
Abstract
Context
Black poplar (Populus nigra L.) is a keystone species of European riparian ecosystems that has been negatively impacted by riverside urbanization for centuries. Consequently, it has become an endangered tree species in many European countries. The establishment of a suitable rescue plan of the remaining black poplar forest stands requires a preliminary knowledge about the distribution of genetic variation among species populations. However, for some parts of the P. nigra distribution in Europe, the genetic resources and demographic history remain poorly recognized.
Aims
Here, we present the first study on identifying and characterizing the genetic resources of black poplar from the Oder valley in Poland. This study (1) assessed the genetic variability and effective population size of populations and (2) examined whether gene flow is limited by distance or there is a single migrant pool along the studied river system.
Methods
A total of 582 poplar trees derived from nine black poplar populations were investigated with nuclear microsatellite markers.
Results
(1) The allelic richness and heterozygosity level were high and comparable between populations. (2) The genetic structure of the studied poplar stands was not homogenous. (3) The signatures of past bottlenecks were detected.
Conclusion
Our study (1) provides evidence for genetic substructuring of natural black poplar populations from the studied river catchment, which is not a frequent phenomenon reported for this species in Europe, and (2) indicates which poplar stands may serve as new genetic conservation units (GCUs) of this species in Europe.
Key message
The genetic resources of black poplar in the Oder River valley are still substantial compared to those reported for rivers in Western Europe. On the other hand, clear signals of isolation by distance and genetic erosion reflected in small effective population sizes and high spatial genetic structure of the analyzed populations were detected. Based on these findings, we recommend the in situ and ex situ conservation strategies for conserving and restoring the genetic resources of black poplar populations in this strongly transformed by human river valley ecosystem.
Similar content being viewed by others


1 Introduction
Black poplar (Populus nigra L.) is a characteristic alluvial forest tree species of great ecological and economic importance in European riparian ecosystems. As a pioneer tree species, it dominates at the first stage of succession in riverine forests and contributes to riverbank restoration and flood control. Natural poplar forests, known as Salici-Populetum (willow-poplar forest) or Populetum albae (poplar forest), are centers of biodiversity and host ecosystems of threatened and common animals, including insects. They also serve as natural corridors that connect larger forest units (Šiler et al. 2014). Economically, black poplar has been used in breeding programs aimed at the development of new highly resistant and fast-growing poplar varieties for years. Moreover, due to its high plasticity, the species plays an important role in phytoremediation projects in postindustrial areas (Koskela et al. 2004; lsebrands et al. 2014).
Due to habitat loss caused by the development of river engineering and intensive land use management of riverside areas over the last few decades, black poplar has become one of the most endangered tree species in Europe, and it was listed in the Strasbourg resolution of 1990 (Arbez and Lefèvre 1997). More recently, the status of black poplar has been specified by the International Union for Conservation of Nature (IUCN) as data deficient, but the species is listed as endangered in many European countries (Holub and Procházka 2000; Dénes 2001; Cheffings and Farrell 2005; Tylkowski 2010). To restore floodplain forests and to conserve the remaining genetic resources of this species, different conservation strategies have been implemented across European countries (Arens et al. 1998; Hughes et al. 2005). In Poland, according to the European Union Habitats Directive 92/43/EEC, the populations of black poplar throughout its wide distribution are protected as units of the Natura 2000 network (91E0). Nevertheless, although this conservation program prevents further devastation of well-preserved and ecologically valuable fragments of river ecosystems, a progressive reduction in black poplar population size is still being observed in Polish river landscapes. Based on ecological data and our own field observations, the most dramatic situation occurs along the most extensively transformed Warta and Oder river valleys and in the upper and lower sections of the Vistula River valley (Boratyński et al. 2001; Tylkowski 2010).
The genetic erosion of black poplar in Poland results from two major factors that can be classified as physical and biological drivers of extinction. The physical factor is associated with severe disturbance of the regeneration niche of the remaining P. nigra populations and results from the lack of suitable habitats for natural regeneration at most sites in which the species survives. For example, when the “Wielka Kępa” nature reserve near Bydgoszcz was created in 1953, much of it was covered by willow-poplar forest, with numerous black poplars. However, since the damming of the Vistula River in Włocławek in 1970 and the resultant flooding regulation, this riparian forest stand has been transformed over time into an elm-ash forest (Ficario-Ulmetum campestris), in which P. nigra does not regenerate (Boratyński et al. 2001).
The biological factor of extinction is directly related to the artificial introduction of fast-growing poplar cultivars consisting of a narrow range of Euramerican hybrids (Populus × canadensis Moench. (syn. P. x euramericana (Dode) Guinier)) and varieties of P. nigra, such as the male Lombardy poplar (P. nigra cv. ‘Italica’ Duroi), between the 1950s and 1970s. Native black poplar trees may hybridize with these taxa (Vanden Broeck et al. 2004; Smulders et al. 2008a; Ziegenhagen et al. 2008; Wójkiewicz et al. 2019). Therefore, there is a real threat that the fast-growing poplar variants, which occur in Polish river landscapes mostly as single trees or groups of individuals spread along the river valleys, endanger native P. nigra populations not only by competing with them for habitats but also by reducing their gene pool integrity via backcrossing and foreign gene introgression.
Taking all these factors into account, the development of a conservation strategy for black poplar is needed to rescue the remaining genetic resources of this species in Poland. Based on the guidelines for genetic conservation of black poplar in Europe proposed in the Forest Genetic Resources Program (IUFRO), the establishment of a suitable rescue plan for this species requires a preliminary evaluation of the background genetic variation in candidate populations to select unique local or regional gene pools that should be protected and which could be classified as the new genetic conservation units (GCUs) of this species at the pan-European level (Vanden Broeck et al. 2004; Koskela et al. 2013). However, little is known about the distribution of genetic variation among the natural P. nigra populations in Poland. Therefore, here, we present a case study on identifying and characterizing the genetic resources of black poplar from the Oder valley in Poland. Despite considerable anthropogenic transformations of this river landscape, it has retained the natural qualities that are crucial for the preservation of the characteristics of riverside vegetation cover. Populus nigra is still very common (frequency >70%) along the Oder valley, in contrast to, e.g., the Warta valley, where this species occurs with a frequency of only 20% relative to other forest tree species (Danielewicz 2008).
