
Abstract
Over time, clinicians have become increasingly comfortable embracing the prescription of biosimilars—highly similar versions of innovator or reference biological agents—for their patients with inflammatory diseases. Although a switch from a reference product to a licensed biosimilar version (or vice versa) is a medical decision robustly supported by the stepwise accumulation of clinical trial evidence concerning comparable safety, immunogenicity, and efficacy between these products, a switch from one biosimilar to another biosimilar of the same reference product, or a cross-switch, is not. Similarity among biosimilars of a reference product is not a regulatory agency concern and therefore is unlikely to be investigated in randomized controlled trials in the foreseeable future. Yet in clinical practice, across a diverse range of patients, the option to cross-switch from one biosimilar to another can and does arise for valid reasons such as convenience or tolerability issues, or driven by third parties (e.g., payers). In the absence of clinical trial data, clinicians must attempt to objectively evaluate the emerging real-world cross-switching evidence within the context of what is known about the science underpinning a designation of biosimilar. That knowledge then needs to be integrated with what clinicians know about their patients and their disease on a case-by-case basis. This review aims to consolidate relevant emerging real-world data and other key information about biosimilar-to-biosimilar cross-switching for prescribing clinicians. In the absence of clear clinical guidelines addressing this topic at present, this review may serve to facilitate discretionary and educated treatment decision making.
Similar content being viewed by others

Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
As an increasing number of more affordable biosimilars enter the marketplace, the decision to switch a patient’s treatment from one biosimilar to another is emerging as a potential practical option. |
Pre-clinical scientific data underpin the evidence for drug biosimilarity, with most evidence established via the early analytical, non-clinical, and comparative clinical pharmacology steps performed prior to the clinical study component. |
In the absence of data from formal clinical trials comparing several distinct biosimilars of the same reference product, early preliminary real-world evidence warrants evaluation in the context of each patient’s and payers’ circumstances, and the scientific principles supporting the utility of biosimilars. |
Currently, there is a lack of clinical guidelines to address the concept of cross-switching, and this educational paper is intended to contribute to bridging the knowledge gap that otherwise fuels prescriber hesitancy when it comes to cross-switching between biosimilars, to facilitate safe and effective ongoing treatment for patients. |
1 Introduction
Biologic medicines have revolutionized the management of chronic inflammatory diseases [1]. A major drawback of biologics is their high cost, which can limit patient access to much needed treatment [2,3,4,5]. To rein in healthcare expenditure and promote greater population-based access to biological medicines, biosimilars—highly similar, reverse-engineered versions of existing innovator biological medicines and their active ingredients (originator or reference products)—have emerged as less expensive treatment options compared with reference products for which market-exclusivity patents and regulatory exclusivities have reached end of term [4, 6, 7].
Across Europe, the USA, and more universally, based on the World Health Organization (WHO) standards, establishing biosimilarity follows a stringent yet abbreviated regulatory pathway compared with that for an originator biologic; one that judiciously exploits the years of knowledge accumulated for the bio-originator [8,9,10,11]. Globally, a biosimilar must be as safe, pure, potent, and efficacious as the reference product based on comprehensive comparability exercises, such that there are no clinically meaningful differences [9, 10, 12, 13].
As the market for biosimilars continues to expand and the number of biosimilar products for each approved biological reference product increases, the likelihood of patients needing to switch from one biosimilar to another (cross-switch), for whatever reason, is also expected to increase [14,15,16]. To date, most of the research conducted on therapeutic exchanges involving biosimilars has focused on the safety, efficacy, and immunogenicity of a rather narrow range of switching scenarios, predominantly in patients new to a reference product or a biosimilar, for which there are registered clinical trial data and emerging extension and post-marketing studies, all capturing longer-term evidence [17, 18]. Indeed, in clinical practice, particularly when patients are treated over a long duration, switching between biosimilars has become a treatment option and in some cases a mandated necessity, as has occurred with respect to originator-to-biosimilar switches [19, 20].
Biosimilars are considered “clinically equivalent” to the reference product, a term adopted by the WHO [21, 22]. Although the clinical equivalence of a biosimilar to its reference product is rigorously tested and well documented, there is no regulatory obligation or industry-driven impetus for approved biosimilars of the same reference product to be evaluated for biosimilarity among themselves [23,24,25]. Attempts to make indirect comparisons between biosimilars of the same reference product can be hampered by the heterogeneity of clinical trial designs between biosimilars and their reference products [26]. Clinical trial components—including, but not limited to, study population, inclusion and exclusion criteria, timing of the primary and secondary endpoints assessed, immunogenicity assays used and the timing of sample collections, equivalence margins, and definitions for adverse events (AEs)—can vary across studies [27]. Consistency with respect to stratification factors (e.g., disease activity, body mass index [BMI]) may impact responses to therapy and warrant careful consideration [28]. Most evidence of biosimilarity is established via the early analytical, non-clinical, and comparative clinical pharmacology steps performed prior to the clinical study component. Randomized clinical trials are then performed to confirm biosimilarity [29,30,31]. However, the ability of clinicians to be able to infer the potential for similarity between biosimilars of a reference product, and the confidence to decide to switch a patient between them, would likely improve if clinical trial designs were better standardized [26, 27].
This review will focus on biosimilar cross-switching or switching from one biosimilar to another biosimilar of the same reference product, with an emphasis on patients with inflammatory diseases. While the regulatory process for determining biosimilarity is scientifically robust, the published evidence for cross-switching is currently sparse. Here we review available real-world data, current recommendations, and consensus statements that may offer clinical practitioners some insights and perspective as they manage their patients’ health and make medical decisions regarding optimal treatment within the context of individual patients.
2 Literature Methods
A search of PubMed was conducted for articles published up to January 2021 using the search terms: switching, cross-switching, and biosimilar. There was no restriction or exclusion related to article type. The reference lists from articles identified in this search were also reviewed and additional publications retrieved if considered within the scope of this review. A separate “Google search” using the same search terms was performed to identify important material relevant to the topic and not captured within the peer-reviewed literature. Articles identified by the authors through their awareness of the topic were also considered for inclusion. All the retained articles were qualitatively assessed and described in this review article.
3 Rationale for Switches Involving Biosimilars
A number of real-world scenarios, of a medical and non-medical nature, may lead to cross-switching between biosimilars of the same reference product.
Medical switching occurs when one medication is exchanged for another at the physician’s discretion [32]. The goal of a medical switch is to optimize the patient’s treatment benefit. The rationale for a medical cross-switch involving biosimilars could be brought about by a change in disease activity (although unlikely, given that similar therapeutic benefits are typically expected from different versions of the same reference product), or more likely be instigated to address tolerability issues and/or AEs (e.g., injection-related reactions), and/or for patient convenience (e.g., device preference, frequency of dosing, storage requirements) [32, 33].
In some instances, cross-switching may be medically prudent or necessary to address intolerance issues, such as the avoidance of an irritating excipient (citrate-free vs citrate-containing biosimilars of adalimumab) or a prefilled delivery device for one biosimilar to which a patient exhibits a hypersensitivity (a needle cover containing a derivative of latex vs a latex-free needle shield) [34, 35]. Other medical reasons for a biosimilar-to-biosimilar cross-switch could relate to differences in the length or gauge of the needle, or the volume in a prefilled syringe if it may improve the patient’s satisfaction [33, 35]. Improving the convenience of drug administration to benefit a patient or their caregiver in the event of manual dexterity issues (e.g., medication delivery via a prefilled pen vs syringe) [35, 36], to facilitate the administration of a lower dose more accurately (e.g., a prefilled syringe to a biosimilar with a single-dose vial option), or to accommodate the preference of one delivery device over another, could also prompt a medical cross-switch [37, 38].
Non-medical switching occurs when a clinically stable patient, whose current therapy is effective and well tolerated, is switched to another therapeutic alternative [39, 40]. This type of switching or cross-switching is not related to improving efficacy, safety, and/or convenience, but rather it is instigated for the purpose of mitigating costs or to ensure that the patient has continued access to the same type of drug [5, 41]. Non-medical cross-switching is typically governed by a third party (e.g., payer who insists that patients align with the particular biosimilar covered by the health-plan drug formulary, or based on an employer-plan offering), initiated by a hospital pharmacist to avert supply-chain issues due to an unreliable manufacturer, or it may be necessary for a traveling or relocating patient whose current biosimilar might not be geographically available [5, 23, 40, 42, 43]. Out-of-pocket expenses, payer incentives, rebates, copays, or fixed reimbursement fees (for hospitals, per inpatient day despite the medication used) may also influence a decision to cross-switch to another biosimilar version, or alternatively, reverse-switch to the reference product when the economic incentives disappear [44]. A reverse-switch to the originator was observed to occur in <10% of patients with inflammatory bowel disease (IBD). The reported reasons for reverse switching were mostly biosimilar-attributed, although logically there is no expectation that AEs, worsening symptoms, or a loss of response should resolve when switching back to an originator. Nevertheless, the vast majority of symptoms (up to 90%) were considered resolved after a reverse switch [45]. This suggests a nocebo effect; however, immunogenicity could have played a role in individual patients with different genetics and varying responses to treatment.
A number of European countries practice tendering via various processes operating at different levels and in several settings (e.g., at the national level and in hospitals or outpatient settings, etc.) [20]. In 2016, the need to comply with single-winner tenders was identified in 12 of 26 (46%) European countries; although physicians are able to opt-out for non-medical reasons for individual patients in all but one-third of those countries where switches are forced (Bulgaria, Poland, Serbia, and Turkey) [20]. Quotas favoring lower-cost biologicals or biosimilars in some countries must be met before opting-out is permitted (mandatory quotas exist in Denmark, Latvia, and Lithuania; indicative quotas exist in Cyprus, Greece, Italy, and some regions of Germany), and physician opposition to pharmacy-level substitution of biologicals can preclude switches in other European countries, which do not include the Czech Republic, Estonia, Latvia, Poland, Serbia, and Turkey [46]. The frequency of single-winner tendering among the 12 countries identified has occurred from every 3 to 36 months, or every 3 to 18 months in the case of the 4 countries in which physicians cannot exercise discretion. It stands to reason that a vast amount of non-medical cross-switching likely occurs based on single-winner tendering as long as competitors exist [46, 47]. If one biosimilar displaces another biosimilar of the same reference product when it becomes the single-tender winner, or one of multiple-tender winners, the switch is likely to be imminent for numerous patients. Procurement via tendering at the national, regional, or hospital level already occurs in numerous European countries. There is a lack of peer-reviewed published literature to indicate whether cross-switches between biosimilars—due to alternating winners of a competitive bidding processes—have caused efficacy or safety issues of concern [48, 49]; however, the stringent pharmacovigilance system used for all biologicals mitigates these safety concerns, as shown in Europe and the USA [50].
4 Switching: Who Decides?
Switching occurs when a treating physician or prescriber “decides to exchange one medicine for another with the same therapeutic intent” [9]. Regardless of the reasons for switching to or between biosimilars, the tendency of many—but not all—prescribers is to adopt a multidisciplinary approach that is patient-centric [51,52,53]. Therefore, when a switch in therapy is imminent for patients with inflammatory conditions, it is often considered that this should involve a decision made by the treating physician and patients together, or at least with the patient’s full awareness [54].
Although the European Medicines Agency encourages any decision made about switching to a biosimilar or among biosimilar medicines to involve the prescriber in consultation with the patient [9, 55], prescribers in European Union (EU) nations fall within the remit of Member States [55]. Therefore, the extent to which this occurs varies by country, region, and hospital. For example, in Norway switching is performed at the discretion of hospitals and/or physicians [56], while in Denmark in 2017, switching from the etanercept reference product to an etanercept biosimilar in patients with inflammatory arthritis was mandated with few exceptions [19]. In the UK, preliminary results of a National Ankylosing Spondylitis Society patient survey revealed that the majority of patients who were switched from adalimumab reference product to an adalimumab biosimilar were never asked to consent to the change, although reasons for this oversight are not described [57, 58].
