
Abstract
Biosimilars have been available in the USA for over a decade, and in Europe for almost two decades. In that time, biosimilars have become established in the treatment landscape for a wide range of diseases, facilitating patient access and affordability of healthcare. However, patients can still struggle to access biological therapies in some markets. There is a need to streamline the process of developing biosimilars without compromising their quality, safety, or efficacy. This opinion piece considers the efficiencies that could be achieved within the biosimilar approval process. In clinical trials for biosimilars, clinical efficacy endpoints have been shown to be less sensitive measures of biosimilarity than biochemical, biophysical, and biological functional assays. Additional clinical efficacy studies comparing potential biosimilars and reference products do not add information that is useful for regulatory purposes. Large clinical studies of biosimilars with immunogenicity endpoints are of limited value, given the quality control processes in place for all biologics, including biosimilars. The expectation for multiple-switch studies for US interchangeability designation should be reconsidered immediately, and the category should be eliminated in the future. As biosimilars are typically approved globally based on a single set of clinical trials, and all subsequent manufacturing changes are already carefully monitored by regulatory authorities, comparative pharmacokinetic testing of EU and US reference products is unnecessary. Manufacturers and regulators could take greater advantage of existing real-world evidence. Streamlining biosimilar development would enable biosimilar development of more and a wider variety of biological drugs, accelerating biosimilar development without impacting patient safety or effectiveness.
Similar content being viewed by others

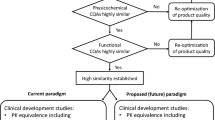

Based on current science and experience gained with biosimilars, it is appropriate and timely to streamline biosimilar development. Such streamlining will accelerate development of biosimilars and will enable biosimilar development of more and a wider variety of biological drugs. |
Comparative efficacy studies, along with clinical pharmacokinetic studies that compare US and EU reference products, do not provide meaningful information that is useful for regulatory decision-making and should not be required. |
Immunogenicity concerns were raised at the inception of the biosimilar industry and have since been shown to be without foundation. If any immunogenicity studies are conducted, they should apply a risk-based approach tailored to each molecule. |
The US designation of interchangeability for biosimilars is not needed, causes confusion, and should be modified immediately and eliminated in the future. |
Science-based regulatory consistency will ensure high global standards for all biosimilars. |
1 Introduction to Biosimilar Development and Use
Biosimilars are subsequent versions of originator biological drugs (“biologics”) that are highly similar to their reference product, and provide patients with products of the same quality, safety, and efficacy. The use of biosimilars in Europe began in 2006 [1], and the USA followed in 2015 [2]. Biosimilars are now improving patient access to life-changing and life-saving biologics for a wide range of disease areas around the world [3,4,5].
When the regulatory pathway for biosimilars was created in the mid-2000s, the analytical requirements reflected the science in routine use at that time. This included methods applied to originator biologics both at their initial approval and to support manufacturing process changes to those products after approval [6]. As implemented in practice, the regulatory approach to biosimilars also reflected the regulatory caution attributable to the uncertainty of implementing a new concept for the development and review of biological drugs. The biosimilar development paradigm proposed was for a stepwise approach, often visualized as a pyramid, based on analytical characterization, and proceeding upwards to animal testing, clinical pharmacology [pharmacokinetics (PK) and/or pharmacodynamic (PD) studies] with clinical efficacy trials as the pinnacle. In the two decades since the pathway was initially implemented, substantial experience has been accrued by biosimilar sponsors and regulators as to how biosimilars are developed in practice [7, 8].
When compared with what is known about the reference biologics, no unusual or unexpected adverse events (including both immunogenicity and loss of effectiveness) have been seen with biosimilars in the highly regulated markets where pharmacovigilance systems are robust and such events would be captured [9,10,11].
The potential for increased or different immunogenicity was one of the foremost concerns when the biosimilar development pathway was first developed. This in turn led to the US Food and Drug Administration (FDA) to recommend performing a multiple-switch study to obtain the additional, optional, US legal designation of “interchangeable biologic” for a given biosimilar [12, 13]. While immunogenicity data should always be collected for both biosimilars and novel biologics, to date, the immunogenicity levels elicited by biosimilars approved by the FDA and European Medicines Agency (EMA) have reliably matched the immunogenicity of their reference products [9,10,11, 14,15,16]. This evidence allows the conclusion that potential immunogenicity issues are less of a concern than initially conjectured.
There are opportunities to revise the current biosimilar development paradigm to make future biosimilar development more efficient by applying current science as well as experience already obtained with the development and marketing of biosimilars to date.
2 Biosimilars Facilitate Patient Access and Affordability, but They Must Be Commercially Viable
The availability of biosimilars has led to increased patient access and a lowering of healthcare costs. Even so, many patients who could benefit from biologics struggle to get timely and affordable access. This applies within the developed markets of Europe and the US, and in many countries elsewhere access is even more limited [5, 17].
While expected to be less expensive and quicker to develop than a wholly new originator medicine, whether a chemical or biological drug, the development of biosimilars is still expensive and can cost as much as US$300 million and take up to 9 years to develop per biosimilar [18]. The most significant development costs are comparative clinical trials [18, 19], which require purchase of the reference biologic and enrollment of sufficient numbers of patients to meet designated endpoints. The cost and time required are increasingly constraining the ability of sponsors to develop and launch biosimilars to many originator biologics. Efficiencies in the process are sought that reflect the experience gained by all stakeholders in the past two decades of biosimilar development. The shared goals in healthcare remain the same, namely greater access and affordability worldwide through competition with no compromise in quality, safety, or efficacy.
It has been predicted that approximately half of biological drugs coming off patent in the next decade may have no biosimilar competitors [20, 21]. This may be due, in part, because their market size is too small to warrant the cost of biosimilar development under the current development paradigm.
The opportunity to streamline biosimilar development has been recognized by multiple stakeholders [14,15,16, 22, 23]. Concurrently, demands for access to biologics at more affordable prices are likely to increase in all regions, both developed and emerging [5, 17, 18]. Viable competition in all regions can only occur and be sustainable if biosimilars can be developed and manufactured more efficiently.
3 Activities Are Already Underway in the US to Enhance the Efficiency of Biosimilar Development
Recognizing the need for more efficient biosimilar development, the reauthorization of the US Biosimilars User Fee Act in 2022 included a Regulatory Science Pilot Program to be paid for by biosimilar developers and administered by the FDA [24]. This acknowledges the value of using regulatory science to progress and support biosimilar development.