For the purpose of this study, the genetic structure and diversity level of nine natural black poplar populations from the Oder valley were investigated with nuclear microsatellite markers. Using different approaches, we investigated the population demographic history and gene flow patterns along the studied fragments of the valley. Specifically, we (1) identified the hybrid variants occurring within the black poplar populations based on the reference samples included in the study, (2) determined the number of multilocus genotypes (MLGs) and clustered them into clonal multilocus lineages (MLLs) to estimate genotypic richness, (3) assessed the effective population size and fine-scale spatial genetic structure (SGS) of the analyzed populations, (4) examined whether there are any signatures of a recent reduction in population size, and (5) established whether gene flow is limited by distance or there is a single migrant pool along the river system. Finally, based on the obtained results and available ecological data, we provide the first guidelines and suggestions on how to conserve and restore the genetic diversity of black poplar in the modern river landscapes of Poland.
2 Material and methods
2.1 The Oder valley
The Oder River is the second largest river in Poland. Its total length is 854.4 km, 742.0 km of which lies in Poland. The riverhead is located in the Odra Mountains (East Sudetes) in the Czech Republic, 634 m above sea level. The river drains a basin of 119,074 km2 and ends in the Roztoka Odrzańska near Police. The riverbed of the Oder is highly regulated along almost the entire length of the valley. According to historical data, the first embankments were already built along the Oder at the end of the 13th century (Hudak et al. 2018). Currently, the flood protection system consists of levees, polder areas, and, since the end of 2019, the Racibórz Dolny water reservoir. One of the most spectacular examples of the performed transformations of the Oder valley is the shortening of the natural river course by approximately 160 km, which constitutes 1/6 of the total river length (Migoń 2006).
Ecologically, despite far-reaching anthropogenic changes in habitats as well as forest and thicket communities, the tree and shrub flora in some parts of the Oder valley still retain its basic riparian character. For this reason, this river valley constitutes one of the major ecological corridors in the European Ecological Network (EECONET) (Liro et al. 1994). Since 2004, approximately 2300 km2 of the valley has been included in the Natura 2000 network, and this area is already legally protected.
With regard to poplar species, a characteristic chorological feature of this group of riparian forest-forming trees in the Oder valley is the much rarer occurrence of Populus alba (frequency 23.5%) than of P. nigra (73.8%) (Danielewicz 2008). This discrepancy in frequency between species may suggest that the white poplar is much more sensitive than the black poplar to changes in riverside environments. On the other hand, there is also some probability that because of the difficulties associated with distinguishing pure black poplars from hybrids on the basis of morphological characters, the true frequency of P. nigra in the Oder valley is overestimated. This hypothesis is supported by the fact that artificially introduced P. × canadensis hybrid trees are very common along the entire Oder valley, with a frequency of 75% (Danielewicz 2008).
2.2 Study sites and plant material
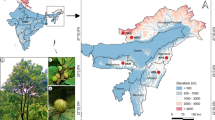
Nine natural black poplar populations located in the Oder valley were analyzed (Fig. 1). Sample sizes ranged from 27 to 99 trees per population (mean of 64.7 individuals), resulting in a total of 582 specimens studied (Table 1). In each study site, we focused on the adult trees that were in good condition. Additionally, the reference individuals of P. × canadensis and four representatives of Populus deltoides were included to eliminate potential hybrids from the analyzed set of black poplar trees in case they had not been recognized based on phenotype. The included reference individuals grow in the Kórnik Arboretum of the Institute of Dendrology, Polish Academy of Sciences. The Euramerican hybrids comprised variants marked as Populus ‘Serotina’ ♂, P. ‘Robusta’ ♂, P. ‘Marilandica’ ♀, and P. ‘Grandis’ ♀. These cultivars represent the most popular male and female poplar variants that have been planted in Poland in the last 100 years as plantations for wood production or singles trees for landscape purposes. Currently, there are no large-area plantations of poplar cultivars in Poland. However, the remnant poplar hybrids occur in the Polish river valleys as single trees or groups of individuals spread along the river valleys with similar frequency as native poplar species (Danielewicz 2008).

Geographical location and genetic structure of the studied black poplar populations from the Oder valley. (a) Map showing the location of the sampled populations (pointed by red points). (b) The distribution of ΔK over K = 1-15. Proportion of the membership coefficient for each black poplar population studied and for each individual of P. nigra tree to the inferred clusters K = 2 (C,C’) and K = 4 (D,D’) from the STRUCTURE analysis
DNA was extracted from young poplar leaves by using the standard CTAB protocol (Dumolin et al. 1995). Qualitative and quantitative assessments of the DNA isolates were conducted by absorbance measurement using an Eppendorf BioPhotometer (Hamburg, Germany).
2.3 Genotype scoring
All samples were genotyped with the 12 nuclear microsatellite loci described by Van der Schoot et al. (2000) and Smulders et al. (2001). Specifically, WPMS01, WPMS04, WPMS06, WPMS07, WPMS08, WPMS09, WPMS10, WPMS11, WPMS12, WPMS16, WPMS18, and WPMS20 were used in the study. Marker amplification was performed according to the methodology described by Wójkiewicz et al. (2019). The products of each polymerase chain reaction (PCR) were analyzed using an ABI 3130 capillary sequencer (Thermo Fisher Scientific, Waltham, Massachusetts, USA) with GeneScan LIZ500 internal size standard. The genotypes were scored using GENEMAPPER vs. 4.0 (Wójkiewicz 2020).