In the USA, switches involving biosimilars are currently physician-led in the absence of biosimilars that have met the additional interchangeability designation set forth by the US Food and Drug Administration (FDA), although payers, vendors, and pharmacy benefit managers continue to influence formulary decisions related to switching a patient’s treatment [59,60,61,62]. Although the FDA encourages patients to ask their doctor, nurse, or pharmacist about what medication they have been prescribed, it has no direct role in therapeutic switching practices in the event that a biosimilar is granted an interchangeable designation. Nevertheless, the FDA recognizes that physicians will know their patients’ condition and specific health risks best [63].
Pharmacovigilance is key when monitoring the use of biosimilars because it allows detection and characterization of adverse drug reactions [64]. Regulatory guidance varies according to the market, for example in Europe, this is provided by the regulatory document entitled “Good Pharmacovigilance Practices for biological medicinal products.” The European Medicines Agency requires close ongoing monitoring of the clinical safety of biosimilars following approval, along with continued benefit-risk assessments. Safety data are periodically assessed and compared with data for the reference product [65].
5 Types of Biosimilar Switching
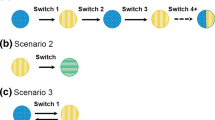
Various switching scenarios involving biosimilars can occur (Fig. 1); however, as the biosimilar landscape has evolved, the definitions for switching are not always uniform or intuitive, they can appear duplicative in some respects, and terminology appears to be lacking when it comes to describing additional potential scenarios.

Potential switching scenarios. The dot (•) end of the arrow indicates the initial biologic. Switch: •A→A1; reverse-switch: •A1→A; multiple switches: •A→A2→A, •B2→B→B1; cross-switch: •A1→A2, •A2→A1; switch within the same drug class: •A→B; •B→A; intraclass cross-switch: •A1→B1, •B1→A1; intraclass switch: •A→B1; intraclass reverse-switch: •B2→A
Switching has been variously defined as “a one-time change from an originator biologic to its biosimilar, or the reverse” [66], and elsewhere, it has been considered as a unidirectional therapy transition, or as “switching from the innovator biologic agent to a biosimilar” [67]. The latter definition can be attributed to earlier times when it was most common for patients to transition from a reference biologic to a biosimilar, rather than be initiated on a biosimilar. In real-world scenarios, prescribers are becoming increasingly inclined to want to introduce biologics earlier during the course of clinical treatment [68], and accordingly prescribe biosimilars to treat inflammatory diseases in biologic-naïve patients when the circumstances are medically appropriate [53, 69,70,71].
Reverse-switching has been defined as “switching from a biosimilar to its reference medical product” [33, 72]. Reverse-switching has also been used to describe both a switch that occurs after prior exposure to an originator product (originator→biosimilar→originator) as well as one that does not (biosimilar→originator) [73, 74]. Intuitively, a reverse-switch might suggest a return to the therapy that was initially prescribed, although it is also used to implicitly denote a single unidirectional switch [67, 75]. Reverse-switching has also been used to describe repetitive switches (repeat changes between a reference product and its biosimilar) and because this description reads like a multiple-switching scenario for an interchangeability designation, it can become imprecise and confusing [76].
Multiple-switching is the term routinely used to describe at least three therapy transitions, or alternating between the reference product and its biosimilar product(s) or between biosimilars [16, 26, 77]. Based on a definition of this nature, multiple-switching may not be mutually exclusive from reverse-switching or cross-switching [72].
Cross-switching has been used to distinguish “a switch from one biosimilar to another [67], switching between two biosimilars” [72], or a transition of therapy from one biosimilar to an alternative biosimilar of the same reference product [76]. The latter definition is more explicit as it excludes another potential scenario that appears to be largely absent in the literature. A biosimilar-to-biosimilar cross-switch in clinical practice could potentially entail the de novo usage of a biosimilar of one reference product followed by a switch to a biosimilar of a different reference product within the same drug class (e.g., a different tumor necrosis factor [TNF] inhibitor) [78]. For the sake of differentiating and improving clarity, that type of switch could be uniquely referred to as an “intraclass cross-switch.” For another switching scenario that is medical in nature and not explicitly defined to date, a switch from a reference biologic to a biosimilar of a different reference product in the same drug class [78] is akin to an “intraclass switch” or vice versa as an “intraclass reverse-switch” (Fig. 1).
Although this review focuses on cross-switching from one biosimilar to an alternative biosimilar of the same reference product, it is noteworthy that current terminology does not always describe switching scenarios in adequate context, which can lead to confusion. Conceivably, a lack of consensus, or a reticence to agree that there are sufficient data in terms of scientific and clinical evidence to support certain biosimilar-related switching scenarios, could be hampering interest in establishing more definitive terminology at this time [6, 54, 75, 79,80,81,82,83]. However, as the real-world utility of biosimilars expands, and as courses of treatment evolve, an agreed-upon convention within the scientific community to describe switching scenarios involving biosimilars might be warranted in the future. Country-specific regulatory agencies will have a major role in defining the scenarios for switching from one biosimilar to another. Moreover, scientific medical societies can share some of this responsibility since they can have an influential role, as already evidenced from their impact on the overall biosimilar debate. Results could vary with contrasting approaches and different levels of pharmacovigilance around the world.
6 Biosimilar Switching Requires an Educated Decision
Switching therapies according to any of the definitions previously provided, including those involving biosimilars, requires potential risks to be balanced against potential benefits [84]. Currently, robust, long-term, and conclusive clinical trial data are lacking with regard to cross-switching between biosimilars, yet, in real-world settings, physicians are required to meet the needs of a more diverse range of patients with chronic inflammation over long periods who are managing their disease under a variety of circumstances.
In December 2020, Medicines for Europe reported that the total clinical experience with biosimilar medicines exceeded two billion treatment days and the cumulative treatment days for patients receiving approved biosimilars in Europe has doubled approximately every 1.5 years over the past decade [85]. Moreover, during more than a decade of use in the EU, no approved biosimilar has needed to be withdrawn or has required additional labeling because it was not equally effective or as safe as the reference product [86]. From a purely practical standpoint, biosimilar cross-switching is already happening, and it is has been suggested to be more likely among patients who receive therapy over a long duration [87].
Biosimilar products in robustly regulated regions such as the USA, Canada, EU, and Japan are considered as efficacious as the reference product when used for approved indications [88,89,90]. The general acceptability of this framework/rationale lends some support to the notion that two biosimilars considered to be highly comparable versions of the same active substance in the reference product—to stringent standards—should be able to be exchanged without unduly compromising efficacy or safety.
Although the scientific and regulatory principles for determining biosimilarity are well established, prescribing physicians generally prefer to make case-by-case, evidence-based healthcare decisions for their patients with inflammatory diseases whenever any type of therapeutic switch involving a biosimilar is warranted [25, 84, 91, 92]. The ability of physicians to do this exemplifies one facet of the art of clinical judgment [93].
Beyond unique patient-specific needs, clinicians must routinely consider—in the event of a biosimilar cross-switch—the fact that head-to-head clinical trials of biosimilars of the same reference product are extremely unlikely to be conducted. In a scenario in which biosimilar B is highly similar to reference product A, and biosimilar C is highly similar to reference product A, the extent of the similarity between biosimilars B and C remains untested and requires logical reasoning when evaluating any potential risks of a switch between biosimilars of a single reference product. This is akin to the situation wherein the second innovator product of two reference products—manufactured and approved in highly regulated yet different jurisdictions (e.g., EU and USA)—is essentially an nth-generation biosimilar of the product approved originally, as they share a common origin (i.e., they are based on the same drug and the same types of clinical trials). Some contend that bridging studies required to formally prove biosimilarity between local and international reference products are unnecessary and should be waived [94, 95]. Understanding the scientific principles underpinning biosimilarity should provide a dependable basis for the intersection of other considerations that must be weighed by physicians exploring the feasibility of a biosimilar cross-switch.
7 What Biosimilarity is Predicated On
Because the amino acid sequence of a biosimilar must show complete fidelity to its reference product, each biosimilar is manufactured so that it contains the same foundational building blocks in addition to the three-dimensional structure. Although all biologics are derived from living cells with inherent variabilities, only minor differences (microheterogeneities) in clinically inactive components of a biosimilar compared with its reference product are expected and permitted [29, 96, 97]. Unlike chemically synthesized drugs for which identical generic versions of the reference product can be routinely produced, the inability to produce large, molecularly complex, identical biological products to the reference product is central to the perceived clinical challenges with biosimilars [86, 98].
Using state-of-the-art methods and technology, analytical biosimilarity must first be demonstrated via extensive in vitro and/or in vivo functional activity characterizations and structural analyses to evaluate product-related variants, stability, and impurities [29, 99,100,101]. In the event that uncertainty exists following in vitro assessments with respect to potential for in vivo toxicity, animal data may be required before clinical pharmacology studies are conducted to test pharmacokinetic and pharmacodynamic equivalence in an adequately sensitive population [12, 60, 75]. However, European regulators de facto do not require animal studies as part of the biosimilarity exercise. As they are still mandated by some regulatory authorities and development programs are global, animal studies still form part of EU submissions even though they are not required. Therefore, there is an increasing need to re-assess the relevance of animal studies to support regulatory approval of biosimilars [102]. The biosimilarity established via the analytical, non-clinical, and comparative clinical pharmacology evaluations is subsequently reaffirmed via adequately powered randomized controlled trials (RCTs) designed to compare—not to establish—therapeutic efficacy, safety, and immunogenicity versus the reference product [29, 30].
Licensure or approval of a biosimilar is based on totality-of-the-evidence and is granted on a case-by-case basis, exclusively with respect to the reference product [29, 98, 103]. Distinct from the approval process for a reference biological product, the regulatory pathway adopted for biosimilars relies more heavily upon comparative analytical data, with less emphasis placed upon multiple, lengthy, large-scale, comparative clinical trials [30, 104].
Overall, for a number of inflammatory diseases, a single switch from a reference product to its biosimilar has generally not been shown to be associated with safety issues and/or anomalous responses [75, 105,106,107]. Nevertheless, exercising diligence in terms of assessing any of the unique needs or challenges for each patient when switching products remains important [32, 84, 108]. For instance, additional patient training may be required as a result of switching [32]. Also, a switch to a biosimilar from its reference product, or a biosimilar cross-switch, becomes unethical if a patient experiences a loss of response [75, 109]. In the EU, clinically important adverse immunogenicity to a reference product precludes the use of any of its approved biosimilar products (and vice versa), given the high degree of similarity among them and the potential for cross-reactivity on a population basis [110, 111]. However, according to the US FDA, expected biosimilarity in terms of immunogenicity results has yet to be proven and approved at the level of “any given patient” via an interchangeability designation [60].