The US FDA has released a Research Roadmap of projects that the FDA believes may be helpful to advance biosimilar development [24]. While advancement of the science supporting biosimilars will continue to be a laudable goal, we can already take risk-based steps to streamline biosimilar development without waiting multiple years for such studies to be proposed, initiated, and completed.
4 The Single Biggest and Most Immediate Opportunity for Greater Efficiency is to Reassess What is Learned from the Comparative Efficacy Trial
The initial regulatory guidances for the development of biosimilars in the USA and the EU used the stepwise approach with the final confirmatory stage of the biosimilarity exercise being a head-to-head clinical efficacy comparison of the proposed biosimilar and its reference product in a single sensitive patient population [7, 8]. Such comparative efficacy studies are commonly modeled on the historic phase 3 trial designs deployed for approval of new drug entities. For several biosimilars, head-to-head efficacy comparisons were larger than the originator phase 3 efficacy studies, especially for oncology drugs [19, 25]. There was a presumption at that time that such studies would be informative, rather than predictable and solely confirmatory, based upon already completed prior sound analytics and the much more sensitive comparative PK study that is also required.
In the USA, the law allows the FDA to waive any studies the Agency believes to be unnecessary, and the FDA’s guidance expresses flexibility too. Indeed, the FDA has often stated that biosimilar clinical studies are not intended to re-establish the safety, purity, and potency of the biosimilar but are instead designed to confirm comparable clinical outcomes to the reference biologic in situations where there is residual uncertainty [8]. However, in the absence of regulatory certainty that such studies are not needed, many sponsors feel that it is pragmatically necessary to propose such studies when they consider the development of any given biosimilar. Waivers for clinical efficacy studies in patients have been granted to some of the less complex biosimilars (e.g., filgrastim, pegfilgrastim, insulin) [26,27,28,29,30] but not to the more complex biosimilars, including monoclonal antibodies, even where analytical comparability and PK have been demonstrated.
It is known that clinical efficacy endpoints are less sensitive measures of biosimilarity than biophysical, biochemical, and functional bioassays [14, 22, 23]. Clinical efficacy studies need to be of sufficient size and duration to detect meaningful differences if such differences exist [7, 8]. There is now extensive published data from European Assessment Reports for EU-approved biosimilars, as well as FDA review summaries of US-licensed biosimilars, confirming that comparative efficacy studies do not provide decisive pivotal biosimilarity data. A review from 2019 revealed that no biosimilar found to be highly similar to their reference product in analytical and PK studies subsequently failed to obtain approval due to failed equivalence observed in a clinical efficacy study [16]. Reviews of approved biosimilars in the EU and USA revealed the comparative efficacy studies always confirmed the efficacy of the biosimilar candidate, such that the comparative efficacy studies provided no new information of actionable value in the approval process [14, 31]. Consequently, such studies lack validity because no human clinical studies should be undertaken unless meaningful new information can be obtained. This experience can be translated into revised regulatory practice.
Recently, a group of regulatory assessors from EU health authorities evaluated the similarity data of seven adalimumab and five bevacizumab biosimilars [23]. They found that although minor differences were often observed in quality attributes, all differences found in quality attributes were justified by quality data alone or by a combination of quality and clinical PK data, the latter including a comparison of safety and immunogenicity. In no instances were data from the comparative clinical study needed to justify differences observed in quality attributes [23].
Overall, experience to date suggests that a more targeted approach to the inclusion of clinical efficacy studies in biosimilar approval applications is needed. Indeed, regulators with extensive biosimilar experience have recommended reducing some routine requirements for comparative clinical efficacy studies [22, 23].
It is possible that, in the future, biosimilars may be developed to reference products that are challenging to characterize analytically or for which PK studies may not yield useful information. Under such circumstances, there might be a degree of residual uncertainty that is best addressed through a comparative efficacy study. But such scenarios are at present purely hypothetical and have not yet been encountered. Indeed, as analytical science advances, the possibility of encountering such biological molecules becomes increasingly remote.
In their revised guidance on the licensing of biosimilar products, implemented in 2021, the UK health authority (Medicines and Healthcare products Regulatory Agency) acknowledges that “each biosimilar development needs to be evaluated on a case-by-case basis,” but explicitly proposes that, “in most cases, a comparative efficacy trial may not be necessary if sound scientific rationale supports this approach” [32]. Similarly, the 2022 World Health Organization (WHO) Guidelines on evaluation of biosimilars also support flexibility, stating that while “clinical data are generally required for any biosimilar,” the type and amount of clinical data required will vary based on the availability of other relevant data [33]. The WHO also notes that “a case-by-case approach will be needed for each class of products,” but also states that “an adequately powered comparative efficacy and safety trial will not be necessary if sufficient evidence of biosimilarity can be drawn from other parts of the comparability exercise… The current data suggest that more-complex products such as mAbs [monoclonal antibodies] can be sufficiently characterized by available suitable analytical methods, plus the structure–function relationships are well known and can be studied by sensitive orthogonal functional assays.”
As of June 2023, the FDA and the EMA have approved multiple biosimilars without a clinical efficacy study when an established PD biomarker was available. This included several biosimilars for filgrastim, pegfilgrastim, and insulin [26,27,28,29,30]. Use of PD biomarkers has been proposed instead of comparative efficacy studies with conventional efficacy endpoints [34, 35]. While undoubtedly informative, this is only feasible for biosimilars if appropriate PD biomarkers are already available for their originator reference products. Such PD markers are rare and only exist for a limited number of biological products. The effort to find and validate a PD biomarker is an exploratory and lengthy endeavor with a high likelihood of failure. Even if successful, the PD biomarker may provide a high variability in responses that would necessitate a large patient sample size. As a result, development of a new PD biomarker will not comport with the timelines necessary for biosimilar development. As such, even when a PD biomarker already has been established with the reference biologic, PD biomarker studies are not expected to contribute to greater efficiencies in biosimilar development [34,35,36].
Given the collective experience of all stakeholders, it is time to rethink the routine expectation for a comparative efficacy study when developing a biosimilar. While conducting comparative efficacy studies may provide reassurance to healthcare professionals (HCPs) and others unfamiliar with the concepts of biosimilarity, conducting additional and unnecessary clinical studies delays biosimilar development and increases development costs, both of which will inevitably have a negative impact on patient access. Separating studies for regulatory decision-making purposes from those for stakeholder education is essential.