2.4 Data analysis
2.4.1 Identification of hybrids and clones
The recognition of hybrid trees was performed based on the microsatellite markers used in study, among which WPMS01, WPMS12, and WPMS18 were previously described as diagnostic and useful for the identification of P. × canadensis cultivars (Smulders et al. 2008a; Jelić et al. 2015; Wójkiewicz et al. 2019). In order to identify the hybrids, the STRUCTURE 2.3.4. software was used (Pritchard et al. 2000). For the 590 sampled trees, which comprised the four reference individuals of P. deltoides, four P. × canadensis representatives (marked as P. ‘Serotina’, P. ‘Robusta’, P. ‘Marilandica’, and P. ‘Grandis’), and 582 investigated trees derived from the nine populations, we set K = 2 (the number of species). Twenty independent runs were performed with a burn-in length of 250,000 and 100,000,000 iterations, with admixture model and correlated allele frequencies, without any prior information. Average admixture coefficients were estimated using the LargeKGreedy algorithm as implemented in the program CLUMPP version 1.1 (Jakobsson and Rosenberg 2007). To assign the detected hybrids to the reference poplar cultivars included in the study, the genotypes of all hybrid individuals were matched using the GenAlEx 6.5 software (Peakall and Smouse 2006). Finally, all identified poplar hybrids were excluded from the data set to analyze clonality.
To determine the genotypic resolution power of the 12 microsatellites used in the study, a test of the reliability of loci was performed (Alberto et al. 2005). The number of distinct multilocus genotypes (MLGs) was assessed, and then the clones were identified as sets of individuals that presented the same MLG using the package RClone (Bailleul et al. 2016). Furthermore, as the number of MLGs can be overestimated due to the occurrence of slightly different MLGs resulting from somatic mutations or scoring errors, discrimination of clonal lineages and assembly of similar MLGs into corresponding MLLs were performed. Discrimination analysis was performed by calculating Rozenfeld’s genetic distance (difference in length between alleles; Rozenfeld et al. 2007) for each pair of unique MLGs in the sample that were initially characterized molecularly and comparing these MLGs to each pair of unique MLGs identified as sexually produced by simulations (Arnaud-Hanod et al. 2007). From this distribution, a threshold was determined, under which genetic distances were considered to be due to somatic mutations or scoring errors, and distinct MLGs belonging to the same MLL were identified. With the aim of assessing the relative importance of asexual reproduction, genotypic richness R was calculated for each population based on the number of detected MLLs (Dorken and Eckert 2001). Finally, for the purpose of population genetic analyses, only one ramet of each genet was left in the data set, as clones do not result from sexual reproduction.
2.4.2 Genetic variation and differentiation
To estimate the frequency of null alleles and detect the loci that deviate from the Hardy–Weinberg equilibrium, we used the exact test based on the Markov Chain Monte Carlo (MCMC) algorithm with Bonferroni correction implemented in GENEPOP v. 4.6 (Rousset 2008). The basic information about the markers used in the study are presented in Table 4 in Appendix. To test for linkage disequilibrium (LD) between the 12 pairs of loci at the individual population level and across the populations, the Fisher’s exact test was used in Arlequin 3.22 (Excoffier et al. 2005). Basic genetic diversity parameters (i.e., A—mean number of alleles, Ae—mean number of effective alleles, PA—number of private alleles, Ho—observed heterozygosity, and He—unbiased expected heterozygosity) were calculated in GenAlEx 6.5. FSTAT 2.9.4 (Goudet 2001) was used to estimate the inbreeding coefficient (Fis) and allelic richness (AR) for the minimum sample size of 19 individuals. A Bayesian approach implemented in the INEST 2.0 software (Chybicki and Burczyk 2009) was applied to estimate the inbreeding coefficient, including ‘null alleles’ correction (Fisnull), according to the individual inbreeding model (IIM). The estimation was run with 500,000 MCMC cycles, with every 200th cycle updated and a burn-in of 50,000. The deviance information criterion (DIC) was used to compare the full model (‘nfb’, when Fis>0) with the random mating model (‘nb’, when Fis = 0) to assess the determinants of homozygosity level. The significance of heterozygote deficiency in the sampled populations was assessed by the U test (Guo and Thompson 1992) in GENEPOP, and p values were obtained with the Markov chain algorithm using default settings.
NeEstimator software ver. 2.01 (Do et al. 2014) was used to estimate the effective population size (Ne^) of each studied population with the LD approach (Waples and Do 2008). To test the sensitivity of the method to the presence of rare alleles, the results obtained with three different allele frequency cut-off thresholds (i.e., Pcrit = 0.01, 0.02, and 0.05) were compared. The 95% confidence intervals (CINe^) were derived using the ‘parametric’ option with χ2 approximation (Waples 2005).
Finally, we assessed interpopulation differentiation by hierarchical analysis of molecular variance (AMOVA) using both FST and RST values computed for all pairs of populations in Arlequin 3.11. To evaluate the influence of stepwise mutations on the differentiation level of populations, RST and permuted RST (pRST—which corresponds to FST) values were compared using the test proposed by Hardy et al. (2003) and implemented in the SPAGeDi ver. 1.4c software (Hardy and Vekemans 2002). Moreover, overall and pairwise FST values were also calculated with a correction for the presence of null alleles (excluding null alleles (ENA), FSTNA) with the use of FreeNA software (Chapuis and Estoup 2007). The bootstrapped 95% confidence intervals (CIs) of FSTNA were calculated using 2000 replicates over the loci. The statistical significance (at the level of p = 0.01) of the estimated pairwise FST values was tested by 10,100 random permutations using the Arlequin software.
2.4.3 Demographic history of the populations
We used two different approaches to elucidate the demographic history of the studied black poplar populations and to test whether past environmental transformations of river landscapes coupled with potential population size variation left detectable signatures of genetic bottlenecks. For each population, we calculated the M ratio (MR, Garza and Williamson 2001) and ran the Wilcoxon test for heterozygote excess (Cornuet and Luikart 1996) using the INEST ver. 2.2 software. The analysis was performed under the two-phase mutation (TPM) model with two parameters: pg = 0.22 (the proportion of multistep mutations) and δg = 0.31 (the mean size of the multistep mutations). The significance of a potential bottleneck was tested using Wilcoxon’s signed-rank test based on 1,000,000 permutations.
