
Abstract
Purpose of Review
To summarize evidence on the potential involvement of the osteoprotegerin (OPG)/receptor activator of nuclear factor-kappa B (NF-κΒ) ligand (RANKL)/receptor activator of NF-κΒ (RANK) axis in the pathogenesis of metabolic diseases.
Recent Findings
The OPG-RANKL-RANK axis, which has been originally involved in bone remodeling and osteoporosis, is now recognized as a potential contributor in the pathogenesis of obesity and its associated comorbidities, i.e., type 2 diabetes mellitus and nonalcoholic fatty liver disease. Besides bone, OPG and RANKL are also produced in adipose tissue and may be involved in the inflammatory process associated with obesity. Metabolically healthy obesity has been associated with lower circulating OPG concentrations, possibly representing a counteracting mechanism, while elevated serum OPG levels may reflect an increased risk of metabolic dysfunction or cardiovascular disease. OPG and RANKL have been also proposed as potential regulators of glucose metabolism and are potentially involved in the pathogenesis of type 2 diabetes mellitus. In clinical terms, type 2 diabetes mellitus has been consistently associated with increased serum OPG concentrations. With regard to nonalcoholic fatty liver disease, experimental data suggest a potential contribution of OPG and RANKL in hepatic steatosis, inflammation, and fibrosis; however, most clinical studies showed reduction in serum concentrations of OPG and RANKL.
Summary
The emerging contribution of the OPG-RANKL-RANK axis to the pathogenesis of obesity and its associated comorbidities warrants further investigation by mechanistic studies and may have potential diagnostic and therapeutic implications.
Similar content being viewed by others

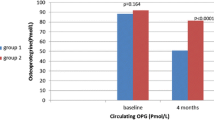
Avoid common mistakes on your manuscript.
Introduction
The prevalence of obesity and its associated comorbidities, mainly type 2 diabetes mellitus (T2DM) and nonalcoholic fatty liver disease (NAFLD), has increased over the last decades [1, 2]. Given their close association with cardiovascular disease (CVD) and all-cause mortality, these metabolic diseases are considered a growing public health burden. Furthermore, except for T2DM, for which a large array of effective and safe pharmacological options have been developed [3], anti-obesity drugs have not meet the desirable expectations [4], while no medications have been approved to-date specifically for the treatment of NAFLD [5]. Better knowledge of the molecular pathways involved in the pathogenesis of these diseases may possibly reveal new molecular targets and may hopefully result in novel therapeutic candidates.
Receptor activator of nuclear factor-kappa B (NF-κΒ) ligand (RANKL), along with its cognate receptor, receptor activator of NF-κΒ (RANK), and osteoprotegerin (OPG), a decoy receptor with high affinity for RANKL, form a molecular system, which has been originally involved in bone remodeling and metabolic bone diseases [6]. Specifically, RANKL, which is produced by osteoblasts, binds to RANK on the surface of osteoclast precursors and promotes osteoclastogenesis and bone resorption. On the other hand, OPG, which is also produced by osteoblasts, attenuates RANKL-RANK interaction through binding to RANKL, thus serving as a negative regulator of osteoclastogenesis and an inhibitor of bone loss. Of note, OPG-expressing osteoblasts have been supported to be a distinct subset of cells from those secreting RANKL, and differentially affect osteoclasts in a paracrine manner [7•]. Disruption of OPG-RANKL-RANK axis in bone has been associated with osteoporosis and other metabolic bone diseases [6].
Recently, a potential role of the OPG-RANKL-RANK axis in metabolic diseases has also emerged. Both obesity and T2DM have been associated with the dysregulation of the OPG-RANKL-RANK axis in bone tissue and subsequently increased risk of low-energy fractures [8••, 9], while a similar association may possibly occur between NAFLD and osteoporosis [10••]. Besides bone, RANKL and OPG are known to be involved also in immune and inflammatory responses, since both are also secreted by T-lymphocytes, and modulate proliferation, activation, and survival of dendritic cells and monocytes/macrophages [11]. In addition to the RANKL-RANK pathway, OPG interferes with the TNF-related apoptosis-inducing ligand (TRAIL)-death receptor (DR) pathway, which is typically involved in inflammation and apoptosis [12]. By binding to TRAIL, OPG interrupts TRAIL-DR interaction and thus, it possibly exerts anti-apoptotic effects. Owing to the biological relationship that links OPG and inflammation through regulating the TRAIL and RANKL pathways, emerging evidence suggests a potential role of these systems in metabolic diseases. It is worth noting that metabolic diseases are considered to be, at least partly, a consequence of a systematic low-grade inflammation, which leads to metabolic aberrations, composing the concept of “metabolic inflammation” [13] or “inflammation-induced dysmetabolism.”
This review aims to summarize evidence regarding the potential involvement of the OPG-RANKL-RANK axis in the pathogenesis of metabolic diseases, which may have potential therapeutic implications. In each section, which consecutively refers to obesity, T2DM, and NAFLD, the first part reports data derived from experimental studies, and the second part focuses on evidence from clinical studies.
Osteoprotegerin/RANKL/RANK in Obesity
Experimental Studies
Previous studies have supported the existence of complex interactions between the adipose tissue and bones [11]. Obesity is a state of low-grade systematic inflammation, in which pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1, IL-6, IL-17 [14], and adipokines, such as leptin [15], released from the dysfunctional adipose tissue into the circulation, may regulate the OPG-RANKL-RANK axis in bones, with main final effect, the inhibition of bone formation and the acceleration of bone resorption. Obesity is also associated with increased adipogenesis in bone marrow, which alters the microenvironment of bone tissue. Accumulation of adipocytes in the bone marrow causes a shift from bone formation to bone resorption via stimulation of the OPG-RANKL-RANK system in favor of osteoclastogenesis [16].
Interestingly, expression and production of RANKL and OPG have also been identified in adipocytes [17]; it has been speculated that these molecules may also contribute to the inflammatory process associated with obesity. Indeed, mice on a high-fat diet (HFD) presented increased expression of OPG in the circulation, adipose tissue, pancreas, and the liver [18]. In addition, OPG administration in normal-weighted mice induced inflammatory changes and metabolic disturbances, i.e., increased macrophage recruitment in the adipose tissue, high circulating and adipose tissue levels of pro-inflammatory cytokines, and glucose intolerance [18], which contrasts with the above mentioned potential anti-apoptotic effect of OPG. In line with these observations, OPG -/- mice on HFD demonstrated lower inflammation in the adipose tissue, as reflected by the reduced macrophage infiltration and decreased pro-inflammatory gene expression compared to controls [19•]. Of note, the latter study also provided further mechanistic insights into the role of RANKL in macrophages infiltrating the adipose tissue. More specifically, macrophages present both RANK and toll-like receptor 4 (TLR4) on their surface. These two receptors partly share intracellular signaling molecules, including the adaptor protein TNF receptor-associated factor 6 (TRAF6). In the presence of RANKL, TRAF6 binds to RANK instead of TLR4, even if lipopolysaccharide (LPS) is present [19•]. This indicates that RANKL reduces inflammation in the adipose tissue, at least partly by inhibiting TLR4 activation in macrophages. If elevated OPG exacerbates inflammation by inhibiting RANKL in the adipose tissue [19•], or exerts anti-apoptotic effects through interacting with the TRAIL-DR [12], needs to be shown in further mechanistic studies. RANKL may also be involved in glucose homeostasis, since it increases energy expenditure and improves glucose metabolism by inducing “beiging” of the white adipocytes in the adipose tissue [20]. These emerging data suggest that OPG and RANKL may serve as mediators potentially involved in the pathogenesis of obesity.
