
Abstract
This review summarizes the available data about genetic factors which can link ischemic stroke and sleep. Sleep patterns (subjective and objective measures) are characterized by heritability and comprise up to 38–46%. According to Mendelian randomization analysis, genetic liability for short sleep duration and frequent insomnia symptoms is associated with ischemic stroke (predominantly of large artery subtype). The potential genetic links include variants of circadian genes, genes encoding components of neurotransmitter systems, common cardiovascular risk factors, as well as specific genetic factors related to certain sleep disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Mendelian randomization analysis showed that genetic liability for short duration and frequent insomnia symptoms are associated with the risk of large artery subtype of ischemic stroke, however, the relation was not found for chronotype. |
Sleep patterns are characterized by high heritability (comprising up to 38–46%), and the genetic factors may underlie the susceptibility to both sleep disturbances and cerebrovascular events. However, the associations might differ with regard to stroke subtypes (with more evidence confirming links between sleep disorders and large artery and cardioembolic subtypes of ischemic stroke). |
The uneven double-peak 24-h distribution of stroke onset can be explained by the involvement of circadian genes and alterations in circadian regulation. Other common genetic factors mediating sleep–stroke interaction can include genes encoding components of neurotransmitter systems, specific genetic factors related to certain sleep disorders and their impact on common cardiovascular risk factors. |
Sleep disorders can also affect post-stroke recovery via modulation of genes involved in inflammatory, oxidative responses, neuroplasticity processes thus affecting the stroke outcomes. |
Genetic studies provide new insights into the association between cerebrovascular disease and sleep patterns as well as into the potential approaches for personalized health care. However, the data of genetic studies should be taken with caution considering multifactorial nature of ischemic stroke and sleep patterns, potential environmental modulation, epigenetic effects, methodology of the studies, etc. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.14769723.
Introduction
The development of genetic studies has broadened our understanding of the underlying causes and factors of various pathologies. Moreover, the integration of data from different databases, implementation of various analysis techniques (fine-mapping FINEMAP, MAGMA, linkage analysis, tissue enrichment, cluster analysis, Mendelian randomization analysis, etc.) allowed the identification of novel gene loci and novel gene associations and unraveled unexpected relations between various physiological and pathological states and processes [1].
However, the interpretation of genetic studies should be careful, in particular, in multifactorial diseases such as stroke. The potential multiple genetic factors can interact with each other as well as with other factors (environmental, behavioral, epigenetic modification). Moreover, there can be population differences in genetic variation, as suggested by several studies [2], therefore, the results generalization should be made with caution.
This review presents some insights into genetic factors potentially linking ischemic stroke and sleep patterns. These include circadian genes, genes encoding components of neurotransmitter systems, as well as specific genetic factors related to certain sleep disorders. This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Heritability of sleep patterns and stroke
The large genome-wide association studies (GWAS) (SiGN [3], METASTROKE [4], NINDS [5], and others) established a number of genes associated with stroke with relation to stroke types and subtypes (Table 1). Some of them can also be linked to sleep–wake regulation and sleep disorders.
By Mendelian randomization analysis, Cai et al. [6] showed an association between large artery stroke and genetic liability for short sleep duration and frequent insomnia symptoms, with no effect of chronotype. On the other hand, Lu et al. [7] analyzing UK Biobank and MEGASTROKE databases did not find any causative association between sleep duration (either total duration or short/long sleep) and stroke risk. The authors of the latter analysis note that there was a weak association between short sleep and the risk of cardioembolic stroke, as well as between long sleep and the risk of large artery stroke, however, these associations were mainly caused by multiple comparisons and disappeared after Bonferroni correction was applied. The differences in the results can be explained by the various methods and instruments applied, as well as by the choice of sleep traits and characteristics and definitions.
Fan et al. [26] analyzed the data of a prospective arm of the large-scale UK Biobank study, which included 385,292 participants free of cardiovascular disease (CVD) at baseline and found that higher values of healthy sleep score correlated with a lower risk of CVD, including stroke (highest vs. lowest score for stroke risk: hazard ratio 0.66 95% confidence interval (CI) (0.58–0.75)). Interestingly, out of five measures of healthy sleep score (early chronotype “morning” or “morning than evening”); sleep 7–8 h per day; reported no or rare insomnia symptoms; no self-reported snoring; and no frequent daytime sleepiness), only sleep duration appeared to be protective against stroke. Using polygenetic score (based on the data of GWAS) the authors established that the combination of high genetic score and low healthy sleep score is characterized by 1.5-fold greater risk of stroke (1.49 (0.90, 2.47)). Subjects with high genetic score and ideal sleep pattern have a lower risk of stroke 1.30 (1.10, 1.54). However, the authors emphasize that there is no direct significant interaction between healthy sleep score and genetic susceptibility to CVD [26].
Although characterized by substantial heterogeneity, tens of cohort cross-sectional and prospective studies demonstrate an association between sleep duration and cardiovascular events. Both short (RR 1.32 (1.18–1.47), p < 0.001) and long sleepers (RR 1.48 (1.31–1.68), p < 0.001) demonstrate a greater risk of stroke compared to subjects with normal sleep duration [27]. Based on a prospective study, this association is present even in subjects without any comorbidities at baseline [28]. Moreover, sleep duration is associated with telomere length considered as a biomarker of aging, as well as a factor associated with higher stroke risk (although the data are controversial and reported mostly in Asian populations) [29]. This association is evident in both children and adults, and shorter telomeres are found in individuals sleeping less [30].
The heritability of sleep patterns has been investigated since the 1990s and comprises up to 38–46%, as shown in the Australian twin study [31]. These include subjective measures—diurnal sleep preferences, usual bedtime, and usual sleep duration, sleepiness, chronotype and objectively measured parameters [32]—total sleep time, sleep efficiency, number of nocturnal sleep episodes, sleep timing, total time of slow-wave sleep, EEG spectral characteristics—delta power, theta power, and sigma power [33].
GWAS studies identified a wide spectrum of candidate genes involved in sleep–wake regulation and heritability of sleep patterns, some of them can play role in stroke occurrence and/or recovery, although populational differences should be noted. Based on UK Biobank data (n = 446,118), Dashti et al. [34] identified 76 independent gene loci associated with habitual subjective and objectively measured (by accelerometer) sleep duration, 27 and eight loci related to self-reported short and long sleep, respectively, which implicate links to multiple signal pathways. The PAX8 locus is the most consistent for various sleep patterns, across various GWAS and different cohorts. PAX8 expression was observed in a number of metastating tumors manifesting as stroke [35,36,37], however no direct evidence linking PAX8 and stroke risks or outcomes is available. Jones et al. [32] analyzed a subset of the UK Biobank database and found 47 loci associated with a number of accelerometer-derived sleep characteristics, some of them overlapping with sleep disorder genetics. One can hypothesize that all the identified genes can mediate the association between sleep patterns, sleep disorders, and cardio/cerebrovascular events. However, the mechanisms are not so evident and the data are controversial.
Circadian regulation, clock genes and stroke
The uneven double-peak 24-h distribution of stroke onset [38] suggests the involvement of circadian system. Although a number of external factors can be responsible for such patterns, the alterations or imbalance in circadian regulation should be considered.
At the molecular level, the circadian system implies a transcription–translation feedback loop that oscillates with 24-h rhythmicity and is regulated by a number of circadian genes. This is a self-regulated system based on the interaction between clock genes (the main are CLOCK and BMAL1 genes forming self-regulatory feedback loop) and their protein products. In mammals, the circadian system comprises plenty of organ- and tissue-specific cellular clocks, which are synchronized by the master pacemaker located in the suprachiasmatic nucleus (SCN) [39]. Circadian genes are involved in multiple metabolic pathways as well as in circadian variations in cardiovascular risk factors (blood pressure, heart rate, coagulation factors, platelet aggregability, hormone concentrations, and insulin sensitivity, etc.) [40], which may be responsible for the day–night distribution and early morning onset of cardio- and cerebrovascular events [41,42,43]. The interpretation of the clock genes studies should be done with care, as many factors can affect the results [44].
The genetic variants [single nucleotide polymorphisms (SNPs)] and haplotypes of the CLOCK gene are associated with various metabolic traits [45], including weight regulation, hyperglycemia, hyperinsulinemia, and higher risk of type 2 diabetes mellitus (T2DM), which can increase the risk of cerebro/cardiovascular diseases. In a longitudinal study by Corella et al. [46], which involved 7098 participants of the PREDIMED trial (PREvención con DIeta MEditerránea), CLOCK gene rs4580704 SNP was associated with stroke risk (G-allele being protective: HR 0.61; 95% CI 0.40–0.94; p = 0.024) in subjects with T2DM [46], but not with myocardial infarction. This association was present in T2DM subjects and was not found in non-diabetics.
Among a number of chronotype-associated gene loci (PER1, CRY1, ARNTL, RGS16, PER2, PER3, PIGK/AK5, INADL, FBXL3, HCRTR2, HTR6, etc.) [47, 48], a relation to cerebrovascular events has been studied only for a few of the genes (PER genes, CRY1, ARNTL/BMAL1, HCRTR2) [49,50,51,52,53].
Not only occurrence but also the size of brain lesion volume can depend on the time of stroke onset as suggested by the experimental data from mice models of ischemic and hemorrhagic stroke [49]. The authors explain the time-of-the-day dependent tolerability to brain injury by the changes in clock gene expression (PER-2, PER-1, CLOCK, BMAL-1, and NPAS-2) in the brain, which are associated with microglia activation and neuroinflammation [50]. Beker et al. [49] showed that stroke induced at night (mid-dark period) was associated with higher levels of AKT and ERK survival kinases correlating with lower infarct area and brain swelling and improved neurological function compared to daytime (early light). According to Lembach et al. [51], both the ischemia vulnerability and the recovery after stroke show sex-dependent association with circadian disruption (Bmal1-deficient mice). The latter can be explained by the interaction between circadian genes and genes involved in the sex hormone regulation [51, 52]. Earlier experimental studies demonstrated selective impact of global ischemia on circadian genes: global ischemia had no effect on Bmal1 or Cry1 circadian expression, while it caused a significant phase advance in the Per1 rhythm in the hippocampus [53].
PER1 and PER2 genes demonstrate a protective role against ischemia. The cytoprotective effects are tightly linked to the adenosine pathway and regulation of hypoxia-inducible factor (HIF). In the model of kidney ischemia–reperfusion injury, the CO-induced Per-2 mediated protection is implemented via up-regulation of EPO (erythropoietin) [54].
Neurotrophic factors (in particular, brain-derived neurotrophic factor, BDNF) which regulate neuronal proliferation and differentiation and neuroplasticity in ischemic brain injury, demonstrate a clear individual circadian rhythmicity in various tissues including brain tissue [55]. In its turn, BDNF can regulate plasticity of SCN and modulate circadian rhythms [56, 57]. Few BDNF polymorphisms are described; in particular, Val66Met is associated with lower BDNF expression, which can be related to alterations in neuronal recovery after injury. Sleep disorders can affect BDNF expression [58, 59]. Acute sleep deprivation was shown to induce an increase in BDNF expression, which can be used for ischemic pre/postconditioning [60] in brain injuries, although this needs further examination.
The genes encoding nuclear receptors Rev‐erbα and RAR-related orphan receptors (RORα) are known as clock genes. They are tightly connected and compete for binding of the promoter of BMAL1 gene, so forming a regulatory loop that enables circadian rhythms and regulates circadian genes. At the same time, they control energy homeostasis, lipid metabolism, fibrinolysis, inflammatory processes in a circadian-dependent manner. Their role is described by Duez and Staels [61]. Moreover, in the experiment, Rev‐erbα regulates mitochondrial function and protects cells against oxidative and environmental stress by upregulating antioxidant enzymes (FoxO1, MnSOD, Hmox1, and catalase) and decreasing ROS production [62]. These effects can be involved in inflammatory response of microglia [63], neuroprotection, and brain recovery after acute damage. However, the evidence regarding the role of Rev‐erbα and RORα in stroke is lacking. This hypothesis is confirmed by experimental observations of RORα-deficient mice that developed greater cerebral infarct size, brain edema, and cerebral apoptosis compared to wild-type animals. The authors explain these effects by the abrogation of RORα-modulating effects on melatonin neuroprotective activity [64].