5 Immunogenicity Assessments Should follow a Risk-Based Approach because Immunogenicity of Biologics, including Biosimilars, is Well Evaluated and Controlled
The primary sources of potential immunogenicity are already well controlled at the quality level for biosimilars developed to the standards of stringent health authorities and switching studies have provided no new direct immunogenicity information [9,10,11, 22, 23].
The amino acid sequence of a protein is the main determinant for immunogenicity of a therapeutic protein. A biosimilar must have the identical amino acid sequence as its reference product [7, 8], and so the linear peptide epitopes presented to T cells of the immune system will be the same, inevitably leading to identical T-cell-mediated immunogenicity of biosimilars and their respective reference products. The primary sources of potential non-T-cell immunogenicity differences are posttranslational modifications and process impurities [37]. Experimental studies have confirmed these expectations [38, 39].
Process impurities can include aggregates, degradants, misfolded proteins, host-cell proteins, and nonhuman glycans. These need to be kept at sufficiently low levels, typically not higher than in the reference product, to further ensure comparable immunogenicity. An orthogonal battery of analytical tests is employed to monitor levels of process impurities in biosimilars [40, 41]. It is permissible to have lower impurity levels in biosimilars compared with their reference products when these lower levels are not clinically relevant [42, 43].
This pivotal quality data package is complemented by a comparative clinical PK study (or if a biomarker is available, a comparative PD study may be provided), typically in healthy volunteers, where in addition to an evaluation of PK per se, a decrease in circulating levels of a biosimilar is also viewed as a sensitive surrogate to detect antidrug antibody formation. These data provide additional evidence for comparable immunogenicity. Direct comparisons of the immunogenicity of the biosimilar and reference product are also conducted within the PK study, although the number of subjects in the study is commonly too low to detect rare immunogenic events [7, 8].
Considering the totality of evidence supporting biosimilars, European regulators, in their personal capacity, concluded that “because of the high similarity, there is no reason to believe that the body’s immune system would react differently to the biosimilar compared with the original biological upon a switch. This view is supported by the current experience with biosimilars on the market and by literature data.” [9, 44]
Historically, many antidrug–antibody assays were unreliable and underestimated the immunogenicity of therapeutic proteins, whereas current state-of-the-art assays have revealed that there is almost always an immune reaction/recognition after administration of a new therapeutic protein, which may or may not be clinically relevant. As a result, it is not unusual for biosimilar developers to detect higher levels of immunogenicity with both the reference biologic and biosimilar than have been reported previously with the reference biologic. However, the difference in absolute levels of immunogenicity across studies is not of concern because it does not distinguish between the biosimilar and its reference product but instead applies equally to both. US product labeling always has a disclaimer that antidrug antibody levels should not be compared across studies.
Historical data that have been obtained over the past two decades with biosimilars shows that there are no unusual or unexpected immunogenicity concerns in practice. Nearly 5 billion patient-days of experience with biosimilars have been accrued to date across the EU and USA [45, 46], regions with strong pharmacovigilance systems that include EudraVigilance in the EU and the US Sentinel system [47, 48]. In the real-world setting encountered post-approval, multiple switches between reference product and biosimilars have already occurred, especially in countries or healthcare networks where patients are switched based on which product has won the most recent tender [7, 10, 11, 49]. Evidence supporting the safety and effectiveness of biosimilar-to-biosimilar switching has also begun to accrue [50,51,52,53]. No differences in efficacy or safety have been detected in any biosimilars marketed in these regions compared with the reference product. Based on these observations, the EMA and Heads of Medicines Agencies (Europe) issued joint statement in 2022 supporting the safety, efficacy, and immunogenicity of both single and multiple switches among reference products and biosimilars [54], and in 2023, the FDA provided similar support in a presentation provided to HCPs [55].
The few observations of greater immunogenicity with marketed follow-on biologics were observed with products that were approved in the late 2000s in developing markets and that were not developed to the biosimilarity standards of the EMA, FDA, WHO, and other advanced health authorities [17, 56]. Such products, whether developed in the past or in the present for markets with less stringent health authorities, should not be considered to be biosimilars, even though they may have been referred to by some using that term [57,58,59,60]. There are various other terms used in the literature to more accurately describe these products, including “non-comparable biotherapeutics,” “intended copies,” “non-innovator biologics,” and “non-regulated copies.” This supports the contention that biosimilars developed to the current high scientific standards are no more immunogenic than their reference products, and there is no evidence that switching multiple times changes the clinical response or has an impact on safety.
There may be exceptional circumstances in which additional clinical immunogenicity data on a product-by-product basis are useful for regulatory decision-making and where uncertainties cannot be resolved before approval without patient exposure. In those circumstances, particular consideration of immunogenicity may be meaningful when the reference product is known to be highly immunogenic and there is an incomplete understanding of the contributing factors [7, 8]. But the specific rationale needs to be identified for such studies before they are conducted.
Both the EMA and FDA require an integrated summary of immunogenicity as part of a marketing authorization application. This summary provides details about what is known about the quality attributes of the molecule that might contribute to immunogenicity, as well as historical immunogenicity data obtained over time with the reference product, and immunogenicity data obtained with both the reference product and proposed biosimilar during biosimilar development. This integrated approach may improve the risk analysis of immunogenicity and helps diminish unnecessary and unwarranted concerns about immunogenicity.
More is known about the immunogenicity of biosimilars at the time of regulatory submission than was known about originators at the time of their approval [61]. We have seen an evolution of science and knowledge about immunogenicity, especially as we have moved from a hypothetical concern to a situation where large amounts of immunogenicity data are available. We suggest that immunogenicity analysis for biosimilars be moved to a risk-based consideration as opposed to a global and routine requirement.
