
Abstract
The synthesis and structural characterization of N-(6-methoxypyridin-3-yl)-4-(pyridin-2-yl)thiazol-2-amine mono-hydrobromide monohydrate (3) and N-(6-methoxypyridin-3-yl)-4-(pyrazin-2-yl)thiazol-2-amine mono-hydrobromide 0.35 methanol solvate (4) are reported. The crystal structures of 3 (monoclinic, space group P21/n, Z = 4) and 4 (monoclinic, space group, C2/c, Z = 8) feature N,4-diheteroaryl 2-aminothiazoles showing similar molecular conformations but different sites of protonation and thus distinctly different intermolecular hydrogen bonding patterns. In 3, Namine–H⋯Br−, N+pyridine–H⋯Owater, and Owater–H⋯Br− hydrogen bonds link protonated N-(6-methoxypyridin-3-yl)-4-(pyridin-2-yl)thiazol-2-amine and water molecules and bromide anions into a three-dimensional hydrogen-bonded network, whereas intermolecular N+methoxypyridine–H⋯Npyrazine hydrogen bonds result in hydrogen-bonded zigzag chains of protonated N-(6-methoxypyridin-3-yl)-4-(pyrazin-2-yl)thiazol-2-amine molecules in 4.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The 2-aminothiazole unit is a synthetically versatile building block, which has been widely used in medicinal chemistry. A number of active pharmaceutical ingredients containing a 2-aminothiazole moiety with different pharmacological properties are on the market. They include, for example, the third-generation cephalosporin anti-infective cefdinir, the β3 adrenergic agonist mirabegron, the tyrosine kinase inhibitor dasatinib, and the recently approved phosphatidylinositol-3-kinase (PI3K) inhibitor alpelisib. Antiproliferative, antidiabetic, antihypertensive, and anti-inflammatory properties as well as antiviral, antitubercular, antifungal, antileishmanial, and antiprion activities of 2-aminothiazoles have been reported [1].
Antileishmanial properties of N,4-diaryl substituted 2-aminothiazoles have been studied based on a hit in a screening of 200,000 compounds [2], and growth inhibition of other microorganisms including plasmodia [3] and mycobacteria [4] by this compound class has also been described. A series of N,4-diaryl 2-aminothiazoles with activity against Mycobacterium tuberculosis were subject of a structure-activity relationship (SAR) study reported by Meissner et al. [5]. Makam and Kannan evaluated N,4-diaryl substituted 2-aminothiazoles for inhibitory potential against M. tuberculosis, H37Rv, and reported minimum inhibitory concentration (MIC) values of 6.25–12.50 μM [6]. In view of these results, N,4-diheteroaryl substituted 2-aminothiazoles attracted our interest in the course of our studies on new antimycobacterial agents. Recently, we have explored the structural chemistry of freebase N,4-diheteroaryl 2-aminothiazoles with a 4-methylpyridin-2-yl group bound to the amino group at the 2-position of the thiazole core [7, 8].
Herein, we report the synthesis and structural characterization of two N,4-diheteroaryl 2-aminothiazole hydrobromides. Bromide is among the anions that are currently available for salt formation of active pharmaceutical ingredients [9], and a number of hydrobromide drugs, for example, the antitussive dextromethorphan hydrobromide, are widely used. To the best of our knowledge and based on the search of the Cambridge Structural Database (CSD) [10] via WebCSD in October 2020 [11], hydrobromide salts of N,4-diaryl 2-aminothiazoles have not been structurally characterized so far. The structure of a related 4-phenyl-2-(2-phenylhydrazinyl)thiazol-3-ium bromide was, however, reported very recently [12]. Structural insight into molecular conformations, preferred sites of protonation, and hydrogen bonding patterns in pharmaceutical salt forms is important for drug design and formulation development.
Experimental section
General
Starting materials were purchased from Sigma-Aldrich and used as received. Solvents were of analytical grade. The synthesis of 2-bromo-1-(pyridin-2-yl)ethanone hydrobromide was published by others [13].
