/BCRP. At least for the most frequently used azole fluconazole, a combination with FXaIs does not appear unlikely. Overall, the clinical relevance of these changes is unclear because large epidemiological studies either found no altered safety profile of apixaban [40] or substantially increased the risks for major bleeding [41]. There is no epidemiologic evidence for any of the other azoles, but given the much larger change in apixaban pharmacokinetics, an effect seems more likely, at least if they are combined for longer periods.
4.2 Edoxaban
Evidence from other DDI studies suggests that changes in edoxaban pharmacokinetics in DDIs are primarily caused by altered P-gp activity in the intestinal wall [42], which is also likely to be responsible for its limited bioavailability of approximately 62% [43]. In contrast, CYP-mediated metabolism is only responsible for a small part of the elimination (< 4%). About 50% of a dose is excreted unchanged in the faeces and approximately 24% is eliminated unchanged in the urine [1].
As expected, no change in edoxaban pharmacokinetics by fluconazole was observed. Furthermore, voriconazole was expected to be without effect because there is no known transporter inhibition. However, our trial revealed a minor, albeit significant, increase in AUC, with a corresponding decrease in CL/F and increase in Ae. Concurrently, CLrenal was unchanged, suggesting that the alteration was likely caused by a change in bioavailability. This is in contrast to an exploratory study in six healthy volunteers, where voriconazole did not alter edoxaban pharmacokinetics despite 1 day of pretreatment with the perpetrator [22]. This might be due to the small number of participants. CYP3A-mediated metabolism of edoxaban is only responsible for a very small percentage of elimination and therefore cannot explain the observed effect size. However, since voriconazole concentrations in the gut are high, inhibition of the intestinal wall CYP3A may be contributing to the minor interaction with edoxaban.
The observed effects of isavuconazole, itraconazole and posaconazole are in line with the hypothesis of increased bioavailability by intestinal P-gp and BCRP inhibition. All three drugs increased edoxaban renal elimination (71%, 94%, and 97%, respectively) but did not increase CLrenal and half-life. Because edoxaban is also actively renally secreted via P-gp/BCRP [1], the unchanged CLrenal is surprising. Similar observations were made in a DDI study using a single dose of rifampicin and cobicistat [22]. Because intestinal perpetrator concentrations are much higher than the subsequent plasma concentrations, intestinal rather than renal P-gp inhibition of edoxaban transport may predominate.
Our results are in line with other interaction studies using cyclosporine, erythromycin, dronedarone and quinidine as P-gp and BCRP inhibitors with 1.7- to 1.9-fold increases in AUC [9, 44]. The label currently recommends a dose reduction from 60 to 30 mg when edoxaban is coadministered with ketoconazole, dronedarone, cyclosporine and erythromycin [13]. On the basis of the observed pharmacokinetic changes, a similar recommendation should also be considered for itraconazole and posaconazole.
The relative contribution of BCRP compared with P-gp has not yet been conclusively determined. In vitro findings suggest an approximately equal contribution of the transporters [45], but P-gp and BCRP generally show a large substrate overlap [46]. Isavuconazole as an inhibitor of P-gp, but likely not BCRP [21], led to an increase in AUC and Cmax by 1.5-fold and 1.6-fold, respectively. Despite the short pre-exposure, itraconazole, and especially posaconazole, led to a similar effect on the pharmacokinetics of edoxaban (doubling of AUC and Cmax) as ketoconazole underlined the high inhibitory potential of both compounds on the transporters.