6 The Unique US Designation of “Interchangeable Biologic” Should be Reconsidered
When the biosimilar pathway was first developed in the USA, hypothetical concerns were raised by some about a potential increase in immunogenicity if the reference product and biosimilar were switched back and forth multiple times. As a result of this purely hypothetical concern, the Biologics Price Competition and Innovation Act of 2009 that established the US biosimilar pathway when it was signed into law in 2010 incorporated a unique designation of interchangeability that is distinct from biosimilarity [62]. The FDA subsequently developed a guidance recommending a multiple switch study to obtain the US designation of interchangeability [12]. Such a designation allows product substitution at a retail or specialty pharmacy by the pharmacist without first obtaining permission from the original prescriber (subject to state law) [12]. Nonetheless, a physician can preclude substitution on any prescription by prescribing by name and explicitly requesting that no substitution be made.
However, as discussed above, there has been no evidence from randomized clinical studies, observational studies, or pharmacovigilance for any biosimilar developed to the standards of the EMA or FDA that biosimilars elicit different immunological responses, either in quality or of a higher magnitude, compared with their reference products.
Given the tight controls already in place on immunogenicity and experience accrued to date, we believe that a multiple-switch study to obtain the unique legal designation of US interchangeability is not warranted [13], and indeed has already been waived by FDA when justified (e.g., insulin glargine and ranibizumab biosimilar products) [63, 64]. Indeed, based on the knowledge and experience already obtained, it is now possible to eliminate multiple-switch PK studies currently required for the separate US category of interchangeability without impacting patient safety or efficacy. As a scientific matter, we support immediate reconsideration of the requirements for US interchangeability and future elimination of this category. All biosimilars are, by definition, of the same quality as interchangeable biologics, and in practical terms, all biosimilars are interchangeable for the purposes of physician prescribing practices. Nonetheless, confusion continues, and misinformation remains a barrier to acceptance and utilization of all biosimilars, whether or not they have a US interchangeability designation [65].
7 There is Minimal Value in Comparative Pharmacokinetic Testing Between US and EU Reference Biologics
Most countries’ biosimilar development regulations specify that the reference product be the locally licensed product. On the face of it, this appears reasonable given that this is the biological product with which the local regulators have experience. However, the originator reference product is often approved globally based on a single development program with a single set of clinical studies [66]. All subsequent manufacturing process changes are always bridged by the sponsor of the originator reference product to this initial dataset [6, 43]. Consequently, by definition, the foreign-sourced reference product is already known to be the same as the locally sourced reference such that no additional studies are necessary. This is readily evident for EU- and US-sourced products, where the EMA and FDA websites carry information on the pivotal clinical trials submitted to each jurisdiction and upon which both the originator products and subsequent biosimilars depend [67, 68]. Furthermore, the EMA website contains an assessment of manufacturing process changes introduced for the reference products approved via the centralized procedure, which includes all biological drugs [67]. To repeat bridging studies between foreign and locally sourced reference products for each subsequent jurisdiction offers no new scientific or clinical information. Such studies significantly inflate the cost and delay biosimilar development and make some markets unviable, limiting patient access in those regions, and so should be eliminated.
The FDA is willing to accept the use of non-US licensed comparator products, but stated in their latest Q&A guidance that, “as a scientific matter, the type of bridging data needed will always include data from analytical studies (e.g., structural and functional data) that directly compare all three products (i.e., the proposed biosimilar product, the US-licensed reference product, and the non-U.S.-licensed comparator product), and is likely to also include bridging clinical PK data or, when appropriate, PD data, for all three products” [69]. As explained above, comparative PK or PD studies of a reference product purchased in different jurisdictions do not provide new scientific information and the value of comparative analytical studies is also questionable. As a result, we strongly suggest that these bridging studies should not be routinely required, and any exceptions should have a specific identified rationale.
8 Good Manufacturing Control is Essential for all Biologics and Prevents “Drift”
The specter of gradual but ultimately significant and clinically meaningful differences in critical quality attributes, commonly referred to as “drift,” has been raised by some as rationale for requiring especially stringent pharmacovigilance of biosimilars or even for periodic reassessment of biosimilarity [70, 71]. As acknowledged in FDA guidance, after approval, both originator biologics and biosimilars have separate life cycles [8], with manufacturing process changes for both subject to International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) Q5E, Comparability of Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process guidance [6]. This consistency in the application of regulatory science to all biologics independent of business models is important and entirely appropriate [15].
In-process controls are a critical part of current Good Manufacturing Practice (cGMP) to help ensure that a consistent drug product is provided. These consist of statistical control limits, specification limits, and safety/efficacy limits (Fig. 1). If, upon routine manufacture, a critical quality attribute of a given batch material approaches or exceeds the statistical process control limits, cGMP dictates that the manufacturer undertakes an investigation into the cause of the excursion, and that corrective action be undertaken to bring the material back to the process mean. As a result, drift over time is exceptionally rare for either a reference product or a biosimilar that is manufactured in adherence to cGMP [43, 72].

Overview of continued process verification. OOE out of expectation, OOS out of specification
Issues of potential drift would most likely apply to originator biologics as they have been available the longest. While there are instances of manufacturing process changes that had an unanticipated clinical impact or of inadequate manufacturing control, they are very rare [73, 74]. That revised pharmacovigilance has not been applied for originator biological drugs in the past half century supports not requiring any additional pharmacovigilance that is specific for biosimilars. Good appropriate pharmacovigilance should be applied to both, along with all other medicines. Science-based regulatory consistency, often termed reliance, is a priority across jurisdictions [75].
9 Use of Collective Experience to Expeditiously Advance Regulatory Decision-Making Will Benefit all Stakeholders
The healthcare community has increasingly become more comfortable with the concepts of biosimilarity and with the use of biosimilars. However, as biosimilars become available for new drug categories, new patient populations and new HCPs are being exposed to them for the first time. As a result, education efforts by regulators, sponsors, and others will continue to be important for the foreseeable future [76,77,78].
When considering the collective experience with biosimilars gained by use of real-world data and real-world evidence, the quality of the data is paramount. Important considerations are the manner in which the data are collected, the reliability of the data including both clinical data collection and the identity of the product used, as well as the nature of the data collected [79, 80]. Several quality pharmacovigilance databases already exist and can be used to provide postapproval confirmation of the safety and effectiveness of biosimilars in a real-world setting [80,81,82,83,84]. These data can be used to provide reassurance to patients and HCPs that biosimilars perform in the same manner as their reference products. For optimal pharmacovigilance, it is important to identify not just the brand name or the nonproprietary name, but the batch number as well [85]. Electronic patient records of hospitals and pharmacies may provide improved real-world data for efficacy and safety in the future. However, the linking of databases is associated with challenges such as confidentiality concerns and technical issues. Increasing the quality of pharmacovigilance is important for all drugs and in all regions to facilitate safety assessment after introduction into the marketplace.
