
Abstract
In a series of salts of ethane-1,2-diamine with acetic (1), trifluoroacetic (2), trichloroacetic (3), and dichloroacetic (4) acids, the amine is doubly protonated so the cation:anion stoichiometry is 2:1. In 1 and 2, dications are lying across the center of symmetry (Z′ = 1/2), in 4 dication is nonsymmetric (Z′ = 1), while the structure of 3 contains both symmetric and nonsymmetric cations (Z′ = 3/2). All dications, either symmetric or not, are in extended (trans) form, by far the most popular among the salts of ethane-1,2-diamine. Strong hydrogen N–H···O bonds constitute the main specific, directional force determining the crystal packing. In 1, one of the N–H hydrogen atoms is involved in bifurcated hydrogen bonds, and in this case, four alresaacceptor oxygen atoms are almost equally involved in accepting the hydrogen bonds; in consequence, the C–O bond lengths are almost equal. In all other cases, the C–O bonds with oxygen atoms involved in more hydrogen bonds are systematically longer than those which accept less such interactions. Halogen C–Cl···O interactions are observed in two (out of five) symmetry-independent anions of 3 and 4, suggesting that at least in this series, the roles of these interactions are only secondary in nature. No significant involvement of fluorine atoms in directional interactions is detected, however. The hydrogen-bond networks are described by means of graph-set method. In addition, Hirshfeld surfaces are used to detect and visualize the differences between similar moieties, and fingerprint plots are presented in order to analyze the mutual importance of different kinds of interactions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The disposition of molecules (or molecular ions) in the molecular crystals is determined by the fine equilibrium between a number of competing factors, among others the tendency toward close packing, the requirements of the space group of symmetry, interplays between different intermolecular interactions, conformational freedom, etc. (e.g., [1, 2]). There are often more than one possible disposition of molecules, which reflect in the phenomenon of polymorphism, and show the level of complication for the attempts of predicting crystal structures ([3], and references therein). It is therefore quite important to gather as much information as possible about the different ways of achieving such an equilibrium state, that is, to determine and analyze different crystal structures. In particular, studying the differences in crystal packing between the compounds that differ slightly can be of certain interest. Also, the structures of multicomponent crystals (co-crystals, solvates, and molecular salts) are widely studied for this very reason. Meanwhile, one can reasonably argue that the simpler the components, the easier the analysis of the hierarchy of factors determining the crystal structure.
For some time, we have studied the packing modes of simple salts of α,ω-diaminoaliphatic alkanes (linear molecules with hydrogen-bond donors located at the ends of the chain) with acetic acid and its mono- di-, and trichloro derivatives, which provide the acceptors for hydrogen bonds as well as the possibility of halogen-bonding interactions (for instance, [4, 5]). In these cases, we have determined that the potential halogen bonds or N–H···Cl hydrogen bonds do not compete successfully with the N–H···O hydrogen bonds of different strengths which—certainly along with the coulombic interactions between the charged species—are the main driving force of the crystal packing along this series of compounds.
In this paper, we present the analyses of the crystal structures and packing modes for a series of salts of the simplest member of this family—ethane-1,2-diamine. We were able to obtain the single crystals of salts with acetic (1), trifluoroacetic (2), trichloroacetic (3), and dichloroacetic (4) acids (Scheme 1). The room-temperature crystal structure of 3 has been previously reported [6], but—as the crystal structure of this compound was not discussed before—we decided to add the low temperature (100 K) structure in this paper. Recently also the structure of 1 has been reported [7], but—as we use it for deeper analysis—we prefer to use our data.

The studied salts
Experimental
General procedure
All chemicals were purchased from Aldrich and were used as received without further purification. Salts were prepared in a single solution or in solvent mixture. All of the salts were obtained in a direct synthesis: 1 mmol of ethane-1,2-diamine was dissolved in 2 cm3 of nitroethane:methane 1:1 mixture in the glass veil and mixed with 2.4 mmol of appropriate acid (20 % of excess for each of amino group to assure complete protonation of both groups). All of the solutions were heated and afterward kept for crystallization at ambient temperature in a well-ventilated room.