2.4.4 Genetic structure and gene flow patterns
To describe the spatial autocorrelation within the populations, the average kinship coefficient over the pairs of studied individuals was computed using the SPAGeDi software. Distance intervals were adjusted by SPAGeDi to obtain approximately the same number of pairs of individuals within each of eight distance classes. The statistical significance of the autocorrelations was tested by 10,000 random permutations, with a 95% CI. Average kinship coefficients between pairs of individuals for each distance interval were plotted against distance classes in a diagram. Significant autocorrelation is shown as an outlier in the observed data from the 95% CIs. Moreover, SGS was also quantified using the Sp statistics (Vekemans and Hardy 2004). Matrices of pairwise spatial physical distances and genetic distances based on kinship coefficients within each population were obtained for simple sequence repeat (SSR) loci using SPAGeDi. Within each population, the relationship between matrices was assessed using the Mantel test implemented in GenAlEx 6. The significance of the Mantel test was evaluated based on 1,000 permutations.
To investigate migration patterns among the analyzed populations, Bayesian assignment testing (Rannala and Mountain 1997) was performed using the Geneclass2 software (Piry et al. 2004). Moreover, the Mantel (1967) test was applied to evaluate whether the distribution of genetic variation was geographically structured and to verify the hypothesis of isolation by distance (IBD) between populations. For this purpose, the GenAlEx software was used, with 1,000 random permutations of the relationship between genetic differentiation, quantified as FST/(1- FST), and corresponding geographical distance matrices between populations.
The substructuring of the black poplar gene pool was evaluated by a nonspatial Bayesian clustering model implemented in STRUCTURE 2.3.4. Twenty independent runs were performed for each K from 1 to 15 (the user-defined number of clusters), with a burn-in length of 250,000 and 100,000,000 iterations. The probability distributions of the data (LnP(D)) and the ΔK values (Evanno et al. 2005) were visualized using the STRUCTURE HARVESTER Web application (Earl and VonHoldt 2012). Average admixture coefficients were estimated for each value of K using the LargeKGreedy algorithm as implemented in the program CLUMPP version 1.1. STRUCTURE plots were generated using the STRUCTURE PLOT v.2.0 Web application (Ramasamy et al. 2014). Furthermore, AMOVA analysis was conducted between the groups of populations defined by STRUCTURE, and significance was tested using 10,000 random permutations in Arlequin 3.11.
3 Results
3.1 Hybrid identification
Out of the 582 poplar individuals analyzed, 34 were classified as hybrids with a probability higher than 90% (Fig. 2 in Appendix). In total, 1, 9, 8, 3, and 13 of the hybrids were found in populations KK, BO, CG, K, and G, respectively (Table 1). Among these individuals and based on the reference P. × canadensis samples, we identified 15 trees with a genotype identical to that of P. ‘Marilandica’ ♀ (i.e., 1 individual in population KK, 9 individuals in population BO, and 5 individuals in population CG), 3 trees with the P. ‘Robusta’ ♂ genotype (population CG), and 3 trees with the P. ‘Serotina’ ♂ genotype (population K). The 13 hybrid individuals found in population G had one unique genotype that could not be assigned to any reference samples. However, taking into account the spatial distribution of these hybrid clones within the population G, and the fact that we sampled old trees during the plant material collection, there is a low probability that those trees are the advanced generation of hybrids (F2 or advance BC) that result from the spontaneous hybridization between black poplar and variants of P. × canadensis trees in nature. The locations of the detected hybrid individuals in the studied populations are marked on the maps (Figs. 3, 8, 9, 10, 11 in Appendix).
3.2 Clonality analysis
The analytical power of the marker set used in this study was high. Based on a box plot describing the genotypic resolution of the microsatellites applied, we conclude that a set of nine loci would be sufficient to accurately determine the number of MLGs in the studied sample (Fig. 12 in Appendix). From the 548 sampling units genotyped, a total of 423 distinct MLGs were detected, and among them, 53 were shared by several individuals (more than two trees). All repeated genotypes were unique to individual populations (i.e., there were no same genotypes detected in different stands). The most frequent genotype, represented by 16 ramets, was found in population G. The maximum distances between the ramets of one genet noted in the studied sample set were 150 m in population G and approximately 80 m in population CI. Based on the pairwise genetic distances between the MLGs identified in each poplar population compared with the MLGs sexually produced according to simulations and taking into account a threshold of 2 different alleles, 403 MLLs were detected. With regard to this result, the estimated genotypic richness of the investigated populations ranged from R = 52% in population CI to R = 87% in population KK, with a mean value of R = 73% (Table 1). The clone positions in each study site are indicated in Figs. 3–11 in Appendix.
3.3 Genetic diversity and effective population size
Genetic diversity estimators calculated per black poplar population estimated based on 12 loci are presented in Table 2. The average number of alleles in a population ranged from A = 6.67 in populations K and G to A = 12.58 in population BD, with a mean of A = 9.99. The number of effective alleles was much smaller, with a mean value of Ae = 5.38. The highest allelic richness estimated for the minimum sample size of 19 individuals (AR19) was observed in population CI (9.34), whereas the lowest was observed in population G (6.20). Private alleles were detected in all studied stands. However, the frequency of all of the private alleles was low. Observed heterozygosity (Ho) was high and comparable between the analyzed populations, with a mean of Ho = 0.71, while expected heterozygosity (He) was only slightly higher, with an average of He = 0.77. Significant positive Fis values, suggesting an excess of homozygotes, were observed in seven populations (with mean Fis = 0.07). However, based on the correction performed for the presence of null alleles (Fisnull) and according to the DIC, in all analyzed populations, the detected inbreeding signals were delusive (i.e., null alleles were most likely responsible for the increased homozygosity level). The effective population size (Ne^) calculated with the lowest allele frequency criterion of 0.02 was comparable among populations KK, B, WR, BD, BO, and CG. The largest effective population size was detected for population CI (133.5), whereas the lowest was detected for two populations from the north, i.e., population K (9.3) and population G (5.2) (Table 2).
