
Abstract
Transthyretin (TTR) transports the retinol-binding protein–vitamin A complex and is a minor transporter of thyroxine in blood. Its tetrameric structure undergoes rate-limiting dissociation and monomer misfolding, enabling TTR to aggregate or to become amyloidogenic. Mutations in the TTR gene generally destabilize the tetramer and/or accelerate tetramer dissociation, promoting amyloidogenesis. TTR-related amyloidoses are rare, fatal, protein-misfolding disorders, characterized by formation of soluble aggregates of variable structure and tissue deposition of amyloid. The TTR amyloidoses present with a spectrum of manifestations, encompassing progressive neuropathy and/or cardiomyopathy. Until recently, the only accepted treatment to halt progression of hereditary TTR amyloidosis was liver transplantation, which replaces the hepatic source of mutant TTR with the less amyloidogenic wild-type TTR. Tafamidis meglumine is a rationally designed, non-NSAID benzoxazole derivative that binds with high affinity and selectivity to TTR and kinetically stabilizes the tetramer, slowing monomer formation, misfolding, and amyloidogenesis. Tafamidis is the first pharmacotherapy approved to slow the progression of peripheral neurologic impairment in TTR familial amyloid polyneuropathy. Here we describe the mechanism of action of tafamidis and review the clinical data, demonstrating that tafamidis treatment slows neurologic deterioration and preserves nutritional status, as well as quality of life in patients with early-stage Val30Met amyloidosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The transthyretin (TTR)-related amyloidoses are rare, fatal protein misfolding and aggregation disorders. Historically, the TTR amyloidoses were grouped into three categories: (1) wild-type TTR amyloidosis (TTRwt), formerly known as senile systemic amyloidosis, presenting as a cardiomyopathy and involving the aggregation of wild-type (WT) TTR; (2) familial amyloid polyneuropathy (FAP); and (3) hereditary TTR amyloid cardiomyopathy, formerly known as familial amyloid cardiomyopathy (FAC). Both hereditary forms involve aggregation of primarily mutant TTR. Current knowledge suggests that the TTR amyloidoses do not fit neatly into these categories, as patients often exhibit a spectrum of phenotypes depending on mutation, genetic background, and extent of progression. Non-fibrillar aggregates and amyloid composed of mutant and/or WT TTR of hepatic origin accumulate in various tissues and organs, particularly the peripheral and autonomic nerves and heart, leading to tissue damage and end-organ failure [1–3]. TTR gene expression by the choroid plexus and retinal pigmented epithelium leads to leptomeningeal and ocular degeneration linked to TTR amyloidogenesis, respectively [4].
Tafamidis (2-(3,5-dichloro-phenyl)-benzoxazole-6-carboxylic acid) is a once-daily, orally bioavailable, non-NSAID small molecule, and specific kinetic stabilizer of TTR [5]. In clinical studies, tafamidis slowed peripheral neurologic impairment and additional clinical manifestations in patients with Val30Met amyloidosis and was associated with an excellent safety profile [6–8]. Tafamidis is the first pharmacotherapy approved to treat TTR-FAP in the European Union and several countries in Latin America and the Asia-Pacific including Argentina and Japan.
The aim of this article is to focus on TTR as a drug target, to provide the rationale used in the development of tafamidis, and review comprehensively the pharmacology of this disease-modifying therapy for TTR amyloidoses. This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Structure and Functions of TTR
Transthyretin, also known as prealbumin, is a soluble, β-sheet rich, 127-amino acid, non-glycosylated protein primarily synthesized and secreted into the blood by the liver. TTR circulates in the blood predominantly as a 55 kDa tetramer, along with a very small amount of dissociated monomer [6].
The monomeric subunits of TTR fold into a β-sandwich tertiary structure that spontaneously assembles into a tetramer within the cellular endoplasmic reticulum. The tetramer exhibits two distinct dimer–dimer interfaces, the less stable of which makes up two highly conserved thyroxine (T4) binding sites [9–13]. The three-dimensional structures of the TTR tetramer bound to T4 and its other known ligand, the vitamin A–retinol-binding protein complex, have been solved [14–16].
Transthyretin is expressed from embryonic development through adult life and its blood concentration over this time reflects those of total body nitrogen and total body potassium levels [17]. Serum TTR concentration so closely mimics the body’s nitrogen balance that it has been widely used as a marker of malnutrition and inflammation in several conditions [17–19]. In fact, TTR is a negative acute phase reactant [20]. In adult humans, plasma TTR concentrations range from 3 to 6 μM; the TTR concentration in cerebrospinal fluid is an order of magnitude lower (approximately 0.36 µM) [21–23]. The half-life of TTR in vivo blood is 24–48 h [22, 24, 25].
In blood, the TTR tetramer binds retinol-binding protein bound to retinol (holo-retinol-binding protein) and a small amount of T4, and transports them to tissues on the minute-to-hour timescale [9, 26, 27]. Although TTR is the only known transporter of circulating holo-retinol-binding protein that facilitates retinol transport, two other proteins, namely, albumin and thyroid binding globulin transport thyroid hormones. Of the three T4 binding proteins, albumin has by far the highest plasma concentration and thyroid binding globulin has the highest affinity for T4 [28, 29]. Thus, given the low level of circulating T4, less than 10% of T4 in blood is bound to TTR and more than 99% of the T4 binding sites in circulating TTR are vacant [21]. Thus, TTR is thought to play only a minor role in the physiological transport of T4. In contrast, TTR is the primary T4 carrier in cerebrospinal fluid [10].
In addition, transthyretin may be a natural detoxifier, as its T4 binding sites bind small molecules with substituted aromatic rings connected by linkers of variable chemical composition [30]. These structures include stilbenes, flavones, benzoxazoles, tetrahydroquinolines, dihydropyridines, benzodiazepines, and phenoxazines [31, 32]. Emerging evidence suggests that WT TTR may also have a protective effect in Alzheimer’s disease through an interaction with the amyloidogenic Aβ-amyloid peptide [30]. TTR has also been shown to influence nerve physiology and to enhance nerve regeneration in the mouse [33]. A role for TTR in nerve biology and repair may explain the preferential deposition of mutant TTR aggregates in the peripheral nervous system of patients with TTR-FAP [33].
Molecular Mechanism of TTR Amyloidogenesis
Experiments with cell lines and Drosophila melanogaster model systems demonstrate the proteotoxicity associated with various TTR sequences linked to pathology [3, 34–39]. Biophysical in vitro studies suggest that tetrameric TTR protein does not form amyloid fibrils and that amyloidogenesis starts with TTR tetramer dissociation into dimers, which then rapidly dissociate to folded monomers (Fig. 1a) [23, 40–45]. Amyloid fibrillogenesis appears to require partial denaturation of released monomers, which exposes stretches of hydrophobic residues that misassemble into spherical aggregates which then undergo conformational conversion into cross-β sheet assemblies [46–49]. In vitro, amyloid formation by TTR monomers proceeds in an energetically downhill polymerization process that is fast compared with the initial tetramer dissociation [50]. Thus, tetramer dissociation is thought to be rate limiting for TTR amyloid formation in the case of the WT protein and most tetramers produced by heterozygotes comprising mutant and WT TTR subunits [45].

TTR amyloidogenesis and TTR stabilization by tafamidis (a). The TTR amyloidogenic cascade. TTR is a tetrameric protein that can be destabilized by mutations in TTR, resulting in its dissociation into monomers that rapidly misfold and misassemble into aggregates, including pathologic structures associated with TTR amyloidosis [23, 40–45]. Also shown are the molecular structure of tafamidis (b) and the co-crystal structure of tafamidis (black, ball-and-stick) bound to a TTR tetramer (ribbon) (c). TTR transthyretin
Most amyloidogenic TTR variants form approximately 6% homotetramers and approximately 87% variant/WT heterotetramers with reduced kinetic or thermodynamic stabilities relative to WT homotetramers (approximately 6%), which renders them more prone to tetramer dissociation and amyloid formation [43, 45]. Oxidative modifications have also been found to energetically destabilize the tetrameric structure and to increase the amyloidogenic potential of TTR protein in vitro [51, 52]. Given the well-established increase in protein oxidation with ageing, this may contribute to senile forms of TTR amyloidosis [52].