4.3 Rivaroxaban
Rivaroxaban has a high bioavailability of 80–100%, which is indicative for low presystemic elimination and very limited involvement of intestinal P-gp/BCRP and CYP3A. Accordingly, DDIs are more likely to change the partial clearances of rivaroxaban, which are mediated by CYP3A (approximately 18% of a dose), CYP2J2 (approximately 14%), non-enzymatic hydrolysis (approximately 14%), and P-gp/BCRP-dependent active renal secretion (approximately 30% of a dose) [3, 10]. The effect of ketoconazole (AUC and Cmax increase of 2.3 and 1.7, respectively) in this study was very similar to published data, with AUC and Cmax increases of 2.6 and 1.7, respectively [10] or 2.3 and 1.5, respectively [24], despite the shorter pre-exposure. Moreover, ketoconazole was the only perpetrator able to reduce the CLrenal of rivaroxaban, which was also observed in all previous studies [10, 24]. The influence of ketoconazole on CYP2J2 seems to be likely due to the magnitude of the interaction and the observed strong in vitro inhibition [47]. Interestingly, the effect of posaconazole and itraconazole as CYP3A and P-gp/BCRP inhibitors was less pronounced, which, in the case of itraconazole, may be due to the fact that it appears to not inhibit CYP2J2 [48], and also because the inhibitors were not administered to steady-state. For posaconazole, this could have resulted from the methodological limitations of the study, which were also reflected in the lower CYP3A inhibition [32].
Based on other interaction studies, only simultaneous strong CYP3A and P-gp/BCRP inhibitors are expected to have clinically relevant interactions with rivaroxaban [4]. Isavuconazole did not alter any pharmacokinetic parameters of rivaroxaban, while the impact of voriconazole was minimal and is in line with recently published data [22]. Fluconazole reduced rivaroxaban CL/F by 20.3%, which is similar to the observed reduction for a therapeutic dose of rivaroxaban (29%) [10]. However, a longer pre-exposure period (4 days) was used in this study, which might also be the reason for the discrepancy in rivaroxaban CLrenal, which was reduced by 20% [10], whereas in our trial it was unchanged.
In clinical practice, an increased frequency in major bleeding during administration of dabigatran, rivaroxaban or apixaban with fluconazole, but not with itraconazole, posaconazole, voriconazole and ketoconazole, was observed based on a retrospective study of 91,330 Taiwanese patients [41]. This observation has already been criticised for methodological deficiencies [49,50,51,52], and the pharmacokinetic results of our trial do not support this observation and other large epidemiological trials. A recent systematic review and meta-analysis of studies that randomised patients with atrial fibrillation to FXaIs or warfarin found that the combination of rivaroxaban with more than one combined P-gp and CYP3A4 inhibitor was associated with a higher bleeding risk compared with warfarin (risk ratio (RR) 1.37, 95% CI 1.01–1.85; p = 0.04), whereas no interaction was found between apixaban and warfarin [40].
4.4 Cytochrome P450 Activity Assessment
Microdosed probe drugs for certain CYP enzymes such as midazolam and yohimbine (CYP3A and CYP2D6) can be used in DDI studies to monitor the effects of perpetrators on selective enzymes. The effects of the azoles on CYP3A activity are already known, but some of the observed differences may be due to the 1 h pre-exposure period of the perpetrators, resulting in a submaximal systemic inhibitor effect. To date, the effect of the different azole antifungals on CYP2D6 activity in vivo has not been studied, except for isavuconazole. A small increase (< 20%) in dextromethorphan exposure with multiple doses of isavuconazole was interpreted to most likely result from the inhibition of CYP3A4 [53]. From the data obtained, it can be concluded that no significant DDIs with any predominant CYP2D6 substrates will be caused by the azole antifungals.
4.5 Limitations
An important limitation of the trial is that the design was not intended to elicit a worst-case scenario or the maximum possible inhibition. Only a single dose of the azole antifungals as perpetrators was administered just 1 h before the microdosed FXaIs. The maximum inhibitory potential in the liver was therefore not achieved, whereas in the gut wall, local concentrations were certainly high enough to exert maximum inhibitor effects. Not all of the azole antifungals have a short tmax; maximum plasma concentrations of posaconazole tablets are reached after 4–5 h, and after 3 h for isavuconazole. Most itraconazole concentrations after the first 200 mg dose were below the LLOQ of the TDM assay, which can be explained by the substantially lower concentrations of itraconazole when administered in a fasting state [54] and the non-linear pharmacokinetics at steady-state usually achieved after 14 days [55]. To acknowledge this limitation, the inhibitory potential of the azoles for CYP3A was monitored by the surrogate of the midazolam CLmet. As anticipated, a smaller extent of CYP3A inhibition, compared with earlier trials, was observed. The extent of the inhibition of other CYPs, such as CYP2J2, and P-gp/BCRP remains unclassified. The consideration of the interaction mechanisms is probably incomplete; other, so far unknown, mechanisms or other transporters might possibly also play a role in these interactions. This study was conducted in healthy volunteers, and extrapolation to patients must consider additional influencing factors.