Immunogenicity pertaining to biosimilars has emerged as a highly controversial topic with the possibility of being misleading if poorly understood [109]. There are opposing views on the importance of immunogenicity in this field, and in general the importance of this issue can be overestimated because most biologicals have low immunogenic potential. Some experts support the view that immunogenicity is an important issue and that patients should be tested prior to switching, while others are of the opinion that immunogenicity is not a concern and testing prior to switching is unnecessary given that immunological overlap exists between a biosimilar and its reference product [112, 113]. At the annual 2019 Biosimilars and Biological Summit in Portugal, João Gonçalves shared expert opinion and pointed out that there are nuances to be considered with respect to immunogenicity when switching to or among biosimilars [109]. Specifically, distinctions often need to be made between the detection of transient versus persistent antidrug antibodies (ADAs; those detected at a single timepoint as opposed to those detected at multiple timepoints). Not surprisingly, epitope-specific antibodies that can signal therapeutic failure tend to be shared between highly similar molecules [9, 12, 109,110,111, 114,115,116]. Other factors that warrant consideration include paying attention to titer kinetics, which have appeared to be better predictors of secondary failure than ADA positivity [109]. Appropriately timed immunogenicity assessments (in advance of a switch and within 12 months of initiating a switch) have been recommended [109, 117]. Guidance is often needed by physicians with regard to the most appropriate use of immunogenicity tests [109]. However, other factors are known to confound the clinical relevance of ADAs. For example, the accuracy of commercial diagnostic tests and reference standards can be variable and the presence of ADAs may not be directly linked to drug tolerance and/or clinical efficacy, such as when the magnitude of drug neutralization is minimal [80]. Drug doses and concomitant use of immunomodulators by patients with rheumatoid arthritis (RA) can differ from patients with IBD, such that immune responses could vary accordingly [110]. Although physicians need to remain mindful of potential immunogenic issues, a review by Strand et al. [118]—which included an assessment of the effect of therapy switching on ADA incidence in 22 studies involving at least 10 biosimilars, multiple cross-over steps in some cases, and patients with rheumatic diseases, plaque psoriasis, or IBD—showed that immunogenicity did not change quantitatively or qualitatively as a result. Across 52 trials (involving 18 biosimilars, including some proposed biosimilars), 14 of which were conducted in healthy subjects, immunogenicity was found to be highly similar between biologic agents within the same class, which is consistent with regulatory findings that govern approvals [118] and bodes well for cross-switching.
8 Evidence of the Safety and Efficacy of Biosimilar Cross-Switching
Currently, the published evidence available in the scientific literature for biosimilar cross-switching is limited [107]. Clinical trial data for cross-switching are largely lacking, primarily because there are no regulatory stipulations in place that require biosimilars of a single reference product to be compared among themselves [119, 120]. In addition, transitioning between biosimilars is such a new concept that published data in this area are scarce. Nevertheless, it can be considered that clinicians experienced in switching from a reference product to a biosimilar will recognize that biosimilar cross-switching poses no additional risks, and they may be more open to introducing such a strategy. To the best of our knowledge, overall, there are seven real-world studies reported in which patients switched from one biosimilar to another. These studies provide some initial and preliminary support for the safety and efficacy of cross-switching, although, overall, the study durations are relatively short, sample sizes are comparatively small, and the existing data are limited to evaluations of infliximab biosimilars [1, 121,122,123,124,125,126]. Nevertheless, it should be considered that several adalimumab biosimilars have now been approved and are on the market in the EU—and presumably switches among them have occurred as matter of practical reality, yet reports of harm associated with adalimumab biosimilar cross-switching are conspicuously scarce [127].
8.1 Ex Vivo Immunogenicity Studies with Implications for Cross-Switching
Two ex vivo studies lend support to biosimilarity among biosimilars of a reference product in terms of immunogenicity and cross-reactivity [114, 128].
In an ex vivo study reported by Magro et al. [128], serum samples collected from patients with IBD receiving infliximab reference product (R-IFX) or biosimilar infliximab CT-P13 were tested for the number of ADAs to R-IFX, CT-P13, or biosimilar infliximab SB2 (Fig. 2). Anti–R-IFX and anti–CT-P13 sera from those samples were subsequently used to assess the immunogenic cross-reactivity via enzyme-linked immunosorbent assay (ELISA) R-IFX-optimized quantification assays [129]. Anti–R-IFX and anti–CT-P13 sera cross-reacted with R-IFX as well as the biosimilars CT-P13 or SB2, thus demonstrating immunogenic similarity among these anti-TNF alpha (TNFα) biologic drugs. This aspect of the study highlighted the futility of biosimilar cross-switching among infliximab biosimilars in the event of an immune response to R-IFX or an infliximab biosimilar. If full cross-reactivity is shown in vitro, this is supportive of the safety of cross-switching. Limitations of this study were that anti-SB2 serum was not tested and another anti-TNFα, besides R-IFX, was not utilized as a control.

Bar chart adapted from Magro et al. [128]. © The Author(s). Reprinted with permission [https://creativecommons.org/licenses/by/4.0/]
Infliximab reference product and its biosimilars (CT-P13 and SB2) demonstrate high cross-reactivity ex vivo [128]. ADA antidrug antibody, CT-P13 biosimilar infliximab, IBD inflammatory bowel disease, ICC intraclass correlation coefficient, LLOQ lower limit of quantification, R-IFX infliximab reference product, SB2 biosimilar infliximab; TNFα tumor necrosis factor alpha.
A retrospective ex vivo study, Fiorino et al. 2018 [114], tested sera samples collected from three cohorts of adult patients with Crohn’s disease (CD) or ulcerative colitis (UC) who participated in the BIOSIM01 study [114] (Fig. 3). Patients in cohort 1 were treated with R-IFX followed by CT-P13 (n = 18), patients in cohort 2 were treated with CT-P13 only (n = 52), and patients in cohort 3 were treated with R-IFX only (n = 30). Among all the 100 treated patients, 76 positive sera samples were collected from 23 (23%) individuals. Another 76 sera samples were collected from 11/28 patients with IBD from an R-IFX–naïve group that served as a negative control. All 152 (76+76) sera samples from 34 (23+11) patients were further exposed to different batches of biologics (R-IFX, or R-IFX and either SB2 or CT-P13) to determine whether there was sera antibody cross-reactivity between R-IFX and CT-P13 or SB2 treatments. Multiple ELISAs, one specific to each biologic, revealed that patients’ antibodies recognized R-IFX and infliximab biosimilars CT-P13 or SB2 equivalently. Although a direct comparison between CT-P13 and SB2 remains untested because anti-SB2 antisera were not available, the uniformity of the cross-reactivity between R-IFX and CT-P13 antibodies and between R-IFX and SB2 antibodies was considered by the authors to suggest that cross-switching between CT-P13 and SB2 may not result in divergent immunogenicity. The same epitopes responsible for mounting an immune response to R-IFX are hypothesized to react in the presence of CT-P13 or SB2, which may indirectly support biosimilarity. At the same time, the authors caution that persistent antibodies to infliximab may be detected at least 1 year after therapy discontinuation, and drug batches could be subtly different (more or less immunogenic). However, considering manufacturers of biosimilars are compelled to vigilantly abide by rigorous quality standards in accordance with regulatory approval specifications, batch-to-batch drifting of the quality attributes of biosimilars is routinely precluded to any extent that is clinically meaningful [130]. For that reason, the proposed drug batch-to-batch–related limitation suggested in relation to the Fiorina et al., study seems improbable.

Antibody cross-reactivity appears equivalent between infliximab reference product and its biosimilars CT-P13 or SB2 [114]. Ab antibody, CD Crohn’s disease, CI confidence interval, CID chronic inflammatory disease, CT-P13 biosimilar infliximab, ELISA enzyme-linked immunosorbent assay, IBD inflammatory bowel disease, IFX infliximab, pts patients, R-IFX infliximab reference product, UC ulcerative colitis, wk week
8.2 In Vivo Cross-Switching Studies
Gisondi et al. [123] evaluated a group of 24 patients (mean age 54 ±13 years, 79% male, mean body mass index [BMI] 27 ± 4) over 6 months, who had chronic plaque psoriasis (mean duration ~ 2 years), to examine the effectiveness and safety of a cross-switch from CT-P13 to SB2 (Fig. 4). Psoriasis Area and Severity Index (PASI) scores were reported at the time of the cross-switch and at 2, 4, and 6 months thereafter. Duration of exposure to CT-P13 and AEs were also reported. The PASI score did not change significantly when measured at 2-monthly intervals; PASI scores at the time of the cross-switch and at 2, 4, and 6 months thereafter were 0.67 ± 1.38, 0.76 ± 1.08, 0.71 ± 0.93, 0.42 ± 0.62, respectively. The previous duration of exposure to CT-P13 was 23.2 ± 7.51 months and the incidence of AEs (two infusion reactions and two upper respiratory bacterial infections) presented no new safety signals. Cross-switching in these patients was considered safe and effective.

Summary of cross-switching study in patients with chronic plaque psoriasis [123]. *6 months after cross-switch (•), SB2 was considered as effective and safe as CT-P13. AE adverse event, CT-P13 biosimilar infliximab, mo months, PASI Psoriasis Area and Severity Index, SB2 biosimilar infliximab, SD standard deviation
In the real-world study conducted by Harris et al. [122], 133 patients with IBD (CD, 78.9%; UC, 21.1%) switched from CT-P13 to SB2 and were assessed for: disease activity (baseline [Week 0] vs Week 16/18), drug persistence (at ≥ 4 months vs an historic CT-P13 cohort), and specific domains of the IBD-control questionnaire (i.e., patient-reported outcome measures [PROMs] at 0 vs 16/18 weeks) (Fig. 5). The treatment discontinuations after cross-switch were not considered unusual, and there were no significant differences in any of the study endpoints over at least 4 months, or for the specific IBD-control PROMs that were assessed (i.e., Question items: 1b, “…your current treatment is useful in controlling your IBD”; 3f, “…think you need a change to your treatment”; 4a, “…would you like to discuss alternative types of drug”; 4b, “…would you like to discuss ways to adjust your own treatment”; and 4c, “…would you like to discuss side effects or difficulties with your medicine”) [131]. These results were published in abstract form and described as preliminary; additional longer-term data in larger cohorts could provide further assurance of the safety and efficacy of cross-switching in the future.

Study flow and key results of real-world biosimilar cross-switching in patients with inflammatory bowel disease [122]. *Mean values. CD Crohn’s disease, CT-P13 biosimilar infliximab, discont’d discontinued, FU follow-up, HBI Harvey Bradshaw Index, IBD inflammatory bowel disease, mo months, pts patients, R-IFX infliximab reference product, SB2 biosimilar infliximab, Tx treatment, UC ulcerative colitis, wk week(s), y year(s)
In a retrospective, single-center study of 31 patients (22 [71%] males) with CD (11 males [35%]) or UC (20 males [65%]), Lovero et al. [121] evaluated the effects of a cross-switch from CT-P13 to SB2 after a median 6 months of follow-up (Fig. 6). Patients had been diagnosed at median age 35.5 years and had a median duration of disease of 11 years. At baseline, 13 patients were already receiving CT-P13, and 18 patients switched to CT-P13 from R-IFX. Overall, patients were treated with CT-P13 for almost 2 years before the cross-switch. There was one treatment suspension and four AEs during CT-P13 therapy, but none during SB2 therapy after the cross-switch. A loss of response (LoR) occurred in 10 patients (32.2%) after switching to SB2 versus in 9 patients (29%) who were treated with CT-P13 (p = 0.10). Among patients whose cross-switch was their only therapy transition, the risk of LoR was higher based on a univariate analysis, but not per the multivariate analysis. Other univariate factors assessed (i.e., age, age at the start of infliximab treatment, combination therapy, previous biologics, CD, CD location, extra-intestinal manifestations, perianal disease, UC location, indication for infliximab, C-reactive protein, severe clinical or endoscopic activity, surgery, or number of CT-P13 infusions) were not associated with LoR. Overall, switching from CT-P13 to SB2 appeared to be safe and effective; there was no additional risk of AEs or LoR when cross-switching was prefaced by an earlier switch from the reference product. Lovero et al. [121] have published these results in an abstract thus far.