Clinical Studies
Few observational studies have evaluated the association between obesity and the OPG-RANKL-RANK axis with the majority of them focusing on OPG, whereas results for RANKL remain limited; these studies are summarized in Table 1. This may be partly attributed to the technical difficulties that were encountered with the previous kits for RANKL measurement, mainly owing to the fact that serum RANKL constitutes only a small part of total RANKL, as it is mainly cell-bounded and thus not detectable in the circulation [21]. Most studies recruited apparently healthy obese children [22,23,24], adolescents [24], or young adults [25,26,27] without other metabolic comorbidities (i.e., metabolically healthy obesity) to show that circulating OPG was lower in obese compared to lean individuals, although some studies showed comparable levels [28, 29] or even increased serum OPG levels in obese than normal-weighted participants [30]. On the other hand, studies which enrolled participants with other metabolic aberrations in the setting of metabolic syndrome (MetS) reported that obese with MetS had higher serum OPG concentrations than controls [18, 31]. Of note, OPG was shown to increase in parallel with the increasing number of metabolic risk factors [31]. Moreover, in a study of 80 elderly overweight or obese adults without diabetes, those with advanced atherosclerosis had higher serum OPG levels than those without it after accounting for potential confounders [32]. Interestingly, there are also some reports, which link specific variants of the OPG [OPG, rs3736228 (AG/AA) variant] and RANK [RANK, rs11664594 (A/T) variant] genes to an increased risk of obesity, which may imply a potential involvement of the OPG-RANKL-RANK axis in the pathogenesis of obesity, but requires validation in independent cohorts of obese individuals [33, 34].
The above considering, we may speculate that obesity per se may be associated with lower circulating OPG concentrations. However, elevated serum OPG levels in obese may reflect an increased risk of metabolic dysfunction or CVD. An appealing hypothesis may be that, in metabolically healthy obesity, OPG is downregulated as a protective mechanism against the potentially adverse effects of OPG. This counteracting mechanism, however, may be dysregulated when metabolic aberrations are accumulated in obese individuals; thus, OPG is increased and possibly exerts adverse effects. Of course, this remains to be shown by studies of different design. With regard to RANKL, limited existing studies indicated comparable circulating RANKL between obese and lean individuals [22, 27], which, however, needs further verification specifically with high sensitivity kits for measuring serum RANKL.
As mentioned above and suggested by experimental studies, OPG and RANKL are also produced by the adipocytes. However, the exact role of these two molecules in the adipose tissue, their contribution to the pathogenesis of obesity, and their interplay with well-established adipokines, such as leptin and adiponectin, is largely unknown. Increased circulating leptin, which characterizes obesity [35], was correlated with decreased circulating OPG [22]. In bones, leptin directly acts to leptin receptors on the surface of osteoblasts, inhibiting OPG production, which results in increased RANKL concentrations and, subsequently, in increased bone resorption. However, the possible interaction of OPG and leptin in the adipose tissue has not yet been displayed. Furthermore, OPG was shown to be positively correlated with adiponectin [27]. Circulating OPG appears to be decreased in obesity, following a similar pattern to that of adiponectin [36].
Osteoprotegerin/RANKL/RANK in T2DM
Experimental Studies
Experimental studies point to an emerging role of the OPG-RANKL-RANK axis not only in the adipose tissue but also in the regulation of glucose homeostasis. Although the molecular mechanisms that link RANKL and OPG with glucose metabolism have not yet been fully elucidated, systemic or hepatic inhibition of RANKL signaling in a mouse model of T2DM ameliorated hepatic insulin resistance (IR), one of the main pathogenic key factors of T2DM, and markedly improved serum glucose concentrations [37]. Moreover, a recent study suggested a potential role of the OPG-RANKL-RANK axis in muscle metabolism, as RANKL promoted IR in muscle cells, while RANKL inhibition, either with denosumab (Dmab) or with OPG immunoglobulin fragment complex (OPG-Fc), resulted in the improvement of muscle strength, insulin sensitivity, and glucose uptake [38]. Of note, Dmab, a human monoclonal IgG2 antibody, which mimics the biological functions of OPG, by blocking RANKL, but not TRAIL, is an established medication for osteoporosis and other metabolic bone diseases [39].
Another potential mechanism by which the OPG-RANKL-RANK system may regulate glucose and insulin metabolism was proposed by a recent study, in which recombinant OPG administration in diabetic mice significantly improved glucose homeostasis by increasing β-cell mass [40]. Notably, in vitro and in vivo studies have identified the expression of OPG, RANKL, and RANK also in the pancreatic human β-cells [41, 42]. The RANKL-RANK pathway was demonstrated to function as an inhibitor of β-cell proliferation in both mice and human islets, an effect that was reversed by OPG, which stimulates β-cell proliferation by inhibiting RANKL-RANK interaction, thus acting as a β-cell mitogen [40]. Additionally, TNF-α, IL-1, and LPS have been shown to induce OPG production by pancreatic β-cells, which, in turn, restricts insulin secretion and improves their survival [42]; this may mean that OPG targets to protect the survival of β-cells with the cost of hypoinsulinemia under inflammatory circumstances, an hypothesis that may warrant further research. Nevertheless, the beneficial effects of OPG on pancreatic β-cells and glucose metabolism were not shown by other studies [43, 44], so this issue warrants further investigation.
Collectively, these findings propose RANKL and OPG as potential regulators of glucose metabolism by acting either in the pancreas or in peripheral tissues. Figure 1 illustrates the potential role of the OPG-RANKL-RANK axis in the regulation of glucose metabolism. In particular, OPG may act locally in the pancreas probably as a protective factor for β-cells, prolonging survival and preventing the exhaustion of their endocrine function, especially under inflammatory conditions, while RANKL signaling appears to have a potential adverse effect on β-cells function. Additionally, RANKL signaling seems to impair insulin sensitivity in peripheral tissues, including the liver and skeletal muscles. Therefore, dysregulation of the OPG-RANKL-RANK system may represent a potential contributor to the pathogenesis of T2DM.