5 Conclusion
This study demonstrates the possibility to elucidate DDIs for three victim drugs with different clearance mechanisms administered as a microdosed cocktail (FXaIs). Using members of the azole fungistatic drug class as perpetrators, a more detailed insight into the underlying interaction mechanisms is possible. The study design was not optimised for maximum possible effects, rather a 24 h dosing of the different perpetrators was chosen, together with a 1 h pre-exposure period before administration of the victim drug. None of the interactions observed was greater than a 2.5-fold exposure increase. Therefore, only preliminary dose recommendations can be suggested for a clinical setting in patients, but most of these studied DDIs might be manageable by a dose reduction.
References
Bathala MS, Masumoto H, Oguma T, He L, Lowrie C, Mendell J. Pharmacokinetics, biotransformation, and mass balance of edoxaban, a selective, direct factor Xa inhibitor, in humans. Drug Metab Dispos. 2012;40(12):2250–5.
Wang L, Zhang D, Raghavan N, Yao M, Ma L, Frost CE, et al. In vitro assessment of metabolic drug-drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab Dispos. 2010;38(3):448–58.
Weinz C, Schwarz T, Kubitza D, Mueck W, Lang D. Metabolism and excretion of rivaroxaban, an oral, direct factor Xa inhibitor, in rats, dogs, and humans. Drug Metab Dispos. 2009;37(5):1056–64.
Mueck W, Schwers S, Stampfuss J. Rivaroxaban and other novel oral anticoagulants: pharmacokinetics in healthy subjects, specific patient populations and relevance of coagulation monitoring. Thromb J. 2013;11(1):10.
Reilly PA, Lehr T, Haertter S, Connolly SJ, Yusuf S, Eikelboom JW, et al. The effect of dabigatran plasma concentrations and patient characteristics on the frequency of ischemic stroke and major bleeding in atrial fibrillation patients: the RE-LY Trial (Randomized Evaluation of Long-Term Anticoagulation Therapy). J Am Coll Cardiol. 2014;63(4):321–8.
Ruff CT, Giugliano RP, Braunwald E, Morrow DA, Murphy SA, Kuder JF, et al. Association between edoxaban dose, concentration, anti-Factor Xa activity, and outcomes: an analysis of data from the randomised, double-blind ENGAGE AF-TIMI 48 trial. Lancet. 2015;385(9984):2288–95.
Foerster KI, Hermann S, Mikus G, Haefeli WE. Drug-drug interactions with direct oral anticoagulants. Clin Pharmacokinet. 2020;59(8):967–80.
Frost CE, Byon W, Song Y, Wang J, Schuster AE, Boyd RA, et al. Effect of ketoconazole and diltiazem on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. Br J Clin Pharmacol. 2015;79(5):838–46.
Parasrampuria DA, Mendell J, Shi M, Matsushima N, Zahir H, Truitt K. Edoxaban drug-drug interactions with ketoconazole, erythromycin, and cyclosporine. Br J Clin Pharmacol. 2016;82(6):1591–600.
Mueck W, Kubitza D, Becka M. Co-administration of rivaroxaban with drugs that share its elimination pathways: pharmacokinetic effects in healthy subjects. Br J Clin Pharmacol. 2013;76(3):455–66.
Bayer AG. Fachinformation Xarelto® 2.5 mg Filmtabletten. Leverkusen; 2019.
Bristol-Myers Squibb GmbH & Co. KGaA, Pfizer EEIG. Fachinformation Eliquis® 2.5 mg Filmtabletten. Dublin (Ireland), Munich, Berlin. 2019.
Daiichi Sankyo Europe GmbH. Fachinformation Lixiana® Filmtabletten. München; 2019.