Melatonin is also a key component and regulator of circadian system; its effects are mediated via melatonin receptors. In experimental stroke, MT2 melatonin receptors activation promotes antioxidative and anti-inflammatory effects [65]. The GWAS studies demonstrated an association between genetic variants of melatonin receptors 2 and the risk of cardiometabolic disorders (T2DM, insulin resistance [66, 67], which may increase the risk of cerebrovascular disease).
Several other factors should be mentioned in relation to circadian regulation and brain injury. The gene of the zinc finger homeobox 3 (Zfhx3), a transcription factor that is highly expressed in the SCN in hypothalamus, is strongly associated with the cardioembolic type of ischemic stroke [4]. On the other hand, it is known to maintain and regulate circadian rhythms confirmed by the experiments in Zfhx3 mutant mice [68]. Mice with the G → T transversion mutation of this gene show significantly shorter circadian periods, altered sleep architectures, more fragmented sleep, and reduced delta power [69].
The disruption in circadian system and circadian misalignment can result from the damage to photoreceptors. Clusterin-associated protein 1 (CLUAP1) is involved in the development of photoreceptors and its mutations are associated with cilial early degeneration [70]. The variants at CLUAP1 locus are associated with the number of nocturnal sleep episodes [32] and the levels of α-synuclein in cerebrospinal fluid, which is known to play role in synaptic plasticity and to be involved in neurodegenerative diseases [71]. However, no clear data on its association with stroke risk and post-stroke recovery are available.
Sleep-regulating neurotransmitter systems and stroke
Several neurotransmitter systems are involved in sleep–wake regulation and include wake-promoting substances [serotonin, norepinephrine (NE), histamine, hypocretins, acetylcholine (ACh), dopamine, glutamate, and gamma-aminobutyric acid (GABA)] and sleep-promoting factors (melanin-concentrating hormone, adenosine, and several cytokines) [72]. The genes encoding the components of these neurotransmitter systems are compelling candidates linking sleep disorders and cerebrovascular disease.
In this context, genes in the dopaminergic signaling pathways are potential candidates including the dopamine receptors DRD1-4 genes, the dopamine transporter gene (DAT), the catechol-O-methyl transferase gene (COMT) and the monoamine oxidase A gene (MAOA). The components of the dopaminergic system are involved in neuroplasticity processes. Experimental studies suggest that changes in expression of the DRD2 gene differ in ipsi- and contralesional motor cortex, can affect post-stroke recovery [73], and (as well as Adora2a and Pde10a-mediated signaling) may be important for spontaneous recovery after stroke. These effects can evolve via DRD2-mediated modulation of astroglia-induced neuroinflammation [74]. However, small human trials provide controversial results and do not confirm the role of DRD(1,2,3) genes in post-stroke neuroplasticity, which can be related to national differences (Korean population, n = 60). COMT gene (Val158Met) was shown to be associated with post-stroke functional recovery, and Met allele has a prognostic value in Korean population with the better prognosis for COMT Met (–) (Val/Val alleles carriers). Substitution of Val to Met leads to the decreased enzyme activity of COMT and accumulation of synaptic dopamine [72].
Dopaminergic system interacts with the circadian PER genes, normal rhythmic expression of PER2 in the striatum depends on the dopamine and dopamine receptors (D2), and can be involved in circadian rhythm disruption in neurological disorders. At the same time, it has no effect on the PER2 expression in SCN [75]. Involvement of circadian genes can explain the association between dopamine-related genes and sleep duration. A large-scale genetic study, which included 25,465 individuals from seven ethnically diverse cohorts, identified an association between SNP rs17601612 (C allele) within intron 1 of DRD2 and shorter sleep duration, as well as polysomnography-measured shorter sleep latency [76]. This was further confirmed in the UK Biobank study, which also found a correlation between sleep duration and another DRD2 variant rs11214607 (G), as well as with two correlated SNPs of DAT1—rs464049 and rs460000 without epistatic interaction between the associated SNPs in DAT1 and DRD2 [77]. COMT genotype is associated with the different increase in sleep duration between workdays and rest days (so-called social jet lag) [78]. The heterozygous Val/Met allele carriers showed lower difference, which can have a protective effect. Although these data cannot be generalized due to the potential ethnic differences, one can speculate that genetic variants in the dopaminergic system components can underlie the association between sleep duration and cardio/cerebrovascular disease.
Moreover, interaction between genetic variants of DRD2 and DAT1 mediates the neurobehavioral consequences of sleep deprivation [79]. The studies led by Prof. H.P. Landolt’s group showed that the DRD2 polymorphism is involved in homeostatic sleep–wake regulation, and the DRD2 C-allele is associated with a greater slow-wave activity after sleep deprivation [80]. As slow-wave sleep is related to neuroprotection, these mechanisms can be involved in post-stroke recovery and can be partly responsible for the individual differences in functional outcomes after stroke.
The dopaminergic system is also involved in stress response and related sleep dysfunction. The homozygous A1A1 carriers of DRD2 can be more susceptible to stress-related sleep dysfunction [81] that hypothetically can result in deleterious consequences in acute events.
In addition, the dopamine system can be responsible for unhealthy behaviors indirectly increasing the risk of cardio-cerebrovascular events. Thus, DRD2-4 genotypes are associated with cigarette smoking and severity of smoking withdrawal symptoms suggesting dopamine-related responses to nicotine [82]. Unhealthy dietary habits, overeating, and higher risk of obesity are related to the genetic variants in DRD2 and ANKK1 (the ANKK1 gene is closely linked to the DRD2 on chromosome band 11q23.1 and can influence the expression of DRD2 receptors) which modulate the dopamine-mediated reward circuitry in the brain and are associated with sleep disorders. In a 7-year prospective study in American children 5–11 years old, the G allele in rs1799978 and the A allele in rs4245149 in DRD2, and the G allele in rs4938012 in ANKK1 were associated with greater association between sleep problems and being overweight reported by children’s mothers. The authors concluded that sleep problems might specifically influence reward-driven eating, which can be targeted by intervention in genetically susceptible individuals [83]. In a rather small study (n = 73), the TaqI DRD2 A1 allele was associated with more severe obesity and carbohydrate preference, although the number of homozygous cases was too low. At the same time, no association with other cardiovascular factors including lipid levels was found [84]. Body mass index (BMI) is also associated with the COMT genotype [78] (Val158Met polymorphism) being the lowest in Val/Met heterozygotes who also demonstrated the most regular circadian rhythm.
The involvement of the dopaminergic system seems to be responsible for the development of diurnal/nocturnal bruxism after brain injury, including stroke [85, 86].
The main genes encoding the components of the serotoninergic system are the serotonin receptor genes 5HT2A and 5HT2C and the serotonin transporter gene (SERT). They exhibit various physiological effects, which have an impact on sleep–wake regulation and sleep disorders (OSA [87] and bruxism [88]), the cardiovascular system, and mental health. At the same time, they interact with the brain-derived neurotrophic factor BDNF gene and are related to the development of depression in acute stroke phase, which can affect post-stroke recovery [89]. Limited data evidences higher risk of ischemic stroke associated with 5HT receptors polymorphisms (102 T/C polymorphism of the 5-HT2A receptors gene) [90].
The GABA-signaling pathway is involved in sleep regulation, as well as neuronal plasticity [91] after stroke via modulation of the endogenous BDNF expression [92] and, on the other hand, can be affected by exogenous BDNF administration [93]. The inhibition of GABAergic neurons and synapses can increase post-stroke neuronal plasticity. This association involves modulation of different types of GABA receptors (GABAA, GABAB), mitogen-activated protein kinase (MAPK) signaling pathways, and the c-Jun N-terminal kinase (JNK) pathway, and can also be dependent on the expression of the ATP2B1 gene, which plays a role in ischemic preconditioning via stabilization of cellular Ca2+ level, and anti-apoptotic gene BCL2L11 [94]. The GABRA2 gene was shown to be associated with daytime sleepiness [95], and the expression of GABAA receptor β1-subunit [96] predicts slow-wave sleep and power spectrum, which is related to clearance (glymphatic) system functioning [97, 98].
GABA-signaling was suggested to mediate ischemic preconditioning effects. Pre-stroke sleep deprivation exhibits ischemic preconditioning effects, providing neuroprotection [99] and reducing brain lesion volume [100] that was associated with favorable effects of sleep rebound and increased REM sleep duration [101]. On the other hand, pre-ischemia acute sleep deprivation was shown to ameliorate inflammatory response by decreasing expression of pro-inflammatory cytokines [tumor necrosis factor alpha (TNF-alpha) and interleukin 6 (IL6)] and increased chaperon Hsp70 expression in heart in the model of myocardial infarction. This effect is mediated by the activation of GABAA receptors in amygdala, which supports the hypothesis that the GABA-signaling pathway is involved in ischemic preconditioning [102].
The orexin/hypocretin system is a wake-promoting system and the dysfunction of orexinergic neurons leads to hypersomnias, first of all narcolepsy. However, recent findings show that orexin system plays important role in other neurologic disease [103]. Transgenic orexin/ataxin-3 mice, which develop degeneration and loss of hypocretin neurons at young age, demonstrate a phenotype similar to narcolepsy including sleep–wake dysregulation, metabolic, electrophysiological, and behavioral patterns [104, 105]. At the same time, these mice develop larger brain lesion volume and worse neurological deficits in the model of ischemic stroke [106]. In clinical studies, a decrease in SNF orexin levels was found in acute stroke, which inversely correlated with brain lesion volume, however, the relation of these changes to genetic variants or altered gene expression needs elucidation.
Orexin system and melanin-concentrating hormone (MCH) are upregulated (an increase in gene expression of MCH precursor Pmch and OxA and their receptors Mchr1, Ox1R, and Ox2R was shown in experimental stroke) during the acute phase of ischemic stroke and might be involved in post-stroke neuroplasticity [101].
The components of adenosine signaling pathways are also involved in both sleep–wake regulation (playing sleep promoting role [107]) and in brain plasticity and strongly interact with dopaminergic and other neurotransmitter systems. The products of the ADORA2A gene modulate endothelial–leukocyte interactions, and ADORA2A suppression inhibits endothelial inflammation. Thus, Adora2a-deficient mice (Adora2a−/−) show smaller infarct lesions and lower neurological deficits suggesting a protective role of Adora2a deletion in acute thromboembolic stroke. Moreover, the local endothelial Adora2a was shown to be a key player inducing neuroinflammation, as animals deficient in endothelial Adora2a (and so lacking endothelial A2A receptors) demonstrated lower post-stroke neuroinflammation that is mediated via suppression of the activity of the NLRP3 inflammasome [108]. On the other hand, the lack of adenosine receptors leads to enhanced anxiety in A2aR-knockout (A2aR−/−) mice [109]. In humans, genetic variants in ADORA1 and ADORA2A genes interacting with DRD2 are also associated with anxiety and panic disorders [110] and can be associated with higher risk of hyperarousal state and insomnia.
Adrenergic neurotransmission should also be mentioned. ADRB2 gene variations are associated with cardiovascular risks. Although the data regarding stroke are scarce and conflicting [111,112,113], a meta-analysis of just three cohort studies confirms [113] that Gln27Glu polymorphism (rs2400707) of ADRB2 gene is associated with increased risk of ischemic stroke. In another study, the A allele of the β2-AR rs2400707 variant was associated with higher risk of non-dipping blood pressure daily profile associated with increased cardiovascular risks and a common feature of OSA and other sleep disorders [114]. We can speculate that genetic variants in adrenergic signaling can mediate higher cardiovascular risks in sleep disturbances, however, more data are needed.
Neurotransmitter systems are interconnected, and the associations and interactions between polymorphisms in these systems can influence clinical manifestations and medication effects in patients with neurological and sleep disorders [115].