Physical methods
Melting points (uncorrected) were determined on a Boëtius hot-stage microscope (VEB Kombinat NAGEMA, Dresden, GDR). NMR spectra were recorded at room temperature on an Agilent Technologies VNMRS 400 and a Varian INOVA 500 NMR spectrometer. The residual solvent signal of DMSO-d6 (δH = 2.50 ppm) was used to reference the spectra (s = singlet, bs = broad singlet, d = doublet, dd = doublet of doublets, td = triplet of doublets). APCI mass spectrometry was carried out on an Advion Expression compact mass spectrometer. High-resolution ESI mass spectra were measured on a Bruker Daltonics Apex III FT-ICR mass spectrometer.
Synthesis and crystallization
N-((6-Methoxypyridin-3-yl)carbamothioyl)benzamide (1) [14]
2.90 g (23.4 mmol) of 2-methoxy-5-aminopyridine were dissolved in 70 mL of acetone and benzoyl isothiocyanate (3.3 mL, 24.5 mmol) was added dropwise with stirring. The reaction mixture was warmed to 40 °C for 10 min, before the solvent was removed under reduced pressure. The crude product was recrystallized from acetone. Yield 4.44 g (15.5 mmol, 66%). 1H NMR (400 MHz, DMSO-d6) δ = 12.30 (s, 1H), 11.66 (s, 1H), 8.28 (d, J = 2.7 Hz, 1H), 7.98 (m, 2H), 7.93 (dd, J = 8.8, 2.7 Hz, 1H), 7.74–7.62 (m, 1H), 7.55 (m, 2H), 6.88 (d, J = 8.8 Hz, 1H), 3.87 (s, 3H) ppm; MS (APCI): m/z [M + H]+ calcd. for C14H14N3O2S+: 288; found 288.
1-(6-Methoxypyridin-3-yl)thiourea (2) [14, 15]
Compound 1 (4.44 g, 15.5 mmol) was suspended in 15 mL of methanol and 1.2 mL of 40% aqueous NaOH (17.2 mmol) was added. The solution turned clear and was heated to reflux for 1 h. Subsequently, the solvent was removed under reduced pressure. The product was washed with water and dried in a vacuum desiccator over P2O5. Yield: 1.61 g (8.8 mmol, 57%). 1H NMR (400 MHz, DMSO-d6) δ = 9.50 (bs, 1H), 8.05 (d, J = 2.7 Hz, 1H), 7.70 (dd, J = 8.8, 2.7 Hz, 1H), 7.49 (bs, 2H), 6.79 (d, J = 8.8 Hz, 1H), 3.83 (s, 3H) ppm; MS (APCI): m/z [M + H]+ calcd. for C7H10N3OS+: 184; found: 184.
N-(6-methoxypyridin-3-yl)-4-(pyridin-2-yl)thiazol-2-amine mono-hydrobromide monohydrate (3)
2-Bromo-1-(pyridine-2-yl)ethanone hydrobromide (696 mg, 2.48 mmol) and 418 mg (2.28 mmol) of 2 were dissolved in 20 mL of ethanol, and triethylamine (0.1 mL) was added. The reaction mixture was heated to reflux for 2 h and subsequently the solvent was removed under reduced pressure. The crude product was recrystallized from methanol. Yield 550 mg (1.44 mmol, 63%); m.p. 152–154 °C. 1H NMR (500 MHz, DMSO-d6) δ = 10.59 (s, 1H), 8.76 (dd, J = 2.9, 0.7 Hz, 1H), 8.75 (dt, J = 5.6 Hz, 1.2 Hz, 1H), 8.50–8.43 (m, 2H), 8.14 (s, 1H), 8.08 (dd, J = 8.8, 2.9 Hz, 1H), 7.81 (td, J = 5.6 Hz, 1H), 6.87 (dd, J = 8.8, 0.7 Hz, 1H), 3.85 (s, 3H) ppm; HRMS (ESI): m/z [M + H]+ calcd. for C14H13N4OS+ 285.0805; found: 285.0800.