3.4 Genetic differentiation and demographic history analysis
Statistically significant differentiation between populations was revealed by AMOVA. The results were similar for FST and RST, with values of 4.43% and 5.27%, respectively. The remaining 95.57% (FST) and 94.73% (RST) of the total molecular variance occurred within populations (Table 3). The similarity of the genetic differentiation estimators used in the investigation was also confirmed by a permutation test. The obtained results revealed no statistically significant difference between RST and permuted pRST values (RST = 0.054; pRST = 0.046; CIpRST95% = 0.031-0.068; pH1: RST > pRST = 0.231), which implied that the rate of migrant exchange by populations was higher than the mutation rate. The global FST values with and without ENA correction were also similar (FST = 4.43% and FSTNA = 4.42%, respectively). Nevertheless, a wider range of differentiation was found with regard to pairwise comparisons of populations. The highest value of FST with ENA correction was noted between the BD and K populations (FSTNA = 0.079), while the lowest was found between the neighboring populations B and WR (FSTNA = 0.025). With regard to pairwise distances evaluated based on the RST method, populations B and G were the most different (RST = 0.140), whereas populations KK and B were the most similar (RST = 0.002) (Table 5 in Appendix).
The Wilcoxon test for heterozygosity, performed under the TPM model, did not indicate recent bottlenecks in any populations analyzed. On the other hand, the MRs were significantly below the mean MR under mutation-drift equilibrium (Mreq) for all black poplar forest stands examined (Table 2).
3.5 Genetic structure and gene flow patterns
The spatial autocorrelation analysis revealed significant SGS in seven of the nine populations analyzed (Fig. 13 in Appendix). Based on the results for the Sp statistic, the strongest SGS was identified in population K (Sp = 0.049), whereas the weakest was identified in population CI (Sp = 0.003). Coancestry coefficients were positive and significant at a distance of approximately 150 m in all analyzed populations. For three to seven distance classes, in most cases, the estimated coancestry coefficients were within the 95% CI, becoming negative and significant, for the last distance class in the populations BO, CG, K, and G (p < 0.05). Significant coancestry coefficients for first distance classes indicate the natural origin of the studied black poplar populations as artificial stands of forest trees were usually established with the use of mixed seed material derived from different populations. In this case, the kinship coefficient between even close neighbors is low and does not increase with distance.
The population assignment test implemented in Geneclass2, performed based on Rannala’s & Mountain’s method, showed that 324/403 (80%) black poplar trees were assigned to their sampled populations (Table 6 in Appendix). Misassignment frequency ranged from 14% in population BD to 41% in population BO. The obtained results showed that gene flow between the poplar populations is bidirectional along the study area and the most effective between neighboring populations. On the other hand, some restrictions on upstream migration from the northernmost populations G and K were observed. No immigrants derived from these populations were identified in the higher-situated poplar stands based on test results (Table 6 in Appendix).
The Mantel test yielded a significant correlation (R2 = 0.63; p = 0.01) between the level of genetic differentiation and geographical distance estimated for each pair of the studied populations (Fig. 14 in Appendix). This result is consistent with an IBD model among the black poplar populations and is in agreement with the detected migration patterns, which suggest that the most effective gene flow occurs between the most geographically proximal poplar stands (Table 6 in Appendix).
The Bayesian assignment of samples by STRUCTURE revealed that the overall gene pool of the analyzed black poplar populations is represented by two (K = 2) or four (K = 4) genetic clusters (Fig. 1). According to this result, and based on the proportion of the membership coefficient estimated by STRUCTURE (Table 7 in Appendix), we classified the analyzed populations to the different groups (two or four) and performed the AMOVA analysis to verify which clustering model better explains the identified patterns of genetic structure. In reference to the two inferred genetic clusters (K = 2), the populations KK, B, WR, and BD were classified to the first group, whereas the populations CI, BO, CG, K, and G were included in the second genetic cluster. With regard to four inferred clusters (K = 4), we assigned populations KK, B, and WR to the first group. The population BD was classified to the second group. The third group comprised the populations CI, BO, and CG. The populations K and G were included in the last fourth group. The AMOVA analysis results showed higher values of parameters of genetic differentiation for the tested variants of groups of populations in the case of population assignment performed based on the four genetic clusters detected, with FST = 1.15% (p = 0.001) and RST = 4.38% (p = 0.006). Moreover, in the case of four genetic clusters, the level of genetic differentiation among groups, estimated based on RST, was higher than the level of differentiation among the populations established within the groups (with RST = 4.38% (p = 0.006) and RST = 1.67% (p < 0.001), respectively). In the case of partition to the two groups (K = 2), the obtained results showed the opposite tendency with RST = 2.21% among groups (P = 0.063) and RST = 3.95% among populations within groups (P < 0.001) (Table 3).
4 Discussion
4.1 Genetic resources and population history of black poplar from the Oder River area
During the last two centuries, the area of riparian forests in Poland has been reduced by approximately 95% (Tylkowski 2010), and many of these natural forest communities have been changed by alien species. However, our results indicate that the analyzed poplar forest stands have maintained their genetic identity. Only 34 P. × canadensis individuals were detected among which 21 were assigned to the reference cultivars of P. × canadensis included in the study. This observation is in agreement with previous findings by different ecologist groups that indicated the high natural qualities of plant formations occasionally occurring in the small fragments of the Oder valley, from which the material for this study was mainly collected (Danielewicz 2008; Wojtkowiak et al. 2013). On the other hand, a relatively large proportion of clones, which ranged from 13% to 48% with an average of 27%, was noted in the studied populations. These results are comparable to those of earlier studies by Smulders et al. (2002) and Barsoum et al. (2005), who reported 42% and 22% replicated genotypes, respectively, among trees sampled along the Rhine and Garonne rivers. Concerning clonal structure, replicate genotypes were mostly found in our study as nearest neighbors and formed relatively small (2-4 trees growing in close proximity to one another) or moderately sized (8-16 ramets distributed in a circle with a maximum diameter of 150 m) clonal units. This finding is in line with the results of Legionnet et al. (1996) and Barsoum et al. (2005), who found mostly small clonal units within the studied populations, and the reports presented by Arens et al. (1998) and Smulders et al. (2002), who found substantially more replicates per P. nigra clone (i.e., up to 24 trees per clone). However, in contrast to Barsoum et al. (2005), we did not find replicate genotypes in separate stands. The difference in the frequency of clones between the studied populations most likely results from the different flooding histories of the studied stands and age structures of the analyzed poplar populations. Our results indicate an important contribution of asexual regeneration strategies to the maintenance of P. nigra populations along the Oder valley and suggest that the actual genetic resources of this species are approximately 30% lower than expected on the basis of the number of black poplar trees observed along the valley.