Efficacy and safety of cross-switching between infliximab biosimilars for patients with IBD [121]. *Infusion reactions. AE adverse event, CD Crohn’s disease, CI confidence interval, CRP C-reactive protein, CT-P13 biosimilar infliximab, discont’d discontinued, LoR loss of response, mo months, OR odds ratio, pts patients, R-IFX infliximab reference product, SB2 biosimilar infliximab, Tx treatment, UC ulcerative colitis, y years
Luber et al. [126] evaluated the effects of a switch from CT-P13 to SB2 on disease activity and trough levels in a 1-year, single-center, prospective observational study of 186 patients with IBD who were stabilized on CT-P13 therapy. Patients undergoing a non-medical switch from CT-P13 to SB2 for the first time (n = 87 [CD, n = 56; UC, n = 31]; mean±SD age, 30.7±12.3 years) or the second time (previously switched from infliximab originator; n = 99 [CD, n = 95; UC, n = 4]; mean±SD age, 33.2±12.5 years) were followed for changes in disease activity indices (Harvey Bradshaw Index [HBI] for CD; Simple Clinical Colitis Activity Index [SCCAI] for UC), infliximab trough levels, and C-reactive protein (CRP) from baseline to the time of the third or fourth infusion of SB2 and 1 year. Time to LoR was also measured. Compared with baseline, there were no statistically significant changes in infliximab trough levels, CRP, HBI, and SCCAI at the early timepoint or at 1 year after the switch for both groups (first-time and second-time switchers). The median time to LoR was not significantly different between patients switching for the first or second time. In this study, in which patients were switched from one infliximab biosimilar to another, no AEs were evident on treatment.
Cross-switching reported for a 3-year prospective, observational, usual-care study conducted by Lauret et al. [1] to assess immunogenicity in patients with various chronic inflammatory diseases provides some longer-term data (Fig. 7). Two cohorts were observed: Cohort-1 comprised patients on R-IFX maintenance therapy who 2 years later were switched to CT-P13 (n = 265) and then later switched to SB2 (n = 140); Cohort-2 comprised patients naïve to biologics (n = 44), 29 of whom initiated treatment with CT-P13 before cross-switching to SB2.

Cross-switching immunogenicity study in patients with chronic inflammatory diseases who were on infliximab maintenance (Cohort-1) or who were biologic-naïve (Cohort-2) [1]. a Study flow and results for Cohort-1. *12/30 ADA-positive pts completed the cross-switch; 7/12 gradually became seronegative and 5/12 remained ADA-positive. †5/30 ADA-positive pts continued R-IFX therapy despite undetectable trough and exhibited a good clinical response. ‡20/60 pts (33%) who discontinued treatment were ADA-positive versus 30/205 (15%) pts who reverse-switched (p = 0.002). §Pts (n) who reverse-switched did so after [no. of CT-P13 infusions]: 19 pts [1], 14 [2], 17 [3], and 5 [> 3]. b Study flow and results for Cohort-2. *Among the patients who discontinued treatment because of LoR (primary or secondary), 4 were ADA-positive and 2 halted treatment due to an infusion-related reaction (both ADA-positive). †Cross-switch groups consisted of AxSpA, n = 11 pts; IBD, n = 8; RA, n = 7; PsA, n = 1; uveitis, 0; and other CID, n = 2. ADA antidrug antibody, AE adverse event, AxSpA axial spondyloarthritis, CD Crohn’s disease, CI confidence interval, CID chronic inflammatory disease, CRP C-reactive protein, CT-P13 biosimilar infliximab, discont’d discontinued, IBD inflammatory bowel disease, LoR loss of response, mo months, MTX methotrexate, PsA psoriatic arthritis, RA rheumatoid arthritis, R-IFX infliximab reference product, SB2 biosimilar infliximab, Tx treatment, UC ulcerative colitis, y year
Positive ADAs (≥ 10 ng/mL at two consecutive tests) were detected in 30/265 patients in Cohort-1 (11.3%, all female) at the first infusion of CT-P13. Among the 235 ADA-free patients at baseline in Cohort-1, 20 (8.5%) developed ADAs (the primary endpoint) over the 3-year observation period at the rate of 3 per 100 patient years (CT-P13, n = 10; R-IFX, n = 4; and SB2, n = 6). Disease, age, gender, methotrexate or concomitant disease-modifying antirheumatic drugs were not considered predictive of immunogenicity in the 140 patients in Cohort-1 who later switched from CT-P13 to SB2. Specifically, cross-switching biosimilars did not increase the development of immunogenicity in these patients who switched from CT-P13 to SB2, as shown in the Kaplan-Meier curve (Fig. 8).

COPYRIGHT OWNER]. ADA antidrug antibody, CT-P13 biosimilar infliximab, pts patients, R-IFX infliximab reference product, SB2 biosimilar infliximab
Immunogenicity-free survival from 24 to 36 months among Cohort-1 patients who were ADA-free at baseline and remained on a biosimilar, reverse-switched, or cross-switched [1]. *ADA-positivity for CT-P13 was only 6/145 pts (4%) from baseline to 12 months and 2/140 pts (1.5%) from 12 to 24 months. Modified with permission from Lauret et al. [1]. Effects of successive switches to different biosimilars infliximab on immunogenicity in chronic inflammatory diseases in daily clinical practice, Issue 6, Pages 1449–1456, Copyright (2020), with permission from Elsevier [OR APPLICABLE SOCIETY
Of the 29 patients in Cohort-2 who cross-switched from CT-IFX to SB2, (25%) developed ADAs within three years (the rate was 14/100 patient years). ADA development was observed to increase the rate of treatment discontinuation in Cohort-2, hazard ratio (HR) = 2.79 (95% confidence interval [CI]: 1.04–7.52; p = 0.042). Furthermore, a baseline BMI >30 mg/m2 and trough concentrations of infliximab < 2 µg/mL were predictive of ADA development: HR = 5.54 (95% CI: 1.30–23.65) and HR = 5.53 (95% CI: 1.30–23.43), respectively. Previously, obesity was shown to modify the response to anti-TNFα biologics [132]. The number of successive biosimilars received appeared to have no influence on ADA development in either cohort.
There were limitations associated with the two-cohort study by Lauret et al. (i.e., observational nature, unblinded physicians, relatively small size of Cohort-2, patients in Cohort-1 were stable on R-IFX before switching to a biosimilar, which may not be generalizable). Nevertheless, the study suggests that the development of ADAs—which diminished infliximab trough concentrations—increased the risk of treatment discontinuations, primarily due to primary or secondary LoR, but overall and importantly, immunogenicity was not increased by cross-switching.
Twelve-month interim analysis results are now available for the biosimilar cross-switching evaluated in the ongoing non-interventional, 24-month French PERFUSE real-world cohort study (N = 1374, comprising 874 patients with a gastroenterological disease and 500 patients with a rheumatic disease) [125, 133]. Thus far, the part of the study conducted across 12 gastroenterology centers evaluated 755 patients with IBD who were either IFX-naïve SB2 initiators (92 patients: CD, n = 59; UC, n = 33) or switchers to SB2 from R-IFX or a Bio-IFX (663 patients: CD, n = 521; UC, n = 142) [125]. No meaningful differences have been reported to date among these switchers in terms of average [95% CI] individual change in disease score (CD: prior R-IFX in 157 patients, − 0.3 [− 0.8 to 0.1] and prior Bio-IFX in 83 patients, − 0.2 [− 0.6 to 0.3]; UC: prior R-IFX in 41 patients, − 0.3 [− 1.1 to 0.4] and prior Bio-IFX in 26 patients, 0.0 [− 0.9 to 0.9]). The average SB2 dose required did not change from baseline over 12 months among patients who were switched from R-IFX or from Bio-IFX (mean range 7.0–7.7 mg/kg; 95% CI: 5.0–10.0 for all groups). The vast majority of patients with IBD (> 90%) were safely and successfully continuing with treatment 12 months after switching to SB2 [125].
Among patients with a rheumatology diagnosis in the PERFUSE multicenter study, evaluations occurred after initiating treatment with SB2 (IFX-naïve, n = 116) or after switching to SB2 following R-IFX (n = 172) or a biosimilar IFX other than SB2 (Bio-IFX, n = 212). There were 99 patients with RA of whom 22 (22.2%) were IFX-naïve and initiated SB2, while 37 (37.4%) and 40 (40.4%) switched from R-IFX and another IFX biosimilar (Bio-IFX) to SB2, respectively [133]. Of the 62 patients with psoriatic arthritis who switched to SB2, 14 (22.6%) were IFX-naïve, 24 (38.7%) switched from R-IFX, and 24 (38.7%) switched from a Bio-IFX. Of the 339 patients with ankylosing spondylitis who switched to SB2, 80 (23.6%) were IFX-naïve, while 111 (32.7%) and 148 (43.5%) switched from R-IFX and a Bio-IFX, respectively. The interim 12-month change in disease scores from baseline were not clinically meaningful by disease type among patients naïve to IFX, and the rate of SB2 persistence (95% CI) was generally highest in groups that switched to SB2 and lowest among the patients who were IFX-naïve and initiated SB2 (Fig. 9). All seven SAEs that were reported in this interim analysis were considered unrelated to SB2. This study showed early evidence that cross-switching can be successfully accomplished from a safety perspective, and it does not result in a loss of disease control or lack of persistence relative to a single switch from a reference product to a biosimilar or when initiating therapy with a biosimilar.

SB2 persistence at 12 months among patients in the PERFUSE study by rheumatological disease and prior therapy [133]. *Persistence over 12 months (95 % confidence interval). Bio-IFX→SB2 switched from a biosimilar IFX to SB2 (cross-switched), IFX infliximab, IFX-naïve initiated treatment on SB2, R-IFX→SB2 switched from IFX reference product to SB2, SB2 biosimilar infliximab
In the Italian SCESICS multicenter study, the safety and effectiveness of cross-switching was evaluated in 52 patients with IBD (CD, n = 39; UC, n = 13; 63% males, average age 41 years) who cross-switched from CT-P13 to SB2 after initial treatment with R-IFX (R-IFX→CT-P13→SB2), and in 66 patients who switched from R-IFX to CT-P13 (R-IFX→CT-P13) [124]. IBD was moderate-to-severe in 50% of patients and steroid-dependent in 25% of patients prescribed R-IFX therapy. Overall, discontinuation rates following cross-switching were 2% (95% CI: 0–6.0%) after 24 weeks and 14% (3–25%) after 48 weeks. Six AEs (two each cutaneous and infectious; one each articular and immunological) were reported among four patients after a median (range) follow-up of 40 (18–48) weeks, and three of these patients (6%) discontinued IFX therapy. Twenty-four weeks after cross-switching, 49/52 patients (94%) achieved clinical remission, and by the end of the follow-up period this decreased to 48/52 (92%) patients after a single patient lost response after 24 weeks. No difference was observed in the rates of biosimilar discontinuation, clinical remissions, and AEs between patients who cross-switched versus those who switched from R-IFX→CT-P13. The safety and effectiveness outcomes in this study were not able to be predicted by any identifiable clinical parameters and cross-switching was considered “non-inferior” to a standard R-IFX→CT-P13 switch.