N-(6-Methoxypyridin-3-yl)-4-(pyrazin-2-yl)thiazol-2-amine mono-hydrobromide 0.35 methanol solvate (4)
Compound 4 was prepared in analogy to 3 from 2 (453 mg, 2.47 mmol) and 2-bromo-1-(pyrazine-2-yl) ethanone hydrobromide [16] (note that the compound is not denoted as hydrobromide therein), which was synthesized from acetylpyrazine (305 mg, 2.50 mmol) using 2-pyrrolidone hydrotribromide (1.36 g, 2.74 mmol) as reagent and used in situ without purification. Yield (based on 2): 344 mg (0.91 mmol, 37%); m.p. 195–197 °C. 1H NMR (500 MHz, DMSO-d6) δ = 10.49 (s, 1H), 9.18 (d, J = 1.5 Hz, 1H), 8.65 (dd, J = 2.6, 1.5 Hz, 1H), 8.63 (d, J = 2.8 Hz, 1H), 8.58 (d, J = 2.5 Hz, 1H), 8.19 (dd, J = 9.0, 2.8 Hz, 1H), 7.68 (s, 1H), 6.97 (d, J = 9.0 Hz, 1H), 3.88 (s, 3H), 3.16 (s, solvate methanol) ppm; HRMS (ESI): m/z [M + H]+ calcd. for C13H12N4OS+ 286.0758; found: 286.0753.
X-ray crystallography
The X-ray intensity data for 3 were measured on a Bruker AXS Apex II diffractometer and those for 4 on an Enraf-Nonius KappaCCD diffractometer, both equipped with a FR591 rotating anode radiation source. Data reductions were performed using the SAINT software [17] for 3 and EvalCCD [18] for 4. In both cases, face-indexed absorption corrections were carried out using SADABS [19]. The crystal structures were solved with SHELXT-2018/1 [20] and refined with SHELXL-2018/3 [21]. The methanol molecule of crystallization in 4 is disordered about a crystallographic twofold rotation axis, and the occupancy was refined freely to yield 0.70(1), using isotropic displacement parameters for the carbon and oxygen atoms. Carbon-bound hydrogen atoms were placed in geometrically calculated positions with Caromatic–H = 0.95 Å and Cmethyl–H = 0.98 Å and refined with the appropriate riding model. Methyl groups (apart from methanol) were allowed to rotate to match the underlying electron density maxima. Hydrogen atoms attached to nitrogen were localized in difference electron density maps and refined with the N–H bond lengths restrained to a target value of 0.88(2) Å. Uiso(H) = 1.2 Ueq(C, N, O) (1.5 for methyl groups) was applied for all hydrogen atoms. We note that the highest difference electron density peak of 1.90 eÅ−3 in 4 is located 0.65 Å from Br1. Crystal data and refinement details for 3 and 4 are summarized in Table 1. Structure pictures were generated with Diamond [22].
Computational methods
DFT calculations were undertaken using the program ORCA (version 4.2) [23] with a B3LYP hybrid functional (20% HF exchange) [24, 25] using a def2-TZVPP basis set [26]. Optimization of the structures used the BFGS method from an initial Hessian according to Almoef’s model with a very tight self-consistent field convergence threshold [27]. Calculations were made on the free cations of 3 and 4 as well as their corresponding free bases. Avogadro was used as a model editor and visualization tool [28]. The results of the DFT calculations are summarized in the Supplementary Material. The optimized structures exhibited no negative frequencies. Natural atomic charges were calculated using NBO analysis [29]. Structure overlay pictures were drawn with Mercury [30].