Stroke risk factors, sleep traits and genes
Recent GWAS identified a number of gene loci associated with main risk factors for stroke (elevated blood pressure (BP), dyslipidemia, glucose metabolism), which interact with various sleep traits. These include sleep duration, sleep timing, day napping, night-shift work, evening chronotype, EEG spectrum power during sleep. In particular, the latter (delta power in the first 6 h of sleep) was shown to be associated with insulin secretion rate in an analysis of a large European pedigree (n = 48) [33]. These findings suggest potential modulation effects of sleep traits on the risk factors of stroke via genetic modification. Some of these effects might be mediated by melatonin and circadian genes [66] and can involve various metabolic pathways.
One analysis, comprising 30 cohorts of five ancestry groups (European, African, Asian, Hispanic and Brazilian), provided evidence on the interactions between sleep duration and BP-regulating genes [116]. It should be noted that the interactions were different for short and long sleep. While short sleep interacted with rs73493041 (SNORA26/C9orf170), which is associated with self-reported chronotype, and rs10406644 (KCTD15/LSM14A), long sleep was associated with rs7955964 (FIGNL2/ANKRD33), as well as other loci near BP genes (NME7, FAM208A, MKLN1, CEP164, and RGL3/ELAVL3). Some of these genes are involved in regulation of neuronal functions, however, currently there is no evidence on direct impact on stroke risk.
In a Swedish study (the Malmö Diet and Cancer Study, MDC), the variant of the phosphodiesterase 11A (PDE11A) gene [stop-codon C → T substitution on position 307 (R307X)] was associated with BP elevation (5 and 3 mmHg higher systolic and diastolic BP, respectively), obesity, and ischemic stroke (OR 1.73 95% CI 1.06–2.82; p = 0.028). The association was stronger for females [117]. At the same time, another PDE11A gene variant (a missense variant p.Tyr727Cys) was shown to be related to objective sleep duration and sleep efficiency. Therefore, the PDE11A gene can mediate the link between sleep characteristics, cerebrovascular risk factors, and stroke risk, in particular, in females.
Over 50 various gene loci associated with lipid metabolism and lipid traits were identified in European and non-European cohorts in relation to sleep duration, once again suggesting the potential role of sleep in modulating cardiovascular risk factors [118]. Sleep duration interacts with the gene variant of CRY2 (rs11605924) modifying HDL-C levels (a 0.010 mmol/l higher HDL-C with each additional hour of sleep in the presence of the effect A allele) [119].
Apolipoprotein E (ApoE) regulates cholesterol deposition and transport. The APOE gene variants (ɛ2, ɛ3, and ɛ4) have been investigated in relation to the cardiovascular risks. Among three alleles, the ɛ4 allele is associated with reduced levels of ApoE, resulting in alterations in cellular membrane stability and increased susceptibility to neuronal injury and other neurological risks [120,121,122]. The data on the relation between APOE and its variants and sleep parameters are rather contradictory [123,124,125]. The results of the UK Biobank GWAS study confirm the association between the APOE ε4 allele with the number of accelerometer-derived indices, e.g., a reduced number of nocturnal sleep episodes (rs429358; − 0.13 sleep episodes; 95% CI − 0.16, − 0.11; p = 4 × 10−8), sleep timing (L5, − 1.8 min per allele, p = 4 × 10−6), sleep midpoint (− 0.6 min per allele; p = 0.002), sleep duration (− 1.1 min per allele, p = 7 × 10−4), and diurnal inactivity (− 1.0 min per allele, p = 2 × 10−5) [32]. In intermittent hypoxia associated with OSA, APOE mutations can be associated with increased risks of large artery atherosclerosis [126].
Circadian genes, and in particular genetic variants of CLOCK gene, were shown to be involved in body weight regulation and metabolic syndrome development [127, 128], although the CLOCK gene haplotypes did not correlate with the levels of adiponectines participating in the metabolism regulation (leptin and adiponectin [45]). However, in GWAS, a variant of CLOCK gene (rs1801260) had no effect on glycemic traits [119].
Kinase Suppressor of Ras 2 (KSR2) (12q24), involved in systemic and local brain metabolism regulation [129] and BP control, is associated with sleepiness and sleep duration [95]. This might be mediated via sleep disorders associated with obesity as some rare KSR2 mutations were reported to be associated with early onset obesity and insulin resistance related to abnormal feeding behaviors in animals [130] and humans [131], while some variants are associated with metabolic syndrome [132].
Several self-reported sleep characteristics independently modify the association between genetic predisposition to obesity (genetic profile risk score for obesity) and BMI and waist circumference, so both short and long sleep duration, day napping, night-shift work, and evening chronotype enhance the association between anthropometric parameters and genetic predisposition to obesity [133]. Moreover, sleep duration is associated with food preferences and consumption (carbohydrates, saturated fatty acids, total fat, etc.) but in an age- and sex-dependent manner, which correlated with the BMI. In addition, dietary consumption weakly correlated with CLOCK gene variants, but specifically to the geographic region of the studied cohort (United States vs. northern Europe vs. Mediterranean [134]). Short and long sleep duration interacts with gene variant of MTNR1B (rs1387153) modifying BMI suggesting 0.25 and 0.60 kg/m2 higher BMI with short (< 7 h) and long sleep (≥ 9 h) durations versus normal sleep duration in the presence of the effect T allele [119]. In another study, sleep duration modified the cardiometabolic effects of the variants of adiponectin-related gene CDH13 (encodes T-cadherin, 16q24, rs4783244) [135], while the GG genotype of the same rs4783244 CDH13 is associated with the higher risk of stroke in the Chinese population [136]. These data suggest that normal sleep duration should be recommended as a factor of healthy lifestyle in order to ameliorate cardiometabolic changes related to the genetic variants.
The onset and progression of carotid atherosclerosis as well as the vulnerability of atherosclerotic plaques can be modulated by gene expression. In a culture of vascular smooth muscle cells derived from the carotid plaques, the levels and rhythms of the clock gene expression (BMAL1, PER, CRY, and Rev-erbα) were attenuated compared with normal human carotid vascular smooth muscle cell culture [137]. Circadian disruption induced by a CLOCK gene mutation (ClkΔ19/Δ19 mice) leads to the development of atherogenic hyperlipidemia with greater levels of total cholesterol, very low-density/low-density lipoproteins and triglycerides, increased ApoB100 and ApoB48, reduced ApoA1 and ApoE, and higher ApoB/ApoAI ratios. As a result, extensive aortic and brachiocephalic atherosclerotic lesions develop characterized by higher amounts of lipids, necrosis, inflammatory factors, smooth muscle cells and collagen independently of the Apoe effects. A clear downregulation of LMOD1 (associated with insomnia, short sleep duration, and sleepiness [95]) was found in advanced carotid plaques obtained during surgery for symptomatic or asymptomatic disease in the Biobank of Karolinska Endarterectomy (BiKE) study [138].
Sleep disorders and risk of stroke
OSA
OSA is a multifactorial disease with the underlying multiple genetic factors, which can at least partly explain the heterogeneity of this pathology [139]. It is considered a risk factor for cardiovascular diseases and is associated with brain damage that manifests as structural (atrophy of amygdala, hippocampus, insula) and functional (cognitive and emotional dysfunction) disorders [140,141,142]. The underlying mechanisms include intermittent hypoxemia-hypercapnia, sleep fragmentation, hemodynamic changes, damages in synaptic plasticity processes. The analysis of the FinnGen study data (a large Biobank study comprising 218,792 samples) showed a strong BMI-independent genetic correlation between OSA and stroke, which seems to be mediated via cardiometabolic genetic correlates [143]) (Table 2).
However, randomized clinical trials (RCT) investigating the effects of CPAP therapy on outcomes in stroke patients showed mixed or negative results [144]. Thus, the routine screening for OSA in acute ischemic stroke for the secondary prevention of cardiovascular events or death is not currently recommended [145]. If passing over methodological issues of the mentioned RCT, a speculative explanation can include several points, i.e., potential preconditioning effect of repetitive apneas-hypopneas associated with intermittent hypoxemia-hypercapnia, and individual variability in susceptibility and vulnerability to ischemia–reperfusion (oxidative) injury. Both of these factors can be mediated by genetic variations, in particular, in the genes and their products involved in redox pathways. The main “coordinators” of the physiological responses to hypoxia are hypoxia-inducible factors 1 and 2 [146]. One of the genes regulated by the activity of HIF-1α is the HO-1 gene, which encodes heme oxygenase-1, which via degradation of heme, producing carbon monoxide, biliverdin, and iron, and inducing various cytokines, provides anti-inflammatory and antioxidative effects. Heme oxygenase 1 interacts with clock genes (BMAL1) [147] and is a component of the system of circadian factors involved in redox pathways (NRF2/ARE) and is activated in ischemia–reperfusion injury [148]. In moderate–severe OSA, peripheral blood levels of HO-1 were decreased and correlated with hypoxemia burden (time with O2 saturation < 90%, mean and lowest O2 saturation, oxygen desaturation index, etc.) as well as with cognitive dysfunction [149]. However, the expression of HO-1 and related effects in response to ischemia–reperfusion can vary in various tissues and organs (and even in different parts of the brain) and depend on the duration and burden of hypoxia exposure [150].
A Chinese study (n = 961) demonstrated a protective role of A allele in rs2071746 polymorphism in the promoter of the HO-1 gene with regard to secondary cardio/cerebrovascular events in patients surviving atherosclerotic ischemic stroke. The authors suggested that the protective factor can be related to a higher expression and levels of HO-1 associated with greater antioxidative effects [151]. The protective effects of higher HO-1 expression were proven in endothelial cell culture exposed to intermittent hypoxia [150] (as a model of OSA) and experimental stroke models with gene modulation [152] and included the suppression of cytotoxicity, cell apoptosis, and inflammation [153], and reduction in brain lesion volume [154,155,156].
The Hp gene (16q22.2) encodes the protein haptoglobin, which binds released hemoglobin (before it is degraded to heme and later converted by HO-1) and possesses antioxidant activities, and can play a protective role in oxidative stress and immunomodulation. Out of three alleles of Hp, Hp 2–2 is associated with the higher risk of cardiovascular disease in OSA patients of younger age (< 55 years old [157]). Although conflicting, several studies [158,159,160] provide evidence on the association of Hp variants and different types of stroke, with Hp 2–2 being more often found in unstable carotid atherosclerotic plaques with higher HO-1 expression [161, 162]. These findings suggest a potential role of the haptoglobin system in cardiovascular risks in susceptible patients with sleep disorders who can benefit more from certain preventive therapies (e.g., antioxidant properties of vitamin E supplementation [163]).
The hypoxia responses are mediated via hypoxia-inducible factor 1, which is activated in OSA associated with intermittent hypoxia/hypoxemia. HIF-1alpha modulates expression of a number of factors, and in particular upregulates the ET-1 gene [164] and increases the expression of endothelin-1, a known strong vasoconstrictor, which can be involved in the regulation and remodeling of cerebrovascular circulation [165, 166].
Another gene involved in hypoxia-induced responses is the EPO gene [167] (encodes erythropoietin which regulates erythrocyte production). Its expression is modulated by HIF-1alpha and the upregulation of EPO and EPOR (EPO receptors) expression is associated with decreased neuronal apoptosis, enhanced neurogenesis (increased number in pyramidal cells), and neuroplasticity (enhanced dendritic spine density and neuronal differentiation) [168]. EPO is also regulated by circadian genes BMAL1 and PER2, showing circadian variation both in normal conditions and in acute cerebrovascular events and may correlate with survival [169]. In its turn, EPO modulates genes involved in synaptic plasticity already at very early stages of brain ischemia (upregulates Arc, BDNF, Ccl7, Dusp5, Egr2, Egr4, Egr1, Fosl2, and Nr4a3 and downregulates Trem1 and Atp7a) when no effect on genes related to inflammation or apoptosis is yet exhibited [170]. Modulation of EPO /EPOR expression or EPO administration in OSA may exhibit neuroprotective effects [171, 172]. At the same time, some authors suggest that the protective effects of EPO are mediated via the FAIM2 gene and upregulation of Faim 2 expression [173] (both in animal models and in humans) and are abrogated in Faim2-deficient mice [174].