For context, it seems important to highlight the difference in discontinuations occurring in real-world studies versus clinical trials. A systematic literature review revealed a 9.7–28.2% discontinuation rate over an approximate 12 (range, 11–15.8)-month period among patients in real-world settings who received CT-P13 after a non-medical switch from R-IFX [134]. This is in contrast to a 7.5% discontinuation rate in patients with immune-mediated inflammatory diseases who discontinued CT-P13 after switching from R-IFX in the randomized controlled Phase IV NOR-SWITCH study [105]. The most frequent reasons reported for real-world discontinuations were a lack of response and AEs, although a considerable proportion of the reasons were subjective and possibly indicative of patients’ negative expectations of switching or the nocebo effect [134]. According to a systematic review of 31 studies, although the development of ADAs and infusion-related reactions have not been observed to differ between patients in open-label and double-blinded studies of infliximab biosimilars, discontinuation rates have been shown to be higher in open-label versus double-blind studies (14.3% vs 6.95%) owing to intolerability, lack of response, and for any reason, which could suggest the possibility of nocebo effects [135].
Overall, the majority of evidence suggests that switching from a reference drug to a biosimilar, or between biosimilars, is not associated with increased immunogenicity, an escalated or clinically meaningful safety risk, or any appreciable loss of effectiveness [136]. Although there exists a single incidental published case of a patient who experienced AEs as a result of cross-switching, this finding is largely discordant with the existing body of evidence [137]. Additional studies of switching between biosimilars other than CT-P13 and SB2 may address remaining clinical concerns by adding to the scientific evidence. However, their perceived importance should not overshadow the credibility of the entire biosimilarity program, which has already demonstrated that these products are essentially similar versions of the same reference product leading to similar clinical results.
9 Obstacles to Biosimilar Switches
Perceived data gaps (e.g., lack of long-term data, generalizability) after the approval of biosimilars can emerge as obstacles to single switches, from the reference product to its biosimilar, and beyond to cross-switching scenarios that involve biosimilars of the same reference product. To date, a paucity of direct evidence for switching to or between biosimilars, particularly for patients with indications that have been extrapolated, has done little to reassure prescribing physicians who often require robust evidence-based data before making treatment decisions [55, 116, 138]. However, the addition of comparative clinical trials of biosimilars in patients with IBD provides much-needed reassurance [119, 139].
For newly approved biosimilars, the available evidence will be limited to data from RCTs conducted to obtain regulatory approval. Gauging the risk or benefits by extrapolating conclusions from RCTs that tend to have less heterogeneous populations than the general patient population in clinical practice, is a challenge for newly approved medicines. Elderly and pediatric populations, as well as those at increased risk because of comorbidities, polypharmacy, and adverse drug interactions, are likely to be under-represented in most RCTs evaluating therapeutic equivalency of biosimilars [54, 75]. Furthermore, monoclonal antibody biosimilar discontinuation rates are generally higher than anticipated in observational studies than in large RCTs [138, 140]. However, this perceived obstacle is largely mitigated given that the profile is already established for the reference product, and once biosimilarity is established for a biosimilar, it can be anticipated that the risk-benefit profile in the general population will be the same; therefore, assessing the behavior of the biosimilar in special populations is not required. In addition, strong pharmacovigilance practices allow for continued monitoring of AEs when extrapolating to a population not studied in RCTs [64].
Among the challenges associated with cross-switching is the time it will take for real-world data to accumulate for more recently approved biosimilars. Although considered important by some, robust immunological data on cross-switching are mostly missing. In rheumatology, ADAs are neither routinely collected nor recommended to be routinely collected [141, 142]. In addition, the evaluation of the potential impact of switching on immunogenicity development, as measured by onset of detection of ADAs, is challenging. Temporal association between ADA at first detection and the switch could lead to misinterpretations, as the switch could coincide with the time when development of ADA detection is naturally expected. The onset of detection of ADAs among patients with RA occurs mostly within 6 months after initiating treatment with infliximab [143, 144]. For other inflammatory diseases such as IBD, ADAs may develop within 2–3 treatments (i.e., within 8 weeks), and have reportedly persisted for up to a year following the discontinuation of therapy [75, 114]. Temporal data of this nature can complicate interpretations, which is why some professional organizations recommend that any switching for non-medical reasons should not occur within 6 months of a therapeutic transition to another biologic [75].
Despite valid reasons for medical and non-medical cross-switching between biosimilars, many of the publications issued by professional organizations, such as medical societies or task forces that provide recommendations, position or consensus statements, and guidance concerning biosimilars, do not elaborate on cross-switching scenarios that can occur, and these must be managed by physicians in clinical practice [145] (Electronic Supplementary Material, Table S1).
However, the foundation for establishing biosimilarity is the detailed comparison of the proposed biosimilar and the originator product at the analytical and functional level; clinical data are used only as confirmation of biosimilarity [146], and the expectation is that the biosimilar will have the same benefit-risk profile as the originator in all disease indications for which the biosimilar is approved. Hence, an over-reliance on the need for clinical data for the biosimilar in disease indications other than for the clinical setting used to support regulatory approval is outdated and unhelpful.
10 Equipping Prescribers of Biosimilars with Necessary Evidence
Although many challenges exist, the ongoing emergence of real-world evidence should serve to address many of the important clinical practice-related uncertainties that may exist after biosimilars are approved [141] (Fig. 10). Large, real-world observational studies that monitor the long-term safety and immunogenicity of biosimilars, retrospective studies, robust post-marketing surveillance, as well as the active maintenance of national databases and registries, can offer additional direct evidence to clarify some of the practical aspects associated with biosimilar use [147,148,149,150,151]. Although variously described, patient registries are essentially databases containing follow-up longitudinal data collected during the course of usual clinical care. The importance of registries is reflected in obligatory licensing stipulations; for example, multiple registries must be maintained until 2026 for infliximab biosimilar CT-P13 in Europe [152]. There are indications that approximately two-thirds of surveillance studies are never initiated or completed [153]. The Danish Registry for Biological Treatment in Rheumatology (DANBIO) is recognized as one of the more effective registries to enhance the knowledge base for biosimilars [19, 154]. The more diverse the range of patients in a registry, the more generalizable those registry data will be in terms of facilitating real-world decision-making [155]. Published case studies describing unique clinical scenarios and off-label outcomes may also provide insights for prescribers to consider.

Key gaps and next steps related to the adoption of cross-switching
11 Conclusions
Robust evidence supports the efficacy and safety for single switches to a biosimilar from its reference product. Comparative studies of biosimilars of the same reference product are not mandated, since the comprehensive biosimilarity exercises that each proposed biosimilar must undergo before regulatory approval is granted suggests that establishing clinical equivalence between the biosimilars ought to be achievable using educated inductive reasoning complimented by robust pharmacovigilance systems. This is supported by the emerging real-world evidence for switching between biosimilars, with no unexpected findings or concerns being raised from the initial data from cross-switching studies published thus far. However, a better understanding is needed of the scientific concept of biosimilarity and the rationale for different regulatory requirements (i.e., comparatively more physicochemical and non-clinical data, and less clinical data, compared with an originator biologic), in order to build the confidence of treating physicians to prescribe biosimilars successively for their patients with immune-related inflammatory diseases. This will avoid the nocebo effect, that lack of information or knowledge from the treating physician could trickle down to the patient.
Nocebo effects are a possible reason for higher discontinuation rates in usual care but lack scientific validity, real-world observational studies, the use of registries, national databases, and pharmacovigilance surveillance findings all help to clarify the risk/benefit profile associated with biosimilar cross-switching for patients with chronic inflammatory diseases. Wider use of ex vivo methods for assessment of cross reactivity of different biosimilars to ADAs of the reference product may also contribute to allaying theoretical concerns about the potential for increased immunogenicity when switching to a biosimilar from the reference product or from one biosimilar to another.
The option to cross-switch from one biosimilar to another is important in clinical practice. In the absence of clinical trial data, clinicians must attempt to objectively evaluate the emerging real-world cross-switching evidence within the context of the science of biosimilarity. This knowledge, combined with a case-by-case awareness of the patient and their disease, will allow the physician to work together with the pharmaceutical industry, patient advocacy groups, and other healthcare professionals to adopt an educated multidisciplinary approach to treatment decision-making, with this review serving that purpose.
References
Lauret A, Molto A, Abitbol V, Gutermann L, Conort O, Chast F, et al. Effects of successive switches to different biosimilars infliximab on immunogenicity in chronic inflammatory diseases in daily clinical practice. Semin Arthritis Rheum. 2020;50(6):1449–56.
Thomas L. Biosimilars: providing more treatment options. 2019. https://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_7071.pdf. Accessed 5 Nov 2019.
Al Sulais E, AlAmeel T. Biosimilars to antitumor necrosis factor agents in inflammatory bowel disease. Biologics Targets Therapy. 2020;14:1–11.
Kim H, Alten R, Avedano L, Dignass A, Gomollon F, Greveson K, et al. The future of biosimilars: maximizing benefits across immune-mediated inflammatory diseases. Drugs. 2020;80(2):99–113.
Smeeding J, Malone DC, Ramchandani M, Stolshek B, Green L, Schneider P. Biosimilars: considerations for payers. P&T Peer-Rev J Formul Manage. 2019;44(2):54–63.
Milassin Á, Fábián A, Molnár T. Switching from infliximab to biosimilar in inflammatory bowel disease: overview of the literature and perspective. Therep Adv Gastroenterol. 2019;12:1–11.
Moots RJ. Biosimilars in the Americas: the future by consensus. Clin Rheumatol. 2019;38(5):1497–9.
US Food and Drug Administration. Advancing health through innovation. New drug therapy approvals 2019. 2020. https://www.fda.gov/media/134493/download. Accessed 20 Mar 2020.
European Medicines Agency, European Commission. Biosimilars in the EU. Information guide for healthcare professionals. 2017. https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf. Accessed 5 Aug 2019.
World Health Organization. Norms and standards. Biotherapeutics and biosimilars. WHO Drug Infomation. 2015;29(2):138–41.
Kay J. Are there benefits and risks to biosimilars from a patient perspective? Rheum Dis Clin North Am. 2019;45(3):465–76.
US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry. 2015. https://www.fda.gov/media/82647/download. Accessed 4 Mar 2020.
van der Martijn PR, Hoefnagel MHN, Hillege HL, Roes KCB. Pragmatic rules for comparability of biological medicinal products. Biologicals. 2020;63:97–100.
Sackman J, Kuchenreuther M. The bullish outlook for biosimilars. BioPharm Int. 2015;2015:38–41.
Dutta B, Huys I, Vulto AG, Simoens S. Identifying key benefits in European off-patent biologics and biosimilar markets: it is not only about price! BioDrugs. 2020;34:159–70.
Blauvelt A, Lacour JP, Fowler JF Jr, Weinberg JM, Gospodinov D, Schuck E, et al. Phase III randomized study of the proposed adalimumab biosimilar GP2017 in psoriasis: impact of multiple switches. Br J Dermatol. 2018;179(3):623–31.
Park W, Yoo DH, Miranda P, Brzosko M, Wiland P, Gutierrez-Ureña S, et al. Efficacy and safety of switching from reference infliximab to CT-P13 compared with maintenance of CT-P13 in ankylosing spondylitis: 102-week data from the PLANETAS extension study. Ann Rheum Dis. 2017;76(2):346–54.
Yoo DH, Prodanovic N, Jaworski J, Miranda P, Ramiterre E, Lanzon A, et al. Efficacy and safety of CT-P13 (biosimilar infliximab) in patients with rheumatoid arthritis: comparison between switching from reference infliximab to CT-P13 and continuing CT-P13 in the PLANETRA extension study. Ann Rheum Dis. 2017;76(2):355–63.
Glintborg B, Sorensen J, Hetland ML. Does a mandatory non-medical switch from originator to biosimilar infliximab lead to increased use of outpatient healthcare resources? A register-based study in patients with inflammatory arthritis. RMD Open. 2018;4(2):e000710.