Results and discussion
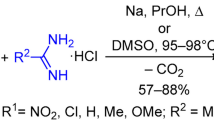
The N,4-diheteroaryl 2-aminothiazole mono-hydrobromide salts investigated in this study were prepared using the Hantzsch thiazole synthesis [31, 32], as summarized in Scheme 1. Reactions of 2-bromo-1-(pyridine-2-yl)ethanone mono-hydrobromide and 2-bromo-1-(pyrazine-2-yl) ethanone mono-hydrobromide with 2 in ethanol afforded 3 and 4 after recrystallization from methanol. The compounds were structurally characterized by X-ray crystallography. Figure 1 depicts the molecular structures in the solid state.

Synthesis of N,4-diheteroaryl 2-aminothiazole hydrobromides 3 and 4 from 2 and the respective α-bromoketone hydrobromide. Solvent molecules of crystallization are not included.

Asymmetric units of 3 (top) and 4 (bottom). Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are shown by small spheres of arbitrary radius. Namine–H⋯Br− and N+pyridine–H···Owater hydrogen bonds are represented by dashed lines
In both 3 and 4, the structure of the central five-membered 1,3-thiazole heterocycle is as expected [33]. The N-(pyridin-3-yl)thiazol-2-amine unit adopts the same conformation in 3 and 4 with the sulfur atom and the pivot atom of the pyridine ring C9 in an antiperiplanar arrangement around the C2–N2 bond. A molecular conformation with a similar orientation of the two aryl rings was observed previously in some crystal structures of N,4-diaryl 2-aminothiazoles [34,35,36]. The thiazole and pyridine rings in 3 and 4 are not entirely coplanar. The angle between the respective mean planes is 10.43(7)° in 3 and 12.6(1)° in 4. The mean planes of the heteroaryl rings attached to C4 of the 1,3-thiazole ring are tilted out of its mean plane by only 1.88(6)° in 3 and 6.8(1)° in 4. Both mono-hydrobromide salts have in common a hydrogen bond formed by the secondary amino group to the bromide anion (Table 2). Moreover, the S1⋯·Br1 short contacts of 3.5891(7) and 3.8344(8) Å as well as the C5–S1⋯Br angles of 177.96(8) and 170.84(9)° in 3 and 4, respectively, provide structural evidence for chalcogen bonding [37, 38]. This arrangement appears not to be unusual. A survey of the CSD (version 5.41 with August 2020 updates) revealed that of 24 crystal structures containing the 2-aminothiazole unit and a bromide anion, where coordinates were available, 17 structures exhibited a short (< 4.0 Å) S⋯Br distance and C–S⋯Br angles in the range of 152–175° (see Supplementary Material).
The protonation sites in both 3 and 4 were identified in difference electron density maps. The observed respective C–N–C bond angles in the six-membered heterocycles (Table S1 in the Supplementary Material) corroborate the assignments made. The C12–N4–C11 and C6–N1–C10 bond angles at the protonated N4 in 3 and N1 in 4, respectively, are significantly larger than 120°. In contrast, the C–N–C bond angles at the unprotonated pyridine and pyrazine nitrogen atoms are within 116–117° and thus significantly smaller than 120°. In 3, the pyridine ring attached to C4 of the 1,3-thiazole ring is protonated, whereas the 2-methoxypyridine ring bonded to the amino group remains unprotonated. Since pyrazine is a weaker base than pyridine, protonation of the 2-methoxypyridine ring is preferred to protonation of the pyrazine ring in 4 as expected.