Increasing evidence indicates that proteolytic cleavage may also play a role in TTR amyloid formation. Analyses of ex vivo TTR amyloid fibrils have revealed that, in addition to full-length protein, amyloid deposits in patients with TTRwt amyloidosis contain a high proportion of C-terminal TTR fragments starting at amino acid residues 46, 49, 52, or 53 [53–55]. Similarly, in patients with hereditary TTR amyloidosis, amyloid fibrils with N-terminally truncated TTR were observed more frequently than amyloid fibrils containing predominantly full-length TTR protein [55–57]. Based on the small N-terminal cleavage products detected at very low amounts in amyloid fibrils, it has been speculated that proteolytic cleavage may occur prior to and play a role in fibrillogenesis [53, 54]. A recent study suggests that, at least for the Ser52Pro (p.Ser72Pro) variant, proteolytic cleavage of the Lys48–Thr49 peptide bond may indeed be a prefibrillar event in vivo which produces a C-terminal fragment that, once released from the TTR tetramer, is highly fibrillogenic at physiological pH in vitro [58].
Clinical Overview of TTR Amyloidoses
The hereditary TTR amyloidoses are rare, autosomal dominant diseases caused by point mutations in the TTR gene [27, 59–62]. More than 100 destabilizing mutations of TTR have been identified [63]. The propensity for a particular TTR mutant to undergo amyloidogenesis in vitro depends on the thermodynamic and kinetic destabilization of the tetramer caused by the substitution [23, 27, 42–45]. However, the most amyloidogenic TTR mutants (e.g., Asp18Gly [p.Asp38Gly] and Ala25Thr [p.Ala45Thr]) do not cause the most severe, early-onset systemic amyloid disease because they are degraded by the quality control system located in the endoplasmic reticulum of liver cells [27, 45]. Thus, for these two mutants, the concentration of destabilized mutant TTR in blood is insufficient to result in systemic amyloidogenesis; however, late-onset central nervous system (CNS) pathology is observed [64, 65].
Although the main pathologic manifestation of hereditary TTR amyloidosis is a progressive ascending polyneuropathy and/or cardiomyopathy, mixed phenotypes, extreme phenotypic variability, and incomplete penetrance often obscure a proper diagnosis for physicians not considering TTR amyloidosis [62, 66–69]. As a consequence, hereditary TTR amyloidosis is under-recognized and often misdiagnosed.
Most known cases of TTR-FAP result from the Val30Met (p.Val50Met) mutation, which is particularly frequent in regions of Portugal, Japan, Sweden, and Brazil [68, 70]. Importantly, age at onset of symptoms and disease penetrance of the Val30Met mutation is very heterogeneous [70–72]. TTR-FAP is characterized by a length-dependent sensory, motor, and autonomic neuropathy [68]. Gastrointestinal symptoms are a common complication of autonomic neuropathy. Amyloid deposits, typically found in ganglia, the endoneurium, and nerve blood vessels, lead to the reduction of nerve fiber density and eventual destruction of unmyelinated nerve fibers, and then small and large myelinated nerve fibers [68, 73]. Distal axonal degeneration is the main pathogenic feature. Possible underlying mechanisms that are not mutually exclusive include: (1) a combination of toxic and mechanical effects of fibrillar amyloid deposits on nerve fibers [68, 74]; and/or (2) the proteotoxicity of non-amyloid TTR aggregates [2, 3]. Clinical manifestations of cardiac involvement may follow later in the course of disease; however, echocardiography in patients with only neurologic symptoms may reveal features of cardiac amyloidosis and electrocardiography can reveal conduction defects early in the course of the condition [75, 76]. In some TTR mutants such as Thr60Ala (p.Thr80Ala), cardiac involvement may be seen early or as the first phenotypic manifestation [77], early and frequent carpal tunnel syndrome, predominant negative sensory symptoms at onset, significant vibration deficits, as well as a non-length-dependent progression of motor deficit are also characteristic [78]; while for patients with the Ser77Tyr mutation, the distinguishable features are late-onset (>50 years) of disease and frequent ataxic phenotype [79]. Neurologic and cardiac deterioration progress relentlessly during active TTR amyloidogenesis, typically leading to death within 5–15 years of symptom onset [66]. It is worth noting that for patients with the Val30Met mutation and whose disease onset occurs at an older age (>50 years), studies of the disease history revealed that the initial manifestation of amyloidosis was somatic neuropathy, and the mean duration of the disease onset to death was 7.3 years [76]. However, for many late-onset cases, due to the inconspicuous nature of the symptoms (i.e., autonomic dysfunctions and sensory dissociation), they are often misdiagnosed as other neuropathies [75]. In addition, distinctive clinical presentation has been found in late-onset cases with the Val30Met mutation from non-endemic areas that differs from that of conventional early-onset cases in endemic foci [79, 80]. Consequently, late-onset cases are still under-recognized and an appropriate diagnosis may benefit from examinations for subclinical cardiac amyloidosis.
In the exclusive or predominant cardiac phenotype, known as hereditary TTR amyloid cardiomyopathy, amyloid is found throughout the cardiovascular system, including the valves, ventricles, atria, and the conduction system, leading to left ventricular wall thickening with a non-dilated left ventricle and impaired longitudinal contraction, and resulting in congestive heart failure and arrhythmias [67, 73, 81]. Survival data for hereditary TTR amyloid cardiomyopathy are not as well documented as for TTR-FAP, but the prognosis is known to be poor with median survival from diagnosis ranging from 25.6 to 36.4 months in three small series [82–84], and with death primarily from heart failure or sudden death [85]. The most common mutation associated with TTR amyloid cardiomyopathy is Val122Ile (p.Val142Ile), which is found in up to 4% of African Americans [86, 87]. The level of penetrance of this mutation has yet to be fully elucidated [87]; 10% of Afro-Caribbean patients with acute decompensated heart failure carry the Val122Ile mutation [88]. Other pathogenic TTR mutations are less prevalent, and often lead to a more ‘mixed’ phenotype of neurologic and multi-organ system involvement [67].
In TTRwt amyloidosis, WT TTR tetramers dissociate into monomers, misfold, and undergo amyloidogenesis. Biopsies from gastrointestinal tract and subcutaneous tissue of TTRwt amyloidosis patients confirmed amyloid deposits in 44% (4/9) of the samples [89]. At autopsy, up to 22–25% of individuals >80 years of age, and 32% of adults over the age of 75 with heart failure and a preserved ejection fraction, have demonstrable TTR amyloid deposits in cardiac tissue (mainly found inside the ventricular wall in a patchy plaque-like shape) albeit usually mild in extent [85, 89, 90]. The process is not well understood but seems to be related to ageing and possibly to destabilizing post-translational modifications. Cardiac amyloidosis is the primary feature of those exhibiting active TTRwt amyloidosis, generally a late-onset sporadic condition primarily affecting men [62]. In two separate observational studies, the prevalence of sensory neuropathy among patients with symptomatic TTRwt amyloidosis was around 28% and nearly half had a history of carpal tunnel syndrome apparently owing to amyloid deposition in the connective tissue of the transverse carpal ligament [69, 84].