Simoens S, Cheung R. Tendering and biosimilars: what role for value-added services? J Mark Access Health Policy. 2020;8(1):1705120.
World Health Organization. Application to add anti-TNFs to the World Health Organization’s essential medicines list. 2021. https://www.who.int/selection_medicines/committees/expert/22/applications/s8.1_TNF-alfa-inhibitors.pdf. Accessed 6 Jul 2021.
Allocati E, Gerardi C. Outcomes of switching from anti-tnf biologic drugs to their biosimilars: a systematic review. Final report November 2020. 2020. https://cdn.who.int/media/docs/default-source/essential-medicines/2021-eml-expert-committee/other-matters/o.7_switching-anti-tnf.pdf?sfvrsn=ddae8024_6. Accessed 7 Jul 2020.
Mehr S. When biosimilars are switched for each other. 2019. https://biosimilarsrr.com/2019/05/23/when-biosimilars-are-switched-for-each-other/. Accessed 20 Sep 2020.
Araújo FC, Fonseca JE, Goncalves J. Switching to biosimilars in inflammatory rheumatic conditions: current knowledge. EMJ Rheumatol. 2018;5(1):66–74.
Numan S, Faccin F. Non-medical switching from originator tumor necrosis factor inhibitors to their biosimilars: systematic review of randomized controlled trials and real-world studies. Adv Ther. 2018;35(9):1295–332.
Isaacs J, Goll GL, Goncalves J, Bosworth A. Biosimilars and switching: what is your perspective? EMJ Rheumatol. 2017;4(1):34–41.
Kay J, Isaacs JD. Clinical trials of biosimilars should become more similar. Ann Rheum Dis. 2017;76(1):4–6.
Lai Z, La Noce A. Key design considerations on comparative clinical efficacy studies for biosimilars: adalimumab as an example. 2018. https://www.syneoshealth.com/sites/default/files/documents/53424_voinv_48_key_design_considerations_case_study_lo4.pdf. Accessed 27 Mar 2020.
McClellan JE, Conlon HD, Bolt MW, Kalfayan V, Palaparthy R, Rehman MI, et al. The “totality-of-the-evidence” approach in the development of PF-06438179/GP1111, an infliximab biosimilar, and in support of its use in all indications of the reference product. Therap Adv Gastroenterol. 2019;12:1756284819852535.
Bhatt V. Current market and regulatory landscape of biosimilars. Am J Manag Care. 2018;24(21 Suppl):S451–6.
Stebbing J, Mainwaring PN, Curigliano G, Pegram M, Latymer M, Bair AH, et al. Understanding the role of comparative clinical studies in the development of oncology biosimilars. J Clin Oncol. 2020;38(10):1070–80.
Azevedo V, Dörner T, Strohal R, Isaacs J, Castañeda-Hernández G, Gonçalves J, et al. Biosimilars: considerations for clinical practice. Considerations Med. 2017;1:13–8.
Regional Medicines Optimisation Committee. Regional Medicines Optimisation Committee briefing best value biologicals: adalimumab update 6. 2019. https://www.sps.nhs.uk/wp-content/uploads/2019/07/Adalimumab-RMOC-Briefing-6.pdf. Accessed 19 Mar 2020.
Houlton S. Benefits and drawbacks of moving to biosimilar medicines. Prescriber. 2019;2019:13–5.
Erskine D, Minshull J. Update on development of biosimilar versions of adalimumab with particular focus on excipients and injection site reactions 2019. https://www.sps.nhs.uk/articles/update-on-development-of-biosimilar-versions-of-adalimumab-with-particular-focus-on-excipients-and-injection-site-reactions/. Accessed 4 Oct 2021.
Ramael S, Van Hoorick B, Tiessen R, van Iersel T, Moschetti V, Lang B, et al. Similar pharmacokinetics of the adalimumab (Humira®) Biosimilar BI 695501 whether administered via subcutaneous autoinjector or prefilled syringe (VOLTAIRE®)-AI and VOLTAIRE®-TAI): phase 1, randomized, open-label, parallel-group trials. Rheumatol Ther. 2018;5(2):403–21.
Sandoz Inc. ZARXIO® (filgrastim-sndz) prescribing information. 2019. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=c0d1c22b-566b-4776-bdbf-00f96dad0cae. Accessed 22 Feb 2021.
Hospira, Inc. (a Pfizer Company). NIVESTYM™ (filgrastim-aafi) prescribing information. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761080s000lbl.pdf Accessed Feb 2021.
Azevedo VF, Babini A, Caballero-Uribe CV, Castañeda-Hernández G, Borlenghi C, Jones HE. Practical guidance on biosimilars, with a focus on Latin America: what do rheumatologists need to know? J Clin Rheumatol. 2019;25(2):91–100.
Bridges SL Jr, White DW, Worthing AB, Gravallese EM, O’Dell JR, Nola K, et al. The science behind biosimilars: entering a new era of biologic therapy. Arthritis Rheumatol. 2018;70(3):334–44.
The Alliance for Safe Biologic Medicines (ASBM). Non-medical switching. 2020. https://safebiologics.org/non-medical-switching/ Accessed 13 Mar 2020.
Mehr S. What happens when switching among biosimilars. 2017. https://biosimilarsrr.com/2017/06/20/what-happens-when-switching-among-biosimilars/ Accessed 13 Mar 2020.
Griffith N, McBride A, Stevenson JG, Green L. Formulary selection criteria for biosimilars: considerations for US health-system pharmacists. Hosp Pharm. 2014;49(9):813–25.
Kim M, Monnard A, da Silva JS. Understanding the opportunity in Japan's biosimilar market. 2019. https://www.mckinsey.com//media/McKinsey/Industries/Pharmaceuticals%20and%20Medical%20Products/Our%20Insights/Understanding%20the%20opportunity%20in%20Japans%20biosimilar%20market/Understanding-the-opportunity-in-Japans-biosimilar-market.pdf?shouldIndex=false. Accessed 18 Nov 2020.
Mahmmod S, Schultheiss JPD, van Bodegraven AA, Dijkstra G, Gilissen LPL, Hoentjen F, et al. Outcome of reverse switching from CT-P13 to originator infliximab in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2021. https://doi.org/10.1093/ibd/izaa364.
Roediger A, Freischem B, Reiland J-B. What pricing and reimbursement policies to use for off-patent biologicals in Europe? Results from the second EBE biological medicines policy survey. GaBI J. 2017;6(2):61–78.
Davio K. Switching among multiple infliximab biosimilars does not cause immunogenicity, study finds. 2019. https://www.centerforbiosimilars.com/conferences/eular-2019/switching-among-multiple-infliximab-biosimilars-does-not-cause-immunogenicity-study-finds. Accessed 28 Jul 2020.
Mack A. Norway, biosimilars in different funding systems. What works? GaBi J. 2015;4:90–2.
Dylst P. The 2017 market review—European biosimilar medicines markets—policy overview. 2017. https://www.medicinesforeurope.com/wp-content/uploads/2017/09/Market-Review-biosimilar-medicines-market-2017.pdf. Accessed 22 Feb 2021.
Singh A, Kalaivani M, Srivastava S, Goyal RK, Gupta SK. Postmarketing safety of biosimilars: current status, challenges, and opportunities in the spontaneous reporting system. Ther Innov Regul Sci. 2019. https://doi.org/10.1177/2168479019872144.2168479019872144.
Frapaise F-X. Biosimilars in the age of patient-centricity. GaBi Online. 2020. http://gabionline.net/Biosimilars/Research/Biosimilars-in-the-age-of-patient-centricity. Accessed 20 Sep 2020.
Krant J. Biosimilar confidence: physician-patient communication sets the tone. 2017. https://www.rheumatologyadvisor.com/home/topics/rheumatoid-arthritis/biosimilar-confidence-physician-patient-communication-sets-the-tone/. Accessed 19 Mar 2020.
Peyrin-Biroulet L, Danese S, Cummings F, Atreya R, Greveson K, Pieper B, et al. Anti-TNF biosimilars in Crohn’s disease: a patient-centric interdisciplinary approach. Expert Rev Gastroenterol Hepatol. 2019;13(8):731–8.
Moots R, Azevedo V, Coindreau JL, Dörner T, Mahgoub E, Mysler E, et al. Switching between reference biologics and biosimilars for the treatment of rheumatology, gastroenterology, and dermatology inflammatory conditions: considerations for the clinician. Curr Rheumatol Rep. 2017;19(6):37.
NHS England and NHS improvement. What is a biosimilar medicine? 2019. https://www.england.nhs.uk/wp-content/uploads/2019/05/what-is-a-biosimilar-medicine-guide-v2.pdf. Accessed 19 Mar 2020.
GaBi Online. Switching approaches to biosimilars in Nordic countries. 2017. http://gabionline.net/Reports/Switching-approaches-to-biosimilars-in-Nordic-countries. Accessed 21 Sep 2020.
Robinson J. Majority of patients not asked for consent before being switched to a biosimilar, survey finds. Pharma J. 2019;303:7928.
GaBi Online. Biosimilars approved in Europe. 2020. http://www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe. Accessed 21 Sep 2020.
AJMC: The Center for Biosimilars. Kathy Oubre: other players complicate biosimilar use for cancer centers. 2020. https://www.centerforbiosimilars.com/interviews/kathy-oubre-other-players-complicate-biosimilar-use-for-cancer-centers. Accessed 21 Sep 2020.
US Food and Drug Administration. Considerations in demonstrating interchangeability with a reference product. Guidance for industry. 2019. https://www.fda.gov/media/124907/download. Accessed 19 Jun 2019.
US Food and Drug Administration. Biosimilar and interchangeable biologics: more treatment choices. 2020. https://www.fda.gov/consumers/consumer-updates/biosimilar-and-interchangeable-biologics-more-treatment-choices. Accessed 27 Jul 2020.
GaBi Online. 45 US states have passed biosimilar substitution laws. 2017. http://gabionline.net/Policies-Legislation/45-US-states-have-passed-biosimilar-substitution-laws#:~:text=To%20date%2C%2045%20US%20states,and%20the%20District%20of%20Columbia. Accessed 21 Sep 2020.
US Food and Drug Administration. Prescribing biosimilar products. 2020. https://www.fda.gov/media/108103/download. Accessed 27 Jul 2020.
Vermeer NS, Giezen TJ, Zastavnik S, Wolff-Holz E, Hidalgo-Simon A. Identifiability of biologicals in adverse drug reaction reports received from European clinical practice. Clin Pharmacol Ther. 2019;105(4):962–9.
European Medicines Agency. Guideline on good pharmacovigilance practices (GVP). Product- or population-specific considerations II: biological medicinal products products. 2016. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-good-pharmacovigilance-practices-gvp-product-population-specific-considerations-ii_en-0.pdf. Accessed 7 Jul 2021.
GI Society: Canadian Society of Intestinal Research. Biologics and biosimilars. 2020. https://badgut.org/information-centre/a-z-digestive-topics/biosimilars-pamphlet/. Accessed 9 Mar 2020.
Danese S, Bonovos S, Peyrin-Biroulet L. Biosimilars in IBD: from theory to practice. Nat Rev Gastroenterol Hepatol. 2017;14:22–31.
Le Berre C, Danese S, Peyrin-Biroulet L. Timely use of biologics in early Crohn’s disease: the return of “hit hard and early”? Digest Dis Sci. 2019;64(11):3035–7.
Sung YK, Jung SY, Kim H, Choi S, Im SG, Lee YS, et al. Factors for starting biosimilar TNF inhibitors in patients with rheumatic diseases in the real world. PLoS ONE. 2020;15(1):e0227960.