US FDA. Drug Development and Drug Interactions. Table of Substrates, Inhibitors and Inducers. 2019. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers. Accessed 12 Mar 2019.
Vermeer LM, Isringhausen CD, Ogilvie BW, Buckley DB. Evaluation of ketoconazole and its alternative clinical CYP3A4/5 inhibitors as inhibitors of drug transporters: the in vitro effects of ketoconazole, ritonavir, clarithromycin, and itraconazole on 13 clinically-relevant drug transporters. Drug Metab Dispos. 2016;44(3):453–9.
Mueck W, Stampfuss J, Kubitza D, Becka M. Clinical pharmacokinetic and pharmacodynamic profile of rivaroxaban. Clin Pharmacokinet. 2014;53(1):1–16.
Lempers VJ, van den Heuvel JJ, Russel FG, Aarnoutse RE, Burger DM, Bruggemann RJ, et al. Inhibitory potential of antifungal drugs on ATP-binding cassette transporters P-glycoprotein, MRP1 to MRP5, BCRP, and BSEP. Antimicrob Agents Chemother. 2016;60(6):3372–9.
Gupta A, Unadkat JD, Mao Q. Interactions of azole antifungal agents with the human breast cancer resistance protein (BCRP). J Pharm Sci. 2007;96(12):3226–35.
Cooper KJ, Martin PD, Dane AL, Warwick MJ, Schneck DW, Cantarini MV. The effect of fluconazole on the pharmacokinetics of rosuvastatin. Eur J Clin Pharmacol. 2002;58(8):527–31.
Townsend R, Dietz A, Hale C, Akhtar S, Kowalski D, Lademacher C, et al. Pharmacokinetic evaluation of CYP3A4-mediated drug-drug interactions of isavuconazole with rifampin, ketoconazole, midazolam, and ethinyl estradiol/norethindrone in healthy adults. Clin Pharmacol Drug Dev. 2017;6(1):44–53.
Yamazaki T, Desai A, Goldwater R, Han D, Lasseter KC, Howieson C, et al. Pharmacokinetic interactions between isavuconazole and the drug transporter substrates atorvastatin, digoxin, metformin, and methotrexate in healthy subjects. Clin Pharmacol Drug Dev. 2017;6(1):66–75.
Mikus G, Foerster KI, Schaumaeker M, Lehmann ML, Burhenne J, Haefeli WE. Application of a microdosed cocktail of 3 oral factor Xa inhibitors to study drug-drug interactions with different perpetrator drugs. Br J Clin Pharmacol. 2020;86(8):1632–41.
Foerster KI, Huppertz A, Muller OJ, Rizos T, Tilemann L, Haefeli WE, et al. Simultaneous quantification of direct oral anticoagulants currently used in anticoagulation therapy. J Pharm Biomed Anal. 2018;148:238–44.
Mikus G, Foerster KI, Schaumaeker M, Lehmann M-L, Burhenne J, Haefeli WE. Microdosed cocktail of three oral factor Xa inhibitors to evaluate drug-drug interactions with potential perpetrator drugs. Clin Pharmacokinet. 2019;58(9):1155–63.
Burhenne J, Halama B, Maurer M, Riedel KD, Hohmann N, Mikus G, et al. Quantification of femtomolar concentrations of the CYP3A substrate midazolam and its main metabolite 1’-hydroxymidazolam in human plasma using ultra performance liquid chromatography coupled to tandem mass spectrometry. Anal Bioanal Chem. 2012;402(7):2439–50.
Vay M, Sauter M, Mikus G, Burhenne J. Quantification of microdosed oral yohimbine and its major metabolite in human plasma in the picogram range. Bioanalysis. 2019;11(16):1459–67.
US FDA. Guidance for industry: bioanalytical method validation. 2018. https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf. Accessed 19 Apr 2021.
European Medicines Agency. Bioanalytical Method Validation. 2011. https://www.emaeuropaeu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_enpdf. Accessed 19 Apr 2021.
Katzenmaier S, Markert C, Mikus G. Proposal of a new limited sampling strategy to predict CYP3A activity using a partial AUC of midazolam. Eur J Clin Pharmacol. 2010;66(11):1137–41.