Rationale for Development of Tafamidis
Three separate but reinforcing clinical observations suggested that molecules stabilizing the TTR tetramer may be effective in treating the TTR amyloidoses. First, in 1990, Holmgren et al. [91] showed that it was possible to halt the progression of polyneuropathy in patients with TTR-FAP by orthotopic liver transplantation using donors carrying two WT TTR genes, which replaces mutant TTR with a native allele, affording a more stable and less amyloidogenic TTR tetramer. Second, a clinical observation showed that a domino recipient of a liver graft from a patient with Val30Met amyloidosis developed amyloidosis [92]. Third, in the 1990s, it was observed that some Portuguese individuals with symptomatic TTR-FAP had uncharacteristically mild symptoms [93]. Gene sequencing studies revealed that these individuals are compound heterozygotes harboring a Val30Met mutation on allele 1 and a Thr119Met (p.Thr139Met) mutation on allele 2, conferring protection against disease development [93]. Incorporation of Thr119Met subunits into tetramers containing disease-associated subunits increases the kinetic stability of the tetramer, slowing tetramer dissociation and suppressing disease [42, 44, 93, 94].
Binding of either T4 or holo-retinol-binding protein stabilizes the TTR tetramer, preventing its dissociation into amyloidogenic monomers [95, 96]. Notably, approximately half of TTR circulating in blood lacks a bound ligand. These observations provided a strong rationale to develop small molecule ligands that bind to the largely unoccupied T4 binding pockets and stabilize the native TTR tetramer as a potential pharmacotherapy to prevent TTR amyloidogenesis [13].
Numerous, structurally distinct, small molecules that bind to the T4 binding sites were found to inhibit amyloid fibril formation in vitro, by kinetically stabilizing the quaternary structure of the TTR tetramer [13, 23, 44]. These TTR kinetic stabilizers included certain non-steroidal anti-inflammatory drugs (NSAIDs) such as diflunisal [23, 97, 98]. However, since NSAIDs are associated with gastrointestinal, renal, and cardiac side effects [99], attention turned to the benzoxazole carboxylic acids. Of these, tafamidis (Fig. 1b, c) was selected for clinical development due to its lack of NSAID-activity, good oral bioavailability, and low toxicity profile.
Clinical Pharmacokinetics of Tafamidis
Tafamidis is highly bound to plasma proteins (binding rate >99.5%) with glucuronidation as the major metabolic pathway, and the major circulating form in plasma is parent compound. In healthy volunteers, tafamidis was rapidly absorbed after a single oral dose of 20 mg. The median time (T max) to reach the maximum concentration (C max) was approximately 2.0 h. Administration of tafamidis with food reduces the rate of absorption, but overall extent of absorption is not affected. Pharmacokinetic (PK) parameters were similar after single and repeated dosing, indicating a lack of metabolic induction or inhibition by tafamidis. Elimination of tafamidis is slow, with a mean half-life of approximately 59 h. Steady state (SS) was achieved by day 14 after once-daily dosing with 20 mg tafamidis for 14 days, with C max(ss) and C min(ss) at 2.7 and 1.6 μg/mL, respectively (Table 1) [100, 101].
Based on population PK analysis, SS clearance of tafamidis was similar in subjects with creatinine clearance <80 and >80 mL/min. However, compared with healthy volunteers, patients with moderate hepatic impairment (Child–Pugh Score of 7–9 inclusive) had decreased systemic exposure (by approximately 40%) of tafamidis. In subjects >60 years old, clearance following repeated tafamidis 20 mg once daily was 19% slower and the C max(ss) was 21% higher compared with those <60 years old [100].
TTR Stabilization by Tafamidis
Nonclinical in vitro and ex vivo experiments have demonstrated that tafamidis binds with selectivity, high affinity, and negative cooperativity (K D1 ~ 2 nM, K D2 ~ 200 nM) to recombinant WT TTR and to TTR in human plasma [5, 102]. Tafamidis exhibits comparable potency against dissociation of WT TTR and heterotetramers of >35 TTR mutants, including the two most common amyloidogenic mutants, Val30Met and Val122Ile [5].
As stated in previous publications [5, 102], an indirect means of detecting TTR monomer formation under physiologic conditions is to employ subunit exchange experiments [42, 103, 104], in which, two homotetramers composed of WT TTR subunits with and without the N-terminal negatively charged FLAG-tag were mixed, and time-dependent subunit exchange was then quantified using ion-exchange chromatography at physiologic pH. Since TTR tetramer dissociation should be slowed by tafamidis, the subunit exchange between the untagged WT TTR homotetramer and the FLAG-tagged WT TTR homotetramer would be slowed in the presence of tafamidis. Experiments showed that subunit exchange was complete in the vehicle control after 96 h of incubation at 25°C when the pH was 7.0; while in the presence of tafamidis, the fraction of subunit exchange was considerably lower. For instance, at a tafamidis:TTR tetramer molar ratio of 1, the subunit exchange was 15% after 24 h compared with 70% in the absence of tafamidis; and when the molar ratio was 1.50, the fraction of exchange was <10% after 11 days [5]. Based on these findings, it has been concluded that tafamidis decreases the rate of tetramer dissociation at physiologic pH in a dose-dependent manner.
By measuring TTR tetramers in plasma, an immunoturbidimetric assay [98] (for details see the supplementary material) was used in nonclinical and clinical studies to measure a key pharmacodynamic endpoint as it demonstrated that tafamidis stabilized TTR mutants ex vivo [5]. In this assay, 4.8 μM urea was added to plasma samples for over 48 h, which was followed by glutaraldehyde crosslinking, and then the remaining tetrameric TTR was quantified using a TTR antibody [5, 98, 102]. In the absence of tafamidis and the presence of urea, TTR tetramers dissociate and irreversibly unfold into monomers, which are extensively modified by glutaraldehyde and thus are not detected by TTR antibody precipitation. In the presence of tafamidis, plasma samples are resistant to TTR tetramer dissociation induced by urea and epitope modification by glutaraldehyde. The immunoturbidity signals are preserved since the TTR tetramer structure is preserved. Therefore, the percentage of TTR stabilization can be calculated as:
where FOI (fraction of initial) is the percentage decrease in TTR immunoturbidity of the plasma samples after 48-h incubation with urea, FOI baseline shows tetramer dissociation at baseline while FOI dosed shows tetramer dissociation after tafamidis dosing. After assessment of random variation in TTR stabilization in placebo-treated healthy volunteers, a cut-off value of 32% was chosen, above which TTR tetramers are considered to have been stabilized by tafamidis. Nonclinical studies suggested that a 1–1.5:1 tafamidis:TTR stoichiometry should be sufficient to stabilize TTR and inhibit tetramer dissociation in human plasma (Pfizer, data on file). In two clinical studies of healthy volunteers with orally administered tafamidis, mean percent TTR stabilization increased with increasing plasma tafamidis:TTR stoichiometry and appeared to reach a plateau (Fig. 2), but at ratios higher than projected from nonclinical data.

Percent change from baseline in TTR tetramer fold stabilization relative to tafamidis:TTR molar ratio in the plasma of healthy volunteers. The data presented originate from two Phase 1, randomized, double-blind, ascending dose tolerance studies. In study Fx-002, healthy adults aged 18–65 years with a body mass index of 19–29 kg/m2 administered single doses of up to 120 mg tafamidis. In study B3461040 (ClinicalTrials.gov identifier, NCT01655511), healthy adults aged 21–55 years with a body mass index of 17.5–30.5 kg/m2 administered single doses of up to 480 mg tafamidis. TTR transthyretin
TTR Tetramer Stabilization in Plasma of TTR Amyloidosis Patients
An ex vivo study on plasma samples from 28 patients with TTR amyloidosis showed that tafamidis can increase the stability of the native tetrameric structure of multiple amyloidogenic TTR variants. Eligible subjects had been diagnosed with TTR amyloidosis and had a documented non-Val30Met, non-Val122Ile TTR mutation, and study protocol and informed consent documentation were approved by the relevant institutional review boards. The stability of tetrameric TTR in the plasma samples [TTR baseline concentration range 4.2–29.6 mg/dL (0.76–5.38 µM)] was compared in the presence or absence of 7.2 µM tafamidis, using the immunoturbidimetric assay. Tafamidis increased the stability of the TTR tetramer in 26 of 27 TTR mutants (Table 2) [105]. One mutant (Pro24Ser) demonstrated equivocal results in vitro. TTR stabilization was also found in patients with TTRwt amyloidosis who were treated with tafamidis (20 mg orally, once daily). Compared with the untreated TTRwt amyloidosis patients and untreated age-matched controls, those treated with tafamidis showed a >73% decrease in the tetramer dissociation rate, and the rate of tetramer dissociation (as measured by subunit exchange) correlated strongly with tafamidis concentration in the plasma (R 2 = 0.929) [102]. Evidence showed that tafamidis binds and stabilizes the functional TTR protein (i.e., the tetrameric form) and the stabilized tetramers are less likely to dissociate into monomers; however, the relationship between increased TTR stabilization and the increase in concentration of circulating functional TTR requires further investigation [7, 8, 102].