The level of genetic variation in the P. nigra populations determined in this work is consistent with previous estimates based on microsatellite markers. The number of alleles and level of observed heterozygosity found in our investigation (mean A = 9.99/Ho = 0.71) were comparable to those reported by Pospíšková and Šálková (2006), with A = 11.00/Ho = 0.80; Smulders et al. (2008b), with A = -(no data)/Ho = 0.74; Rathmacher et al. (2010), with A = 11.57/Ho = 0.70; Jelić et al. (2015), with A = 8.96/Ho = 0.70; Lewandowski and Litkowiec (2016), with A = 14.7/Ho = 0.73; Čortan and Tubić (2017), with A = 8.5/Ho = 0.76; and Wójkiewicz et al. (2019), with A = 12.17/Ho = 0.70. The Fis values estimated with correction for the presence of null alleles in all populations were positive but close to zero, which suggests random gene exchange among distantly related poplar individuals. The assignment analysis of individuals indicates bidirectional gene flow along the Oder valley. A similar mode of gene transmission was described by Imbert and Lefèvre (2003), who studied dispersal and gene flow among P. nigra populations from the Drome River area in France. Asymmetry in gene transfer was noted only in the lower reaches of the Oder River, where migration in the downstream direction was predominant. Nevertheless, this mating pattern may result from the largest gap between sampled stands, which occurred between the population from the middle section of the Oder valley (population CG) and the first population from the upper section of the river (population K), which reached approximately 150 km. Furthermore, it may result from the smaller sample size of populations from the north than of populations from the middle section of the valley. The assignment analysis also revealed that the most effective gene exchange occurred between neighboring poplar stands. This observation is consistent with expectations of a one-dimensional stepping-stone gene flow model which assumes that an individual can migrate at most one step in either direction between populations (Slatkin 1993). This is also in line with the results presented by Rathmacher et al. (2010), who reported short-distance gene flow between poplar populations near the Eder River. According to the authors’ conclusions, the majority of gene transfer (i.e., 70%) among the studied poplar populations from this river valley took place over a distance of less than 1 km. Limited gene transmission promotes the formation of small-scale SGS, which we detected in all black poplar populations analyzed.
The Oder valley populations were not genetically homogenous. The clustering analysis showed that two (K = 2) or four (K = 4) genetic clusters can be distinguished. Nevertheless, the estimated differentiation level was low (FST = 4.43%; RST = 5.27%) and comparable to levels presented in previous studies on black poplar populations originating from one river catchment (Imbert and Lefèvre 2003; Pospíšková and Bartáková 2004; Jiang et al. 2015; Čortan et al. 2016). Based on these findings (high spatial genetic structure and low overall genetic differentiation level), we hypothesize that the interplay of different demographic forces during the last several generations has driven the subdivision process of the once homogenous genetic structure of the analyzed populations. This indication is supported by the results of the Mantel test which showed that the detected patterns of genetic differentiation among the studied populations fit the IBD model well. The correlation between genetic and geographic distances was significant (R2 = 0.63; p = 0.01). Comparable results were also reported by, e.g., Imbert and Lefèvre (2003) who studied gene flow among black poplar populations from the Drôme river (France). This finding supports our conclusions about the gene exchange model (one-dimensional stepping-stone model) and indicates that the detected disturbances in gene flow among the studied populations seem to be more significant than those shown in previous studies for this species. Moreover, it seems that the observed gene pool substructuring of the studied poplar populations and detected geographical patterns of genetic variation along the Oder River have their background also in the demographic history of these forest stands. Specific tests for bottleneck effects using microsatellites yielded interesting results, with the MR test suggesting a bottleneck in all populations, although the heterozygote excess test, performed under the TPM model, showed no evidence of a bottleneck in any of the populations analyzed. Detection of a bottleneck using MRs but not heterozygote excess is more likely when the bottleneck is older and more severe. Strong support for this hypothesis is provided by the effective population size results for the studied poplar stands. In two cases, we found that the Ne^ was less than 10, and in only three populations, it was greater than 50. In contrast to these findings, the mean effective population size of two old black poplar populations from the Vistula valley in Poland estimated based on microsatellite markers was Ne^ = 263.75 (Wójkiewicz et al. 2019). Taking all these results into account, we speculate that the genetic structure of black poplar was homogenous along the Oder catchment in the distant past. The currently observed genetic subdivision of the gene pool of populations of this species is most likely a consequence of genetic drift, intensified due to a strong reduction in population size in the past, restriction in gene flow in consequence of transformation and fragmentation of natural riparian habitats, and diminishment of the success of sexual reproduction for vegetative propagation due to a lack of suitable places for seedling establishment.