It is worth noting that in the crystal structure of 3, the bromide anion is involved in a hydrogen bond with H2 at the amine nitrogen atom; although H4 attached to the pyridinium nitrogen atom, the site of protonation of the free base exhibits the larger natural charge in the cation calculated by DFT methods and subsequent NBO analysis (Table 3). Similarly, in the crystal structure of 4, the bromide anion is involved in a hydrogen bond with H2; although H1 attached to the methoxypyridinium nitrogen atom, here the site of protonation has the larger natural charge in the calculated cation. Natural atomic charge or the site of protonation of the free base does therefore not appear to be an indicator of why the bromide anion is hydrogen-bonded to the amine H atom. In the crystal structures of both 3 and 4, the Namine–H⋯Br angles are close to 180° (Table 2), indicating that the main interaction is with the amine H atom, although it is worth noting that the second highest positively charged atom in both cases is the sulfur atom (3 cation 0.43867 e and 4 cation 0.42585 e). In contrast, the amine nitrogen atom is the atom with the most negative natural atomic charge of all atoms in the calculated free cations of 3 (− 0.55164 e) and 4 (− 0.52947 e). For the calculated free bases, the largest positive natural atomic charge resides on the amine H atom (3 free base 0.39616 e and 4 free base 0.39634 e) closely followed by the sulfur atom (3 free base 0.34563 e, 4 free base 0.34574 e), whereas the atom with the most negative natural atomic charge is the amine nitrogen atom in both cases (3 free base − 0.54077 e, 4 free base − 0.53978 e), as for the calculated cations.
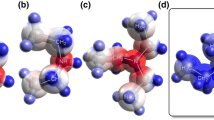
In 4, the methoxy group in ortho-position to N1 is rotated by approximately 180° compared with 3, which appears to be associated with the protonation state of N1. This observation is mirrored in the DFT-optimized structures of the free cations and free bases of 3 and 4 (Fig. 2), whereby the methoxy group in the ortho-position to N1 only points away from N1 when it is protonated (cation of 4).

Structure overlays of the thiazole units of the DFT-optimized structures of the cations (green) and free bases (orange) of 3 (top) and 4 (bottom). Sulfur and nitrogen atoms are highlighted with yellow and blue, respectively
As shown in Fig. 3, Namine–H⋯Br−, N+pyridine–H⋯Owater, and Owater–H⋯Br− hydrogen bonds dominate the supramolecular structure of 3 in the solid state. Two solvate water molecules and two bromide anions form a centrosymmetric \( {\mathrm{R}}_4^2(8) \) hydrogen bond motif [39], which is surrounded by protonated N-(6-methoxypyridin-3-yl)-4-(pyridin-2-yl)thiazol-2-amine molecules. The protonated pyridin-2-yl group forms a hydrogen bond to the solvate water molecule and, as mentioned above, the amino group to a bromide anion, affording a three-dimensional hydrogen-bonded structure. In addition, the unprotonated pyridine nitrogen atom N1 appears to accept a weak C–H⋯N hydrogen bond from C14 of an adjacent molecule (Figure S1 in the Supplementary Material). The centroid-centroid distance between face-to-face stacked methoxypyridine rings and thiazole rings is 3.51 Å. The hydrogen bonding scheme in 4 is distinctly different from that in 3. In 4, protonated N-(6-methoxypyridin-3-yl)-4-(pyrazin-2-yl)thiazol-2-amine molecules are joined via N+–H⋯N hydrogen bonds between the protonated methoxypyridine ring and the pyrazine nitrogen atom N5 of an adjacent molecule, resulting in zigzag chains extending in the [101] direction in the crystal (Fig. 4). The second pyrazine nitrogen atom remains without a hydrogen bond donor in the crystal structure. The disordered solvate methanol molecule is also not involved in significant hydrogen bonding interactions. Geometric parameters of hydrogen bonds in 3 and 4, as summarized in Table 2, are within expected ranges [40].