Therapeutic Use
Transthyretin stabilization, efficacy, and safety of treatment with oral tafamidis 20 mg once daily have to date been evaluated in four completed clinical trials in polyneuropathy and cardiomyopathy (Table 3) [7, 8, 106, 107]:
-
1.
The pivotal Fx-005 study, a randomized, placebo-controlled, 18-month trial in 128 patients with early-stage Val30Met-FAP, showed that tafamidis successfully increased TTR tetramer stability in 98% of the tafamidis group throughout the 18-month treatment period and this increase in TTR stability was associated with a reduction in neurologic deterioration, preservation of nerve function (large and small fibers), improved nutritional status, and maintained quality of life after 18 months compared with placebo group (Fig. 3) [7].
Fig. 3 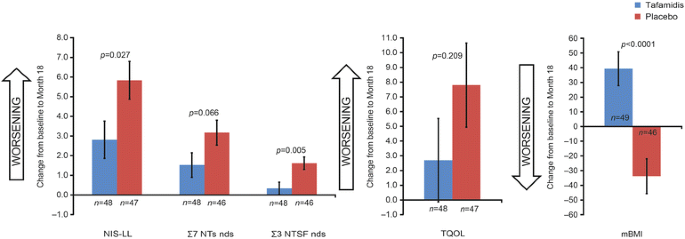
Least square mean (±standard error) change from baseline to Month 18 in efficacy measures (Fx-005, ITT population) [7]. Least square mean estimates and p values are based on a repeated measures analysis of variance model using observed values with change from baseline as the dependent variable. Σ7 NTs nds summated 7 nerve tests normal deviates, Σ3 NTSF nds summated 3 nerve tests small-fiber normal deviates, ITT intention to treat, mBMI modified body mass index, NIS-LL neuropathy impairment score—lower limb, TQOL total quality of life
-
2.
In Study Fx-006, a 12-month, open-label extension study to Study Fx-005, where 86 of the 91 patients who had completed the Month 18 visit of Fx-005 either continued to receive tafamidis (tafamidis–tafamidis) or were switched to tafamidis (placebo–tafamidis) for 12 months, tafamidis conferred increased TTR tetramer stability in >93% of participants. Moreover, the observed slowing of disease progression among the tafamidis-treated patients of Fx-005 was extended to 30 months in the tafamidis–tafamidis cohort and the rate of disease progression was reduced in the placebo–tafamidis group over the 12-month active treatment period (Fig. 4) [8].
-
3.
Fx1A-201, a Phase 2, open-label study in 21 patients with TTR-FAP due to eight different non-Val30Met TTR mutations, demonstrated general tolerability and efficacy of tafamidis on TTR tetramer stability in >94% of the polyneuropathy patients with other TTR genotypes. These patients were older, had longer disease duration, and more severe disease—frequently including cardiac abnormalities—at baseline than the Fx-005 population [106].
-
4.
Fx1B-201, a Phase 2, open-label, 12-month study (ClinicalTrials.gov identifier, NCT00694161), found that tafamidis was associated with an acceptable safety profile and successfully increased the kinetic stability of TTR tetramers in the vast majority (96.8%) of 35 patients with Val122Ile or TTRwt amyloidosis [107].


Patients in these trials had variable characteristics and severity of symptoms at baseline. Therefore, the differences observed in clinical efficacy and safety across these trials may be confounded by the patients’ underlying disease and comorbid conditions.
Safety
Adverse Events
The risk of adverse events (AEs) is difficult to evaluate based on the small number of patients who received tafamidis to date. The available clinical trial data indicate that tafamidis was generally well tolerated, with the type and frequency of the most commonly reported AEs partly dependent on the primary phenotype and clinical status of the patient in addition to any drug effects. Many of the AEs with an overall incidence >5% are of a similar nature as the symptoms of the underlying disease.
In the Fx-005 study of patients with early-stage Val30Met-FAP, the overall incidences of any AE, AE leading to drug discontinuation, and serious AEs were similar in the tafamidis and placebo treatment groups [7]. Furthermore, the overall incidence rates of AEs among the polyneuropathy patients in studies Fx-005, Fx-006, and Fx1A-201 were similar [7, 8, 106]. Differences in incidence rates of individual AEs across these studies were generally not clinically meaningful. One exception was treatment-emergent urinary tract infection (UTI): 15 of 65 tafamidis-treated patients (23%) in Fx-005 reported a UTI compared with 8 of 63 placebo-treated patients (13%) [7]. UTIs were also reported by 12 of 85 tafamidis-treated patients (14%) in Fx-006 [8], and by 3 of 35 patients (9%) in Fx1B-201 [107]. In contrast, none of the 21 patients who received tafamidis in the open-label Fx1A-201 study developed a UTI [106]. Additionally, a higher incidence of diarrhea was reported in the tafamidis group than in the placebo group in the pivotal Fx-005 study (17 of 65 [26.2%] vs. 11 of 63 [17.5%]) [7]. Diarrhea events were also reported by 7 of 85 patients (8.2%) in Fx-006 [8], by 5 of 21 patients (23.8%) in Fx1A-201 [106], and by 7 of 35 patients (20.0%) in Fx1B-201 [107]. Also of note was the incidence of patients experiencing a fall in Fx-005 (0 of 65 tafamidis-treated patients vs. 1 of 63 placebo-treated patients), Fx-006 (2 of 85 patients), Fx1A-201 (5 of 21 patients), and Fx1B-201 (3 of 35 patients).
In the Fx1B-201 study of TTR amyloid cardiomyopathy, 15 of 35 patients had a serious AE, the most common being cardiac disorders (12 patients) including heart failure (9 patients) and atrial fibrillation (3 patients) [107]. Four patients reported six SAEs that were considered possibly related to tafamidis, namely, syncope, ataxia, falls (reported by two patients), hemorrhagic stroke, and cardiac heart failure [107].
Deaths
In the Fx-005 study, five Val30Met-FAP patients (two receiving tafamidis, three receiving placebo) died, all after liver transplant [7]. These deaths were considered unrelated to the study drug. No deaths were reported in Fx-006 or Fx1A-201 [8, 106].
Two TTRwt amyloidosis patients died during the Fx1B-201 study (causes of death were hemorrhagic stroke and amyloid light-chain [AL] amyloidosis) [107]. These deaths were considered unrelated to the study drug. The patient with AL amyloidosis had been enrolled in the study based on positive amyloid biopsy, absence of evidence of a plasma cell dyscrasia, and positive TTR staining by immunogold electron microscopy. After an unexplained, rapid decline at 11 months, a re-biopsy demonstrated amyloid positive for immunoglobulin light chain and no evidence of TTR deposition, after which the patient withdrew from the study, and died from complications due to pleurodesis treatment for AL amyloidosis. Subsequent re-evaluation of the biopsies using mass spectrometry demonstrated that both had amyloid deposits derived from light chains, without evidence of TTR.
Discussion
The nonclinical and clinical studies described here demonstrate that tafamidis kinetically stabilizes TTR tetramers thereby slowing TTR tetramer dissociation. TTR tetramer disassembly is the rate-limiting step of amyloidogenesis and its inhibition is expected to inhibit the amyloidogenesis cascade and, in turn, to slow disease progression. Strong evidence for this concept was that tafamidis treatment slowed neurologic disease progression in patients with Val30Met amyloidosis compared with placebo [7], and by the fact that modest deterioration in neurologic function was observed in patients with TTR-FAP caused by non-Val30Met mutations and who were treated with tafamidis [106].