Katzenmaier S, Markert C, Riedel KD, Burhenne J, Haefeli WE, Mikus G. Determining the time course of CYP3A inhibition by potent reversible and irreversible CYP3A inhibitors using A limited sampling strategy. Clin Pharmacol Ther. 2011;90(5):666–73.
Halama B, Hohmann N, Burhenne J, Weiss J, Mikus G, Haefeli WE. A nanogram dose of the CYP3A probe substrate midazolam to evaluate drug interactions. Clin Pharmacol Ther. 2013;93(6):564–71.
Rohr BS, Mikus G. Proposal of a safe and effective study design for CYP3A-mediated drug-drug interactions. J Clin Pharmacol. 2020;60(10):1294–303.
Yamaguchi Y, Akiyoshi T, Kawamura G, Imaoka A, Miyazaki M, Guengerich FP, et al. Comparison of the inhibitory effects of azole antifungals on cytochrome P450 3A4 genetic variants. Drug Metab Pharmacokinet. 2021;38:100384.
Isoherranen N, Kunze KL, Allen KE, Nelson WL, Thummel KE. Role of itraconazole metabolites in CYP3A4 inhibition. Drug Metab Dispos. 2004;32(10):1121–31.
Luo X, Li T, Yu Z, Xue X, Zhao H, Li N, et al. The impact of azole antifungal drugs on imatinib metabolism in human liver microsomes. Xenobiotica. 2019;49(7):753–61.
Bhatnagar S, Mukherjee D, Salem AH, Miles D, Menon RM, Gibbs JP. Dose adjustment of venetoclax when co-administered with posaconazole: clinical drug-drug interaction predictions using a PBPK approach. Cancer Chemother Pharmacol. 2021;87(4):465–74.
Byon W, Garonzik S, Boyd RA, Frost CE. Apixaban: a clinical pharmacokinetic and pharmacodynamic review. Clin Pharmacokinet. 2019;58(10):1265–79.
Garonzik S, Byon W, Myers E, Li X, Marchisin D, Murthy B. The effects of clarithromycin on the pharmacokinetics of apixaban in healthy volunteers: a single-sequence crossover study. Am J Cardiovasc Drugs. 2019;19(6):561–7.
Frost C, Shenker A, Gandhi MD, Pursley J, Barrett YC, Wang J, et al. Evaluation of the effect of naproxen on the pharmacokinetics and pharmacodynamics of apixaban. Br J Clin Pharmacol. 2014;78(4):877–85.
Harskamp RE, Teichert M, Lucassen WAM, van Weert H, Lopes RD. Impact of polypharmacy and P-glycoprotein- and CYP3A4-modulating drugs on safety and efficacy of oral anticoagulation therapy in patients with atrial fibrillation. Cardiovasc Drugs Ther. 2019;33(5):615–23.
Chang SH, Chou IJ, Yeh YH, Chiou MJ, Wen MS, Kuo CT, et al. Association between use of non-vitamin K oral anticoagulants with and without concurrent medications and risk of major bleeding in nonvalvular atrial fibrillation. JAMA. 2017;318(13):1250–9.
Parasrampuria DA, Truitt KE. Pharmacokinetics and pharmacodynamics of edoxaban, a non-vitamin K antagonist oral anticoagulant that inhibits clotting factor Xa. Clin Pharmacokinet. 2016;55(6):641–55.
Mikkaichi T, Yoshigae Y, Masumoto H, Imaoka T, Rozehnal V, Fischer T, et al. Edoxaban transport via P-glycoprotein is a key factor for the drug’s disposition. Drug Metab Dispos. 2014;42(4):520–8.
Mendell J, Zahir H, Matsushima N, Noveck R, Lee F, Chen S, et al. Drug-drug interaction studies of cardiovascular drugs involving P-glycoprotein, an efflux transporter, on the pharmacokinetics of edoxaban, an oral factor Xa inhibitor. Am J Cardiovasc Drugs. 2013;13(5):331–42.