Part of the crystal structure of 3. Namine–H···Br–, N+pyridine–H⋯Owater and Owater–H⋯Br− hydrogen bonds (dashed lines), viewed down the a axis direction. Carbon-bound hydrogen atoms are omitted for clarity. Symmetry codes: (a) x – 1/2, −y + 1/2, z – 1/2; (b) –x + 3/2, y + 1/2, −z + 3/2

N+methoxypyridine–H⋯Npyrazine hydrogen-bonded zigzag chain in the crystal structure of 4, viewed towards the (−101) plane. Carbon-bound hydrogen atoms and disordered methanol solvent molecules of crystallization are omitted for clarity. Symmetry code: (a) x + 1/2, −y + 1/2, z + 1/2
Conclusions
We have prepared the two related N,4-diheteroaryl 2-aminothiazoles 3 and 4 using the Hantzsch thiazole synthesis and structurally characterized their mono-hydrobromide salts. The N,4-diheteroaryl 2-aminothiazole cations exhibit similar molecular conformations in the solid state but different sites of protonation. The intermolecular hydrogen bonding patterns in the crystal structures are markedly different. Despite the different solid-state supramolecular structures, a similar Namine–H⋯Br−⋯S association is encountered in both 3 and 4, indicating a preferred interaction of these groups, even though the site of protonation of the free base is elsewhere. With regard to the hitherto limited knowledge of N,4-diheteroaryl 2-aminothiazole mono-hydrobromide salts, structural information gained from the present study should be conducive for further investigations of this compound class in medicinal chemistry.
Data availability
Results of the DFT calculations have been placed in the electronic Supplementary Material. CCDC 2043373 (3) and 2043374 (4) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
References
Das D, Sikdar P, Bairagi M (2016) Recent developments of 2-aminothiazoles in medicinal chemistry. Eur J Med Chem 109:89–98. https://doi.org/10.1016/j.ejmech.2015.12.022
Bhuniya D, Mukkavilli R, Shivahare R, Launay D, Dere RT, Deshpande A, Verma A, Vishwakarma P, Moger M, Pradhan A, Pati H, Gopinath VS, Gupta S, Puri SK, Martin D (2015) Aminothiazoles: hit to lead development to identify antileishmanial agents. Eur J Med Chem 102:582–593. https://doi.org/10.1016/j.ejmech.2015.08.013
Paquet T, Gordon R, Waterson D, Witty MJ, Chibale K (2012) Antimalarial aminothiazoles and aminopyridines from phenotypic whole-cell screening of a SoftFocus® library. Future Med Chem 4(18):2265–2277. https://doi.org/10.4155/fmc.12.176
Kesicki EA, Bailey MA, Ovechkina Y, Early JV, Alling T, Bowman J, Zuniga ES, Dalai S, Kumar N, Masquelin T, Hipskind PA, Odingo JO, Parish T (2016) Synthesis and evaluation of the 2-aminothiazoles as anti-tubercular agents. PLoS One 11(5):e0155209. https://doi.org/10.1371/journal.pone.0155209
Meissner A, Boshoff HI, Vasan M, Duckworth BP, Barry 3rd CE, Aldrich CC (2013) Structure-activity relationships of 2-aminothiazoles effective against Mycobacterium tuberculosis. Bioorg Med Chem 21(21):6385–6397. https://doi.org/10.1016/j.bmc.2013.08.048
Makam P, Kannan T (2014) 2-Aminothiazole derivatives as antimycobacterial agents: synthesis, characterization, in vitro and in silico studies. Eur J Med Chem 87:643–656. https://doi.org/10.1016/j.ejmech.2014.09.086
Beuchel A, Goddard R, Imming P, Seidel RW (2020) A solid solution of ethyl and d3-methyl 2-[(4-methylpyridin-2-yl)amino]-4-(pyridin-2-yl)thiazole-5-carboxylate. Acta Crystallogr E Crystallogr Commun 76(8):1255–1259. https://doi.org/10.1107/S2056989020008956
Böck D, Beuchel A, Goddard R, Imming P, Seidel RW (2020) Structural characterization of two polymorphs of 1-(4-methylpyridin-2-yl)thiourea and two derived 2-aminothiazoles. J Chem Crystallogr. https://doi.org/10.1007/s10870-020-00863-0