Tafamidis increases the kinetic stability of a large number of TTR variants, as well as for TTRwt, suggesting that the tetramer stabilization effect may extend to many of the >100 TTR genotypes associated with amyloidosis. Furthermore, tafamidis proved effective in stabilizing TTR throughout the treatment period in a number of study populations, differing in severity of disease, phenotype(s), and TTR genotype. While TTR tetramer stabilization along with patient- and disease-related factors may predict clinical response, placebo data in Val30Met-FAP patients suggest that lack of TTR tetramer stabilization predicts clinical nonresponse.
In patients with early-stage Val30Met amyloidosis who were treated with tafamidis, the rate of neurologic deterioration was slowed, quality of life was maintained, and nutritional status was improved, compared with placebo-treated patients [7, 108]; and TTR stabilization was associated with slower disease progression [8]. There was also some indication that 12 months of open-label tafamidis might improve some signs and symptoms of TTR-FAP associated with non-Val30Met mutations in older patients with more advanced disease [106]. Considering the mechanism of action and the available results on TTR stabilization, the tafamidis effect on disease progression is expected to extend to TTR amyloid polyneuropathy due to non-Val30Met mutations [106].
Long-term study demonstrated that the clinical benefits experienced by Val30Met-FAP patients receiving tafamidis were sustained for the duration of the study (30 months for Study Fx-006) [8]. Furthermore, there was an advantage of initiating treatment earlier, or at an earlier stage of disease, although those starting treatment later still experienced patterns of slowed disease progression [8], suggesting that earlier initiation of tafamidis therapy may result in improved clinical outcomes. These results underscore the need to identify subjects early and treat Val30Met-FAP soon after the diagnosis is made. Moreover, the summary of product characteristics in Europe states: “Treatment should be initiated by and remain under the supervision of a physician knowledgeable in the management of patients with transthyretin amyloid polyneuropathy” [100].
Noticeably, across the different groups of tafamidis-treated patients with TTR amyloidosis, including those with cardiomyopathy and a small number of patients with renal impairment, the AEs (e.g., UTI, respiratory infections, diarrhea, headache, pain, and vomiting) were generally mild to moderate in severity, with no major safety concerns identified [7, 8, 106, 107]. Overall, tafamidis 20 mg once daily is safe and well tolerated.
In cardiomyopathy patients receiving open-label tafamidis, measures of disease progression did not deteriorate markedly during 12 months [107] warranting further evaluation of tafamidis in TTR amyloid cardiomyopathy. In addition, it has been reported that increasing the dose of tafamidis appears to be safe [109, 110]. Currently, a multicenter, double-blind, placebo-controlled, randomized, Phase 3 study is being conducted (ClinicalTrials.gov identifier, NCT01994889) to investigate the efficacy, safety, and tolerability of tafamidis 20 or 80 mg once daily for patients with TTR cardiomyopathy.
While tafamidis treatment may present benefits to patients with polyneuropathy or cardiomyopathy, it is unlikely that CNS manifestations of TTR amyloidosis, such as leptomeningeal or ocular amyloidosis, would be effectively treated with oral tafamidis therapy, as the drug does not penetrate extensively into the brain or eye in rats. Rather, tafamidis accumulates preferentially in the bloodstream and in the liver, which is the primary source of mutant TTR.
Tafamidis is the only currently approved pharmacotherapy for the treatment of early-stage TTR-FAP, with ongoing clinical investigation for TTR-FAC. The favorable tolerability profile and suitability for long-term use, further underscore its benefits in patients with TTR amyloidosis. Diagnosing patients as early as possible for the initiation of tafamidis therapy before substantial accumulation of amyloid in the nerves and organs is of paramount importance.
Following regulatory approvals in the European Union and several countries in Latin America and the Asia-Pacific, tafamidis has emerged as a new standard of care for patients with TTR-FAP. Prior to tafamidis, the only disease-modifying treatment available for TTR amyloidosis was liver transplantation [111–117]. However, liver transplantation is an invasive procedure requiring lifelong immunosuppressive therapy and has limited applicability to a carefully selected cohort of primarily Val30Met patients (non-elderly) with early-stage disease, and good nutritional status [111, 117–119].
Summary
Tafamidis is a well-tolerated and efficacious pharmacotherapy for the treatment of early-stage TTR-FAP. It effectively stabilizes TTR across a number of amyloidogenic TTR genotypes, including WT TTR, and slows disease progression. In patients with early-stage Val30Met amyloid polyneuropathy, tafamidis delayed neurological deterioration, preserved nutritional status and quality of life. A Phase 3, randomized, placebo-controlled trial (ClinicalTrials.gov identifier, NCT01994889) is currently underway to investigate the efficacy, safety, and tolerability of tafamidis in patients with TTR amyloid cardiomyopathy. The long-term efficacy and safety of tafamidis for periods up to 5–8 years appears favorable, based on extension studies from the tafamidis clinical trial program and real-world experience in hundreds of patients to date.
References
Cohen AS, Calkins E. Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature. 1959;183:1202–3.
Sousa MM, Cardoso I, Fernandes R, Guimarães A, Saraiva MJ. Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: evidence for toxicity of nonfibrillar aggregates. Am J Pathol. 2001;159:1993–2000.
Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci USA. 2004;101:2817–22.
Petersen RB, Goren H, Cohen M, Richardson SL, Tresser N, Lynn A, Gali M, Estes M, Gambetti P. Transthyretin amyloidosis: a new mutation associated with dementia. Ann Neurol. 1997;41:307–13.
Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, Packman J, Powers ET, Wiseman RL, Foss TR, Wilson IA, Kelly JW, Labaudinière R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA. 2012;109:9629–34.
Buxbaum JN. Evaluation of tafamidis as first-line therapeutic agent for transthyretin familial amyloidotic polyneuropathy. Degener Neurol Neuromuscul Dis. 2012;2:165–73.
Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Planté-Bordeneuve V, Lozeron P, Suhr OB, Campistol JM, Conceição IM, Schmidt HH, Trigo P, Kelly JW, Labaudinière R, Chan J, Packman J, Wilson A, Grogan DR. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79:785–92.
Coelho T, Maia LF, da Silva AM, Cruz MW, Planté-Bordeneuve V, Suhr OB, Conceição I, Schmidt HH, Trigo P, Kelly JW, Labaudinière R, Chan J, Packman J, Grogan DR. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260:2802–14.
Blake CC, Geisow MJ, Oatley SJ, Rérat B, Rérat C. Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 Å. J Mol Biol. 1978;121:339–56.
Schreiber G, Southwell BR, Richardson SJ. Hormone delivery systems to the brain-transthyretin. Exp Clin Endocrinol Diabetes. 1995;103:75–80.
Foss TR, Kelker MS, Wiseman RL, Wilson IA, Kelly JW. Kinetic stabilization of the native state by protein engineering: implications for inhibition of transthyretin amyloidogenesis. J Mol Biol. 2005;347:841–54.
Foss TR, Wiseman RL, Kelly JW. The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry (Mosc). 2005;44:15525–33.
Johnson SM, Wiseman RL, Sekijima Y, Green NS, Adamski-Werner SL, Kelly JW. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidoses. Acc Chem Res. 2005;38:911–21.
Raz A, Goodman DS. The interaction of thyroxine with human plasma prealbumin and with the prealbumin-retinol-binding protein complex. J Biol Chem. 1969;244:3230–7.
Andrea TA, Cavalieri RR, Goldfine ID, Jorgensen EC. Binding of thyroid hormones and analogues to the human plasma protein prealbumin. Biochemistry (Mosc). 1980;19:55–63.
Monaco HL. Three-dimensional structure of the transthyretin-retinol-binding protein complex. Clin Chem Lab Med. 2002;40:1229–36.