X-ray diffraction
Diffraction data were collected on an Agilent Technologies Xcalibur diffractometer equipped with an EOS CCD area detector (fine-focus tube, MoKα, λ = 0.71073 Å) for 3, 4 at 100(1) K; and for 1 at room temperature, and for 2 at room temperature on Agilent Technologies SuperNova diffractometer equipped with an Atlas CCD area detector (microfocus tube, CuKα, λ = 1.54178 Å). The data collections for the crystals were performed using CrysAlisPro [8]. Unit cell parameters were determined by the least-squares procedure for the reflections (1693 for 1, 1193 for 2, 11,249 for 3, and 3722 for 4) chosen from the whole datasets on the basis of their intensities. The data were corrected for Lorentz and polarization effects, as well as for absorption, by means of empirical correction using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm. All structures were determined by direct methods using SIR92 [9] and refined by the full-matrix least-squares method using SHELXL–97 [10] (these procedures were performed within WinGX suite of programs [11]). The crystals of 1 turned out to be twins; this fact was taken into account in the data reduction [8] and in refinement [10]. The BASF parameter, which describes the ratio of two components, refined at 0.625(2). In 3 I turn, one of the trichloroacetate anions was refined as disordered over two positions with site occupation factors of 0.813(9) and 0.187(9). In the room-temperature study [6], similar disorder was detected but the s.o.f.s were simply fixed at 0.5.
All non-hydrogen atoms were refined with anisotropic displacement parameters. Positions of hydrogen atoms were located from different electron density maps and isotropically refined for 1, 2, and 3, while for 4, the isotropic displacement parameters were set at 1.2 (1.5 for NH3 hydrogens) times the U eq of the parent non-hydrogen atom. Table 1 lists the relevant crystallographic data together with the refinement details.
Results and discussion
Geometry of ions
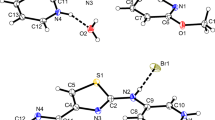
Figures 1, 2, 3, 4 show the perspective views of the salts. In three out of four structures (1–3), the cations occupy the special positions (on the center of symmetry). The structure of 3 contains also another, nonsymmetric cation in the asymmetric part of the unit cell (Z′ = 3/2 in this case, while it is 1/2 in 1 and 2). The C i —symmetric cations are—by the virtue of symmetry—fully extended (trans), N–C–C–N torsion angles are 180°.

Perspective views of the salt 1 (a), 2 (b), 3 (c), and 4 (d) with the labeling scheme. The ellipsoids are drawn at the 50 % probability level; hydrogen atoms are shown as spheres of arbitrary radii. Blue dashed lines show the hydrogen bonds. The primed atoms are related to nonprimed ones by the symmetry operation a 1 − x, −y, 2 − z (center of inversion at ½ 0 1) b 1 − x, 1 − y, 1 − z (center of inversion at ½ ½ ½) c 1 − x, 2 − y, 1 − z (center of inversion at ½ 1 ½). For the disordered anion in 3 (C), only the higher occupied part is shown

A number of the hits in the CSD [12] as a function of the N–C–C–N torsion angle for ethane-1,2-diamine cation

Hirshfeld surfaces for dication in 1 (left) and 2 (right); red spots in the surface show the close intermolecular contacts. The latter one is typical for all other examples. The difference in the hydrogen-bond donor capability of the NH group on the right-hand side of the cation is clearly visible (bifurcated for 1, single for 2)

A comparison of Hirshfeld surfaces for two symmetry-independent cations in 4; one of it (left) is involved in halogen bond, while the other is not
Also in the two nonsymmetric cations, these torsion angles are close to 180°: 168.93(12)° in 3 and −177.86(19)° in 4. Such conformation is strongly preferred; Fig. 2 shows the distribution of the N–C–C–N torsion angles for 238 cations found in the CSD ([12], CSD version 5.35, last updated Nov. 2013). One can infer that there is approximately 3:1 preference of trans conformation with respect to the gauche one (almost half of cations are C i -symmetric).