It is not likely that real-world evidence will replace detailed analytical comparisons or PK clinical studies when developing a preapproval data package to support licensure of a biosimilar. However, as the use of real-world evidence increases in regulatory decision-making actions for originator biologic approvals, its value for biosimilars may be important to consider as well.
10 Conclusions
Having a fair and consistent approach to science-based regulatory decision-making demands that the same principles be applied to all products independently of the sponsor’s business model. Science is global and regulations for originator products are increasingly harmonized globally, and this offers further opportunities to extend the experience with biosimilars in Europe and the USA to global development and availability of biosimilars.
As a scientific matter, several options are available to streamline biosimilar development immediately without compromising patient safety or product quality (Table 1). These include limiting clinical studies to comparative PK, using a risk-based approach when evaluating immunogenicity, elimination of bridging PK studies through acceptance of global comparator reference product, and immediate reconsideration of the requirement for multiple-switch studies to obtain US interchangeability designation and future elimination of that category. Evolution of biosimilar guidelines to reflect more streamlined biosimilar development will create greater regulatory certainty, and also help ensure a sustainable future market for biosimilars, which in turn will help free monies in healthcare budgets for newer medications.
Access to the current biological drugs, including biosimilars is not optimal in the EU, USA, and other advanced markets, and very limited in low- and middle-income countries. The development of new biological therapies to serious diseases is more intensive than ever. Unfortunately, it is likely that these products will be very costly, perhaps more than the current products. It is clear that the healthcare systems will not be able provide these therapies to all patients in a timely manner, even in the wealthiest countries.
Recent estimates project that no biosimilars are being developed to more than half of the current biological drugs because, given the current extensive data requirements, the market sizes of those biologics are too low to justify the investment for developing biosimilars to these biological drugs [20, 21]. Streamlining biosimilar development is very important as this will make it feasible for more biosimilars to be developed to more reference biologics, all to the benefits of patients and healthcare systems.
By reducing the need for redundant clinical data with less sensitive endpoints and increasing the use of publicly available information, the feasibility of developing biosimilars will be enhanced by lowering development costs, which will lead to increased competition as more biosimilars are brought to the market. This can increase patient access and allow earlier treatment. The proposed steps will expand worldwide access to these increasingly essential medicines in a manner that does not compromise the quality, safety, and efficacy of biologics for any patients, whatever jurisdiction in which they happen to reside.
References
European Medicines Agency. Omnitrope. 2023. https://www.ema.europa.eu/en/medicines/human/EPAR/omnitrope. Accessed 19 July 2023.
US Food & Drug Administration. ZarxioTM highlights of prescribing information. 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125553s001lbl.pdf. Accessed 19 July 2023.
IQVIA. The global use of medicines 2023. 2023. https://www.iqvia.com/insights/the-iqvia-institute/reports/the-global-use-of-medicines-2023. Accessed 19 July 2023.
Association for Accessible Medicines. 2021 U.S. generic and biosimilar medicines savings report. 2021. https://accessiblemeds.org/resources/reports/2021-savings-report. Accessed 19 July 2023.
Kvien TK, Patel K, Strand V. The cost savings of biosimilars can help increase patient access and lift the financial burden of health care systems. Semin Arthritis Rheum. 2022;52:151939. https://doi.org/10.1016/j.semarthrit.2021.11.009.
US Food & Drug Administration. Guidance for industry. Q5E comparability of biotechnological/biological products subject to changes in their manufacturing process. 2005. https://www.fda.gov/media/71489/download. Accessed 19 July 2023.
European Medicines Agency. Biosimilars in the EU: Information guide for healthcare professionals. 2019. https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf. Accessed 19 July 2023.
US Food & Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry. 2015. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/scientific-considerations-demonstrating-biosimilarity-reference-product. Accessed 19 July 2023.
Kurki P, Barry S, Bourges I, Tsantili P, Wolff-Holz E. Safety, immunogenicity and interchangeability of biosimilar monoclonal antibodies and fusion proteins: a regulatory perspective. Drugs. 2021;81:1881–96. https://doi.org/10.1007/s40265-021-01601-2.
Cohen HP, Blauvelt A, Rifkin RM, Danese S, Gokhale SB, Woollett G. Switching reference medicines to biosimilars: a systematic literature review of clinical outcomes. Drugs. 2018;78:463–78. https://doi.org/10.1007/s40265-018-0881-y.
Barbier L, Ebbers HC, Declerck P, Simoens S, Vulto AG, Huys I. The efficacy, safety, and immunogenicity of switching between reference biopharmaceuticals and biosimilars: a systematic review. Clin Pharmacol Ther. 2020;108:734–55. https://doi.org/10.1002/cpt.1836.
US Food & Drug Administration. Guidance for industry. Considerations in demonstrating interchangeability with a reference product. 2019. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-demonstrating-interchangeability-reference-product-guidance-industry. Accessed 19 July 2023.
Park JP, Jung B, Park HK, Shin D, Jung JA, Ghil J, et al. Interchangeability for biologics is a legal distinction in the USA, not a clinical one. BioDrugs. 2022;36:431–6. https://doi.org/10.1007/s40259-022-00538-6.
Schiestl M, Ranganna G, Watson K, Jung B, Roth K, Capsius B, et al. The path towards a tailored clinical biosimilar development. BioDrugs. 2020;34:297–306. https://doi.org/10.1007/s40259-020-00422-1.
Webster CJ, George KL, Woollett GR. Comparability of biologics: global principles, evidentiary consistency and unrealized reliance. BioDrugs. 2021;35:379–87. https://doi.org/10.1007/s40259-021-00488-5.
Webster CJ, Wong AC, Woollett GR. An efficient development paradigm for biosimilars. BioDrugs. 2019;33:603–11. https://doi.org/10.1007/s40259-019-00371-4.
Klein K, Gencoglu M, Heisterberg J, Acha V, Stolk P. The global landscape of manufacturers of follow-on biologics: an overview of five major biosimilar markets and 15 countries. BioDrugs. 2023;37:235–45. https://doi.org/10.1007/s40259-022-00568-0.