The proposed role of the OPG-RANKL-RANK axis in main organs contributing to glucose metabolism. In the pancreas, the RANKL-RANK signaling pathway inhibits β-cell proliferation, while OPG, by blocking this interaction, stimulates pancreatic β-cell proliferation. In the liver, the RANKL-RANK pathway potentiates hepatic insulin resistance through activating the NF-κB. In contrast, inhibition of hepatic RANKL by OPG or anti-RANKL treatment may ameliorate hepatic insulin resistance and improve serum glucose levels. Similarly, in muscle cells, RANKL promotes insulin resistance, while RANKL inhibition by OPG or anti-RANKL treatment may result in improvement of insulin sensitivity, glucose uptake, and muscle strength. Abbreviations: IR, insulin resistance; NF-κB, nuclear factor-kappa B; OPG, osteoprotegerin; RANK, receptor activator of nuclear factor-kappa B; RANKL, receptor activator of nuclear factor-kappa B ligand
Clinical Studies
Clinical studies on OPG-RANKL-RANK axis in T2DM patients are summarized in Table 2. In clinical terms, T2DM has been consistently associated with increased serum OPG concentrations [45], while some anti-diabetic medications, i.e., rosiglitazone, but not metformin, have been shown to reduce them [46]. Circulating OPG levels increased gradually from healthy controls to patients with pre-diabetes [47, 48] or early onset T2DM [49, 50] and even more in diabetic patients with longer disease duration [51, 52]. Among patients with established T2DM, circulating OPG was elevated in those with poor glycemic control compared to those with adequate glucose control [53]. Interestingly, increased circulating OPG has been proposed as a potentially useful biomarker for predicting loss of glycemic control among patients with T2DM [54].
Furthermore, increased circulating OPG was associated with diabetic complications and increased in parallel with their severity [51]. More specifically, several studies have shown association of increased circulating OPG with worsened macrovascular complications in T2DM, including coronary artery disease [55,56,57,58], carotid artery disease [58,59,60,61], and peripheral artery disease [59, 62, 63]. Of note, one study reported that increased circulating OPG could predict all-cause mortality in patients with T2DM [64]. In addition, microvascular complications of T2DM, such as diabetic nephropathy [65,66,67], diabetic neuropathy [68, 69], and diabetic retinopathy [70, 71], have also been associated with increased plasma OPG concentrations.
The source of the observed increase in circulating OPG in T2DM remains largely unknown. In fact, OPG may be derived from several sources, e.g., bone, pancreas, and blood vessels. Hyperglycemia has been speculated to increase circulating OPG [72] and reduce RANKL concentrations [73]. Moreover, IR has been also associated with increased serum OPG levels [74]. Intriguingly, studies including non-diabetic individuals suggest that circulating OPG is negatively associated with IR, possibly implying that insulin may reduce serum OPG concentrations, or that OPG may reduce insulin concentrations. However, this association seems to be reversed, since their association becomes positive in certain conditions with advanced IR, such as T2DM [27], which, however, requires studies of different design to be validated. It is also important to underline that increased circulating OPG may originate from the atherosclerotic vessels, given the consistent positive association of OPG with vascular calcification (VC), a process that is accelerated in diabetic patients [75]. OPG is produced in large quantities by endothelial and vascular smooth muscle cells, and possibly acts locally, since its tissue concentrations are 500 times greater than plasma concentrations [27]. It should be also highlighted that RANK and RANKL expression are also observed in atherosclerotic lesions, but not in healthy vessels [76]. Actually, elevated circulating OPG and RANKL may reflect an active calcifying process, which is propagated in the setting of T2DM, raising the possibility of the existence of a bone-vascular axis. However, contrary to the known functions of OPG and RANKL at bone metabolism, RANKL seems to increase calcification in the vasculature, whereas OPG blocks this effect [77, 78], indicating a differential effect of the OPG-RANKL-RANK signaling in vascular compared to bone metabolism.
Findings on the association between RANKL and T2DM are less conclusive since some studies have reported decreased circulating RANKL in diabetic patients in comparison to non-diabetic individuals [49, 61], whereas other authors failed to demonstrate any difference [79]. In addition, although an observational cross-sectional study did not show an association between circulating RANKL and peripheral artery disease in T2DM patients [80], the same authors demonstrated that circulating RANKL, but not circulating OPG, was associated with the progression of lower limb arterial calcification in a prospective observational study [81]. Intriguingly, it has also been suggested that increased circulating RANKL may precede T2DM onset and possibly serve as a predictor of T2DM development, an hypothesis needing to be validated, and that OPG concentrations may not precede T2DM, but rather emerge as T2DM occurs, potentially as a compensatory mechanism, which is consistent with the findings from experimental studies [37]. Beyond this hypothesis, circulating OPG may only reflect diabetic vascular complications, and thus are increasingly higher as diabetes worsens overtime. Further studies are needed to clarify the role of the OPG-RANKL-RANK axis in T2DM.
Osteoprotegerin/RANKL/RANK in NAFLD
Experimental Studies
Another emerging topic is the potential implication of the OPG-RANKL-RANK axis in the pathogenesis of NAFLD [82]. Mice on HFD, which represents an experimental model of obesity and NAFLD, were initially shown to have not only increased circulating OPG but also increased OPG gene expression in the liver [18]. However, other authors showed that mice on HFD have lower circulating ORG and higher RANKL than control mice [83]. In line, another group reported that HFD caused a gradual increase in circulating RANKL levels and hepatic RANKL expression from controls to mice with simple nonalcoholic fatty liver (NAFL) and then to mice with nonalcoholic steatohepatitis (NASH), regarded as a more severe than NAFL phenotype of the disease [84]. This study, importantly, provided some interesting mechanistic insights, as it showed in vitro that the expression of runt-related transcription factor 2 (Runx2) regulated the production of RANKL in hepatic stellate cells (HSCs), which could subsequently mediate macrophage infiltration in the liver [84]. Intriguingly, a transgenic mouse model of osteoporosis (TgHuRANKL), which overexpresses human RANKL, may develop NAFLD [85].
These experimental data suggest that hepatic expression of RANKL may potentially be upregulated in NAFLD, while relevant data on hepatic OPG are still contradictory; however, the exact source, role, and regulation of these molecules in the liver are largely unknown. HSCs and more specifically their activated type (myofibroblasts) have been proposed as the main source of OPG in the liver, linking OPG to hepatic fibrogenesis [86]. Notably, transforming growth factor-β (TGF-β) and IL-13, two important mediators of hepatic fibrogenesis were shown to induce OPG production in murine liver tissue [87] and, vice versa, OPG was shown to enhance fibrosis by stimulating TGF-β production in the liver, thus creating a local vicious loop, possibly contributing to hepatic fibrinogenesis [86]. In addition, OPG seems to affect hepatic steatosis, as its overexpression triggers the signal-regulated kinase (ERK)-peroxisome proliferator-activated receptor-γ (PPAR-γ)-cluster of differentiation (CD36) pathway and, therefore, resulted in increased hepatic lipid accumulation, highlighting the potential pleiotropic effects of OPG in liver disease [88].