The bond lengths and angles in the cations are quite typical. The anions show the characteristic equalization of C–O bond lengths, due to the deprotonation; the smallest difference in lengths is observed in 1 (vide infra), and it seems to correlate with the smallest value of O–C–O bond angle. Conformation of the anion might be described by the dihedral angles between the COO and CCH (CCF, CCCl) planes. For 1–3, the conformation is almost identical, even taking into account the disorder of one of the trichloroacetates in 3: the dihedral angles are around 20°, 40°, and 80°. Similar conformation is observed for 4A anion (with CCO/CCH angle of ca. 28°), while for 4B, it is a little bit different: around 0°, 60°, 60° (CCO/CCH angles of ca. 54°).
Intermolecular interactions
It is obvious—just by looking at the structures of components of the salts—that the principal interactions, responsible for the details of the crystal packing, in every case should be hydrogen bonding. Coulombic interactions between charged species are of course also present and strong, but they are less specific and directional. In 3 and 4, some halogen bonds are also possible, as well as—in all cases—weaker hydrogen-bond-type interactions like C–H···O, C–H···F or C–H···Cl.
In fact, for this case, Table 2 lists the hydrogen-bond data for all compounds. Interestingly, one can detect quite fine but an important difference between acetate (1) on one side, and all halogeno derivatives on the other. Only in the case of 1, there are both strong, directional N–H···O hydrogen bonds and weaker, bifurcated one (H1B2). This can be visualized in the Hirshfeld surfaces [13–15] of these cations (Fig. 3). All calculations were performed by means of the CrystalExplorer software [16].
In 1, also the H-atoms from the anion are involved in relatively short and directional C–H···O anion-to-anion hydrogen bonds. In effect, in 1, each oxygen atom is involved in one strong N–H···O hydrogen bond and two weaker (but comparable) interactions,—cf. Table 2, and both C–O bonds are almost equal [1.2514(17) Å and 1.2530(17) Å]. In all other derivatives, there are only strong, directional, nonbifurcated hydrogen bonds, and—as there are three donors per two acceptors—the oxygen atoms accept different numbers of bonds. These differences correlate well with the C–O bond lengths: those with O atoms accepting two h-bonds are systematically longer [mean value of 1.250(4) Å] than those with O atoms accepting only one hydrogen bond [1.231(5) Å].
In both structures containing chlorine atoms (3 and 4), one can identify one (only one!) contact which can be regarded as a weak C–Cl···O halogen bond [Cl···O distances of 3.0673(12) Å in 3 and 3.0745(10) in 4, C–Cl···O angles: 154.69(6)° and 170.62(5)°, respectively], and in fact, this can be also observed in Hirshfeld surfaces (Fig. 4). These interactions should be probably regarded as secondary ones (similar to few C–H···O interactions in 2–4, listed in Table 2). No Cl···Cl contacts shorter than 3.503 Å are observed.
Fluorine atoms in 2 seemingly are not involved in any specific interactions; shortest H···F distance is 2.76 Å and that of F···F is 2.96 Å.
To analyze the hydrogen-bond networks, we applied the graph-set method [17, 18], which allows to systematize different packing motifs. To simplify analysis, we are taking into account only N–H···O hydrogen bonds; as the other interactions are almost certainly of secondary nature—the consequence of the stronger interaction rather than the structure-defining ones.
Of course, strictly speaking, all the simplest motifs are noncyclic dimers (D) as the hydrogen bonds connect different (charged) species—cations and anions.