Restless leg syndrome (RLS)/periodic limb movements (PLMS)
The genes regulating dopaminergic neurotransmission and iron metabolism can mediate the observed association between RLS/PLMS and cardiovascular risks [175,176,177,178,179]. However, the direct evidence is rather scarce. GWASs have identified gene variants of the following genes associated with RLS/PLMs: MEIS1 (2p), LBXCOR1/MAP2K5 (15q), BTBD9 (6p), neuronal nitric oxide synthase (NOS1) (12q) and protein tyrosine phosphatase receptor type delta (9p) genes [180]. BTBD9 (rs9369062), associated with sleep duration and MEIS1 associated with objective sleep efficiency and sleep duration (rs113851554) [32], seem to be the most potent candidates linking RLS/PLMs and cerebrovascular risks. MEIS1 (rs2300481) was also shown to be related to metabolic changes (body mass index), which can be an additional linking factor [181].
Proteomic analysis studies elucidated the potential role of several molecules in severe symptomatic RLS. Among these, downregulation of alpha-1-antitrypsin (A1AT) and upregulation of kininogen-1 (KNG1) are associated with increased cardiovascular risks [182, 183]. The variants in the KNG1 gene were shown to affect the susceptibility to ischemic stroke in a Chinese cohort [184]. This can be mediated by the genetic interactions with plasma coagulation factors (FXI), activated partial thromboplastin time and increased risk of thrombosis [185, 186].
As for alpha-1-antitrypsin (A1AT), MM homozygote of A1AT producing gene (SERPINA1) is more prevalent among patients with acute ischemic stroke [187]. SERPINE1 activation can exacerbate apoptosis and inflammation, contributing to progression of stroke injury [188] and can also be associated with arterial remodeling and atherosclerosis progression. It is supposed to be specific to large-artery subtype of ischemic stroke [189], and patients with large-artery ischemic stroke demonstrate higher expression of the SERPINE1 gene compared to healthy controls [190].
Insomnia
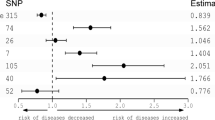
Larsson and Markus [191] applied Mendelian randomization analysis and found that the SNPs related to insomnia complaints are associated with significantly increased odds ratios for ischemic stroke subtypes: large artery stroke 1.13 (95% CI 1.03–1.24; p = 0.010), small artery stroke 1.08 (95% CI 0.99–1.17; p = 0.080), and cardioembolic stroke 1.06 (95% CI 0.99–1.14; p = 0.080).
Bruxism
As mentioned before, the development of bruxism related to brain injury appears to depend on dopaminergic dysfunction. Resistance to common treatment and high response to D2 dopamine receptor blockade (by metoclopramide) confirms the dopaminergic involvement as the underlying cause of this disorder and can be explained by the recovery of dopaminergic flow due to a selective blockade of the hypersensitive presynaptic dopamine receptors [86]. Additional mechanisms include modulation of various neurotransmitters, e.g., NMDA, glutamate, and GABA. Genetic variants in the dopaminergic system can be associated with circadian differences in manifestations (diurnal/sleep/combined) of bruxism [85], with the G allele (vs. A allele) of DRD2 rs1800497 polymorphism being protective against combined bruxism. The C allele of the DRD3 rs6280 SNP (Gly⇒Ser) was associated with an increased risk of sleep bruxism. The C allele of the DRD5 rs6283 polymorphism was associated with decreased risk of diurnal and combined bruxism. However, the association between genetic variants and different response to treatment is not yet established.
Gene Modulation in stroke: Evidence and Prospects
An important clinical issue is the potential modulation of genetic factors by external and behavioral factors in order to influence the prognosis. The evidence is rather scarce.
Corella et al. [46] demonstrated the modulation effect of the Mediterranean diet on the CLOCK gene and its association with glucose metabolism (lower fasting glucose, lower T2D incidence) and prognosis.
Among non-pharmacological interventions, pre-ischemia sleep deprivation was shown to play a neuroprotective role, leading to smaller brain infarct size [99] that can be mediated via the GABA signaling pathway [192] and modulation of gene expression of inflammatory factors.
Genetic modulation effects of pharmacological agents and chemicals are also being investigated. Sleep-promoting substances, which targeting various neurotransmitter systems, draw attention, as sleep may play a neuroprotective role in post-stroke patients. Exogenous modulation of adenosine receptors enhancing the A2AR signaling pathway promotes slow-wave sleep in experiments, which can [193] be favorable in the post-stroke period [194].
The upregulation of the signaling pathway of peroxisome proliferator-activated receptor (PPAR)α and PPARγ has a protective effect against deleterious consequences of ischemia–reperfusion brain and other tissue injury. The activation of peroxisome proliferator activated receptor (PPAR)γ coactivator-1α (PGC-1α) by glucagon-like peptide-1 (GLP-1) receptor agonists was shown to prevent cell apoptosis, reduce brain damage, and improve outcomes [195, 196]. These effects can be even more favorable in conditions of chronic intermittent hypoxia observed in OSA.
Melatonin is a promising neuroprotective agent [197] and can target a number of genes in hypoxia-mediated signaling pathways [198]. In experiments, exogenous melatonin supplementation is associated with the modulation of circadian gene expression and gene expression of redox enzymes (in particular, NOS-2 and HO-1, but also NOS-1, HO-2, GPx, GSR, Mn-SOD) in the pituitary gland [199]. Although, there were no changes in pituitary lipid peroxidation, hypothetically, the changes in expression of redox enzymes can modulate post-stroke ischemia–reperfusion injury in other brain regions and need further investigation. Similar changes were also observed in vehicle-treated animals, suggesting that physical training can be another behavioral approach for gene modification.
In experimental models of ischemia–reperfusion, pretreatment with carbon monoxide CO (inhaled, orally, or as a prodrug) led to lower inflammation (decreased TNF, MCP-1, and IL-1β, increased IL-10, inhibited ROS generation) and tissue injury, which are mediated via up-regulation of CD39 (ectonucleotidase) gene and modulation of adenosine receptors [54] and Per2-signaling pathway. Thus, clock gene Per2 might be involved in ischemic preconditioning and providing neuroprotection in acute cerebrovascular events.
Gene therapy is a promising approach and is being investigated in stroke models. Modulation of HO-1 gene has a neuroprotective effect in mice model of ischemic stroke [152] and is associated with the reduction of infarct volume [154,155,156].
Conclusions
The data of genetic studies provide new insights into association of sleep patterns and brain injury, recovery, and outcomes after ischemic stroke.
One of the clinical applications of these results implies the selection of higher-risk groups as well as identification of those who are at lower risk of unfavorable outcomes in similar circumstances and to implement preventive or treatment strategies [46]. However, the results of genetic studies should be interpreted carefully considering their heterogeneity, gene interactions, environmental influences, and multifactorial nature of cerebrovascular disease and sleep disorders.
Therefore, genetic screening can be implemented in personalized medicine for life-style modification, professional choices, work schedule planning, individual sleep–wake schedule, and bedtime recommendations, etc., in order to reduce cardio/cerebrovascular risks. Healthy sleep (achieved by healthy lifestyle and sleep hygiene, as well as by treating sleep disorders) may play a crucial role in the primary and potentially secondary prevention of cardiometabolic and cerebrovascular disease.
References
Durmaz AA, Karaca E, Demkow U, Toruner G, Schoumans J, Cogulu O. Evolution of genetic techniques: past, present, and beyond. BioMed Res Int. 2015. https://doi.org/10.1155/2015/461524.
Erhart G, Lamina C, Lehtimäki T, et al. Genetic factors explain a major fraction of the 50% lower lipoprotein(a) concentrations in Finns. Arterioscler Thromb Vasc Biol. 2018;38(5):1230–41. https://doi.org/10.1161/ATVBAHA.118.310865.
NINDS Stroke Genetics Network (SiGN) and International Stroke Genetics Consortium. Loci associated with ischaemic stroke and its subtypes (SiGN): a genome-wide association study. Lancet Global Health. 2015;4422(15):4–7. https://doi.org/10.1016/S1474-4422(15)00338-5.
Traylor M, Farrall M, Holliday EG, et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE Collaboration): a meta-analysis of genome-wide association studies. Lancet Neurol. 2012;11(11):951–62. https://doi.org/10.1016/S1474-4422(12)70234-X.
Pulit SL, McArdle PF, Wong Q, et al. The NINDS Stroke Genetics Network: a genome-wide association study of ischemic stroke and its subtypes. Lancet Neurol. 2016;15(2):174–84. https://doi.org/10.1016/S1474-4422(15)00338-5.The.
Cai H, Liang J, Liu Z, et al. Causal effects of sleep traits on ischemic stroke and its subtypes: a Mendelian randomization study. Nat Sci Sleep. 2020;12:783–90. https://doi.org/10.2147/NSS.S265946.
Lu H, Wu PF, Li RZ, Zhang W, Huang GX. Sleep duration and stroke: a Mendelian randomization study. Front Neurol. 2020;11(October):1–6. https://doi.org/10.3389/fneur.2020.00976.
Duan R, Liu X, Wang T, Wu L, Gao X, Zhang Z. Histone acetylation regulation in sleep deprivation-induced spatial memory impairment. Neurochem Res. 2016;41(9):2223–32. https://doi.org/10.1007/s11064-016-1937-6.
Zheng Q, Ma Y, Chen S, Che Q, Zhou Z, Chen D. Identification of genetic loci jointly influencing coronary artery disease risk and sleep traits of insomnia, sleep duration, and chronotype. Sleep Med. 2020;74:116–23. https://doi.org/10.1016/j.sleep.2020.06.027.
Foo JC, Trautmann N, Sticht C, et al. Longitudinal transcriptome-wide gene expression analysis of sleep deprivation treatment shows involvement of circadian genes and immune pathways. Transl Psychiatry. 2019. https://doi.org/10.1038/s41398-019-0671-7.
Sandford AJ, Ha A, Ngan DA, et al. Adhesion molecule gene variants and plasma protein levels in patients with suspected obstructive sleep apnea. PLoS ONE. 2019;14(1):1–12. https://doi.org/10.1371/journal.pone.0210732.
Zhao W, Yuan T, Fu Y, et al. Seasonal differences in the transcriptome profile of the Zhedong white goose (Anser cygnoides) pituitary gland. Poult Sci. 2021;100(2):1154–66. https://doi.org/10.1016/j.psj.2020.10.049.
Reis LM, Tyler RC, Volkmann Kloss BA, et al. PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur J Hum Genet. 2012;20(12):1224–33. https://doi.org/10.1038/ejhg.2012.80.
Milićević N, Duursma A, ten Asbroek ALMA, Felder-Schmittbuhl MP, Bergen AA. Does the circadian clock make RPE-mediated ion transport “tick” via SLC12A2 (NKCC1)? Chronobiol Int. 2019;36(11):1592–8. https://doi.org/10.1080/07420528.2019.1653317.
Chauhan G, Arnold CR, Chu AY, et al. Identification of additional risk loci for stroke and small vessel disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2016;15(7):695–707. https://doi.org/10.1016/S1474-4422(16)00102-2.
Lane JM, Liang J, Vlasac I, et al. Genome-wide association analyses of sleep disturbance traits identify new loci and highlight shared genetics with neuropsychiatric and metabolic traits. Nat Genet. 2017;49(2):274–81. https://doi.org/10.1038/ng.3749.
Traylor M, Andreson C, Rutten-Jacobs LCA, et al. The subtype specificity of genetic loci associated with stroke in 16,664 cases and 32,792 controls. Circ Genom Precis Med. 2019;12(7): e002338. https://doi.org/10.1161/CIRCGEN.118.002338.The.
Buck D, Diefenbach K, Penzel T, Malzahn U, Roots I, Fietze I. Genetic polymorphisms in endothelin-receptor-subtype-a-gene as susceptibility factor for obstructive sleep apnea syndrome. Sleep Med. 2010;11(2):213–7. https://doi.org/10.1016/j.sleep.2009.06.009.
Chatsuriyawong S, Gozal D, Kheirandish-Gozal L, et al. Polymorphisms in nitric oxide synthase and endothelin genes among children with obstructive sleep apnea. BMC Med Genom. 2013;6(1):1. https://doi.org/10.1186/1755-8794-6-29.