Leonard E, Wascovich M, Oskouei S, Gurz P, Carpenter D. Factors affecting health care provider knowledge and acceptance of biosimilar medicines: a systematic review. J Manag Care Spec Pharm. 2019;25(1):102–12.
Barker J, Girolomoni G, Egeberg A, Goncalves J, Pieper B, Kang T. Anti-TNF biosimilars in psoriasis: from scientific evidence to real-world experience. J Dermatolog Treat. 2020;31(8):794–800.
Zheng MK, Shih DQ, Chen GC. Insights on the use of biosimilars in the treatment of inflammatory bowel disease. World J Gastroenterol. 2017;23(11):1932–43.
Gonczi L, Ilias A, Szanto K, Kurti Z, Golovics PA, Farkas K, et al. Mo1892—non-medical reverse switch between the originator infliximab and its biosimilar in patients with inflammatory bowel disease: clinical outcomes and therapeutic drug monitoring. Gastroenterology. 2019;156:S876–7.
Davio K. Switching back to reference infliximab from biosimilar appears effective for patients with IBD. 2019. https://www.centerforbiosimilars.com/news/switching-back-to-reference-infliximab-from-biosimilar-appears-effective-for-patients-with-ibd. Accessed 9 Mar 2020.
Danese S, Fiorino G, Raine T, Ferrante M, Kemp K, Kierkus J, et al. ECCO position statement on the use of biosimilars for inflammatory bowel disease-an update. J Crohns Colitis. 2017;11(1):26–34.
Toussirot E, Marotte H. Switching from originator biological agents to biosimilars: what is the evidence and what are the issues? RMD Open. 2017;3(2):e000492.
Danese S, Peyrin-Biroulet L. IBD: to switch or not to switch: that is the biosimilar question. Nat Rev Gastroenterol Hepatol. 2017;14(9):508–9.
Schulze-Koops H, Skapenko A. Biosimilars in rheumatology: a review of the evidence and their place in the treatment algorithm. Rheumatol (Oxf). 2017;56(4 Suppl):iv30–48.
Feagan BG, Lam G, Ma C, Lichtenstein GR. Systematic review: efficacy and safety of switching patients between reference and biosimilar infliximab. Aliment Pharmacol Ther. 2019;49(1):31–40.
Murdaca G, Negrini S, Greco M, Schiavi C, Giusti F, Borro M, et al. Immunogenicity of infliximab and adalimumab. Expert Opin Drug Safety. 2019;18(5):343–5.
Goncalves J, Myung G, Park M, Jeong D, Ghil J. SB5 shows cross-immunogenicity to adalimumab but not infliximab: results in patients with inflammatory bowel disease or rheumatoid arthritis. Therap Adv Gastroenterol. 2019;12:1756284819891081.
Gisbert JP, Chaparro M. Switching from an originator anti-TNF to a biosimilar in patients with inflammatory bowel disease: can it be recommended? A systematic review. Gastroenterol Hepatol. 2018;41(6):389–405.
Kurti Z, Gonczi L, Lakatos PL. Progress with infliximab biosimilars for inflammatory bowel disease. Expert Opin Biol Ther. 2018;18(6):633–40.
Chu R, Tortensson D, Pugatch M. Patient safety and comfort: the challenges of switching medicine. Stockholm Netw. 2010;2010.
Medicines for Europe. The total clinical experience with biosimilar medicines exceeds 2 billion patient treatment days. 2020. https://www.medicinesforeurope.com/wp-content/uploads/2020/12/BIOS5.pdf. Accessed 7 Jul 2021.
Weise M. From bioequivalence to biosimilars: how much do regulators dare? Z Evid Fortbild Qual Gesundhwes. 2019;140:58–62.
D’Amore C, Da Cas R, Rossi M, Traversa G. Switching between epoetins: a practice in support of biosimilar use. BioDrugs. 2016;30(1):27–32.
Guerra Veloz MF, Arguelles-Arias F, Castro Laria L, Maldonado Perez B, Benitez Roldan A, Perea Amarillo R, et al. Loss of efficacy and safety of the switch from infliximab original to infliximab biosimilar (CT-P13) in patients with inflammatory bowel disease. World J Gastroenterol. 2018;24(46):5288–96.
Arato T. Japanese regulation of biosimilar products: past experience and current challenges. Br J Clin Pharmacol. 2016;82(1):30–40.
Health Canada. Biosimilar biologic drugs. 2020. https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/biosimilar-biologic-drugs.html. Accessed 18 Nov 2020.
Bielsky MC, Cook A, Wallington A, Exley A, Kauser S, Hay JL, et al. Streamlined approval of biosimilars: moving on from the confirmatory efficacy trial. Drug Discov Today. 2020. https://doi.org/10.1016/j.drudis.2020.09.006.
Doevendans E, Schellekens H. Immunogenicity of innovative and biosimilar monoclonal antibodies. Antibodies (Basel). 2019;8(1).
Groopman J, Hartzband P. Framing risks, benefits perilous for physicians and patients. 2012. https://www.acpinternist.org/archives/2012/01/gray.htm. Accessed 11 Mar 2020.
Webster CJ, Woollett GR. Comment on “Analysis of pharmacokinetic and pharmacodynamic parameters in EU-versus US-licensed reference biological products: are in vivo bridging studies justified for biosimilar development?” BioDrugs. 2019;33(5):581–2.
Tu C-L, Wang Y-L, Hu T-M, Hsu L-F. Analysis of pharmacokinetic and pharmacodynamic parameters in EU- versus US-licensed reference biological products: are in vivo bridging studies justified for biosimilar development? BioDrugs. 2019;33(4):437–46.
Webster CJ, Wong AC, Woollett GR. An efficient development paradigm for biosimilars. BioDrugs. 2019;06(33):603–11.
Gerrard TL, Johnston G, Gaugh DR. Biosimilars: extrapolation of clinical use to other indications. GaBi J. 2015;4(3):118–24.
Markus R, Liu J, Ramchandani M, Landa D, Born T, Kaur P. Developing the totality of evidence for biosimilars: regulatory considerations and building confidence for the healthcare community. BioDrugs. 2017;31(3):175–87.
US Food and Drug Administration. Biosimilar and interchangeable products. 2017. https://www.fda.gov/drugs/biosimilars/biosimilar-and-interchangeable-products. Accessed 29 Sep 2019.
US Food and Drug Administration. Development of therapeutic protein biosimilars: comparative analytical assessment and other quality-related considerations. Guidance for industry [DRAFT GUIDANCE] 2019. https://www.fda.gov/media/125484/download. Accessed 27 Mar 2020.
Yuan J, Xu WW, Poon H. Analytical approach for biosimilar development: special focus on monoclonal antibody biosimilars. Int J Biopharm Sci. 2018;1(2):1–4.
Pipalava P, Patel R, Mehta M, Dahiya M, Singh I, Jose V. An update on the animal studies conducted for biosimilar approvals—regulatory requirement vs actual scenario. Regul Toxicol Pharmacol. 2019;107:104415.
Beyer B, Walch N, Jungbauer A, Lingg N. How similar is biosimilar? A comparison of infliximab therapeutics in regard to charge variant profile and antigen binding affinity. Biotechnol J. 2019;14(4):e1800340.
Isaacs J, Gonçalves J, Strohal R, Castañeda-Hernández G, Azevedo V, Dörner T, et al. The biosimilar approval process: how different is it? Considerations Med. 2017;1(1):3–6.
Jørgensen KK, Olsen IC, Goll GL, Lorentzen M, Bolstad N, Haavardsholm EA, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet. 2017;389(10086):2304–16.
Cohen HP, Blauvelt A, Rifkin RM, Danese S, Gokhale SB, Woollett G. Switching reference medicines to biosimilars: a systematic literature review of clinical outcomes. Drugs. 2018;78(4):463–78.
Ebbers HC, Schellekens H. Are we ready to close the discussion on the interchangeability of biosimilars? Drug Discov Today. 2019;24(10):1963–7.
Kaplan GG, Ma C, Seow CH, Kroeker KI, Panaccione R. The argument against a biosimilar switch policy for infliximab in patients with inflammatory bowel disease living in Alberta. J Can Assoc Gastroenterol. 2020;3(5):234–42.
Davio K. An expert view on immunogenicity and biosimilars. 2019. https://www.centerforbiosimilars.com/conferences/biotech-pharma-summit-biosimilars/an-expert-view-on-immunogenicity-and-biosimilars. Accessed 29 Jul 2020.
Ben-Horin S, Yavzori M, Benhar I, Fudim E, Picard O, Ungar B, et al. Cross-immunogenicity: antibodies to infliximab in Remicade-treated patients with IBD similarly recognise the biosimilar Remsima. Gut. 2016;65(7):1132–8.
Ruiz-Argüello MB, Maguregui A, del Ruiz AA, Pascual-Salcedo D, Martínez-Feito A, Jurado T, et al. Antibodies to infliximab in Remicade-treated rheumatic patients show identical reactivity towards biosimilars. Ann Rheum Dis. 2016;75(9):1693–6.
Kurki P, van Aerts L, Wolff-Holz E, Giezen T, Skibeli V, Weise M. Interchangeability of biosimilars: a European perspective. BioDrugs. 2017;31(2):83–91.
Fleischmann RM, Alten R, Pileckyte M, Lobello K, Hua SY, Cronenberger C, et al. A comparative clinical study of PF-06410293, a candidate adalimumab biosimilar, and adalimumab reference product (Humira®) in the treatment of active rheumatoid arthritis. Arthritis Res Ther. 2018;20(1):178.
Fiorino G, Ruiz-Arguello MB, Maguregui A, Nagore D, Correale C, Radice S, et al. Full interchangeability in regard to immunogenicity between the infliximab reference biologic and biosimilars CT-P13 and SB2 in inflammatory bowel disease. Inflamm Bowel Dis. 2018;24(3):601–6.
Ooi CJ, Hilmi I, Banerjee R, Chuah SW, Ng SC, Wei SC, et al. Best practices on immunomodulators and biologic agents for ulcerative colitis and Crohn’s disease in Asia. Intest Res. 2019;17(3):285–310.
Mayoral-Zavala A, Esquivel-Aguilar A, Del Real-Calzada CM, Gutierrez-Grobe Y, Ramos-Garcia J, Rocha-Ramirez JL, et al. Update on biosimilars in inflammatory bowel disease: position and recommendations in Mexico. Rev Gastroenterol Mex. 2018;83(4):414–23.
Pineda C, Castañeda Hernández G, Jacobs IA, Alvarez DF, Carini C. Assessing the immunogenicity of biopharmaceuticals. BioDrugs. 2016;30(3):195–206.
Strand V, Gonçalves J, Hickling TP, Jones HE, Marshall L, Isaacs JD. Immunogenicity of biosimilars for rheumatic diseases, plaque psoriasis, and inflammatory bowel disease: a review from clinical trials and regulatory documents. BioDrugs. 2020;34(1):27–37.
de Ridder L, Assa A, Bronsky J, Romano C, Russell RK, Afzal NA, et al. Use of biosimilars in pediatric inflammatory bowel disease: an updated position statement of the Pediatric IBD Porto Group of ESPGHAN. J Pediatr Gastroenterol Nutr. 2019;68(1):144–53.
Declerck P, Bakalos G, Zintzaras E, Barton B, Schreitmüller T. Monoclonal antibody biosimilars in oncology: critical appraisal of available data on switching. Clin Ther. 2018;40(5):798–809.e2.