In 1, there are infinite chains of hydrogen-bonded cations and anions (Fig. 5a); this motif might be described as \( {\text{C}}_{2}^{2} \left( 6\right), \) which are connected into \( {\text{R}}_{4}^{4} \left( { 1 4} \right) \) hydrogen-bonded centrosymmetric rings. In connection with bifurcated N–H···O hydrogen bonds, the bi-layers of anions connected by perpendicularly oriented cations are formed (Fig. 5b). The bi-layers are tightly packed, probably by electrostatic interactions. Interestingly, such highly ordered packing leads to very high packing coefficients as calculated by PLATON [19] of 76.2 % compared with ca. 68–69 % in other three structures). Also the void volume [20]—as calculated by means of Crystal Explorer [16] with the default threshold value of electron density equal to 0.002e au−3—is very small in 1, equal to 6.07 Å3, which amounts to 2.8 % of the unit cell volume. In the remaining structures, the voids are larger: in 2: 92.3 Å3 (16.2 %), in 3: 301.6 Å3 (13.4 %), and in 4: 161.3 Å3 (12.4 %).

a The hydrogen-bonded bilayer in the structure of 1; b fragment of crystal packing of 1 as seen along a direction
In 2–4, there are no such prominent and structure-defining chains as described above; instead, the dominating features are symmetric or even unsymmetric rings. In 2, the main motif is the \( {\text{R}}_{4}^{4} \left( { 1 2} \right) \) ring, as well as more complicated, larger rings, e.g., \( {\text{R}}_{6}^{8} \left( { 2 0} \right) \) and \( {\text{R}}_{8}^{8} \left( { 3 0} \right) \).
In 3, simple \( {\text{R}}_{2}^{4} \left( 8\right) \) rings are interconnected by larger \( {\text{R}}_{3}^{4} \left( { 1 3} \right) \) and \( {\text{R}}_{4}^{4} \left( { 1 8} \right) \) ones, while in 4, there are only larger rings: \( {\text{R}}_{3}^{4} \left( { 1 3} \right) \) and \( {\text{R}}_{8}^{6} \left( { 2 2} \right) \).
The mutual importance of different intermolecular contacts/interactions might be analyzed/visualized by the so-called fingerprint graphs [21]. In the study case, we compared the fingerprints for cations and anions. Figure 6 shows the appropriate plots for all cations in the analyzed series. Obviously, the kind of anion influences the shape of the fingerprint, but always there is a sharp spike extending toward ca. 1.05, 0.7 point. These spikes are related to the H···O hydrogen-bond interactions; remaining part of the plot is connected with other interactions. Table 3 lists the percentage amounts of different contacts in the plots. It might be stressed that all these contacts are related with hydrogen atoms in the cations—these atoms make all the available surface. Almost exclusively, the shortest contacts are between cations and anions; the share of H···H contacts, quite popular in many organic structures, is very small in structures where the anions have no hydrogen atoms (2 and 3).

Fingerprint plots for anions in the structures 1–4
The analysis of the shapes of fingerprint plots for the anions (Fig. 7) is even more interesting. Table 4 lists the shares of the most important kinds of contacts.

Fingerprint plots for cations in the structures 1–4
Here also, in all cases the lower spike (at ca. 0.7, 1.1) is related to the O···H contacts, so in these cases, to the accepting of strong hydrogen bonds. Other features, specific for each structure, depend on the structure of the anion. In 1, the additional spike (at 1.3, 1.0) is built by H···O contacts and is related to the C–H···O anion···anion hydrogen bonds (cf. discussion above). In 2, the structures above main spike (1.7, 1.5) are due to the different F···F, H, O contacts. In both 3 and 4, one can observe similar structures caused by contacts with Cl atoms. It might be noted, however, that the most pronounced spikes are observed in 3A and 4B anions—exactly those which were identified as the acceptors of C–Cl···O halogen bonds.