Krohn M, Bracke A, Avchalumov Y, et al. Accumulation of murine amyloid-β mimics early Alzheimer’s disease. Brain. 2015;138(8):2370–82. https://doi.org/10.1093/brain/awv137.
Nishiyama T, Nakatochi M, Goto A, et al. Genome-wide association meta-analysis and Mendelian randomization analysis confirm the influence of ALDH2 on sleep duration in the Japanese population. Sleep. 2019;42(6):1–11. https://doi.org/10.1093/sleep/zsz046.
Lin CY, Yu RL, Wu RM, Tan CH. Effect of ALDH2 on sleep disturbances in patients with Parkinson’s disease. Sci Rep. 2019;9(1):1–8. https://doi.org/10.1038/s41598-019-55427-w.
Lahtinen A, Puttonen S, Vanttola P, et al. A distinctive DNA methylation pattern in insufficient sleep. Sci Rep. 2019;9(1):1–9. https://doi.org/10.1038/s41598-018-38009-0.
Fu X-J, Li H-X, Yang K, Chen D, Tang H. The important tumor suppressor role of PER1 in regulating the cyclin–cDK–cKi network in scc15 human oral squamous cell carcinoma cells. Oncol Targets Ther. 2016;9:2237–45.
Bouchard-Cannon P, Mendoza-Viveros L, Yuen A, Kærn M, Cheng HYM. The circadian molecular clock regulates adult hippocampal neurogenesis by controlling the timing of cell-cycle entry and exit. Cell Rep. 2013;5(4):961–73. https://doi.org/10.1016/j.celrep.2013.10.037.
Fan M, Sun D, Zhou T, et al. Sleep patterns, genetic susceptibility, and incident cardiovascular disease: a prospective study of 385,292 UK biobank participants. Eur Heart J. 2020;41(11):1182–9. https://doi.org/10.1093/eurheartj/ehz849.
Krittanawong C, Tunhasiriwet A, Wang Z, et al. Association between short and long sleep durations and cardiovascular outcomes: a systematic review and meta-analysis. Eur Heart J Acute Cardiovasc Care. 2019;8(8):762–70. https://doi.org/10.1177/2048872617741733.
Cai H, Shu XO, Xiang YB, et al. Sleep duration and mortality: a prospective study of 113,138 middle-aged and elderly Chinese men and women. Sleep. 2015;38(4):529–36. https://doi.org/10.5665/sleep.4564.
Jin X, Pan B, Dang X, Wu H, Xu D. Relationship between short telomere length and stroke: a meta-analysis. Medicine (Baltimore). 2018;97(39):e12489. https://doi.org/10.1097/MD.0000000000012489.
James S, McLanahan S, Brooks-Gunn J, et al. Sleep duration and telomere length in children. J Pediatr. 2017;187:247–52. https://doi.org/10.1016/j.jpeds.2017.05.014.Sleep.
Heath AC, Kendler KS, Eaves LJ, Martin NG. Evidence for genetic influences on sleep disturbance and sleep pattern in twins. Sleep. 1990;13(4):318–35. https://doi.org/10.1093/sleep/13.4.318.
Jones SE, van Hees VT, Mazzotti DR, et al. Genetic studies of accelerometer-based sleep measures yield new insights into human sleep behaviour. Nat Commun. 2019;10(1):1–12. https://doi.org/10.1038/s41467-019-09576-1.
Morselli LL, Gamazon ER, Tasali E, Cox NJ, Van Cauter E, Davis LK. Shared genetic control of brain activity during sleep and insulin secretion: a laboratory-based family study. Diabetes. 2018;67(1):155–64. https://doi.org/10.2337/db16-1229.
Dashti HS, Jones SE, Wood AR, et al. Genome-wide association study identifies genetic loci for self-reported habitual sleep duration supported by accelerometer-derived estimates. Nat Commun. 2019;10(1):1–12. https://doi.org/10.1038/s41467-019-08917-4.
Nesbitt AL, Lim ZLT, Chan KJ, Zardawi I, Pridgeon SW. Metastatic renal cell carcinoma presenting with both acute stroke and an oral lesion. Urol Case Rep. 2018;2019(23):75–7. https://doi.org/10.1016/j.eucr.2019.01.010.
Prayson RA. Metastatic chromophobe renal cell carcinoma to the brain. J Clin Neurosci. 2016;26:152–3. https://doi.org/10.1016/j.jocn.2015.10.007.
Jang AI, Bernstock JD, Segar DJ, Distasio M, Matulonis U, Bi WL. Case report: frontoparietal metastasis from a primary fallopian tube carcinoma. Front Surg. 2021;8(February):1–6. https://doi.org/10.3389/fsurg.2021.594570.
Manfredini R, Boari B, Smolensky M, et al. Circadian variation in stroke onset: identical temporal pattern in ischemic and hemorrhagic events. Chronobiol Int. 2005;22(3):417–53. https://doi.org/10.1081/CBI-200062927.
Lincoln GA, Clarke IJ, Hut RAHD. Characterizing a mammalian circannual pacemaker. Science. 2006;314:1941–4.
Christou S, Wehrens SMT, Isherwood C, et al. Circadian regulation in human white adipose tissue revealed by transcriptome and metabolic network analysis. Sci Rep. 2019;9(1):1–12. https://doi.org/10.1038/s41598-019-39668-3.
Takeda N, Maemura K. The role of clock genes and circadian rhythm in the development of cardiovascular diseases. Cell Mol Life Sci. 2015. https://doi.org/10.1007/s00018-015-1923-1.
Iii EM, Adams HP, Marler JR, Hulbert JR, Love BB, Gordon DL. Circadian variation in onset of acute ischemic stroke. Arch Neurol. 1990;47:1178–80.
Zhuang Z, Gao M, Yang R, et al. Association of physical activity, sedentary behaviours and sleep duration with cardiovascular diseases and lipid profiles: a Mendelian randomization analysis. Lipids Health Dis. 2020;19(1):1–11. https://doi.org/10.1186/s12944-020-01257-z.
Hughes ME, Abruzzi KC, Allada R, et al. Guidelines for genome-scale analysis of biological rhythms. J Biol Rhythms. 2017;32(5):380–93. https://doi.org/10.1177/0748730417728663.
Riestra P, Gebreab SY, Xu R, et al. Circadian CLOCK gene polymorphisms in relation to sleep patterns and obesity in African Americans: findings from the Jackson heart study. BMC Genet. 2017;18(1):1–10. https://doi.org/10.1186/s12863-017-0522-6.
Corella D, Asensio EM, Coltell O, et al. CLOCK gene variation is associated with incidence of type-2 diabetes and cardiovascular diseases in type-2 diabetic subjects: dietary modulation in the PREDIMED randomized trial. Cardiovasc Diabetol. 2016;15(1):1–12. https://doi.org/10.1186/s12933-015-0327-8.
Jones SE, Lane JM, Wood AR, et al. Genome-wide association analyses of chronotype in 697,828 individuals provides insights into circadian rhythms. Nat Commun. 2019. https://doi.org/10.1038/s41467-018-08259-7.
Veatch OJ, Keenan BT, Gehrman PR, Malow BA, Pack AI. Pleiotropic genetic effects influencing sleep and neurological disorders. Lancet Neurol. 2017;16(2):158–70. https://doi.org/10.1016/S1474-4422(16)30339-8.
Beker MC, Caglayan B, Yalcin E, et al. Time-of-day-dependent neuronal injury after ischemic stroke: implication of circadian clock transcriptional factor bmal1 and survival kinase AKT. Mol Neurobiol. 2018;55(3):2565–76. https://doi.org/10.1007/s12035-017-0524-4.
Schallner N, Lieberum J-L, Gallo D, et al. Carbon monoxide preserves circadian rhythm to reduce the severity of subarachnoid hemorrhage in mice. Stroke. 2017;48(9):2565–73. https://doi.org/10.1161/STROKEAHA.116.016165.Carbon.
Lembach A, Stahr A, Ali AAH, Ingenwerth M, von Gall C. Sex-dependent effects of bmal1-deficiency on mouse cerebral cortex infarction in response to photothrombotic stroke. Int J Mol Sci. 2018;19(10):1–16. https://doi.org/10.3390/ijms19103124.
Boden MJ, Varcoe TJ, Voultsios A, Kennaway DJ. Reproductive biology of female Bmal1 null mice. Reproduction. 2010;139(6):1077–90. https://doi.org/10.1530/REP-09-0523.
Tischkau SA, Cohen JA, Stark JT, Gross DR, Bottum KM. Time-of-day affects expression of hippocampal markers for ischemic damage induced by global ischemia. Exp Neurol. 2007;208(2):314–22. https://doi.org/10.1016/j.expneurol.2007.09.003.
Correa-Costa M, Gallo D, Csizmadia E, et al. Carbon monoxide protects the kidney through the central circadian clock and CD39. Proc Natl Acad Sci USA. 2018;115(10):E2302–10. https://doi.org/10.1073/pnas.1716747115.
Cain SW, Chang A, Vlasac I, et al. Circadian rhythms in plasma brain-derived neurotrophic factor differ in men and women. J Biol Rhythms. 2017;32(1):75–82. https://doi.org/10.1177/0748730417693124.
Liang F, Walline R, Earnest DJ. Circadian rhythm of brain-derived neurotrophic factor in the rat suprachiasmatic nucleus. Neurosci Lett. 1998;242:89–92.
Girardet C, Lebrun B, Cabirol-Pol M-J, et al. Brain-derived neurotrophic factor/TrkB signaling regulates daily astroglial plasticity in the suprachiasmatic nucleus: electron-microscopic evidence in mouse. Glia. 2013;61:1172–7. https://doi.org/10.1002/glia.22509.
Xie H, Yung WH. Chronic intermittent hypoxia-induced deficits in synaptic plasticity and neurocognitive functions: a role for brain-derived neurotrophic factor. Acta Pharmacol Sin. 2012;33(1):5–10. https://doi.org/10.1038/aps.2011.184.
Flores KR, Viccaro F, Aquilini M, et al. Protective role of brain derived neurotrophic factor (BDNF) in obstructive sleep apnea syndrome (OSAS) patients. PLoS ONE. 2020;15(1):1–11. https://doi.org/10.1371/journal.pone.0227834.
Schmitt K, Holsboer-Trachsler E, Eckert A. BDNF in sleep, insomnia, and sleep deprivation. Ann Med. 2016;48(1–2):42–51. https://doi.org/10.3109/07853890.2015.1131327.
Duez H, Staels B. The nuclear receptors Rev-erbs and RORs integrate circadian rhythms and metabolism. Diab Vasc Dis Res. 2008;5(2):82–8. https://doi.org/10.3132/dvdr.2008.0014.
Sengupta S, Yang G, Donnell JCO, et al. The circadian gene Rev-erbα improves cellular bioenergetics and provides preconditioning for protection against oxidative stress. Free Radic Biol Med. 2016;93:177–89. https://doi.org/10.1016/j.freeradbiomed.2016.02.004.The.
Wolff SEC, Wang XL, Jiao H, et al. The effect of Rev-erbα agonist SR9011 on the immune response and cell metabolism of microglia. Front Immunol. 2020. https://doi.org/10.3389/fimmu.2020.550145.
Zang M, Zhao Y, Gao L, et al. The circadian nuclear receptor RORα negatively regulates cerebral ischemia-reperfusion injury and mediates the neuroprotective effects of melatonin. BBA Mol Basis Dis. 2020. https://doi.org/10.1016/j.bbadis.2020.165890.
Chern CM, Liao JF, Wang YH, Shen YC. Melatonin ameliorates neural function by promoting endogenous neurogenesis through the MT2 melatonin receptor in ischemic-stroke mice. Free Radical Biol Med. 2012;52(9):1634–47. https://doi.org/10.1016/j.freeradbiomed.2012.01.030.
Andersson EA, Holst B, Sparsø T, et al. MTNR1B G24E variant associates with BMI and fasting plasma glucose in the general population in studies of 22,142 Europeans. Diabetes. 2010;59(June):1539–48. https://doi.org/10.2337/db09-1757.