Gupta D, Bhatia D, Dave V, Sutariya V, Varghese Gupta S (2018) Salts of therapeutic agents: chemical, physicochemical, and biological considerations. Molecules 23(7). https://doi.org/10.3390/molecules23071719
Groom CR, Bruno IJ, Lightfoot MP, Ward SC (2016) The Cambridge Structural Database. Acta Crystallogr Sect B: Struct Sci Cryst Eng Mater 72(Pt 2):171–179. https://doi.org/10.1107/S2052520616003954
Thomas IR, Bruno IJ, Cole JC, Macrae CF, Pidcock E, Wood PA (2010) WebCSD: the online portal to the Cambridge Structural Database. J Appl Crystallogr 43:362–366. https://doi.org/10.1107/s0021889810000452
Hassan AA, Mohamed NK, Aly AA, Tawfeek HN, Bräse S, Nieger M (2020) Synthesis and structure confirmation of 2,4-disubstituted thiazole and 2,3,4-trisubstituted thiazole as thiazolium bromide salts. Monatsh Chem 151(7):1143–1152. https://doi.org/10.1007/s00706-020-02640-3
Cuconati A, Xu X, Block TM (2013) Preparation of substituted aminothiazoles as inhibitors of cancers, including hepatocellular carcinoma, and as inhibitors of hepatitis virus replication. WO2013052613A1,
Kretschmer MSB, Woltersdorf S, Rödl CB, Vogt D, Häfner A-K, Steinhilber D, Stark H, Hofmann B (2016) Development of novel aminothiazole-comprising 5-LO inhibitors. Future Med Chem 8(2):149–164. https://doi.org/10.4155/fmc.15.174
Sarkis GY, Faisal ED (1985) Synthesis and spectroscopic properties of some new N,N’-disubstituted thiorueas of potential biological interest. J Heterocyclic Chem 22(1):137–140. https://doi.org/10.1002/jhet.5570220134
Dicks JP, Zubair M, Davies ES, Garner CD, Schulzke C, Wilson C, McMaster J (2015) Synthesis, structure and redox properties of asymmetric (cyclopentadienyl)(ene-1,2-dithiolate)cobalt(III) complexes containing phenyl, pyridyl and pyrazinyl units. Eur J Inorg Chem 2015(21):3550–3561. https://doi.org/10.1002/ejic.201500138
SAINT (2012) Bruker AXS Inc. Madison, Wisconsin, USA
Duisenberg AJM, Kroon-Batenburg LMJ, Schreurs AMM (2003) An intensity evaluation method: EVAL-14. J Appl Crystallogr 36(2):220–229. https://doi.org/10.1107/S0021889802022628
SADABS (2012) Bruker AXS Inc. Madison, Wisconsin
Sheldrick GM (2015) SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr A Found Adv 71(Pt 1):3–8. https://doi.org/10.1107/S2053273314026370
Sheldrick GM (2015) Crystal structure refinement with SHELXL. Acta Crystallogr C Struct Chem 71(Pt 1):3–8. https://doi.org/10.1107/S2053229614024218
Brandenburg K (2018) Diamond. 3.2k3 edn. Crystal Impact GbR, Bonn
Neese F (2018) Software update: the ORCA program system, version 4.0. WIREs Comput Mol Sci 8(1):e1327. https://doi.org/10.1002/wcms.1327
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98(7):5648–5652. https://doi.org/10.1063/1.464913
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37(2):785–789. https://doi.org/10.1103/PhysRevB.37.785
Weigend F, Ahlrichs R (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys Chem Chem Phys 7(18):3297–3305. https://doi.org/10.1039/B508541A
Fletcher R (2000) Practical methods of optimization, 2nd edn. John Wiley & Sons, Chichester
Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminformat 4(1):17. https://doi.org/10.1186/1758-2946-4-17
Glendening JED, Badenhoop K, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Karafiloglou P, Landis CR, Weinhold F (2018) NBO 7.0. Theoretical Chemistry Institute, University of Wisconsin, Madison
Macrae CF, Sovago I, Cottrell SJ, Galek PTA, McCabe P, Pidcock E, Platings M, Shields GP, Stevens JS, Towler M, Wood PA (2020) Mercury 4.0: from visualization to analysis, design and prediction. J Appl Crystallogr 53(Pt 1):226–235. https://doi.org/10.1107/S1600576719014092