4.2 Implications for the conservation management strategy of black poplar
The effective maintenance of existing genetic resources is crucial for forest species survival and adaptation in changing environments. For this purpose, population genetic studies provide key insights to assist in the development of suitable management and conservation strategies. The present study is by far the most extensive report describing the distribution of genetic variability and underlying processes among natural black poplar populations in the Oder River region. Our results revealed significant differentiation among the black poplar populations studied. However, the level of differentiation was low, suggesting that the black poplar populations from the Oder valley should be treated as a single panmictic unit, which in the past was characterized by a homogenous genetic structure. Nowadays, based on the STRUCTURE and AMOVA analysis results, it seems that four (K = 4) genetic clusters most likely represent the overall genetic structure of the studied populations. Therefore, we recommend establishing one clone archive in which 200 representatives of the four genetic clusters detected are grown together in one location. More precisely, we suggest collecting at least 50 cuttings (with a 1:1 ratio of female:male trees) from each of the genetically distinct groups of populations, which include the following stands: Group 1—populations KK, B, and WR; Group 2—populations BD; Group 3—populations CI, BO, and CG; and Group 4—populations K and G. This sampling strategy, based on the assumptions presented by Brown and Hardner (2000), should ensure to capture at least one copy of alleles with a frequency of 0.05 from each defined group with a certainty of 95%. Moreover, in the future, this stable clone archive population with effective population size Ne^ > 50 (so that it can maintain a balance between genetic drift and mutation (Franklin 1980)) can be classified as the new seed source population. To avoid close relatedness among the selected P. nigra individuals as well as duplicate genotypes in the archive, the distance between the sampled trees in each site should be at least 200 m. Moreover, as hybrid trees occur among natural black poplar trees along the Oder valley as single or grouped individuals, genetic assessment of the trees selected and planted in the clone archive should be performed in the final stage. As shown in our study, there are molecular tools suitable for the precise identification of hybrid individuals. Locally, the black poplar gene pools may be protected by artificial plantings of black poplars along the Oder River with the use of seeds originating from the studied poplar stands. Only in the case of populations from the north, i.e., populations K and G, which had the smallest effective population sizes as well as quite a large proportion of clones, should additional paternity analyses with the aim of establishing the number of male trees that contribute to the pollination of selected maternal trees be performed to assess the genetic value of the obtained offspring population. During the selection of maternal black poplar trees from which the seeds will be collected, we suggest focusing on the locations of the studied trees reported in the Electronic Supplementary Material. This should help avoid sampling the clones and hybrid trees detected in the studied areas. Moreover, as P. ‘Marilandica’ is one of the most common female cultivars along the Oder valley, seeds from black poplar trees should be collected directly from the selected maternal trees and not from the ground. We also recommend focusing on trees that are in good condition and that grow in close neighborhoods of groups of pure male black poplar trees. Targeting close neighborhoods of male black poplar trees may help avoid sampling seeds of female black poplar trees that could be potentially pollinated by males of hybrid poplar cultivars such as P. ‘Robusta’ and P. ‘Serotina’, which were detected in some of the studied populations.
Finally, based on the obtained results and assumptions presented by Koskela et al. (2013) concerning pan-European minimum requirements for dynamic conservation units of forest tree genetic diversity (GCUs), we recommend two candidate populations to establish new GCUs of this species in the studied area of black poplar distribution in Europe. The black poplar stands which meet the genetic requirements are populations from the area near Brzeg (pop. B) and area near Ciechanów (pop. CI) (Figs. 4 and 7 in Appendix). These populations are still genetically stable, at least in the short-term predictions, and were classified to different genetic clusters taking both K = 2 and K = 4 structure assumptions. However, before the final decision will be made, in both cases, the sex ratio should be estimated to establish the number of female trees in candidate populations which should not be lower than 50 in the case of dioecious tree species. Currently, although the genetic resources of black poplar seem to be large along the Oder and Vistula river valleys, only two GCUs of this species from forty registered across Europe exist in the territory of Poland. Moreover, both are located in close neighborhood along the Vistula river valley that suggests their homogenous genetic structure which, however, remains unstudied.
5 Conclusion
Clear evidence of negative genetic changes in the studied black poplar populations from the Oder valley reflected in the high spatial genetic structure and low effective population sizes was presented in this study. The obtained findings suggest a small-scale isolation by distance of black poplar populations which results most likely from the alteration of natural dynamics of the riparian ecosystems rather than from the presence of physical barriers for gene flow. The disturbance in gene flow among the natural black poplar populations noted in this study is not a frequent phenomenon observed for this species along the river systems of Europe as it is known as a very good disperser through both seeds and pollen. Nevertheless, the genetic resources of black poplar in the Oder River valley are still substantial compared to those reported for rivers in Western Europe. However, due to the lack of places for natural regeneration of this species along the modern river landscapes in Poland as well as taking into account assumptions for the development plans of inland waterways in Poland for 2016-2020 with a 2030 perspective, the genetic erosion of black poplar populations will likely progress, and the number of genetically unique specimens will be lower in the future. Considering the ongoing environmental changes and degradation of places where black poplar naturally occurs, this seems to be the last chance to introduce conservation strategies in order to save the natural genetic diversity of this endangered riparian forest species.