Lovero R, Losurdo G, Biscaglia G, Valvano MR, Biancofiore A, Martino G, et al. P.07.13 Safety and efficacy of infliximab biosimilar SB2 in patients with inflammatory bowel disease who underwent a switch from infliximab biosimilar CT-P13. Dig Liver Dis. 2019;51:e227.
Harris C, Harris R, Young D, McDonnell M, Harvey J, Felwick R, et al. IBD biosimilar to biosimilar infliximab switching study: preliminary results. United Eur Gastroenterol J. 2019;7(8):361.
Gisondi P, Virga C, Girolomoni G. P049. Cross-switch from CT-P13 to SB2 infliximab biosimilars in patients with chronic plaque psoriasis. J Eur Acad Dermatol Venereol. 2019;33(3 Suppl):29.
Mazza S, Fascì A, Casini V, Ricci C, Munari F, Pirola LL, et al. P360 Safety and clinical efficacy of double switch from originator infliximab to biosimilars CT-P13 and SB2 in patients with inflammatory bowel diseases (SCESICS); a multicentre study. J Crohn’s Colitis. 2020;14(1 Suppl):S342.
Bouhnik Y, Fautrel B, Desjeux G, Freudensprung U, Brigui A, Addison J. P637 PERFUSE: a French non-intervential cohort study of infliximab-naive and trnasitioned patients receiving infliximab biosimilar SB2; an interim analysis. J Crohn’s Colitis. 2020;14:S529.
Luber RP, O’Neill R, Singh S, Sharma E, Cunningham G, Honap S, et al. An observational study of switching infliximab biosimilar: no adverse impact on inflammatory bowel disease control or drug levels with first or second switch. Aliment Pharmacol Ther. 2021. https://doi.org/10.1111/apt.16497.
Davio K. Switching to biosimilars in the NHS provides savings, presents unique challenges. 2018. https://www.centerforbiosimilars.com/conferences/smi-biosimilars/switching-to-biosimilars-in-the-nhs-provides-savings-presents-unique-challenges. Accessed 16 Mar 2020.
Magro F, Rocha C, Vieira AI, Sousa HT, Rosa I, Lopes S, et al. The performance of Remicade®-optimized quantification assays in the assessment of Flixabi® levels. Therap Adv Gastroenterol. 2018;11:1756284818796956.
Magro F, Rocha C, Vieira AI, Sousa HT, Rosa I, Lopes I, et al. P229 The new biosimilar of infiximab SB2 can be quantifed by IFX-optimised therapeutic drug monitoring assays. J Crohn’s Colitis. 2018;12(1 Suppl):S216.
Ebbers HC, Fehrmann B, Ottosen M, Hvorslev N, Hoier P, Hwang JW, et al. Batch-to-batch consistency of SB4 and SB2, etanercept and infliximab biosimilars. BioDrugs. 2020;34(2):225–33.
Bodger K, Ormerod C, Shackcloth D, Harrison M, Collaborative IBDC. Development and validation of a rapid, generic measure of disease control from the patient’s perspective: the IBD-control questionnaire. Gut. 2014;63(7):1092–102.
Singh S, Facciorusso A, Singh AG, Vande Casteele N, Zarrinpar A, Prokop LJ, et al. Obesity and response to anti-tumor necrosis factor-alpha agents in patients with select immune-mediated inflammatory diseases: a systematic review and meta-analysis. PLoS ONE. 2018;13(5):e0195123.
Fautrel B, Bouhnik Y, Desjeux G, Brigui A, Freudensprung U, Addison J. THU0164 PERFUSE: a French prospective/retrospective non-interventional cohort study of infliximab-naiive and transitioned patients receiving infliximab biosimilar SB2; an interim analysis. Ann Rheum Dis. 2020;79(1 Suppl):297.
Bakalos G, Zintzaras E. Drug discontinuation in studies including a switch from an originator to a biosimilar monoclonal antibody: a systematic literature review. Clin Ther. 2019;41(1):155–73.e13.
Odinet JS, Day CE, Cruz JL, Heindel GA. The biosimilar nocebo effect? A systematic review of double-blinded versus open-label studies. J Manag Care Spec Pharm. 2018;24(10):952–9.
Barbier L, Ebbers HC, Declerck P, Simoens S, Vulto AG, Huys I. The efficacy, safety, and immunogenicity of switching between reference biopharmaceuticals and biosimilars: a systematic review. Clin Pharmacol Ther. 2020;108(4):734–55.
Pagnini C, Di Paolo MC, De Angelis G, Torcolacci F, Milano M, Trinca D, et al. Similar but not identical: plaque psoriasis exacerbation in a patient with Crohn’s disease after switching from ct-p13 to sb2 infliximab biosimilar. Inflamm Bowel Dis. 2020;26(8):e83–4.
Cantini F, Benucci M. Focus on biosimilar etanercept—bioequivalence and interchangeability. Biologics. 2018;12:87–95.
Ye BD, Pesegova M, Alexeeva O, Osipenko M, Lahat A, Dorofeyev A, et al. Efficacy and safety of biosimilar CT-P13 compared with originator infliximab in patients with active Crohn’s disease: an international, randomised, double-blind, phase 3 non-inferiority study. Lancet. 2019;393(10182):1699–707.
Souto A, Maneiro JR, Gomez-Reino JJ. Rate of discontinuation and drug survival of biologic therapies in rheumatoid arthritis: a systematic review and meta-analysis of drug registries and health care databases. Rheumatol (Oxf). 2016;55(3):523–34.
Ebbers HC, Pieper B, Issa A, Addison J, Freudensprung U, Rezk MF. Real-world evidence on etanercept biosimilar SB4 in etanercept-naive or switching patients: a systematic review. Rheumatol Ther. 2019;6(3):317–38.
Kay J, Schoels MM, Dorner T, Emery P, Kvien TK, Smolen JS, et al. Consensus-based recommendations for the use of biosimilars to treat rheumatological diseases. Ann Rheum Dis. 2018;77(2):165–74.
Bendtzen K, Geborek P, Svenson M, Larsson L, Kapetanovic MC, Saxne T. Individualized monitoring of drug bioavailability and immunogenicity in rheumatoid arthritis patients treated with the tumor necrosis factor alpha inhibitor infliximab. Arthritis Rheum. 2006;54(12):3782–9.
Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, et al. Anti-infliximab and anti-adalimumab antibodies in relation to response to adalimumab in infliximab switchers and anti-tumour necrosis factor naive patients: a cohort study. Ann Rheum Dis. 2010;69(5):817.
Faccin F, Tebbey P, Alexander E, Wang X, Cui L, Albuquerque T. The design of clinical trials to support the switching and alternation of biosimilars. Expert Opin Biol Ther. 2016;16(12):1445–53.
Wolff-Holz E, Tiitso K, Vleminckx C, Weise M. Evolution of the EU biosimilar framework: past and future. BioDrugs. 2019;33(6):621–34.
Fiorino G, Caprioli F, Daperno M, Mocciaro F, Principi M, Viscido A, et al. Use of biosimilars in inflammatory bowel disease: a position update of the Italian Group for the Study of Inflammatory Bowel Disease (IG-IBD). Dig Liver Dis. 2019;51(5):632–9.
Ebbers HC, Muenzberg M, Schellekens H. The safety of switching between therapeutic proteins. Expert Opin Biol Ther. 2012;12(11):1473–85.
El Zorkany B, Al Ani N, Al Emadi S, Al Saleh J, Uthman I, El Dershaby Y, et al. Biosimilars in rheumatology: recommendations for regulation and use in Middle Eastern countries. Clin Rheumatol. 2018;37(5):1143–52.
Wiland P, Batko B, Brzosko M, Kucharz EJ, Samborski W, Swierkot J, et al. Biosimilar switching—current state of knowledge. Reumatologia. 2018;56(4):234–42.
Kay J, Dorner T, Emery P, Kvien TK, Breedveld FC. Clinical trial and “real-world” data support switching from a bio-originator to its biosimilar. Ann Rheum Dis. 2020;79(4):e44.
Uhlig T, Goll GL. Reviewing the evidence for biosimilars: key insights, lessons learned and future horizons. Rheumatol (Oxf). 2017;56(4):iv49–62.
Sutka R, Pec J, Pecova T. Biosimilar medicines and patient registries—expectations, limitations, and opportunities. Acta Med Martiniana. 2018;17(3):39–51.
Edwards C, Hetland ML, Kristensen LE, Cuadrado M. Switching patients from originator to biosimilar medications in rheumatoid arthritis: limiting the “nocebo” effect. Eur Med J. 2017;2017:42–8.
Yoshida K, Radner H, Kavanaugh A, Sung YK, Bae SC, Kishimoto M, et al. Use of data from multiple registries in studying biologic discontinuation: challenges and opportunities. Clin Exp Rheumatol. 2013;31(4 Suppl 78):S28–32.
Acknowledgements
Medical writing support was provided by Sue Reinwald of Engage Scientific Solutions and was funded by Pfizer.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was funded by Pfizer.
Conflicts of interest
BI, DA, and MM are full-time employees of and own stock or options in Pfizer. NI was a full-time employee of Pfizer at the time the manuscript was developed and owns stock or options in Pfizer. EM received or has pending grants from Roche, Pfizer, Bristol-Myers Squibb and Novartis received honorarium from Eli Lilly, Pfizer, GlaxoSmithKline, Roche, Sanofi, AstraZeneca, Sandoz, Amgen, Gemmene, and AbbVie provided writing assistance, medicines, equipment, or administrative support to Pfizer, AbbVie, and Roche, and received payment for lectures including service on speakers’ bureaus to Eli Lilly, Pfizer, GlaxoSmithKline, Roche, Sanofi, AstraZeneca, Sandoz, Amgen, Gema Biotech, and AbbVie. VFA has received grants to conduct trials on biosimilars from Boehringer Ingelheim, consulting fees related to advisory boards from Pfizer, Amgen, and Sandoz, and payment for lectures provided to Pfizer, Amgen, Sandoz, Celltrion, and Janssen. SD reports consultancy fees from AbbVie, Allergan, Amgen, AstraZeneca, Athos Therapeutics, Biogen, Boehringer Ingleheim, Celgene, Celltrion, Eli Lilly, Enthera, Ferring Pharmaceuticals Inc., Gilead, Hospira, Inotrem, Janssen, Johnson & Johnson, MSD, Mundipharma, Mylan, Pfizer, Roche, Sandoz, Sublimity Therapeutics, Takeda, TiGenix, UCB Inc., and Vifor. LP-B served as a speaker, consultant, and advisory member for AbbVie, Allergan, Alma, Amgen, Applied Molecular Transport, Arena, Biogen, Bristol-Myers Squibb, Boehringer Ingelheim, Celgene, Celltrion, Enterome, Enthera, Ferring, Fresenius Kabi SwissBioSim GmbH, Genentech, Gilead, Hikma, Index Pharmaceuticals, Janssen, Eli Lilly, MSD, Mylan, Nestle, Norgine, Oppilan Pharma, OSE Immunotherapeutics, Pfizer, Pharmacosmos, Roche, Samsung Bioepis, Sandoz, Sterna, Sublimity Therapeutics, Takeda, Tillots, and Vifor, received grants from AbbVie, MSD, and Takeda, and owns stock options from CTMA.
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent to participate
Not applicable.
Data availability
Not applicable.
Authors’ contributions
All authors participated in the structuring of this review, contributed to drafting and reviewing the work critically for important intellectual content, and approved the final submitted version.
Consent for publication
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Mysler, E., Azevedo, V.F., Danese, S. et al. Biosimilar-to-Biosimilar Switching: What is the Rationale and Current Experience?. Drugs 81, 1859–1879 (2021). https://doi.org/10.1007/s40265-021-01610-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01610-1