Hantzsch A, Weber JH (1887) Ueber Verbindungen des Thiazols (Pyridins der Thiophenreihe). Ber Dtsch Chem Ges 20(2):3118–3132. https://doi.org/10.1002/cber.188702002200
Wang Z (2010) Hantzsch thiazole synthesis. In: Comprehensive organic name reactions and reagents. pp. 1330–1334. https://doi.org/10.1002/9780470638859.conrr296
Eicher TH, Siegfried, Speicher A (2013) The chemistry of heterocycles: structures, reactions, synthesis, and applications, 3rd, completely revised and enlarged edition. Wiley-VCH, Weinheim
Kuleshova LN, Averkiev BB, Gusev DV, Suponitskii KY, Antipin MY (2004) Conformational polymorphism of N-(4-butoxyphenyl)-4-(4′-nitrophenyl)-2-thiazolamine. Crystallogr Rep 49(5):798–806. https://doi.org/10.1134/1.1803309
He L-M, Hu A-X, Cao G, Peng J-J (2007) Ethyl 2-methyl-1-(4-phenylthiazol-2-yl)-1H-benzimidazole-6-carboxylate. Acta Crystallogr E Crystallogr Commun 63(8):o3643. https://doi.org/10.1107/S1600536807036483
Shao L, Zhou X, Fang J-X (2006) 4-(4-Chlorophenyl)-N-phenyl-5-(1H-1,2,4-triazol-1-yl)thiazol-2-amine. Acta Crystallogr E Crystallogr Commun 62(1):o91–o93. https://doi.org/10.1107/S1600536805040146
Scilabra P, Terraneo G, Resnati G (2019) The chalcogen bond in crystalline solids: a world parallel to halogen bond. Acc Chem Res 52(5):1313–1324. https://doi.org/10.1021/acs.accounts.9b00037
Vogel L, Wonner P, Huber SM (2019) Chalcogen bonding: an overview. Angew Chem Int Ed 58(7):1880–1891. https://doi.org/10.1002/anie.201809432
Bernstein J, Davis RE, Shimoni L, Chang NL (1995) Patterns in hydrogen bonding - functionality and graph set analysis in crystals. Angew Chem Int Edit 34(15):1555–1573. https://doi.org/10.1002/anie.199515551
Thakuria R, Sarma B, Nangia A (2017) 7.03 - Hydrogen bonding in molecular crystals. In: Atwood JL (ed) Comprehensive supramolecular chemistry II. Elsevier, Oxford, pp 25–48. https://doi.org/10.1016/B978-0-12-409547-2.12598-3
Acknowledgments
Professor Christian W. Lehmann is gratefully acknowledged for providing access to the X-ray diffraction facility. RWS would like to thank Jan Henrik Halz for helpful discussions.
Code availability
Not applicable.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization: PI and RWS; data curation: RWS, RG, and AR; formal analysis: RWS, RG, and AR; investigation: DB, AB, and RG; methodology: RG and RWS; project administration: PI and RWS; supervision: PI; validation: RWS, RG, and AR; visualization: RWS and RG; writing—original draft: RWS and RG; writing, review, and editing: RWS, RG, and PI.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All co-authors have seen and approved the manuscript.
Additional information
Dedicated to Dr. Dietrich Seidel on the occasion of his 65th birthday
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
Additional figures and a table of selected bond lengths, bond angles and torsion angles for the crystal structures of 3 and 4, results of a survey of the CSD for crystal structures containing a 2-aminothiazole unit and a bromide anion and results of the DFT calculations can be found in the Supplementary Material. CCDC 2043373 (3) and 2043374 (4) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures. (DOCX 2765 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Böck, D., Beuchel, A., Goddard, R. et al. Protonation sites and hydrogen bonding in mono-hydrobromide salts of two N,4-diheteroaryl 2-aminothiazoles. Struct Chem 32, 989–996 (2021). https://doi.org/10.1007/s11224-021-01730-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-021-01730-0