McKinsey & Company. Three imperatives for R&D in biosimilars. 2022. https://www.mckinsey.com/industries/life-sciences/our-insights/three-imperatives-for-r-and-d-in-biosimilars. Accessed 19 July 2023.
Moore TJ, Mouslim MC, Blunt JL, Alexander GC, Shermock KM. Assessment of availability, clinical testing, and US food and drug administration review of biosimilar biologic products. JAMA Intern Med. 2021;181:52–60. https://doi.org/10.1001/jamainternmed.2020.3997.
European Commission. Biosimilar medicines—multistakeholder event. 2022. https://health.ec.europa.eu/events/biosimilar-medicines-multistakeholder-event-2022-12-13_en. Accessed 19 July 2023.
IQVIA. Biosimilars in the United States 2023–2027. 2023. https://www.iqvia.com/insights/the-iqvia-institute/reports/biosimilars-in-the-united-states-2023-2027. Accessed 19 July 2023.
Bielsky M, Cook A, Wallington A, Exley A, Kauser S, Hay JL, et al. Streamlined approval of biosimilars: moving on from the confirmatory efficacy trial. Drug Discov Today. 2020;25:1910–8. https://doi.org/10.1016/j.drudis.2020.09.006.
Guillen E, Ekman N, Barry S, Weise M, Wolff-Holz E. A data driven approach to support tailored clinical programs for biosimilar monoclonal antibodies. Clin Pharmacol Ther. 2023;113:108–23. https://doi.org/10.1002/cpt.2785.
US Food & Drug Administration. BsUFA III regulatory research pilot program: research roadmap. 2023. https://www.fda.gov/media/164751/download. Accessed 19 July 2023.
Bloomfield D, D’Andrea E, Nagar S, Kesselheim A. Characteristics of clinical trials evaluating biosimilars in the treatment of cancer: a systematic review and meta-analysis. JAMA Oncol. 2022;8:537–45. https://doi.org/10.1001/jamaoncol.2021.7230.
Jung EH, Sarpatwari A, Kesselheim AS, Sinha MS. FDA and EMA biosimilar approvals. J Gen Intern Med. 2020;35:1908–10. https://doi.org/10.1007/s11606-019-05408-6.
US Food & Drug Administration. Biosimilar multi-disciplinary evaluation and review (BMER) BLA 761111 (Nyvepria). 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761111Orig1s000MultidisciplineR.pdf. Accessed 19 July 2023.
US Food & Drug Administration. Biosimilar multi-disciplinary evaluation and review (BMER) BLA 761173 (Stimufend). 2021. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2022/761173Orig1s000MultidisciplineR.pdf. Accessed 19 July 2023.
US Food & Drug Administration. Biosimilar multi-disciplinary evaluation and review (BMER) BLA 761084 (Fylnetra). 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2022/761084Orig1s000MultidisciplineR.pdf. Accessed 19 July 2023.
US Food & Drug Administration. Biosimilar clinical review BLA 761082 (Releuko). 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2022/761082Orig1s000MedR.pdf. Accessed 19 July 2023.
Wolff-Holz E, Tiitso K, Vleminckx C, Weise M. Evolution of the EU biosimilar framework: past and future. BioDrugs. 2019;33:621–34. https://doi.org/10.1007/s40259-019-00377-y.
Medicines & Healthcare products Regulatory Agency. Guidance on the licensing of biosimilar products. 2022. https://www.gov.uk/government/publications/guidance-on-the-licensing-of-biosimilar-products/guidance-on-the-licensing-of-biosimilar-products. Accessed 19 July 2023.
World Health Organization. Guidelines on evaluation of biosimilars. Replacement of annex 2 of WHO technical report series, no. 977. 2022. https://cdn.who.int/media/docs/default-source/biologicals/bs-documents-(ecbs)/annex-3---who-guidelines-on-evaluation-of-biosimilars_22-apr-2022.pdf?sfvrsn=e127cbf4_1&download=true. Accessed 19 July 2023.
Duke-Margolis Center for Health Policy. Public workshop. Pharmacodynamic biomarkers for biosimilar development and approval. 2021. https://healthpolicy.duke.edu/sites/default/files/2021-09/Presentation%20Slides_PD%20Biomarkers%20for%20Biosimilar%20Development%20and%20Approval_2021-09-27_0.pdf. Accessed 19 July 2023.
van der Graaf PH, Giacomini K, Cascorbi I, Chung C, Holstein S, Joshi A, et al. Innovations in biosimilars. Clin Pharmacol Ther. 2023;113:1–195. https://doi.org/10.1002/cpt.2653.
Woollett GR, Park JP, Han J, Jung B. The role of PD biomarkers in biosimilar development—to get the right answer one must first ask the right question. Clin Pharmacol Ther. 2023;113:50–4. https://doi.org/10.1002/cpt.2753.
Jawa V, Terry F, Gokemeijer J, Mitra-Kaushik S, Roberts BJ, Tourdot S, et al. T-cell dependent immunogenicity of protein therapeutics pre-clinical assessment and mitigation-updated consensus and review 2020. Front Immunol. 2020;11:1301. https://doi.org/10.3389/fimmu.2020.01301.
Ben-Horin S, Yavzori M, Benhar I, Fudim E, Picard O, Ungar B, et al. Cross-immunogenicity: antibodies to infliximab in Remicade-treated patients with IBD similarly recognise the biosimilar Remsima. Gut. 2016;65:1132–8. https://doi.org/10.1136/gutjnl-2015-309290.
Ruiz-Argüello MB, Maguregui A, Ruiz del Agua A, Pascual-Salcedo D, Martínez-Feito A, Jurado T, et al. Antibodies to infliximab in Remicade-treated rheumatic patients show identical reactivity towards biosimilars. Ann Rheum Dis. 2016;75:1693–6. https://doi.org/10.1136/annrheumdis-2015-208684.
Nupur N, Joshi S, Gulliarme D, Rathore AS. Analytical similarity assessment of biosimilars: Global regulatory landscape, recent studies and major advancements in orthogonal platforms. Front Bioeng Biotech. 2022;10:832059. https://doi.org/10.3389/fbioe.2022.832059.
Vulto AG, Jaquez OA. The process defines the product: what really matters in biosimilar design and production? Rheumatology. 2017;56:iv14–29. https://doi.org/10.1093/rheumatology/kex278.