Hodin S, Basset T, Jacqueroux E, Delezay O, Clotagatide A, Perek N, et al. In vitro comparison of the role of P-glycoprotein and breast cancer resistance protein on direct oral anticoagulants disposition. Eur J Drug Metab Pharmacokinet. 2018;43(2):183–91.
Mao Q, Unadkat JD. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport: an update. AAPS J. 2015;17(1):65–82.
Lee CA, Jones JP 3rd, Katayama J, Kaspera R, Jiang Y, Freiwald S, et al. Identifying a selective substrate and inhibitor pair for the evaluation of CYP2J2 activity. Drug Metab Dispos. 2012;40(5):943–51.
Shon JH, Yeo CW, Liu KH, Lee SS, Cha IJ, Shin JG. Itraconazole and rifampin alter significantly the disposition and antihistamine effect of ebastine and its metabolites in healthy participants. J Clin Pharmacol. 2010;50(2):195–204.
Linnebur SA, Hanlon JT. Drug interactions with non-vitamin K oral anticoagulants. JAMA. 2018;319(8):828–9.
Sennesael AL, Henrard S, Spinewine A. Drug interactions with non-vitamin K oral anticoagulants. JAMA. 2018;319(8):829.
Vazquez SR, Allen A. Drug interactions with non-vitamin K oral anticoagulants. JAMA. 2018;319(8):829–30.
Li Y, Dong S, Soria-Saucedo R. Drug interactions with non-vitamin K oral anticoagulants. JAMA. 2018;319(8):827–8.
Yamazaki T, Desai A, Goldwater R, Han D, Howieson C, Akhtar S, et al. Pharmacokinetic effects of isavuconazole coadministration with the cytochrome P450 enzyme substrates bupropion, repaglinide, caffeine, dextromethorphan, and methadone in healthy subjects. Clin Pharmacol Drug Dev. 2017;6(1):54–65.
Heykants J, Van Peer A, Lavrijsen K, Meuldermans W, Woestenborghs R, Cauwenbergh G. Pharmacokinetics of oral antifungals and their clinical implications. Br J Clin Pract Suppl. 1990;71:50–6.
Accord UK. SMPC Itraconazole 100mg Capsules, hard. 2009. https://www.medicines.org.uk/emc/product/5914/smpc#DOCREVISION. Accessed 16 May 2020.
Acknowledgements
The authors would like to thank Janina Bittmann for monitoring the study; Florian Michel and Marlies Stützle‐Schnetz for their excellent assistance during the study performance; and Magdalena Longo and Andrea Deschlmayr for their support in the analyses of the samples.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL. This trial was funded by the Department’s internal resources. No medical writing support was used.
Conflict of interest
Brit Silja Rohr, Kathrin Isabelle Foerster, Antje Blank, Jürgen Burhenne, Mazyar Mahmoudi, Walter Emil Haefeli and Gerd Mikus declare they have no potential conflicts of interest that might be relevant to the contents of this manuscript.
Ethical approval and informed consent
All procedures performed in studies involving human participants were in accordance with Good Clinical Practice (ICH-GCP) guidelines and the ethical standards of the current Declaration of Helsinki. All included volunteers gave their written informed consent prior to any study procedures. The trial protocol (EudraCT 2017-004453-16) was approved by the responsible Ethics Committee of the Medical Faculty of Heidelberg University and the competent authority (BfArM, Germany).
Consent for publication
Not applicable.
Trial registration
The trial was registered at the European Union Drug Regulating Authorities for Clinical Trials (EudraCT No. 2017-004453-16).
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author contributions
All authors participated in the drafting, critical revision, and approval of the final version of the manuscript. BSR, WEH and GM designed the study; BSR, AB, MM and GM conducted the study; KIF and JB determined all drug concentrations; BSR, KIF and GM performed the data analysis; and all authors performed the interpretation of the results.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Rohr, B.S., Foerster, K.I., Blank, A. et al. Perpetrator Characteristics of Azole Antifungal Drugs on Three Oral Factor Xa Inhibitors Administered as a Microdosed Cocktail. Clin Pharmacokinet 61, 97–109 (2022). https://doi.org/10.1007/s40262-021-01051-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-021-01051-9