Collectively, limited data support that hepatic OPG may favor hepatic steatosis, NASH, and fibrosis, while hepatic RANKL upregulation may be related to persistent hepatic inflammation and hepatocellular injury. However, more data are needed to consolidate or not these findings.
Clinical Studies
Clinical studies on the OPG-RANKL-RANK axis in NAFLD patients are summarized in Table 3. Contrary to the above mentioned experimental studies, NASH, but not NAFL, was associated with lower circulating OPG compared to non-NAFLD participants [89], whereas two subsequent case-control studies with biopsy-proven NAFLD supported a gradual decrease of serum OPG levels from controls to patients with NAFL and then to NASH patients [90, 91]. In another case-control study of patients with T2DM, those with concomitant ultrasound-defined NAFLD had lower circulating OPG than those without [92]. Similarly, OPG was shown to be lower in obese children with NAFLD compared to non-NAFLD obese children [93]. Additionally, a more recent study showed reduced circulating OPG together with reduced mRNA and serum levels of RANKL in NAFLD patients compared to healthy controls [94]. Consistent with this, gene expression and plasma concentration of RANK were also reported to be downregulated in NAFLD patients in comparison to healthy subjects [95]. However, few studies have also supported either comparable circulating OPG levels between patients with and without NAFLD [23, 96] or increased serum OPG levels in NAFLD patients when compared to non-NAFLD individuals [97]. Of note, one study including participants with at least one MetS criterion, but not exclusively NAFLD patients, supported the existence of a positive association between circulating OPG and hepatic fat content, which is in line with experimental studies linking OPG with hepatic steatosis [98].
Taken together, most studies showed reduction in serum concentrations of OPG and RANKL in patients with NAFLD, whose pathophysiological explanation, if any, remains obscure. Moreover, to-date, any effort to explain the discrepancy in OPG and RANKL between experimental and clinical studies in NAFLD is considered to be insecure. It seems that OPG follows the pattern observed in T2DM in animal NAFLD, i.e., it increases with the disease severity, whereas OPG follows the pattern observed in obesity in human NAFLD, i.e., it decreases with disease severity. As mentioned above, apart from being a decoy receptor for RANKL, OPG also operates as a trap receptor for TRAIL, a major apoptotic factor for hepatocytes. Consequently, OPG depletion potentiates apoptosis, which is a hallmark of NASH. However, this speculation remains to be shown specifically for NASH. In agreement with this scenario, high serum OPG and low serum RANKL levels have been reported in advanced fibrosis or cirrhosis of various etiologies [99•], including also NAFLD-related fibrosis [100]. In this regard, OPG has been proposed as a promising biomarker of liver fibrosis [99•], which also remains to be validated. Hepatic RANKL may probably follow an opposite direction than OPG, but it also remains to be definitely established in NAFLD.
Conclusion
OPG and RANKL, traditionally included in osteokines and playing an important role in bone metabolism, are now increasingly recognized to be involved in the pathogenesis of chronic metabolic diseases, based on emerging experimental evidence. In the clinical setting, most observational studies showed low OPG concentrations in metabolically healthy obesity and NAFLD, whereas high concentrations in T2DM, in which higher OPG was also associated with the severity of disease and diabetic complications. In addition, RANKL seems to adversely affect glucose metabolism and may be positively associated with NAFL and NASH.
This topic has certain challenges, perspectives, and clinical implications. Firstly, determination of circulating OPG and RANKL remains challenging, since they may originate from different tissues and most importantly, their circulating concentrations may largely differ from those on distinct tissue levels (e.g., bone, adipose tissue, liver, vessels). A recent study suggested that OPG and RANKL functions are restricted exclusively at their production sites [7•], highlighting the importance of tight local control of the OPG-RANKL-RANK network in various tissues, while measurements of circulating OPG or RANKL may be not clinically relevant, since they may possibly be an epiphenomenon, which, however, needs to be verified by subsequent studies. Furthermore, commercially available kits for circulating RANKL or OPG do not always provide optimal results, especially older ELISA kits for RANKL, which creates the need for newer kits for more accurate measurement of circulating OPG and RANKL concentrations.
In therapeutic terms, Dmab, an anti-RANKL medication approved for the treatment of osteoporosis, may also prove suitable in the future for the treatment of metabolic diseases, e.g., obesity, T2DM, and NAFLD. One-year administration of Dmab in T2DM patients with osteoporosis improved glycated hemoglobin (HbA1c), homeostasis model assessment – IR (HOMA-IR), an index of IR, and liver function tests [101]. Another prospective study demonstrated that a single dose of Dmab was effective in reducing HbA1c and hepatic insulin resistance index in postmenopausal women with osteoporosis [102]. Similarly, a recent meta-analysis reported that Dmab improved glycemic parameters, mainly in patients with impaired glucose tolerance, such as those with pre-diabetes or diabetes [103••]. Moreover, administration of Dmab to a woman with osteoporosis and concomitant NASH improved her liver function tests, which deserves further investigation [104]. We previously hypothesized Dmab repurposing in NAFLD [105•], and, to this aim, we are currently running a non-sponsored clinical study with Dmab administration in patients with osteoporosis and NAFLD (clinicaltrials.gov identifier: 88235).
In conclusion, this review summarizes current evidence on the potential contribution of the OPG-RANKL-RANK axis to the pathogenesis of metabolic diseases (obesity, T2DM, and NAFLD). Although the topic seems to be challenging, further mechanistic studies are needed to shed light in the definite implication of OPG and RANKL in the pathophysiology of metabolic diseases. Diagnostic accuracy studies are also warranted to show whether OPG and RANKL may serve as predictors of metabolic diseases or their severity, as well as clinical trials to show the efficacy of anti-RANKL treatment in metabolic diseases beyond the bone.
Data Availability
No datasets were generated or analyzed during the current review article.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Younossi ZM. Non-alcoholic fatty liver disease - a global public health perspective. J Hepatol. 2019;70:531–44.
Polyzos SA, Mantzoros CS. Diabetes mellitus: 100 years since the discovery of insulin. Metabolism. 2021;118:154737.
Upadhyay J, Polyzos SA, Perakakis N, Thakkar B, Paschou SA, Katsiki N, et al. Pharmacotherapy of type 2 diabetes: an update. Metabolism. 2018;78:13–42.
Pilitsi E, Farr OM, Polyzos SA, Perakakis N, Nolen-Doerr E, Papathanasiou A-E, et al. Pharmacotherapy of obesity: available medications and drugs under investigation. Metabolism. 2019;92:170–92.
Polyzos SA, Kang ES, Boutari C, Rhee E-J, Mantzoros CS. Current and emerging pharmacological options for the treatment of nonalcoholic steatohepatitis. Metabolism. 2020;111S:154203.
Anastasilakis AD, Polyzos SA, Makras P. Therapy of endocrine disease: denosumab vs bisphosphonates for the treatment of postmenopausal osteoporosis. Eur J Endocrinol. 2018;179:R31-45.