Ingenbleek Y, Young VR. Significance of transthyretin in protein metabolism. Clin Chem Lab Med. 2002;40:1281–91.
Potter MA, Luxton G. Transthyretin measurement as a screening tool for protein calorie malnutrition in emergency hospital admissions. Clin Chem Lab Med. 2002;40:1349–54.
Myron Johnson A, Merlini G, Sheldon J, Ichihara K. Clinical indications for plasma protein assays: transthyretin (prealbumin) in inflammation and malnutrition. Clin Chem Lab Med. 2007;45:419–26.
Ritchie RF, Palomaki GE, Neveux LM, Navolotskaia O, Ledue TB, Craig WY. Reference distributions for the negative acute-phase serum proteins, albumin, transferrin and transthyretin: a practical, simple and clinically relevant approach in a large cohort. J Clin Lab Anal. 1999;13:273–9.
Purkey HE, Dorrell MI, Kelly JW. Evaluating the binding selectivity of transthyretin amyloid fibril inhibitors in blood plasma. Proc Natl Acad Sci USA. 2001;98:5566–71.
Beck FK, Rosenthal TC. Prealbumin: a marker for nutritional evaluation. Am Fam Physician. 2002;65:1575–8.
Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: from delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012;421:185–203.
Ando Y, Tanaka Y, Nakazato M, Ericzon BG, Yamashita T, Tashima K, Sakashita N, Suga M, Uchino M, Ando M. Change in variant transthyretin levels in patients with familial amyloidotic polyneuropathy type I following liver transplantation. Biochem Biophys Res Commun. 1995;211:354–8.
Schmidt HH, Nashan B, Pröpsting MJ, Nakazato M, Flemming P, Kubicka S, Böker K, Pichlmayr R, Manns MP. Familial amyloidotic polyneuropathy: domino liver transplantation. J Hepatol. 1999;30:293–8.
Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science. 1995;268:1039–41.
Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr Pharm Des. 2008;14:3219–30.
Bartalena L, Robbins J. Variations in thyroid hormone transport proteins and their clinical implications. Thyroid. 1992;2:237–45.
Bartalena L, Robbins J. Thyroid hormone transport proteins. Clin Lab Med. 1993;13:583–98.
Buxbaum JN, Reixach N. Transthyretin: the servant of many masters. Cell Mol Life Sci. 2009;66:3095–101.
Klabunde T, Petrassi HM, Oza VB, Raman P, Kelly JW, Sacchettini JC. Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol. 2000;7:312–21.
Connelly S, Choi S, Johnson SM, Kelly JW, Wilson IA. Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr Opin Struct Biol. 2010;20:54–62.
Fleming CE, Saraiva MJ, Sousa MM. Transthyretin enhances nerve regeneration. J Neurochem. 2007;103:831–9.
Andersson K, Olofsson A, Nielsen EH, Svehag SE, Lundgren E. Only amyloidogenic intermediates of transthyretin induce apoptosis. Biochem Biophys Res Commun. 2002;294:309–14.
Hou X, Richardson SJ, Aguilar MI, Small DH. Binding of amyloidogenic transthyretin to the plasma membrane alters membrane fluidity and induces neurotoxicity. Biochemistry (Mosc). 2005;44:11618–27.
Pokrzywa M, Dacklin I, Hultmark D, Lundgren E. Misfolded transthyretin causes behavioral changes in a Drosophila model for transthyretin-associated amyloidosis. Eur J Neurosci. 2007;26:913–24.
Pokrzywa M, Dacklin I, Vestling M, Hultmark D, Lundgren E, Cantera R. Uptake of aggregating transthyretin by fat body in a Drosophila model for TTR-associated amyloidosis. PLoS One. 2010;5:e14343.
Bourgault S, Choi S, Buxbaum JN, Kelly JW, Price JL, Reixach N. Mechanisms of transthyretin cardiomyocyte toxicity inhibition by resveratrol analogs. Biochem Biophys Res Commun. 2011;410:707–13.
Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, Rappley I, Vogel H, Liedtke M, Witteles RM, Powers ET, Reixach N, Chan WK, Wilson IA, Kelly JW, Graef IA, Alhamadsheh MM. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc Natl Acad Sci USA. 2013;110:9992–7.
Colon W, Kelly JW. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry (Mosc). 1992;31:8654–60.
Lai Z, Colón W, Kelly JW. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry (Mosc). 1996;35:6470–82.
Hammarström P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293:2459–62.
Hammarström P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci USA. 2002;99(Suppl 4):16427–32.
Hammarström P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713–6.
Sekijima Y, Wiseman RL, Matteson J, Hammarström P, Miller SR, Sawkar AR, Balch WE, Kelly JW. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85.
Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–8.
DuBay KF, Pawar AP, Chiti F, Zurdo J, Dobson CM, Vendruscolo M. Prediction of the absolute aggregation rates of amyloidogenic polypeptide chains. J Mol Biol. 2004;341:1317–26.
Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22:1302–6.
Tartaglia GG, Pawar AP, Campioni S, Dobson CM, Chiti F, Vendruscolo M. Prediction of aggregation-prone regions in structured proteins. J Mol Biol. 2008;380:425–36.
Hurshman AR, White JT, Powers ET, Kelly JW. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry (Mosc). 2004;43:7365–81.
Saito S, Ando Y, Nakamura M, Ueda M, Kim J, Ishima Y, Akaike T, Otagiri M. Effect of nitric oxide in amyloid fibril formation on transthyretin-related amyloidosis. Biochemistry (Mosc). 2005;44:11122–9.
Zhao L, Buxbaum JN, Reixach N. Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry (Mosc). 2013;52:1913–26.
Gustavsson A, Jahr H, Tobiassen R, Jacobson DR, Sletten K, Westermark P. Amyloid fibril composition and transthyretin gene structure in senile systemic amyloidosis. Lab Invest. 1995;73:703–8.
Westermark P, Sletten K, Johnson KH. Ageing and amyloid fibrillogenesis: lessons from apolipoprotein AI, transthyretin and islet amyloid polypeptide. Ciba Found Symp. 1996;199:205–18 (discussion 218–22).
Bergström J, Gustavsson A, Hellman U, Sletten K, Murphy CL, Weiss DT, Solomon A, Olofsson BO, Westermark P. Amyloid deposits in transthyretin-derived amyloidosis: cleaved transthyretin is associated with distinct amyloid morphology. J Pathol. 2005;206:224–32.
Thylen C, Wahlqvist J, Haettner E, Sandgren O, Holmgren G, Lundgren E. Modifications of transthyretin in amyloid fibrils: analysis of amyloid from homozygous and heterozygous individuals with the Met30 mutation. EMBO J. 1993;12:743–8.
Ihse E, Rapezzi C, Merlini G, Benson MD, Ando Y, Suhr OB, Ikeda S, Lavatelli F, Obici L, Quarta CC, Leone O, Jono H, Ueda M, Lorenzini M, Liepnieks J, Ohshima T, Tasaki M, Yamashita T, Westermark P. Amyloid fibrils containing fragmented ATTR may be the standard fibril composition in ATTR amyloidosis. Amyloid. 2013;20:142–50.
Mangione PP, Porcari R, Gillmore JD, Pucci P, Monti M, Porcari M, Giorgetti S, Marchese L, Raimondi S, Serpell LC, Chen W, Relini A, Marcoux J, Clatworthy IR, Taylor GW, Tennent GA, Robinson CV, Hawkins PN, Stoppini M, Wood SP, Pepys MB, Bellotti V. Proteolytic cleavage of Ser52Pro variant transthyretin triggers its amyloid fibrillogenesis. Proc Natl Acad Sci USA. 2014;111:1539–44.
Benson MD. Familial amyloidotic polyneuropathy. Trends Neurosci. 1989;12:88–92.
Saraiva MJ. Transthyretin mutations in health and disease. Hum Mutat. 1995;5:191–6.
Schulze PC, Maurer MS. Transthyretin Val30Met mutation in an African American with cardiac amyloidosis. Congest Heart Fail. 2010;16:73–6.
Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation. 2011;124:1079–85.
Connors LH, Lim A, Prokaeva T, Roskens VA, Costello CE. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid. 2003;10:160–84.
Hammarström P, Sekijima Y, White JT, Wiseman RL, Lim A, Costello CE, Altland K, Garzuly F, Budka H, Kelly JW. D18G transthyretin is monomeric, aggregation prone, and not detectable in plasma and cerebrospinal fluid: a prescription for central nervous system amyloidosis? Biochemistry (Mosc). 2003;42:6656–63.
Sekijima Y, Hammarström P, Matsumura M, Shimizu Y, Iwata M, Tokuda T, Ikeda S, Kelly JW. Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology. Lab Invest. 2003;83:409–17.
Rapezzi C, Perugini E, Salvi F, Grigioni F, Riva L, Cooke RM, Ferlini A, Rimessi P, Bacchi-Reggiani L, Ciliberti P, Pastorelli F, Leone O, Bartolomei I, Pinna AD, Arpesella G, Branzi A. Phenotypic and genotypic heterogeneity in transthyretin-related cardiac amyloidosis: towards tailoring of therapeutic strategies? Amyloid. 2006;13:143–53.
Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M, Ciliberti P, Biagini E, Salvi F, Branzi A. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010;7:398–408.
Planté-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10:1086–97.
Coelho T, Maurer MS, Suhr OB. THAOS—The Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29:63–76.
Hellman U, Alarcon F, Lundgren HE, Suhr OB, Bonaiti-Pellié C, Planté-Bordeneuve V. Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid. 2008;15:181–6.
Planté-Bordeneuve V, Carayol J, Ferreira A, Adams D, Clerget-Darpoux F, Misrahi M, Said G, Bonaiti-Pellie C. Genetic study of transthyretin amyloid neuropathies: carrier risks among French and Portuguese families. J Med Genet. 2003;40:e120.
Suhr OB, Svendsen IH, Andersson R, Danielsson A, Holmgren G, Ranløv PJ. Hereditary transthyretin amyloidosis from a Scandinavian perspective. J Intern Med. 2003;254:225–35.
Koike H, Misu K, Sugiura M, Iijima M, Mori K, Yamamoto M, Hattori N, Mukai E, Ando Y, Ikeda S, Sobue G. Pathology of early- vs late-onset TTR Met30 familial amyloid polyneuropathy. Neurology. 2004;63:129–38.
Said G, Ropert A, Faux N. Length-dependent degeneration of fibers in Portuguese amyloid polyneuropathy: a clinicopathologic study. Neurology. 1984;34:1025–32.
Koike H, Hashimoto R, Tomita M, Kawagashira Y, Iijima M, Tanaka F, Sobue G. Diagnosis of sporadic transthyretin Val30Met familial amyloid polyneuropathy: a practical analysis. Amyloid. 2011;18:53–62.
Koike H, Tanaka F, Hashimoto R, Tomita M, Kawagashira Y, Iijima M, Fujitake J, Kawanami T, Kato T, Yamamoto M, Sobue G. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: analysis of late-onset cases from non-endemic areas. J Neurol Neurosurg Psychiatry. 2012;83:152–8.
Sattianayagam PT, Hahn AF, Whelan CJ, Gibbs SD, Pinney JH, Stangou AJ, Rowczenio D, Pflugfelder PW, Fox Z, Lachmann HJ, Wechalekar AD, Hawkins PN, Gillmore JD. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J. 2012;33:1120–7.
Carr AS, Pelayo-Negro AL, Evans MR, Laura M, Blake J, Stancanelli C, Iodice V, Wechalekar AD, Whelan CJ, Gilmore JD, Hawkins PN, Reilly MM. A study of the neuropathy associated with transthyretin amyloidosis (ATTR) in the UK. J Neurol Neurosurg Psychiatry. 2015. doi:10.1136/jnnp-2015-310907 [Epub ahead of print].
Adams D, Lozeron P, Theaudin M, Mincheva Z, Cauquil C, Adam C, Signate A, Vial C, Maisonobe T, Delmont E, Franques J, Vallat JM, Sole G, Pereon Y, Lacour A, Echaniz-Laguna A, Misrahi M, Lacroix C. Regional difference and similarity of familial amyloidosis with polyneuropathy in France. Amyloid. 2012;19(Suppl 1):61–4.
Koike H, Misu K, Ikeda S, Ando Y, Nakazato M, Ando E, Yamamoto M, Hattori N, Sobue G. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early- vs late-onset form. Arch Neurol. 2002;59:1771–6.
Falk RH, Dubrey SW. Amyloid heart disease. Prog Cardiovasc Dis. 2010;52:347–61.
Connors LH, Prokaeva T, Lim A, Théberge R, Falk RH, Doros G, Berg A, Costello CE, O’Hara C, Seldin DC, Skinner M. Cardiac amyloidosis in African Americans: comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. Am Heart J. 2009;158:607–14.
Givens RC, Russo C, Green P, Maurer MS. Comparison of cardiac amyloidosis due to wild-type and V122I transthyretin in older adults referred to an academic medical center. Aging Health. 2013;9:229–35.
Pinney JH, Whelan CJ, Petrie A, Dungu J, Banypersad SM, Sattianayagam P, Wechalekar A, Gibbs SD, Venner CP, Wassef N, McCarthy CA, Gilbertson JA, Rowczenio D, Hawkins PN, Gillmore JD, Lachmann HJ. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc. 2013;2:e000098.
Coelho T, Ericzon BG, Falk RH, Grogan DR, Ikeda S, Maurer M, Planté-Bordeneuve V, Suhr O, Trigo P (2008) A Physician’s Guide to Transthyretin Amyloidosis. Amyloidosis Foundation. http://www.amyloidosis.org/pdf/TTR%2008.pdf. Accessed 2 April 2013.
Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, Buxbaum JN. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336:466–73.
Yamashita T, Hamidi Asl K, Yazaki M, Benson MD. A prospective evaluation of the transthyretin Ile122 allele frequency in an African-American population. Amyloid. 2005;12:127–30.
Dungu J, O’Donnell MS, Hawkins PN, Anderson LJ. Systematic review of 1142 admissions with acute heart failure reveals high frequency of transthyretin V122i cardiac amyloidosis in Afro-Caribbean patients. J Am Coll Cardiol. 2012;59:E993.
Ueda M, Horibata Y, Shono M, Misumi Y, Oshima T, Su Y, Tasaki M, Shinriki S, Kawahara S, Jono H, Obayashi K, Ogawa H, Ando Y. Clinicopathological features of senile systemic amyloidosis: an ante- and post-mortem study. Mod Pathol. 2011;24:1533–44.
Mirzoyev SA, Edwards WD, Mohammed SF, Donovan JL, Roger VL, Grogan DR, Redfield MM. Abstract 17926: Cardiac amyloid deposition is common in elderly patients with heart failure and preserved ejection fraction. Circulation. 2010;122:Abstract A17926.
Holmgren G, Ericzon BG, Groth CG, Steen L, Suhr O, Andersen O, Wallin BG, Seymour A, Richardson S, Hawkins PN, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet. 1993;341:1113–6.
Stangou AJ, Heaton ND, Hawkins PN. Transmission of systemic transthyretin amyloidosis by means of domino liver transplantation. N Engl J Med. 2005;352:2356.
Coelho T, Chorão R, Sousa A, Alves I, Torres MF, Saraiva MJM. Compound heterozygotes of transthyretin Met30 and transthyretin Met119 are protected from the devastating effects of familial amyloid polyneuropathy. Neuromuscul Disord. 1996;6(Suppl 1):Abstract S20.
Longo Alves I, Hays MT, Saraiva MJ. Comparative stability and clearance of [Met30]transthyretin and [Met119]transthyretin. Eur J Biochem. 1997;249:662–8.
Miroy GJ, Lai Z, Lashuel HA, Peterson SA, Strang C, Kelly JW. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc Natl Acad Sci USA. 1996;93:15051–6.