Lamanna WC, Mayer RE, Rupprechter A, Fuchs M, Higel F, Fritsch C, et al. The structure-function relationship of disulfide bonds in etanercept. Sci Rep. 2017;7:3951. https://doi.org/10.1038/s41598-017-04320-5.
Lamanna WC, Holzmann J, Cohen HP, Guo X, Schweigler M, Stangler T, et al. Maintaining consistent quality and clinical performance of biopharmaceuticals. Expert Opin Biol Ther. 2018;18:369–79. https://doi.org/10.1080/14712598.2018.1421169.
Kurki P, van Aerts L, Wolff-Holz E, Giezen T, Skibeli V, Weise M. Interchangeability of biosimilars: a European perspective. BioDrugs. 2017;31:83–91. https://doi.org/10.1007/s40259-017-0210-0.
Medicines for Europe. Billions more euros to re-invest in better healthcare thanks to biosimilar medicines. 2022. https://www.medicinesforeurope.com/wp-content/uploads/2022/12/20221213-Press-Release-Bios-stakeholder-workshop.pdf#:~:text=Biosimilar%20medicines%20have%20now%20generated%20over%2030%20billion,4.5%20billion%20patient%20treatment%20days%20of%20positive%20experience. Accessed 19 July 2023.
Biosimilars Council. Biosimilars are a prescription for better health. 2022. https://biosimilarscouncil.org/. Accessed 19 July 2023.
European Medicines Agency. EudraVigilance. 2023. https://www.ema.europa.eu/en/human-regulatory/research-development/pharmacovigilance/eudravigilance. Accessed 19 July 2023.
US Food & Drug Administration. FDA's Sentinel initiative. 2023. https://www.fda.gov/safety/fdas-sentinel-initiative. Accessed 19 July 2023.
Xue X, Truong B, Qian J. Adverse event reporting of marketed biosimilar and biological monoclonal antibody cancer treatments in the United States. Expert Opin Biol Ther. 2023. https://doi.org/10.1080/14712598.2023.2189007.
Allocati E, Godman B, Gobbi M, Garattini S, Banzi R. Switching among biosimilars: a review of clinical evidence. Front Pharmacol. 2022;13:917814. https://doi.org/10.3389/fphar.2022.917814.
Cohen HP, Hachaichi S, Bodenmueller W, Kvien TK, Danese S, Blauvelt A. Switching from one biosimilar to another biosimilar of the same reference biologic: a systematic review of studies. BioDrugs. 2022;36:625–37. https://doi.org/10.1007/s40259-022-00546-6.
Lasala R, Abrate P, Zovi A, Santoleri F. Safety and effectiveness of multiple switching between originators and biosimilars: literature review and status report on interchangeability. Ther Innov Regul Sci. 2023;57:352–64. https://doi.org/10.1007/s43441-022-00473-2.
Mysler E, Azevedo VF, Danese S, Alvarez D, Iikuni N, Ingram B, et al. Biosimilar-to-biosimilar switching: what is the rationale and current experience? Drugs. 2021;81:1859–79. https://doi.org/10.1007/s40265-021-01610-1.
European Medicines Agency. Statement on the scientific rationale supporting interchangeability of biosimilar medicines in the EU. 2023. https://www.ema.europa.eu/en/documents/public-statement/statement-scientific-rationale-supporting-interchangeability-biosimilar-medicines-eu_en.pdf. Accessed 19 July 2023.
Brahme N, Skibinski S, Ikenberry S. FDA drug topics—biosimilars: a review of scientific, regulatory, and clinical considerations for health care providers. 2023. (Slide 27) https://www.fda.gov/media/169599/download. Accessed 19 July 2023.
Praditpornsilpa K, Tiranathanagul K, Kupatawintu P, Jootar S, Intragumtornchai T, Tungsanga K, et al. Biosimilar recombinant human erythropoietin induces the production of neutralizing antibodies. Kidney Int. 2011;80:88–92. https://doi.org/10.1038/ki.2011.68.
Cazap E, Jacobs I, McBride A, Popovian R, Sikora K. Global acceptance of biosimilars: importance of regulatory consistency, education, and trust. Oncologist. 2018;23:1188–98. https://doi.org/10.1634/theoncologist.2017-0671.
International Federation of Pharmaceutical Manufacturers & Associations. Policy statement: non-comparable biotherapeutic products. 2014. https://www.ifpma.org/wp-content/uploads/2023/01/i2023_Non-comparable_Biotherapeutic_Products__English__02.pdf. Accessed 19 July 2023.
Mysler E, Pineda C, Horiuchi T, Singh E, Mahgoub E, Coindreau J, et al. Clinical and regulatory perspectives on biosimilar therapies and intended copies of biologics in rheumatology. Rheumatol Int. 2016;36:613–25. https://doi.org/10.1007/s00296-016-3444-0.
Weise M, Bielsky MC, De Smet K, Ehmann F, Ekman N, Narayanan G, et al. Biosimilars—why terminology matters. Nat Biotechnol. 2011;29:690–3. https://doi.org/10.1038/nbt.1936.
Strand V, Gonçalves J, Hickling TP, Jones HE, Marshall L, Isaacs JD. Immunogenicity of biosimilars for rheumatic diseases, plaque psoriasis, and inflammatory bowel disease: a review from clinical trials and regulatory documents. BioDrugs. 2020;34:27–37. https://doi.org/10.1007/s40259-019-00394-x.
Section 7002 of the Affordable Care Act of 2010 that incorporated the Biologics Price Competition and Innovation Act of 2009, and that added Section 351k to the Public Health and Safety Act (42 U.S.C. 262(k)). 2010. https://www.govinfo.gov/content/pkg/PLAW-111publ148/pdf/PLAW-111publ148.pdf. Accessed 19 July 2023.
Sharma A, Kumar N, Parachuri N, Regillo C, Bandello F, Kuppermann BD. Biosimilar ranibizumab interchangeability: what does it mean to retinal physicians? Eye (London). 2022. https://doi.org/10.1038/s41433-022-02287-w.
US Food & Drug Administration. Guidance for industry. Clinical immunogenicity considerations for biosimilar and interchangeable insulin products. 2019. https://www.fda.gov/media/133014/download. Accessed 19 July 2023.
Cohen HP, McCabe D. The importance of countering biosimilar disparagement and misinformation. BioDrugs. 2020;34:407–14. https://doi.org/10.1007/s40259-020-00433-y.