Sparsø T, Andersson E, Bouatia-naji N, et al. G-allele of intronic rs10830963 in MTNR1B confers increased risk of impaired fasting glycemia and type 2 diabetes through an impaired glucose-stimulated insulin release: studies involving 19605 Europeans. Diabetes. 2009;58(June):1450–6. https://doi.org/10.2337/db08-1660.T.S.
Wilcox AG, Vizor L, Parsons MJ, Banks G, Nolan PM. Inducible knockout of mouse Zfhx3 emphasizes its key role in setting the pace and amplitude of the adult circadian clock. J Biol Rhythms. 2017;32(5):433–43. https://doi.org/10.1177/0748730417722631.
Balzani E, Lassi G, Maggi S, et al. The Zfhx3-mediated axis regulates sleep and interval timing in mice. Cell Rep. 2016;16(3):615–21. https://doi.org/10.1016/j.celrep.2016.06.017.The.
Lee C, Wallingford JB, Gross JM, Gross J. Cluap1 is essential for ciliogenesis and photoreceptor maintenance in the vertebrate eye. Investig Opthalmol Vis Sci. 2014;55(7):4585.
Zhong X, Li JQ, Sun L, et al. A Genome-wide association study of α-synuclein levels in cerebrospinal fluid. Neurotox Res. 2019;35(1):41–8. https://doi.org/10.1007/s12640-018-9922-2.
Falup-Pecurariu C, Diaconu S, Tînt D, Falup-Pecurariu O. Neurobiology of sleep (review). Exp Ther Med. 2021;21(3):1–4. https://doi.org/10.3892/etm.2021.9703.
Ito M, Aswendt M, Lee AG, Ishizaka S, Cao Z, Wang EH, et al. RNA sequencing analysis revealed a distinct motor cortex transcriptome in spontaneously-recovered mice after stroke Masaki. Stroke. 2018;49(9):2191–9. https://doi.org/10.1161/STROKEAHA.118.021508.RNA.
Qiu J, Yan Z, Tao K, et al. Sinomenine activates astrocytic dopamine D2 receptors and alleviates neuroinflammatory injury via the CRYAB/STAT3 pathway after ischemic stroke in mice. J Neuroinflammation. 2016;13(1):1–13. https://doi.org/10.1186/s12974-016-0739-8.
Hood S, Cassidy P, Cossette MP, et al. Endogenous dopamine regulates the rhythm of expression of the clock protein PER2 in the rat dorsal striatum via daily activation of D2 dopamine receptors. J Neurosci. 2010;30(42):14046–58. https://doi.org/10.1523/JNEUROSCI.2128-10.2010.
Cade BE, Gottlieb DJ, Lauderdale DS, et al. Common variants in DRD2 are associated with sleep duration: the CARe Consortium. Hum Mol Genet. 2015;25(1):167–79.
Rhodes JA, Lane JM, Vlasac IM, Rutter MK, Czeisler CA, Saxena R. Association of DAT1 genetic variants with habitual sleep duration in the UK Biobank. Sleep. 2019;42(1):1–6. https://doi.org/10.1093/sleep/zsy193.
Valomon A, Holst SC, Bachmann V, et al. Genetic polymorphisms of DAT1 and COMT differentially associate with actigraphy-derived sleep-wake cycles in young adults. Chronobiol Int. 2014;31(5):705–14. https://doi.org/10.3109/07420528.2014.896376.
Holst SC, Müller T, Valomon A, Seebauer B, Berger W, Landolt HP. Functional Polymorphisms in dopaminergic genes modulate neurobehavioral and neurophysiological consequences of sleep deprivation. Sci Rep. 2017;7(April):1–11. https://doi.org/10.1038/srep45982.
Holst SC, Bersagliere A, Bachmann V, Berger W, Achermann P, Landolt HP. Dopaminergic role in regulating neurophysiological markers of sleep homeostasis in humans. J Neurosci. 2014;34(2):566–73. https://doi.org/10.1523/JNEUROSCI.4128-13.2014.
Jiang Y, Liu B, Wu C, et al. Dopamine receptor d2 gene (Drd2) polymorphisms, job stress, and their interaction on sleep dysfunction. Int J Environ Res Public Health. 2020;17(21):1–11. https://doi.org/10.3390/ijerph17218174.
Vandenbergh DJ, O’Connor RJ, Grant MD, et al. Dopamine receptor genes (DRD2, DRD3 and DRD4) and gene-gene interactions associated with smoking-related behaviors. Addict Biol. 2007;12(1):106–16. https://doi.org/10.1111/j.1369-1600.2007.00054.x.
Chan T, Bates J, Lansford J, et al. Impulsivity and genetic variants in DRD2 and ANKK1 moderate longitudinal associations between sleep problems and overweight from ages 5 to 11. Int J Obes (Lond). 2014;38(3):404–10. https://doi.org/10.1038/ijo.2013.123.Impulsivity.
Noble EP, Noble RE, Ritchie T, et al. D2 dopamine receptor gene and obesity. Int J Eat Disord. 1994;15(3):205–17. https://doi.org/10.1002/1098-108X(199404)15:3%3c205::AID-EAT2260150303%3e3.0.CO;2-P.
Oporto GH, Bornhardt T, Iturriaga V, Salazar LA. Single nucleotide polymorphisms in genes of dopaminergic pathways are associated with bruxism. Clin Oral Invest. 2018;22(1):331–7. https://doi.org/10.1007/s00784-017-2117-z.
Yi HS, Kim HS, Seo MR. Trial of oral metoclopramide on diurnal bruxism of brain injury. Ann Rehabil Med. 2013;37(6):871–4. https://doi.org/10.5535/arm.2013.37.6.871.
Xu H, Guan J, Yi H, Yin S. A systematic review and meta-analysis of the association between serotonergic gene polymorphisms and obstructive sleep apnea syndrome. PLoS ONE. 2014;9(1):1–8. https://doi.org/10.1371/journal.pone.0086460.
Oporto GH, Bornhardt T, Iturriaga V, Salazar LA. Genetic polymorphisms in the serotonergic system are associated with circadian manifestations of bruxism. J Oral Rehabil. 2016;43(11):805–12. https://doi.org/10.1111/joor.12436.
Kim JM, Stewart R, Bae KY, et al. Serotonergic and BDNF genes and risk of depression after stroke. J Affect Disord. 2012;136(3):833–40. https://doi.org/10.1016/j.jad.2011.09.029.
Olesen OF, Bennike B, Dam H, Mellerup E. Association of the 5-HT2A receptor gene polymorphism 102T/C with ischemic stroke. J Mol Neurosci. 2006;30(3):323–8. https://doi.org/10.1385/JMN:30:3:323.
Bai X, Kirchhoff F, Scheller A. Oligodendroglial GABAergic signaling: more than inhibition! Neurosci Bull. 2021. https://doi.org/10.1007/s12264-021-00693-w.
Okamura M, Inoue T, Takamatsu Y, Maejima H. Low-level inhibition of GABAergic synapses enhances gene expressions crucial for neuronal plasticity in the hippocampus after ischemic stroke. J Stroke Cerebrovasc Dis. 2020;29(12): 105316. https://doi.org/10.1016/j.jstrokecerebrovasdis.2020.105316.
Müller HD, Neder A, Sommer C, Schäbitz WR. Different postischemic protein expression of the GABAA receptor α2 subunit and the plasticity-associated protein MAP1B after treatment with BDNF versus G-CSF in the rat brain. Restor Neurol Neurosci. 2009;27(1):27–39. https://doi.org/10.3233/RNN-2009-0459.
Gao J, Qin Z, Qu X, et al. Endogenous neuroprotective mechanism of ATP2B1 in transcriptional regulation of ischemic preconditioning. Am J Transl Res. 2021;13(3):1170–83.
Wang H, Lane JM, Jones SE, et al. Genome-wide association analysis of self-reported daytime sleepiness identifies 42 loci that suggest biological subtypes. Nat Commun. 2019;10(1):1–12. https://doi.org/10.1038/s41467-019-11456-7.
Xu X, Qu W, Bian M, Huang F, Fei J, Urade Y. Essential roles of GABA transporter-1 in controlling rapid eye movement sleep and in increased slow wave activity after sleep deprivation. PLoS ONE. 2013;8(10): e75823. https://doi.org/10.1371/journal.pone.0075823.
Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain lulu. Science. 2014;342(6156):1–11. https://doi.org/10.1126/science.1241224.Sleep.
Kim Y-K, Nam KI, Song J. The glymphatic system in diabetes-induced dementia. Front Neurol. 2018;9(October):1–10. https://doi.org/10.3389/fneur.2018.00867.
Cam E, Gao B, Imbach L, Hodor A, Bassetti CL. Sleep deprivation before stroke is neuroprotective: a pre-ischemic conditioning related to sleep rebound. Exp Neurol. 2013;247:673–9. https://doi.org/10.1016/j.expneurol.2013.03.003.
Moldovan M, Constantinescu AO, Balseanu A, Oprescu N, Zagrean L, Popa-Wagner A. Sleep deprivation attenuates experimental stroke severity in rats. Exp Neurol. 2010;222(1):135–43. https://doi.org/10.1016/j.expneurol.2009.12.023.
Pace M, Adamantidis A, Facchin L, Bassetti C. Role of REM sleep, melanin concentrating hormone and orexin/hypocretin systems in the sleep deprivation pre-ischemia. PLoS ONE. 2017;12(1):1–22. https://doi.org/10.1371/journal.pone.0168430.
Jaskova K, Pavlovicova M, Jurkovicova D. Electrophysiological variability in the SH-SY5Y cellular line. Gen Physiol Biophys. 2014;31(4):375–82. https://doi.org/10.4149/gpb.
Berteotti C, Liguori C, Pace M. Dysregulation of the orexin/hypocretin system is not limited to narcolepsy but has far-reaching implications for neurological disorders. Eur J Neurosci. 2021;53(4):1136–54. https://doi.org/10.1111/ejn.15077.
Beuckmann CT, Sinton CM, Williams SC, et al. Expression of a poly-glutamine-Ataxin-3 transgene in orexin neurons induces narcolepsy-cataplexy in the rat. J Neurosci. 2004;24(18):4469–77. https://doi.org/10.1523/JNEUROSCI.5560-03.2004.
Hara J, Beuckmann CT, Nambu T, et al. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30(2):345–54. https://doi.org/10.1016/S0896-6273(01)00293-8.
Xiong X, White RE, Xu L, et al. Mitigation of murine focal cerebral ischemia by the hypocretin/ orexin system is associated with reduced inflammation. Stroke. 2013;44(3):764–70. https://doi.org/10.1161/STROKEAHA.112.681700.Mitigation.
Wei CJ, Li W, Chen JF. Normal and abnormal functions of adenosine receptors in the central nervous system revealed by genetic knockout studies. Biochim Biophys Acta. 2011;1808(5):1358–79. https://doi.org/10.1016/j.bbamem.2010.12.018.
Zhou Y, Zeng X, Li G, et al. Inactivation of endothelial adenosine A2A receptors protects mice from cerebral ischaemia-induced brain injury. Br J Pharmacol. 2019;176(13):2250–63. https://doi.org/10.1111/bph.14673.
Ledent C, Vaugeoist JM, Schiffmann SN, et al. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A(2a) receptor. Nature. 1997;388(6643):674–8. https://doi.org/10.1038/41771.
Hohoff C, Mullings EL, Heatherley SV, et al. Adenosine A2A receptor gene: evidence for association of risk variants with panic disorder and anxious personality. J Psychiatr Res. 2010;44(14):930–7. https://doi.org/10.1016/j.jpsychires.2010.02.006.
Stanzione R, Di Angelantonio E, Evangelista A, et al. β2-adrenergic receptor gene polymorphisms and risk of ischemic stroke. Am J Hypertens. 2007;20(6):657–62. https://doi.org/10.1016/j.amjhyper.2007.01.006.
Schürks M, Kurth T, Ridker PM, Buring JE, Zee RYL. Association between polymorphisms in the β2-adrenergic receptor gene with myocardial infarction and ischaemic stroke in women. Thromb Haemost. 2009;101(2):351–8. https://doi.org/10.1160/TH08-05-0284.