White JT, Kelly JW. Support for the multigenic hypothesis of amyloidosis: the binding stoichiometry of retinol-binding protein, vitamin A, and thyroid hormone influences transthyretin amyloidogenicity in vitro. Proc Natl Acad Sci USA. 2001;98:13019–24.
Miller SR, Sekijima Y, Kelly JW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Invest. 2004;84:545–52.
Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13:236–49.
Dharmarajan K, Maurer MS. Transthyretin cardiac amyloidoses in older North Americans. J Am Geriatr Soc. 2012;60:765–74.
European Medicines Agency. Vyndaqel 20 mg soft capsules. Summary of product characteristics. 2011 http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002294/WC500117862.pdf. Accessed 18 Nov 2014.
Schmidt HH-J. Tafamidis for the treatment of transthyretin-associated familial amyloid polyneuropathy. Expert Opin Orphan Drugs. 2013;1:837–45.
Rappley I, Monteiro C, Novais M, Baranczak A, Solis G, Wiseman RL, Helmke S, Maurer MS, Coelho T, Powers ET, Kelly JW. Quantification of transthyretin kinetic stability in human plasma using subunit exchange. Biochemistry (Mosc). 2014;53:1993–2006.
Schneider F, Hammarstrom P, Kelly JW. Transthyretin slowly exchanges subunits under physiological conditions: a convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein Sci. 2001;10:1606–13.
Wiseman RL, Green NS, Kelly JW. Kinetic stabilization of an oligomeric protein under physiological conditions demonstrated by a lack of subunit exchange: implications for transthyretin amyloidosis. Biochemistry (Mosc). 2005;44:9265–74.
Coelho T, Grogan DR, Packman J, Labaudiniere R, Fleming J, Souther E, Bulawa CE, Kelly J, Wang L, Subramaniam K. Tafamidis: a potent small molecule stabilizer of tetrameric transthyretin in patients with transthyretin amyloidosis. Paper presented at the 37th Lorne Conference on Protein Structure and Function, 2012 Feb 5–9, Lorne, Victoria, Australia. 2012.
Merlini G, Planté-Bordeneuve V, Judge DP, Schmidt H, Obici L, Perlini S, Packman J, Tripp T, Grogan DR. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res. 2013;6:1011–20.
Maurer MS, Grogan DR, Judge DP, Mundayat R, Packman J, Lombardo I, Quyyumi AA, Aarts J, Falk RH. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Heart Fail. 2015;8:519–26.
Suhr OB, Conceicao IM, Karayal ON, Mandel FS, Huertas PE, Ericzon BG. Post hoc analysis of nutritional status in patients with transthyretin familial amyloid polyneuropathy: impact of tafamidis. Neurol Ther. 2014;3:101–12.
Klamerus KJ, Watsky E, Moller R, Wang R, Riley S. The effect of tafamidis on the QTc interval in healthy subjects. Br J Clin Pharmacol. 2015;79:918–25.
Cho Y, Baranczak A, Helmke S, Teruya S, Horn EM, Maurer MS, Kelly JW. Personalized medicine approach for optimizing the dose of tafamidis to potentially ameliorate wild-type transthyretin amyloidosis (cardiomyopathy). Amyloid. 2015;22:175–80.
Suhr OB, Holmgren G, Steen L, Wikström L, Norden G, Friman S, Duraj FF, Groth CG, Ericzon BG. Liver transplantation in familial amyloidotic polyneuropathy. Follow-up of the first 20 Swedish patients. Transplantation. 1995;60:933–8.
Bergethon PR, Sabin TD, Lewis D, Simms RW, Cohen AS, Skinner M. Improvement in the polyneuropathy associated with familial amyloid polyneuropathy after liver transplantation. Neurology. 1996;47:944–51.
Parrilla P, Ramirez P, Andreu LF, Bueno SF, Robles R, Miras M, Acosta F, Viader-Farre D, de Mingo P, Bermejo J, Garrido SF, Munar-Ques M. Long-term results of liver transplantation in familial amyloidotic polyneuropathy type I. Transplantation. 1997;64:646–9.
Takei Y, Ikeda S, Hashikura Y, Ikegami T, Kawasaki S. Partial-liver transplantation to treat familial amyloid polyneuropathy: follow-up of 11 patients. Ann Intern Med. 1999;131:592–5.
Adams D, Samuel D, Goulon-Goeau C, Nakazato M, Costa PM, Feray C, Planté V, Ducot B, Ichai P, Lacroix C, Metral S, Bismuth H, Said G. The course and prognostic factors of familial amyloid polyneuropathy after liver transplantation. Brain. 2000;123(Pt 7):1495–504.
de Carvalho M, Conceição I, Bentes C, Luís ML. Long-term quantitative evaluation of liver transplantation in familial amyloid polyneuropathy (Portuguese V30M). Amyloid. 2002;9:126–33.
Ericzon BG, Wilczek HE, Larsson M, Wijayatunga P, Stangou A, Pena JR, Furtado E, Barroso E, Daniel J, Samuel D, Adam R, Karam V, Poterucha J, Lewis D, Ferraz-Neto BH, Cruz MW, Munar-Ques M, Fabregat J, Ikeda S, Ando Y, Heaton N, Otto G, Suhr O. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99:1847–54.
Okamoto S, Hörnsten R, Obayashi K, Wijayatunga P, Suhr OB. Continuous development of arrhythmia is observed in Swedish transplant patients with familial amyloidotic polyneuropathy (amyloidogenic transthyretin Val30Met variant). Liver Transpl. 2011;17:122–8.
Ando Y, Ueda M. Diagnosis and therapeutic approaches to transthyretin amyloidosis. Curr Med Chem. 2012;19:2312–23.
Ruberg FL, Maurer MS, Judge DP, Zeldenrust S, Skinner M, Kim AY, Falk RH, Cheung KN, Patel AR, Pano A, Packman J, Grogan DR. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the Transthyretin Amyloidosis Cardiac Study (TRACS). Am Heart J. 2012;164(222–8):e1.
Acknowledgments
Editorial support for the development of this manuscript was provided by Malcolm Darkes and Dave Cornick of Engage Scientific Solutions and was funded by Pfizer. The article processing charges for this publication were funded by Pfizer. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Dr. Coelho’s institution received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010; she has served on the speakers bureau of Pfizer Inc. and received funding from Pfizer Inc. for scientific meeting expenses (travel, accommodation, and registration), and currently serves on the scientific advisory board of THAOS (natural history disease registry) without any financial compensation. Dr. Merlini received speaker honoraria from Pfizer Inc. Dr. Bulawa was a full-time employee of FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010, and is now an employee of Pfizer Inc. Dr. Fleming was a full-time employee of FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010, and is now an employee of Pfizer Inc. Dr. Judge received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010, as principal investigator and served as a member of the Global Commercial Advisory Board at the London Vyndaqel meeting 18–19 October 2012. Dr. Kelly is a founder, shareholder (and option holder), and paid consultant for FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010. Dr. Maurer has received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010, as a clinical investigator and for scientific meeting expenses. His institution has received grant support from Pfizer Inc. Dr. Planté-Bordeneuve received support from FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010, as a clinical investigator, and serves on the scientific advisory board of THAOS (natural history disease registry) but did not receive compensation for her involvement with this study. Dr. Labaudinière, as former chief executive officer, is an option holder of FoldRx Pharmaceuticals, which was acquired by Pfizer Inc. in October 2010. Mr. Mundayat is an employee of Pfizer Inc. Dr. Riley is an employee of Pfizer Inc. Dr. Lombardo is a former employee of Pfizer Inc. Dr. Huertas is a former employee of Pfizer Inc.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Coelho, T., Merlini, G., Bulawa, C.E. et al. Mechanism of Action and Clinical Application of Tafamidis in Hereditary Transthyretin Amyloidosis. Neurol Ther 5, 1–25 (2016). https://doi.org/10.1007/s40120-016-0040-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-016-0040-x