Conclusions
N–H···O hydrogen bonds, relatively short and directional, are the principal factors in the determination of the crystal structures of four 1:2 salts of ethane-1,2-diamine with acetic (1), trifluoroacetic (2), trichloroacetic (3), and dichloroacetic acids (4). The dications—either C i -symmetric (1, 2, one of symmetry-independent cations in 3) or nonsymmetric (the other one in 3 and 4)—are all in extended conformation, with N–C–C–N torsion angles being equal or close to 180°. The C–O bond lengths in the anions are correlated with the number of hydrogen bonds accepted by oxygen atoms. In 1, both O atoms are involved in the same number of similar interactions, and C–O bonds are almost equal; in all other cases (six anions altogether), the oxygen atom involved in more hydrogen bonds is longer than the other one [1.250(4) Å vs. 1.231(5) Å]. The graph-set analysis allowed us to pinpoint the differences in hydrogen-bond networks between 1—with both chain and ring motifs—and the other structures where the ring structures of different sizes are dominant. These differences reflect in the small void volume (2.8 % of the unit cell) and large packing index (76.2 %) in 1 compared with 2–4 (voids 12–16 %, packing index 68–69 %). The fingerprint analysis, based on the Hirshfeld surface calculations, allowed us to visualize and quantitate the roles of different intermolecular interactions, not only specific and directional, in crystal packing.
References
G.R. Desiraju, Angew. Chem. Int. Ed. Engl. 34, 2311 (1995)
J.D. Dunitz, A. Gavezzotti, Angew. Chem. Int. Ed. Engl. 44, 1766 (2005)
C.R. Groom, A.M. Reilly, Acta Cryst. B70, 776 (2014)
A. Paul, M. Kubicki, J. Mol. Struct. 938, 238 (2009)
A. Paul, M. Kubicki, J. Mol. Struct. 966, 107 (2010)
J.H. Buttery, Effendy, P.C. Junk, S. Mutrofin, B.W. Skelton, C.R. Whitaker, A.H. White, Z. Anorg. Allg. Chem. 632, 1326 (2006)
M. Luo, L. Wang, J.-J. Qiu, J.-H. Zhang, Q.-R. Li, H. Yin, Jiegou Huaxue (Chin. J. Struct. Chem.) 32, 1835 (2014)
Agilent Technologies (2012) CrysAlisPro
A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M.C. Burla, G. Polidori, M. Camalli, J. Appl. Cryst. 27, 435 (1994)
G.M. Sheldrick, Acta Cryst. C71, 3 (2015)
L.J. Farrugia, J. Appl. Cryst. 45, 849 (2012)
F.H. Allen, Acta Cryst. B58, 380 (2002)
J.J. McKinnon, F.P.A. Fabbiani, M.A. Spackman, Cryst. Growth Des. 7, 755 (2007)
M.A. Spackman, D. Jayatilaka, CrystEngComm 11, 19 (2009)
M.A. Spackman, Phys. Scr. 87, 048103 (2013)
S.K. Wolff, D.J. Grimwood, J.J. McKinnon, M.J. Turner, D. Jayatilaka, M.A. Spackman, M.A. CrystalExplorer (Version 3.1), University of Western Australia (2012)
M.C. Etter, J.C. MacDonald, J. Bernstein, Acta Cryst. B46, 256 (1990)
J. Bernstein, R.E. Davis, L. Shimoni, N.-L. Chang, Angew. Chem. Int. Ed. Engl. 34, 1555 (1995)
A.L. Spek, Acta Cryst. D65, 148 (2009)
M.J. Turner, J.J. McKinnon, D. Jayatilaka, M.A. Spackman, CrystEngComm 13, 1804 (2011)
M.A. Spackman, J.J. McKinnon, CrystEngComm 4, 378 (2002)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Poulain, A., Kubicki, M. Crystal packing modes in the salts of ethane-1,2-diamine with acetic acid and its halogeno derivatives. J IRAN CHEM SOC 12, 2115–2123 (2015). https://doi.org/10.1007/s13738-015-0688-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-015-0688-7