• Tsukasaki M, Asano T, Muro R, Huynh NC-N, Komatsu N, Okamoto K, et al. OPG production matters where it happened. Cell Rep. 2020;32:108124. OPG-expressing osteoblasts are a distinct subset of cells from those secreting RANKL, while locally produced OPG, rather than circulating OPG, may be crucial for bone and immune homeostasis.
•• Gkastaris K, Goulis DG, Potoupnis M, Anastasilakis AD, Kapetanos G. Obesity, osteoporosis and bone metabolism. J Musculoskelet Neuronal Interact. 2020;20:372–81. Obesity may have a negative impact on bone microarchitecture through low-grade systemic inflammation and increased bone marrow adipogenesis, possibly leading to a higher risk of certain fractures, albeit with increased bone mineral density.
Napoli N, Chandran M, Pierroz DD, Abrahamsen B, Schwartz AV, Ferrari SL, et al. Mechanisms of diabetes mellitus-induced bone fragility. Nat Rev Endocrinol. 2017;13:208–19.
•• Vachliotis ID, Anastasilakis AD, Goulas A, Goulis DG, Polyzos SA. Nonalcoholic fatty liver disease and osteoporosis: a potential association with therapeutic implications. Diabetes Obes Metab. 2022;24:702–1720. NAFLD and osteoporosis may be pathogenically associated, resulting in important treatment considerations and potential therapeutic implications.
Musso G, Paschetta E, Gambino R, Cassader M, Molinaro F. Interactions among bone, liver, and adipose tissue predisposing to diabesity and fatty liver. Trends Mol Med. 2013;19:522–35.
Bernardi S, Bossi F, Toffoli B, Fabris B. Roles and clinical applications of OPG and TRAIL as biomarkers in cardiovascular disease. Biomed Res Int. 2016;2016:1752854.
Wu H, Ballantyne CM. Metabolic inflammation and insulin resistance in obesity. Circ Res Ovid Technologies (Wolters Kluwer Health). 2020;126:1549–64.
Polyzos SA, Kountouras J, Mantzoros CS. Adipose tissue, obesity and non-alcoholic fatty liver disease. Minerva Endocrinol. 2017;42:92–108.
Cheng M, Li T, Li W, Chen Y, Xu W, Xu L. Leptin can promote mineralization and up-regulate RANKL mRNA expression in osteoblasts from adult female SD rats. Int J Clin Exp Pathol. 2018;11:1610–9.
Halade GV, El Jamali A, Williams PJ, Fajardo RJ, Fernandes G. Obesity-mediated inflammatory microenvironment stimulates osteoclastogenesis and bone loss in mice. Exp Gerontol. 2011;46:43–52.
An J-J, Han D-H, Kim D-M, Kim S-H, Rhee Y, Lee E-J, et al. Expression and regulation of osteoprotegerin in adipose tissue. Yonsei Med J. 2007;48:765–72.
Bernardi S, Fabris B, Thomas M, Toffoli B, Tikellis C, Candido R, et al. Osteoprotegerin increases in metabolic syndrome and promotes adipose tissue proinflammatory changes. Mol Cell Endocrinol. 2014;394:13–20.
• Mota RF, Cavalcanti de Araújo PH, Cezine MER, Matsuo FS, Metzner RJM, Oliveira de Biagi Junior CA, et al. RANKL impairs the TLR4 pathway by increasing TRAF6 and RANK interaction in macrophages. Biomed Res Int. 2022; 2022:7740079. RANKL-RANK interaction inhibits TLR4 activation in adipose tissue macrophages, reducing the expression of pro-inflammatory mediators, whereas increased OPG exacerbates inflammation by inhibiting RANKL.
Matsuo FS, Cavalcanti de Araújo PH, Mota RF, Carvalho AJR, Santos de Queiroz M, Baldo de Almeida B, et al. RANKL induces beige adipocyte differentiation in preadipocytes. Am J Physiol Endocrinol Metab. 2020;318:E866–77.
Anastasilakis AD, Goulis DG, Polyzos SA, Gerou S, Koukoulis G, Kita M, et al. Serum osteoprotegerin and RANKL are not specifically altered in women with postmenopausal osteoporosis treated with teriparatide or risedronate: a randomized, controlled trial. Horm Metab Res. 2008;40:281–5.
Dimitri P, Wales JK, Bishop N. Adipokines, bone-derived factors and bone turnover in obese children; evidence for altered fat-bone signalling resulting in reduced bone mass. Bone. 2011;48:189–96.
Erol M, Bostan Gayret O, Tekin Nacaroglu H, Yigit O, Zengi O, Salih Akkurt M, et al. Association of osteoprotegerin with obesity, insulin resistance and non-alcoholic fatty liver disease in children. Iran Red Crescent Med J. 2016;18: e41873.
Kotanidou EP, Kotanidis CP, Giza S, Serbis A, Tsinopoulou V-R, Karalazou P, et al. Osteoprotegerin increases parallel to insulin resistance in obese adolescents. Endocr Res. 2019;44:9–15.
Ugur-Altun B, Altun A, Gerenli M, Tugrul A. The relationship between insulin resistance assessed by HOMA-IR and serum osteoprotegerin levels in obesity. Diabetes Res Clin Pract. 2005;68:217–22.
Ugur-Altun B, Altun A. Circulating leptin and osteoprotegerin levels affect insulin resistance in healthy premenopausal obese women. Arch Med Res. 2007;38:891–6.
Ashley DT, O’Sullivan EP, Davenport C, Devlin N, Crowley RK, McCaffrey N, et al. Similar to adiponectin, serum levels of osteoprotegerin are associated with obesity in healthy subjects. Metabolism. 2011;60:994–1000.
Gannagé-Yared M-H, Yaghi C, Habre B, Khalife S, Noun R, Germanos-Haddad M, et al. Osteoprotegerin in relation to body weight, lipid parameters insulin sensitivity, adipocytokines, and C-reactive protein in obese and non-obese young individuals: results from both cross-sectional and interventional study. Eur J Endocrinol. 2008;158:353–9.
Ayina Ayina CN, Sobngwi E, Essouma M, Noubiap JJN, Boudou P, Etoundi Ngoa LS, et al. Osteoprotegerin in relation to insulin resistance and blood lipids in sub-Saharan African women with and without abdominal obesity. Diabetol Metab Syndr. 2015;7:47.
Suliburska J, Bogdanski P, Gajewska E, Kalmus G, Sobieska M, Samborski W. The association of insulin resistance with serum osteoprotegerin in obese adolescents. J Physiol Biochem. 2013;69:847–53.
Pérez de Ciriza C, Moreno M, Restituto P, Bastarrika G, Simón I, Colina I, et al. Circulating osteoprotegerin is increased in the metabolic syndrome and associates with subclinical atherosclerosis and coronary arterial calcification. Clin Biochem. 2014;47:272–8.