Webster CJ, Woollett GR. A 'global reference’ comparator for biosimilar development. BioDrugs. 2017;31:279–86. https://doi.org/10.1007/s40259-017-0227-4.
European Medicines Agency. Medicines. 2023. https://www.ema.europa.eu/en/medicines. Accessed 19 July 2023.
US Food & Drug Administration. Drugs@FDA: FDA-Approved Drugs. 2023. https://www.accessdata.fda.gov/scripts/cder/daf/. Accessed 19 July 2023.
US Food & Drug Administration. Guidance for industry. Questions and answers on biosimilar development and the BPCI Act. 2021. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/questions-and-answers-biosimilar-development-and-bpci-act-guidance-industry. Accessed 19 July 2023.
Ramanan S, Grampp G. Drift, evolution, and divergence in biologics and biosimilars manufacturing. BioDrugs. 2014;28:363–72. https://doi.org/10.1007/s40259-014-0088-z.
Matar P. Biosimilarity is not a transitive property: implication for interchangeability, naming and pharmacovigilance. GaBI J. 2022;11:36–40. https://doi.org/10.5639/gabij.2022.1101.006.
Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29:310–2. https://doi.org/10.1038/nbt.1839.
Bennett CL, Luminari S, Nissenson AR, Tallman MS, Klinge SA, McWilliams N, et al. Pure red-cell aplasia and epoetin therapy. N Engl J Med. 2004;351:1403–8. https://doi.org/10.1056/NEJMoa040528.
Kim S, Song J, Park S, Ham S, Paek K, Kang M, et al. Drifts in ADCC-related quality attributes of Herceptin®: Impact on development of a trastuzumab biosimilar. MAbs. 2017;9:704–14. https://doi.org/10.1080/19420862.2017.1305530.
National Academies of Sciences, Engineering, and Medicine. Regulating medicines in a globalized world. The need for increased reliance among regulators. 2020. https://nap.nationalacademies.org/catalog/25594/regulating-medicines-in-a-globalized-world-the-need-for-increased. Accessed 19 July 2023.
US Food & Drug Administration. Biosimilars info sheet. Comparative clinical studies. 2022. https://www.fda.gov/media/154916/download. Accessed 19 July 2023.
US Food & Drug Administration. Biosimilars—review and approval. 2022. https://www.fda.gov/drugs/biosimilars/review-and-approval. Accessed 19 July 2023.
Vulto AG, Vanderpuye-Orgle J, van der Graaff M, Simoens SRA, Dagna L, Macaulay R, et al. Sustainability of biosimilars in Europe: a Delphi panel consensus with systematic literature review. Pharmaceuticals (Basel). 2020;13:400. https://doi.org/10.3390/ph13110400.
Califf RM. Now is the time to fix the evidence generation system. Clin Trials. 2023;20:3–12. https://doi.org/10.1177/17407745221147689.
Lockhart CM, McDermott CL, Felix T, Lin ND, Cziraky MJ, Mendelsohn AB, et al. Barriers and facilitators to conduct high-quality, large-scale safety and comparative effectiveness research: the Biologics and Biosimilars Collective Intelligence Consortium experience. Pharmacoepidemiol Drug Saf. 2020;29:811–3. https://doi.org/10.1002/pds.4885.
Grampp G, Felix T. Pharmacovigilance considerations for biosimilars in the USA. BioDrugs. 2015;29:309–21. https://doi.org/10.1007/s40259-015-0137-2.
Glintborg B, Loft AG, Omerovic E, Hendricks O, Linauskas A, Espesen J, et al. To switch or not to switch: results of a nationwide guideline of mandatory switching from originator to biosimilar etanercept. One-year treatment outcomes in 2061 patients with inflammatory arthritis from the DANBIO registry. Ann Rheum Dis. 2019;78:192–200. https://doi.org/10.1136/annrheumdis-2018-213474.
Nabi H, Hendricks O, Jensen DV, Loft AG, Pedersen JK, Just SA, et al. Infliximab biosimilar-to-biosimilar switching in patients with inflammatory rheumatic disease: clinical outcomes in real-world patients from the DANBIO registry. RMD Open. 2022. https://doi.org/10.1136/rmdopen-2022-002560.
Scavone C, Sessa M, Clementi E, Corrao G, Leone R, Mugelli A, et al. Real world data on the utilization pattern and safety profile of infliximab originator versus biosimilars in Italy: a multiregional study. BioDrugs. 2018;32:607–17. https://doi.org/10.1007/s40259-018-0313-2.
Vermeer NS, Giezen TJ, Zastavnik S, Wolff-Holz E, Hidalgo-Simon A. Identifiability of biologicals in adverse drug reaction reports received from European clinical practice. Clin Pharmacol Ther. 2019;105:962–9. https://doi.org/10.1002/cpt.1310.
Acknowledgements
The input of Martin Schiestl (an employee of Sandoz) is appreciated. Medical writing support was provided by Dan Hami of OPEN Health Communications, in accordance with Good Publication Practice (GPP) guidelines (www.ismpp.org/gpp-2022).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The Biosimilars Forum paid for medical writing support and the Open Access charges.
Conflicts of interest
HPC is an employee of Sandoz Inc. MT is an employee of Fresenius Kabi. DM is an employee of Boehringer Ingelheim. GRW is an employee of Samsung Bioepis. The authors are all members of the Science and Education Committee of the Biosimilars Forum. HPC and DM are co-chairs of this committee. The Biosimilars Forum is a nonprofit biosimilars trade association whose mission is to promote education and policy advancement on biosimilars in the USA, with the intent of expanding access and availability of biological medicines and improving healthcare. The views expressed here are those of the authors in their roles with the Biosimilars Forum and not of their respective companies. Furthermore, the contents may not reflect the positions of all member companies of the Biosimilars Forum.
Availability of data and material
Not applicable.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Author contributions
All authors contributed to the drafting and review of the manuscript. The final manuscript was approved by all authors who take responsibility for the content.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Cohen, H.P., Turner, M., McCabe, D. et al. Future Evolution of Biosimilar Development by Application of Current Science and Available Evidence: The Developer’s Perspective. BioDrugs 37, 583–593 (2023). https://doi.org/10.1007/s40259-023-00619-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-023-00619-0