Kumar A, Prasad M, Kumar P, Yadav AK, Pandit AK, Kathuria P. Association between beta adrenergic receptor polymorphism and ischemic stroke: a meta-analysis. J Stroke. 2015;17(2):138–43. https://doi.org/10.5853/jos.2015.17.2.138.
Vardeny O, Peppard PE, Finn LA, Faraco JH, Mignot E, Hla KM. β2 adrenergic receptor polymorphisms and nocturnal blood pressure dipping status in the Wisconsin Sleep Cohort Study. J Am Soc Hypertens. 2011;5(2):114–22. https://doi.org/10.1016/j.jash.2011.01.005.
Proitsi P, Lupton MK, Reeves SJ, et al. Association of serotonin and dopamine gene pathways with behavioral subphenotypes in dementia. Neurobiol Aging. 2012;33(4):791–803. https://doi.org/10.1016/j.neurobiolaging.2010.06.011.
Wang H, Noordam R, Cade BE, Schwander K, Thomas W. Multi-ancestry genome-wide gene-sleep interactions identify novel loci for blood pressure. Mol Psychiatry. 2021. https://doi.org/10.1038/s41380-021-01087-0.
Ohlsson T, Lindgren A, Engström G, Jern C, Melander O. A stop-codon of the phosphodiesterase 11A gene is associated with elevated blood pressure and measures of obesity. J Hypertens. 2016;34(3):445–51. https://doi.org/10.1097/HJH.0000000000000821.
Noordam R, Bos MM, Wang H, et al. Multi-ancestry sleep-by-SNP interaction analysis in 126,926 individuals reveals lipid loci stratified by sleep duration. Nat Commun. 2019;10(1):1–13. https://doi.org/10.1038/s41467-019-12958-0.
Dashti HS, Follis JL, Smith CE, et al. Gene-environment interactions of circadian-related genes for cardiometabolic traits. Diabetes Care. 2015;38(8):1456–66. https://doi.org/10.2337/dc14-2709.
Fallaize R, Carvalho-Wells AL, Tierney AC, et al. APOE genotype influences insulin resistance, apolipoprotein CII and CIII according to plasma fatty acid profile in the Metabolic Syndrome. Sci Rep. 2017;7(1):1–10. https://doi.org/10.1038/s41598-017-05802-2.
Martínez-Martínez AB, Torres-Perez E, Devanney N, Del Moral R, Johnson LA, Arbones-Mainar JM. Beyond the CNS: the many peripheral roles of APOE. Neurobiol Dis. 2019;2020(138): 104809. https://doi.org/10.1016/j.nbd.2020.104809.
Yesavage JA, Friedman L, Kraemer H, et al. Sleep/wake disruption in Alzheimer’s disease: APOE status and longitudinal course. J Geriatr Psychiatry Neurol. 2004;17(1):20–4. https://doi.org/10.1177/0891988703261994.
Pellegrino R, Mazzotti DR, Guindalini C, Santos-Silva R, Bittencourt LRA, Tufik S. Apolipoprotein E polymorphisms and sleep quality in obstructive sleep apnea syndrome. Clin Chim Acta. 2011;412(23–24):2223–7. https://doi.org/10.1016/j.cca.2011.08.007.
Yang L, Ma L, Guo W, Fang Q, Lai X, Zhang X. Interaction of polymorphisms in APOA4-APOA5-ZPR1-BUD13 gene cluster and sleep duration on 5-year lipid changes in middle aged and older Chinese. Sleep. 2019;42(8):1–9. https://doi.org/10.1093/sleep/zsz115.
Hwang JY, Byun MS, Choe YM, et al. Moderating effect of APOE ∈4 on the relationship between sleep-wake cycle and brain β-amyloid. Neurology. 2018;90(13):e1167–73. https://doi.org/10.1212/WNL.0000000000005193.
Xue J, Zhou D, Poulsen O, et al. Intermittent hypoxia and hypercapnia accelerate atherosclerosis, partially via trimethylamine-oxide. Am J Respir Cell Mol Biol. 2017;57(5):581–8. https://doi.org/10.1165/rcmb.2017-0086OC.
Maury E, Ramsey KM, Bass J. Circadian rhythms and metabolic syndrome: from experimental genetics to human disease. Circ Res2. 2010;106(3):447–62. https://doi.org/10.1161/CIRCRESAHA.109.208355.Circadian.
Gamble KL, Berry R, Frank SJ, Young ME. Circadian clock control of endocrine factors. Nat Rev Endocrinol. 2014;10(8):466–75. https://doi.org/10.1038/nrendo.2014.78.
Guo L, Costanzo-Garvey DL, Smith DR, Neilsen BK, MacDonald RG, Lewis RE. Kinase Suppressor of Ras 2 (KSR2) expression in the brain regulates energy balance and glucose homeostasis. Mol Metab. 2017;6(2):194–205. https://doi.org/10.1016/j.molmet.2016.12.004.
Revelli JP, Smith D, Allen J, et al. Profound obesity secondary to hyperphagia in mice lacking kinase suppressor of ras 2. Obesity. 2011;19(5):1010–8. https://doi.org/10.1038/oby.2010.282.
Pearce LR, Atanassova N, Banton MC, et al. KSR2 mutations are associated with obesity, insulin resistance, and impaired cellular fuel oxidation. Cell. 2013;155(4):765. https://doi.org/10.1016/j.cell.2013.09.058.
Tekola-Ayele F, Doumatey AP, Shriner D, et al. Genome-wide association study identifies African-ancestry specific variants for metabolic syndrome. Mol Genet Metab. 2015;116(4):305–13. https://doi.org/10.1016/j.ymgme.2015.10.008.Genome-wide.
Celis-Morales C, Lyall DM, Guo Y, et al. Sleep characteristics modify the association of genetic predisposition with obesity and anthropometric measurements in 119,679 UK Biobank participants. Am J Clin Nutr. 2017;105(4):980–90. https://doi.org/10.3945/ajcn.116.147231.
Dashti HS, Follis JL, Smith CE, et al. Habitual sleep duration is associated with BMI and macronutrient intake and may be modified by CLOCK genetic variants. Am J Clin Nutr. 2015;101(1):135–43. https://doi.org/10.3945/ajcn.114.095026.
Li G, Feng D, Wang Y, et al. Loss of Cardio-protective effects at the CDH13 locus due to gene-sleep interaction: the BCAMS Study. EBioMedicine. 2018;32:164–71. https://doi.org/10.1016/j.ebiom.2018.05.033.
Chung CM, Lin TH, Chen JW, et al. A genome-wide association study reveals a quantitative trait locus of adiponectin on CDH13 that predicts cardiometabolic outcomes. Diabetes. 2011;60(9):2417–23. https://doi.org/10.2337/db10-1321.
Lin C, Tang X, Zhu Z, et al. The rhythmic expression of clock genes attenuated in human plaque-derived vascular smooth muscle cells. Lipids Health Dis. 2014;13(1):1–9. https://doi.org/10.1186/1476-511X-13-14.
Perisic L, Aldi S, Sun Y, et al. Gene expression signatures, pathways and networks in carotid atherosclerosis. J Intern Med. 2016;279(3):293–308. https://doi.org/10.1111/joim.12448.
Kaparianos A, Sampsonas F, Karkoulias K, Spiropoulos K. Obstructive sleep apnoea syndrome and genes. Neth J Med. 2006;64(8):280–9.
Shahar E, Whitney CW, Redline S, et al. Sleep-disordered breathing and cardiovascular disease: cross-sectional results of the Sleep Heart Health Study. Am J Respir Crit Care Med. 2001;163(1):19–25. https://doi.org/10.1164/ajrccm.163.1.2001008.
Caples SM, Garcia-Touchard A, Somers VK. Sleep-disordered breathing and cardiovascular risk. Sleep. 2007;30(3):291–303.
Linz D, Woehrle H, Bitter T, et al. The importance of sleep-disordered breathing in cardiovascular disease. Clin Res Cardiol. 2015;104(9):705–18. https://doi.org/10.1007/s00392-015-0859-7.
Strausz S, Ruotsalainen S, Ollila HM, et al. Genetic analysis of obstructive sleep apnoea discovers a strong association with cardiometabolic health. Eur Respir J. 2021;57(5):2003091.
Parra O, Sánchez-Armengol A, Bonnin M, et al. Early treatment of obstructive apnoea and stroke outcome: a randomised controlled trial. Eur Respir J. 2011;37(5):1128–36. https://doi.org/10.1183/09031936.00034410.
Powers WJ, Rabinstein AA, Ackerson T, et al. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke a guideline for healthcare professionals from the American Heart Association/American Stroke A. Stroke. 2019;50(12):E344–418. https://doi.org/10.1161/STR.0000000000000211.
Prabhakar NR, Semenza GL. Responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev. 2012;92(3):967–1003. https://doi.org/10.1152/physrev.00030.2011.ADAPTIVE.
Minegishi S, Sagami I, Negi S, Kano K, Kitagishi H. Circadian clock disruption by selective removal of endogenous carbon monoxide. Sci Rep. 2018;8(1):1–12. https://doi.org/10.1038/s41598-018-30425-6.
Sun Q, Zeng C, Du L, Dong C. Mechanism of circadian regulation of the NRF2/ARE pathway in renal ischemia-reperfusion. Exp Ther Med. 2021;21(3):1–9. https://doi.org/10.3892/etm.2021.9622.
Zhou L, Ouyang R, Luo H, et al. Dysfunction of Nrf2-ARE signaling pathway: potential pathogenesis in the development of neurocognitive impairment in patients with moderate to severe obstructive sleep apnea-hypopnea syndrome. Oxid Med Cell Longev. 2018. https://doi.org/10.1155/2018/3529709.
Han Q, Yeung SC, Ip MSM, Mak JCW. Intermittent hypoxia-induced NF-κB and HO-1 regulation in human endothelial EA.hy926 Cells. Cell Biochem Biophys. 2013;66(3):431–41. https://doi.org/10.1007/s12013-012-9491-6.
Cao L, Zhang Z, Cai B, et al. Association of heme oxygenase-1 gene rs2071746 polymorphism with vascular outcomes in patients with atherosclerotic stroke. J Neurol Sci. 2014;344(1–2):154–7. https://doi.org/10.1016/j.jns.2014.06.046.
Oh J, Lee MS, Jeong JH, Lee M. Deoxycholic acid–conjugated polyethylenimine for delivery of heme oxygenase-1 gene in rat ischemic stroke model. J Pharm Sci. 2017;106(12):3524–32. https://doi.org/10.1016/j.xphs.2017.07.020.
Hyun H, Won YW, Kim KM, Lee J, Lee M, Kim YH. Therapeutic effects of a reducible poly (oligo-d-arginine) carrier with the heme oxygenase-1 gene in the treatment of hypoxic-ischemic brain injury. Biomaterials. 2010;31(34):9128–34. https://doi.org/10.1016/j.biomaterials.2010.08.038.
Hyun H, Lee J, Hwang DW, et al. Combinational therapy of ischemic brain stroke by delivery of heme oxygenase-1 gene and dexamethasone. Biomaterials. 2011;32(1):306–15. https://doi.org/10.1016/j.biomaterials.2010.08.116.
Lee Y, Lee J, Kim M, Kim GY, Choi JS, Lee M. Brain gene delivery using histidine and arginine-modified dendrimers for ischemic stroke therapy. J Control Release. 2021;330(October):907–19. https://doi.org/10.1016/j.jconrel.2020.10.064.
Oh J, Kim SM, Lee EH, et al. Messenger RNA/polymeric carrier nanoparticles for delivery of heme oxygenase-1 gene in the post-ischemic brain. Biomater Sci. 2020;8(11):3063–71. https://doi.org/10.1039/d0bm00076k.
Lavie L, Lotan R, Hochberg I, Herer P, Lavie P, Levy AP. Haptoglobin polymorphism is a risk factor for cardiovascular disease in patients with obstructive sleep apnea syndrome. Sleep. 2003;26(5):592–5. https://doi.org/10.1093/sleep/26.5.592.