Del Toro R, Cavallari I, Tramontana F, Park K, Strollo R, Valente L, et al. Association of bone biomarkers with advanced atherosclerotic disease in people with overweight/obesity. Endocrine. 2021;73:339–46.
Zhao L-J, Guo Y-F, Xiong D-H, Xiao P, Recker RR, Deng H-W. Is a gene important for bone resorption a candidate for obesity? An association and linkage study on the RANK (receptor activator of nuclear factor-kappaB) gene in a large Caucasian sample. Hum Genet. 2006;120:561–70.
Jiang X-Y, Chen H-H, Cao F-F, Li L, Lin R-Y, Wen H, et al. A polymorphism near osteoprotegerin gene confer risk of obesity in Uyghurs. Endocrine. 2010;37:383–8.
Moon H-S, Dalamaga M, Kim S-Y, Polyzos SA, Hamnvik O-P, Magkos F, et al. Leptin’s role in lipodystrophic and nonlipodystrophic insulin-resistant and diabetic individuals. Endocr Rev The Endocrine Society. 2013;34:377–412.
Polyzos SA, Kountouras J, Mantzoros CS. Obesity and nonalcoholic fatty liver disease: from pathophysiology to therapeutics. Metabolism. 2019;92:82–97.
Kiechl S, Wittmann J, Giaccari A, Knoflach M, Willeit P, Bozec A, et al. Blockade of receptor activator of nuclear factor-κB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat Med. 2013;19:358–63.
Bonnet N, Bourgoin L, Biver E, Douni E, Ferrari S. RANKL inhibition improves muscle strength and insulin sensitivity and restores bone mass. J Clin Invest. 2019;129:3214–23.
Polyzos SA, Makras P, Tournis S, Anastasilakis AD. Off-label uses of denosumab in metabolic bone diseases. Bone. 2019;129:115048.
Kondegowda NG, Fenutria R, Pollack IR, Orthofer M, Garcia-Ocaña A, Penninger JM, et al. Osteoprotegerin and denosumab stimulate human beta cell proliferation through inhibition of the receptor activator of NF-κB ligand pathway. Cell Metab. 2015;22:77–85.
Kutlu B, Kayali AG, Jung S, Parnaud G, Baxter D, Glusman G, et al. Meta-analysis of gene expression in human pancreatic islets after in vitro expansion. Physiol Genomics Am Physiol Soc. 2009;39:72–81.
Schrader J, Rennekamp W, Niebergall U, Schoppet M, Jahr H, Brendel MD, et al. Cytokine-induced osteoprotegerin expression protects pancreatic beta cells through p38 mitogen-activated protein kinase signalling against cell death. Diabetologia. 2007;50:1243–7.
Toffoli B, Bernardi S, Candido R, Sabato N, Carretta R, Corallini F, et al. Osteoprotegerin induces morphological and functional alterations in mouse pancreatic islets. Mol Cell Endocrinol. 2011;331:136–42.
Ominsky MS, Stolina M, Li X, Corbin TJ, Asuncion FJ, Barrero M, et al. One year of transgenic overexpression of osteoprotegerin in rats suppressed bone resorption and increased vertebral bone volume, density, and strength. J Bone Miner Res Wiley. 2009;24:1234–46.
Barchetta I, Ceccarelli V, Cimini FA, Bertoccini L, Fraioli A, Alessandri C, et al. Impaired bone matrix glycoprotein pattern is associated with increased cardio-metabolic risk profile in patients with type 2 diabetes mellitus. J Endocrinol Invest. 2019;42:513–20.
Nybo M, Preil SR, Juhl HF, Olesen M, Yderstraede K, Gram J, et al. Rosiglitazone decreases plasma levels of osteoprotegerin in a randomized clinical trial with type 2 diabetes patients. Basic Clin Pharmacol Toxicol. 2011;109:481–5.
Mashavi M, Menaged M, Shargorodsky M. Circulating osteoprotegerin in postmenopausal osteoporotic women: marker of impaired glucose regulation or impaired bone metabolism. Menopause. 2017;24:1264–8.
Bilgir O, Yavuz M, Bilgir F, Akan OY, Bayindir AG, Calan M, et al. Relationship between insulin resistance, hs-CRP, and body fat and serum osteoprotegerin/RANKL in prediabetic patients. Minerva Endocrinol. 2018;43:19–26.
Secchiero P, Corallini F, Pandolfi A, Consoli A, Candido R, Fabris B, et al. An increased osteoprotegerin serum release characterizes the early onset of diabetes mellitus and may contribute to endothelial cell dysfunction. Am J Pathol. 2006;169:2236–44.
Niu Y, Yang Z, Li X, Zhang W, Lu S, Zhang H, et al. Association of osteoprotegerin with impaired glucose regulation and microalbuminuria: the REACTION study. BMC Endocr Disord. 2015;15:75.
Bjerre M. Osteoprotegerin (OPG) as a biomarker for diabetic cardiovascular complications. Springerplus. 2013;2:658.
Nabipour I, Kalantarhormozi M, Larijani B, Assadi M, Sanjdideh Z. Osteoprotegerin in relation to type 2 diabetes mellitus and the metabolic syndrome in postmenopausal women. Metabolism. 2010;59:742–7.
Altinova AE, Toruner F, Akturk M, Bukan N, Yetkin I, Cakir N, et al. Relationship between serum osteoprotegerin, glycemic control, renal function and markers of atherosclerosis in type 2 diabetes. Scand J Clin Lab Invest. 2011;71:340–3.
Moh AMC, Pek SLT, Liu J, Wang J, Subramaniam T, Ang K, et al. Plasma osteoprotegerin as a biomarker of poor glycaemic control that predicts progression of albuminuria in type 2 diabetes mellitus: a 3-year longitudinal cohort study. Diabetes Res Clin Pract. 2020;161:107992.
Avignon A, Sultan A, Piot C, Elaerts S, Cristol JP, Dupuy AM. Osteoprotegerin is associated with silent coronary artery disease in high-risk but asymptomatic type 2 diabetic patients. Diabetes Care. 2005;28:2176–80.
Anand DV, Lahiri A, Lim E, Hopkins D, Corder R. The relationship between plasma osteoprotegerin levels and coronary artery calcification in uncomplicated type 2 diabetic subjects. J Am Coll Cardiol. 2006;47:1850–7.
Reinhard H, Nybo M, Hansen PR, Wiinberg N, Kjær A, Petersen CL, et al. Osteoprotegerin and coronary artery disease in type 2 diabetic patients with microalbuminuria. Cardiovasc Diabetol. 2011;10:70.
Ishiyama M, Suzuki E, Katsuda J, Murase H, Tajima Y, Horikawa Y, et al. Associations of coronary artery calcification and carotid intima-media thickness with plasma concentrations of vascular calcification inhibitors in type 2 diabetic patients. Diabetes Res Clin Pract. 2009;85:189–96.
Poulsen MK, Nybo M, Dahl J, Hosbond S, Poulsen TS, Johansen A, et al. Plasma osteoprotegerin is related to carotid and peripheral arterial disease, but not to myocardial ischemia in type 2 diabetes mellitus. Cardiovasc Diabetol. 2011;10:76.
Akinci B, Demir T, Celtik A, Baris M, Yener S, Ozcan MA, et al. Serum osteoprotegerin is associated with carotid intima media thickness in women with previous gestational diabetes. Diabetes Res Clin Pract. 2008;82:172–8.
Gaudio A, Privitera F, Pulvirenti I, Canzonieri E, Rapisarda R, Fiore CE. Relationships between osteoprotegerin, receptor activator of the nuclear factor kB ligand serum levels and carotid intima-media thickness in patients with type 2 diabetes mellitus. Panminerva Med. 2014;56:221–5.
Esteghamati A, Aflatoonian M, Rad MV, Mazaheri T, Mousavizadeh M, Nakhjavani M, et al. Association of osteoprotegerin with peripheral artery disease in patients with type 2 diabetes. Arch Cardiovasc Dis. 2015;108:412–9.
Niu Y, Zhang W, Yang Z, Li X, Wen J, Wang S, et al. Association of plasma osteoprotegerin levels with the severity of lower extremity arterial disease in patients with type 2 diabetes. BMC Cardiovasc Disord. 2015;15:86.
Reinhard H, Lajer M, Gall M-A, Tarnow L, Parving H-H, Rasmussen LM, et al. Osteoprotegerin and mortality in type 2 diabetic patients. Diabetes Care Am Diabetes Assoc. 2010;33:2561–6.
Chang Y-H, Lin K-D, He S-R, Hsieh M-C, Hsiao J-Y, Shin S-J. Serum osteoprotegerin and tumor necrosis factor related apoptosis inducing-ligand (TRAIL) are elevated in type 2 diabetic patients with albuminuria and serum osteoprotegerin is independently associated with the severity of diabetic nephropathy. Metabolism. 2011;60:1064–9.
Xiang GD, Pu JH, Zhao LS, Sun HL, Hou J, Yue L. Association between plasma osteoprotegerin concentrations and urinary albumin excretion in type 2 diabetes. Diabet Med. 2009;26:397–403.
Wang S-T, Zhang C-Y, Zhang C-M, Rong W. The plasma osteoprotegerin level and osteoprotegerin expression in renal biopsy tissue are increased in type 2 diabetes with nephropathy. Exp Clin Endocrinol Diabetes. 2015;123:106–11.
Terekeci HM, Senol MG, Top C, Sahan B, Celik S, Sayan O, et al. Plasma osteoprotegerin concentrations in type 2 diabetic patients and its association with neuropathy. Exp Clin Endocrinol Diabetes. 2009;117:119–23.
Nybo M, Poulsen MK, Grauslund J, Henriksen JE, Rasmussen LM. Plasma osteoprotegerin concentrations in peripheral sensory neuropathy in type 1 and type 2 diabetic patients. Diabet Med. 2010;27:289–94.
Abu El-Asrar AM, Struyf S, Mohammad G, Gouwy M, Rytinx P, Siddiquei MM, et al. Osteoprotegerin is a new regulator of inflammation and angiogenesis in proliferative diabetic retinopathy. Invest Ophthalmol Vis Sci. 2017;58:3189–201.
Yu G, Ji X, Jin J, Bu S. Association of serum and vitreous concentrations of osteoprotegerin with diabetic retinopathy. Ann Clin Biochem. 2015;52:232–6.
Hygum K, Starup-Linde J, Harsløf T, Vestergaard P, Langdahl BL. Mechanisms in Endocrinology: Diabetes mellitus, a state of low bone turnover - a systematic review and meta-analysis. Eur J Endocrinol. 2017;176:R137–57.
Niță G, Niță O, Gherasim A, Arhire LI, Herghelegiu AM, Mihalache L, et al. The role of RANKL and FGF23 in assessing bone turnover in type 2 diabetic patients. Acta Endocrinol. 2021;17:51–9.
Tavintharan S, Pek LTS, Liu JJ, Ng XW, Yeoh LY, Su Chi L, et al. Osteoprotegerin is independently associated with metabolic syndrome and microvascular complications in type 2 diabetes mellitus. Diab Vasc Dis Res. 2014;11:359–62.
Ndip A, Wilkinson FL, Jude EB, Boulton AJM, Alexander MY. RANKL-OPG and RAGE modulation in vascular calcification and diabetes: novel targets for therapy. Diabetologia. 2014;57:2251–60.
Aoki A, Murata M, Asano T, Ikoma A, Sasaki M, Saito T, et al. Association of serum osteoprotegerin with vascular calcification in patients with type 2 diabetes. Cardiovasc Diabetol. 2013;12:11.
Harper E, Forde H, Davenport C, Rochfort KD, Smith D, Cummins PM. Vascular calcification in type-2 diabetes and cardiovascular disease: integrative roles for OPG, RANKL and TRAIL. Vascul Pharmacol. 2016;82:30–40.
Forde H, Davenport C, Harper E, Cummins P, Smith D. The role of OPG/RANKL in the pathogenesis of diabetic cardiovascular disease. Cardiovasc Endocrinol Metab. 2018;7:28–33.
O’Sullivan EP, Ashley DT, Davenport C, Devlin N, Crowley R, Agha A, et al. Osteoprotegerin and biomarkers of vascular inflammation in type 2 diabetes. Diabetes Metab Res Rev. 2010;26:496–502.
Bourron O, Aubert CE, Liabeuf S, Cluzel P, Lajat-Kiss F, Dadon M, et al. Below-knee arterial calcification in type 2 diabetes: association with receptor activator of nuclear factor κB ligand, osteoprotegerin, and neuropathy. J Clin Endocrinol Metab. 2014;99:4250–8.
Bourron O, Phan F, Diallo MH, Hajage D, Aubert C-E, Carlier A, et al. Circulating receptor activator of nuclear factor kB ligand and triglycerides are associated with progression of lower limb arterial calcification in type 2 diabetes: a prospective, observational cohort study. Cardiovasc Diabetol. 2020;19:140.
Pacifico L, Andreoli GM, D’Avanzo M, De Mitri D, Pierimarchi P. Role of osteoprotegerin/receptor activator of nuclear factor kappa B/receptor activator of nuclear factor kappa B ligand axis in nonalcoholic fatty liver disease. World J Gastroenterol. 2018;24:2073–82.
Karmakar S, Majumdar S, Maiti A, Choudhury M, Ghosh A, Das AS, et al. Protective role of black tea extract against nonalcoholic steatohepatitis-induced skeletal dysfunction. J Osteoporos. 2011;2011:426863.
Zhong L, Yuan J, Huang L, Li S, Deng L. RANKL is involved in Runx2-triggered hepatic infiltration of macrophages in mice with NAFLD induced by a high-fat diet. Biomed Res Int. 2020;2020:6953421.