Milman U, Blum S, Shapira C, et al. Vitamin E supplementation reduces cardiovascular events in a subgroup of middle-aged individuals with both type 2 diabetes mellitus and the haptoglobin 2–2 genotype: a prospective double-blinded clinical trial. Arterioscler Thromb Vasc Biol. 2008;28(2):341–7. https://doi.org/10.1161/ATVBAHA.107.153965.
Staals J, Bons J, van Oostenbrugge R, et al. A SELDI-TOF-MS Study in Lacunar Stroke with Subsequent Haptoglobin Phenotyping. Curr Neurovasc Res. 2008;5(2):93–8. https://doi.org/10.2174/156720208784310187.
Staals J, Pieters B, Knottnerus I, et al. Haptoglobin polymorphism and lacunar stroke. Curr Neurovasc Res. 2008;5(3):153–8. https://doi.org/10.2174/156720208785425675.
Merkler A, Sertić J, Bazina Martinović A, et al. Haptoglobin genotype 2–2 associated with atherosclerosis in patients with ischemic stroke. Gene. 2020. https://doi.org/10.1016/j.gene.2020.144786.
Ijäs P, Saksi J, Soinne L, et al. Haptoglobin 2 allele associates with unstable carotid plaque and major cardiovascular events. Atherosclerosis. 2013;230(2):228–34. https://doi.org/10.1016/j.atherosclerosis.2013.07.008.
Bale BF, Doneen AL, Vigerust DJ. Precision healthcare of type 2 diabetic patients through implementation of haptoglobin genotyping. Front Cardiovasc Med. 2018;5(October):1–8. https://doi.org/10.3389/fcvm.2018.00141.
Belaidi E, Joyeux-Faure M, Ribuot C, Launois SH, Levy P, Godin-Ribuot D. Major Role for hypoxia inducible factor-1 and the endothelin system in promoting myocardial infarction and hypertension in an animal model of obstructive sleep apnea. J Am Coll Cardiol. 2009;53(15):1309–17. https://doi.org/10.1016/j.jacc.2008.12.050.
Abeysinghe HCS, Bokhari L, Dusting GJ, Roulston CL. Brain remodelling following endothelin-1 induced stroke in conscious rats. PLoS ONE. 2014;9(5):1–14. https://doi.org/10.1371/journal.pone.0097007.
Ziv I, Fleminger G, Djaldetti R, Achiron A, Melamed E, Sokolovsky M. Increased plasma endothelin-1 in acute ischemic stroke. Stroke. 1992;23(7):1014–6. https://doi.org/10.1161/01.STR.23.7.1014.
Li J, Xu J, Liu Z, Zou Z, Jin M, Tao T. HIF-1α attenuates neuronal apoptosis by upregulating EPO expression following cerebral ischemia-reperfusion injury in a rat MCAO model. Int J Mol Med. 2020;45(4):1027–36. https://doi.org/10.3892/ijmm.2020.4480.
Wakhloo D, Scharkowski F, Curto Y, et al. Functional hypoxia drives neuroplasticity and neurogenesis via brain erythropoietin. Nat Commun. 2020;11(1):1–12. https://doi.org/10.1038/s41467-020-15041-1.
Tani N, Ikeda T, Aoki Y, Shida A, Oritani S, Ishikawa T. Pathophysiological significance of clock genes BMAL1 and PER2 as erythropoietin-controlling factors in acute blood hemorrhage. Hum Cell. 2019. https://doi.org/10.1007/s13577-019-00248-2.
Mengozzi M, Cervellini I, Villa P, et al. Erythropoietin-induced changes in brain gene expression reveal induction of synaptic plasticity genes in experimental stroke. Proc Natl Acad Sci USA. 2012;109(24):9617–22. https://doi.org/10.1073/pnas.1200554109.
Lam SY, Tipoe GL, Fung ML. Upregulation of erythropoietin and its receptor expression in the rat carotid body during chronic and intermittent hypoxia. Adv Exp Med Biol. 2009;648:207–14. https://doi.org/10.1007/978-90-481-2259-2_24.
Dayyat EA, Zhang SX, Wang Y, Cheng ZJ, Gozal D. Exogenous erythropoietin administration attenuates intermittent hypoxia-induced cognitive deficits in a murine model of sleep apnea. BMC Neurosci. 2012;13(1):1. https://doi.org/10.1186/1471-2202-13-77.
Chen Q, Lin G, Huang J, et al. Inhibition of miR-193a-3p protects human umbilical vein endothelial cells against intermittent hypoxia-induced endothelial injury by targeting FAIM2. Aging. 2020;12(2):1899–909. https://doi.org/10.18632/aging.102729.
Komnig D, Gertz K, Habib P, et al. Faim2 contributes to neuroprotection by erythropoietin in transient brain ischemia. J Neurochem. 2018;145(3):258–70. https://doi.org/10.1111/jnc.14296.
Kendzerska T, Kamra M, Murray BJ, Boulos MI. Incident cardiovascular events and death in individuals with restless legs syndrome or periodic limb movements in sleep: a systematic review. Sleep. 2017;40(3):1–14. https://doi.org/10.1093/sleep/zsx013.
Cholley-Roulleau M, Chenini S, Béziat S, Guiraud L, Jaussent I, Dauvilliers Y. Restless legs syndrome and cardiovascular diseases: a case-control study. PLoS ONE. 2017;12(4):1–13. https://doi.org/10.1371/journal.pone.0176552.
Ferini-Strambi L, Walters AS, Sica D. The relationship among restless legs syndrome (Willis-Ekbom Disease), hypertension, cardiovascular disease, and cerebrovascular disease. J Neurol. 2014;261(6):1051–68. https://doi.org/10.1007/s00415-013-7065-1.
Gao X, Ba DM, Bagai K, Liu G, Ma C, Walters AS. Treating restless legs syndrome was associated with low risk of cardiovascular disease: a cohort study with 3.4 years of follow-up. J Am Heart Assoc. 2021;10(4):1–15. https://doi.org/10.1161/JAHA.120.018674.
Winkelman JW, Blackwell T, Stone K, Ancoli-Israel S, Redline S. Associations of Incident cardiovascular events with restless legs syndrome and periodic leg movements of sleep in older men, for the outcomes of sleep disorders in older men study (MrOS Sleep Study). Sleep. 2017;40(4):11. https://doi.org/10.1093/sleep/zsx023.
Winkelmann J, Schormair B, Lichtner P, et al. Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat Genet. 2007;39(8):1000–6. https://doi.org/10.1038/ng2099.
Graff M, Scott RA, Justice AE, et al. Genome-wide physical activity interactions in adiposity—a meta-analysis of 200,452 adults. PLoS Genet. 2017;13(4):1–26. https://doi.org/10.1371/journal.pgen.1006528.
Bellei E, Bergamini S, Monari E, et al. Evaluation of potential cardiovascular risk protein biomarkers in high severity restless legs syndrome. J Neural Transm. 2019;126(10):1313–20. https://doi.org/10.1007/s00702-019-02051-7.
Bellei E, Monari E, Ozben S, et al. Discovery of restless legs syndrome plasmatic biomarkers by proteomic analysis. Brain Behavior. 2018;8(10):1–9. https://doi.org/10.1002/brb3.1062.
Hu Z, Liu J, Song Z, Hou Q, Fan X, Hou D. Variants in the atherogenic ALOX5AP, THBD, and KNG1 genes potentiate the risk of ischemic stroke via a genetic main effect and epistatic interactions in a Chinese population. J Stroke Cerebrovasc Dis. 2015;24(9):2060–8. https://doi.org/10.1016/j.jstrokecerebrovasdis.2015.04.036.
Rohmann JL, de Haan HG, Algra A, Vossen CY, Rosendaal FR, Siegerink B. Genetic determinants of activity and antigen levels of contact system factors. J Thromb Haemost. 2019;17(1):157–68. https://doi.org/10.1111/jth.14307.
Sabater-Lleal M, Martinez-Perez A, Buil A, et al. A genome-wide association study identifies KNG1 as a genetic determinant of plasma factor XI level and activated partial thromboplastin time. Arterioscler Thromb Vasc Biol. 2012;32(8):2008–16. https://doi.org/10.1161/ATVBAHA.112.248492.
Mahta A, Yaghi S, Reznik ME, et al. Serum alpha-1 antitrypsin in acute ischemic stroke: a prospective pilot study. J Clin Neurosci. 2020;76:20–4. https://doi.org/10.1016/j.jocn.2020.04.074.
Wang T, Lu H, Li D, Huang W. Tgf-β1-mediated activation of serpine1 is involved in hemin-induced apoptotic and inflammatory injury in ht22 cells. Neuropsychiatr Dis Treat. 2021;17:423–33. https://doi.org/10.2147/NDT.S293772.
Malik R, Dau T, Gonik M, et al. Common coding variant in SERPINA1 increases the risk for large artery stroke. Proc Natl Acad Sci USA. 2017;114(14):3613–8. https://doi.org/10.1073/pnas.1616301114.
Liu Q, Cui P, Zheng K, et al. SERPINA1 gene expression in whole blood links the rs6647 variant G allele to an increased risk of large artery atherosclerotic stroke. FASEB J. 2020;34(8):10107–16. https://doi.org/10.1096/fj.201903197R.
Larsson SC, Markus HS. Genetic liability to insomnia and cardiovascular disease risk. Circulation. 2019;140(9):796–8. https://doi.org/10.1161/CIRCULATIONAHA.119.041830.
Parsa H, Imani A, Faghihi M, et al. Acute sleep deprivation preconditions the heart against ischemia/reperfusion injury: the role of central GABA-A receptors. Iran J Basic Med Sci. 2017;20:1232–41. https://doi.org/10.22038/IJBMS.2017.9539.
Tao L, Wang L, Yang X, Jiang X, Hua F. Recombinant human glucagon-like peptide-1 protects against chronic intermittent hypoxia by improving myocardial energy metabolism and mitochondrial biogenesis. Mol Cell Endocrinol. 2018;2019(481):95–103. https://doi.org/10.1016/j.mce.2018.11.015.
Korkutata M, Saitoh T, Cherasse Y, et al. Enhancing endogenous adenosine A2A receptor signaling induces slow-wave sleep without affecting body temperature and cardiovascular function. Neuropharmacology. 2018;2019(144):122–32. https://doi.org/10.1016/j.neuropharm.2018.10.022.
Li Y, Perry TA, Kindy MS, et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci USA. 2009;106(4):1285–90. https://doi.org/10.1073/pnas.0806720106.
Fang X, Tong KZ, Wang X, Ni HF. Two mixed-ligand coordination polymers: Crystal structures and protective effect on ischemic stroke by increasing glp1r expression. J Oleo Sci. 2020;69(7):743–50. https://doi.org/10.5650/jos.ess20047.
Shinozuka K, Staples M, Borlongan CV. Melatonin-based therapeutics for neuroprotection in stroke. Int J Mol Sci. 2013;14(5):8924–47. https://doi.org/10.3390/ijms14058924.
Rhim T, Lee DY, Lee M. Hypoxia as a target for tissue specific gene therapy. J Control Release. 2013;172(2):484–94. https://doi.org/10.1016/j.jconrel.2013.05.021.
Jiménez-Ortega V, Barquilla PC, Pagano ES, Fernández-Mateos P, Esquifino AI, Cardinali DP. Melatonin supplementation decreases prolactin synthesis and release in rat adenohypophysis: correlation with anterior pituitary redox state and circadian clock mechanisms. Chronobiol Int. 2012;29(8):1021–35. https://doi.org/10.3109/07420528.2012.705936.
Acknowledgements
Compliance with ethics guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Funding
No Rapid Service Fee was received by the journal for the publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author contributions
LK: concept and design, drafting and revision of the manuscript.
Disclosures
Lyudmila Korostovtseva has nothing to disclose.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Korostovtseva, L. Ischemic Stroke and Sleep: The Linking Genetic Factors. Cardiol Ther 10, 349–375 (2021). https://doi.org/10.1007/s40119-021-00231-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40119-021-00231-9