Rinotas V, Niti A, Dacquin R, Bonnet N, Stolina M, Han C-Y, et al. Novel genetic models of osteoporosis by overexpression of human RANKL in transgenic mice. J Bone Miner Res. 2014;29:1158–69.
Adhyatmika A, Beljaars L, Putri KSS, Habibie H, Boorsma CE, Reker-Smit C, et al. Osteoprotegerin is more than a possible serum marker in liver fibrosis: a study into its function in human and Murine liver. Pharmaceutics. 2020;12:471.
Adhyatmika A, Putri KSS, Gore E, Mangnus KA, Reker-Smit C, Schuppan D, et al. Osteoprotegerin expression in liver is induced by IL13 through TGFβ. Cell Physiol Biochem. 2022;56:28–38.
Zhang C, Luo X, Chen J, Zhou B, Yang M, Liu R, et al. Osteoprotegerin promotes liver steatosis by targeting the ERK-PPAR-γ-CD36 pathway. Diabetes. 2019;68:1902–14.
Yilmaz Y, Yonal O, Kurt R, Oral AY, Eren F, Ozdogan O, et al. Serum levels of osteoprotegerin in the spectrum of nonalcoholic fatty liver disease. Scand J Clin Lab Invest. 2010;70:541–6.
Yang M, Xu D, Liu Y, Guo X, Li W, Guo C, et al. Combined serum biomarkers in non-invasive diagnosis of non-alcoholic steatohepatitis. PLoS ONE. 2015;10: e0131664.
Yang M, Liu Y, Zhou G, Guo X, Zou S, Liu S, et al. Value of serum osteoprotegerin in noninvasive diagnosis of nonalcoholic steatohepatitis. Zhonghua Gan Zang Bing Za Zhi. 2016;24:96–101.
Niu Y, Zhang W, Yang Z, Li X, Fang W, Zhang H, et al. Plasma osteoprotegerin levels are inversely associated with nonalcoholic fatty liver disease in patients with type 2 diabetes: a case-control study in China. Metabolism. 2016;65:475–81.
El Amrousy D, El-Afify D. Osteocalcin and osteoprotegerin levels and their relationship with adipokines and proinflammatory cytokines in children with nonalcoholic fatty liver disease. Cytokine. 2020;135:155215.
Nikseresht M, Azarmehr N, Arya A, Alipoor B, Fadaei R, Khalvati B, et al. Circulating mRNA and plasma levels of osteoprotegerin and receptor activator of NF-κB ligand in nonalcoholic fatty liver disease. Biotechnol Appl Biochem. 2021;68:1243–9.
Hadinia A, Doustimotlagh AH, Goodarzi HR, Arya A, Jafarinia M. Plasma levels and gene expression of RANK in non-alcoholic fatty liver disease. Clin Lab. 2020;66.
Oğuz D, Ünal HÜ, Eroğlu H, Gülmez Ö, Çevik H, Altun A. Aortic flow propagation velocity, epicardial fat thickness, and osteoprotegerin level to predict subclinical atherosclerosis in patients with nonalcoholic fatty liver disease. Anatol J Cardiol. 2016;16:974–9.
Ayaz T, Kirbas A, Durakoglugil T, Durakoglugil ME, Sahin SB, Sahin OZ, et al. The relation between carotid intima media thickness and serum osteoprotegerin levels in nonalcoholic fatty liver disease. Metab Syndr Relat Disord. 2014;12:283–9.
Monseu M, Dubois S, Boursier J, Aubé C, Gagnadoux F, Lefthériotis G, et al. Osteoprotegerin levels are associated with liver fat and liver markers in dysmetabolic adults. Diabetes Metab. 2016;42:364–7.
• Habibie H, Adhyatmika A, Schaafsma D, Melgert BN. The role of osteoprotegerin (OPG) in fibrosis: its potential as a biomarker and/or biological target for the treatment of fibrotic diseases. Pharmacol Ther. 2021; 228:107941. OPG has been linked to fibrogenesis in various tissues, including the liver, and may prove suitable as a non-invasive biomarker, probably in combination with other markers, for detecting fibrosis and/or monitoring the efficacy of anti-fibrotic therapy.
Mantovani A, Sani E, Fassio A, Colecchia A, Viapiana O, Gatti D, et al. Association between non-alcoholic fatty liver disease and bone turnover biomarkers in post-menopausal women with type 2 diabetes. Diabetes Metab. 2019;45:347–55.
Abe I, Ochi K, Takashi Y, Yamao Y, Ohishi H, Fujii H, et al. Effect of denosumab, a human monoclonal antibody of receptor activator of nuclear factor kappa-B ligand (RANKL), upon glycemic and metabolic parameters: effect of denosumab on glycemic parameters. Medicine. 2019;98: e18067.
Passeri E, Benedini S, Costa E, Corbetta S. A single 60 mg dose of denosumab might improve hepatic insulin sensitivity in postmenopausal nondiabetic severe osteoporotic women. Int J Endocrinol. 2015;2015:352858.
•• Pacheco-Soto BT, Elguezabal-Rodelo RG, Porchia LM, Torres-Rasgado E, Pérez-Fuentes R, Gonzalez-Mejia ME. Denosumab improves glucose parameters in patients with impaired glucose tolerance: a systematic review and meta-analysis. J Drug Assess. 2021;10:97–105. Denosumab, an established anti-osteoporotic medication, significantly improved parameters of glucose metabolism and insulin resistance, especially in patients with pre-diabetes and diabetes mellitus.
Takeno A, Yamamoto M, Notsu M, Sugimoto T. Administration of anti-receptor activator of nuclear factor-kappa B ligand (RANKL) antibody for the treatment of osteoporosis was associated with amelioration of hepatitis in a female patient with growth hormone deficiency: a case report. BMC Endocr Disord. 2016;16:66.
• Polyzos SA, Goulas A. Treatment of nonalcoholic fatty liver disease with an anti-osteoporotic medication: a hypothesis on drug repurposing. Med Hypotheses. 2021; 146:110379. Hepatic upregulation of RANKL has been hypothesized to contribute to the pathogenesis of NAFLD. In this context, denosumab, an established anti-osteoporotic medication with anti-RANKL activity, may be repurposed for the treatment of some patients with NASH.
Funding
Open access funding provided by HEAL-Link Greece.
Author information
Authors and Affiliations
Contributions
IDV and SAP: concept and design; IDV: acquisition of data; IDV and SAP: interpretation of data; IDV: drafting the manuscript; IDV and SAP: critically revising the manuscript; SAP: study supervision; IDV and SAP: approval of the version to be submitted.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vachliotis, I.D., Polyzos, S.A. Osteoprotegerin/Receptor Activator of Nuclear Factor-Kappa B Ligand/Receptor Activator of Nuclear Factor-Kappa B Axis in Obesity, Type 2 Diabetes Mellitus, and Nonalcoholic Fatty Liver Disease. Curr Obes Rep 12, 147–162 (2023). https://doi.org/10.1007/s13679-023-00505-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13679-023-00505-4