Data availability
The datasets analyzed during the current study are available online https://doi.org/10.18150/EIGSVA
References
Alberto F, Gouveia L, Arnaud-Haond S, Pérens-Lloréns JL, Duarte CM, Serrão EA (2005) Within-population spatial genetic structure, neighbourhood size and clonal subrange in the seagrass Cymodocea nodosa. Mol Ecol 14:2669–2681. https://doi.org/10.1111/j.1365-294X.2005.02640.x
Arbez M, Lefèvre F (1997) The European forest genetic resource programme. Objectives and general concept. A case study concerning the black poplar. Bocconea 7:389–398
Arens P, Coops H, Jansen J, Vosman B (1998) Molecular genetic analysis of black poplar (Populus nigra L.) along Dutch rivers. Mol Ecol 7:11–18. https://doi.org/10.1046/j.1365-294x.1998.00316.x
Arnaud-Hanod S, Duarte CM, Alberto F, Serrão EA (2007) Standardizing methods to address clonality in population studies. Mol Ecol 16:5115–5139. https://doi.org/10.1111/j.1365-294X.2007.03535.x
Bailleul D, Stoeckel S, Arnaud-Haond S (2016) RClone: a package to identify multi locus clonal lineages and handle clonal data sets in R. Methods Ecol Evol 7:966–970. https://doi.org/10.1111/2041-210X.12550
Barsoum N, Muller E, Skot L (2005) Variations in levels of clonality among Populus nigra L. stands of different ages. Evol Ecol 18:601–624. https://doi.org/10.1007/s10682-004-5146-4
Boratyński A, Bartczak A, Przybylińska J, Figaj J (2001) Skuteczność ochrony starych drzew topoli czarnej i białej na przykładzie rezerwatu Wielka Kępa Ostromecka [The efficiency of the protection program of the old black poplar and white poplar trees on the example of the Wielka Kępa Ostromecka reserve]. Przegl Przyrodn 12:141–147
Brown AHD, Hardner CM (2000) Sampling the gene pools of forest trees for ex situ conservation. In: Young AG, Boshier DH, Boyle TJ (eds) Forest Conservation Genetics: Principles and Practice. CABI Publishing, Wallingford, pp 185–196
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631. https://doi.org/10.1093/molbev/msl191
Cheffings CM, Farrell L (2005) The vascular plant red data list for Great Britain. Species status no. 7. Joint Nature Conservation Committee, Peterborough
Chybicki IJ, Burczyk J (2009) Simultaneous estimation of null alleles and inbreeding coefficients. J Hered 100:106–113. https://doi.org/10.1093/jhered/esn088
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Čortan D, Tubić B (2017) Viability and genetic diversity of Populus nigra population from riparian forest in SNR Gornje Podunavlje. Dendrobiology 78:157–167. https://doi.org/10.12657/denbio.078.015
Čortan D, Schroeder H, Šijačić-Nikolić M, Wehenkel C, Fladung M (2016) Genetic structure of remnant black poplar (Populus nigra L.) populations along biggest rivers in Serbia assessed by SSR markers. Silvae Genet 65:12–19. https://doi.org/10.1515/sg-2016-0002
Danielewicz W (2008) Ekologiczne uwarunkowania zasięgów drzew i krzewów na aluwialnych obszarach doliny Odry [Ecological determinants of the range of trees and shrbs in the alluvial areas of the Oder valley]. Wyd. UP, Poznań
Dénes B (2001) Veszélyeztetett erdőtársulások magyarországon [Endangered forest associations in Hungary] WWF füzetek 18. WWF, Budapest
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Res 14:209–214. https://doi.org/10.1111/1755-0998.12157
Dorken ME, Eckert CG (2001) Severely reduced sexual reproduction in northern populations of a clonal plant, Decodonverticillatus (Lythraceae). J Ecol 89:339–350. https://doi.org/10.1046/j.1365-2745.2001.00558.x
Dumolin S, Demesure B, Petit RJ (1995) Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor Appl Genet 91:1253–1256
Earl DA, Vonholdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform 1:47–50
Franklin IR (1980) Evolutionary changes in small populations. In: Soulé ME, Wilcox BA (eds) Conservation Biology: An Evolutionary-Ecological Perspective. Sinauer, Sunderland, MA, pp 135–150
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318. https://doi.org/10.1046/j.1365-294X.2001.01190.x
Gaudet M, Jorge V, Paolucci I, Beritognolo I, Scarascia Mugnozza G, Sabatti M (2008) Genetic linkage maps of Populus nigra L. including AFLPs, SSRs, SNPs, and sex trait. Tree Genet Genom 4:25–36. https://doi.org/10.1007/s11295-007-0085-1
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices, Version 2.9.3. http://www2.unil.ch/popgen/softwares/fstat.htm.
Guo SW, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48:361–372
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620. https://doi.org/10.1046/j.1471-8286.2002.00305.x
Hardy OJ, Charbonnel N, Freville H, Heuertz M (2003) Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics 163:1467–1482
Holub J, Procházka F (2000) Red list of vascular plants of the Czech Republic. Preslia 72:187–230
Hudak M, Karczmar C, Kołodziejczyk U, Kostecki J (2018) Flood protection on the Odra River in the segment between Nowa Sól and Cigacice. CEER 28:54–63. https://doi.org/10.2478/ceer-2018-0005
Hughes FMR, Colston A, Mountford OJ (2005) Restoring riparian ecosystems: the challenge of accommodating variability and designing restoration trajectories. Ecol Soc 10:12 http://www.ecologyandsociety.org/vol10/iss1/art12/
Imbert E, Lefèvre F (2003) Dispersal and gene flow of Populus nigra (Salicaceae) along a dynamic river system. J Ecol 91:447–456. https://doi.org/10.1046/j.1365-2745.2003.00772.x
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. https://doi.org/10.1093/bioinformatics/btm233
Jelić M, Patenković A, Skorić M, Mišić D, Novičić ZK, Bordács S, Várhidi F, Vasić I, Benke A, Frank G, Šiler B (2015) Indigenous forests of European black poplar along the Danube River: genetic structure and reliable detection of introgression. Tree Genet Genom 11:89. https://doi.org/10.1007/s11295-015-0915-5
Jiang D, Wu G, Mao K, Feng J (2015) Structure of genetic diversity in marginal populations of black poplar (Populus nigra L.). Biochem. Syst Ecol 61:297–302. https://doi.org/10.1016/j.bse.2015.06.014
Koskela J, de Vries SGM, Kajba D, von Wühlisch G (2004) Populus nigra Network, report of the seventh (25-27 October 2001, Osijek, Croatia) and eighth meetings (22-24 May 2003, Treppeln, Germany). International Plant Genetic Resources Institute, Rome, Italy
Koskela J, Lefèvre F, Schüler S, Kraigher H, Olrik DC, Hubert J, Longauer R, Bozzano M, Yrjänä L, Alizoti P, Rotach P, Vietto L, Bordács S, Myking T, Eysteinsson T, Souvannavong O, Fady B, De Cuyper B, Heinze B, von Wühlisch G, Ducousso A, Ditlevsen B (2013) Translating conservation genetics into management: pan-European minimum requirements for dynamic conservation units of forest tree genetic diversity. Biol Conserv 157:39–49. https://doi.org/10.1016/j.biocon.2012.07.023
Legionnet A, Faivre-Rampant P, Villar M, Lefèvre F (1996) Sexual and asexual reproduction in natural stands of Populus nigra. Bot Acta 110:257–263. https://doi.org/10.1111/j.1438-8677.1997.tb00638.x
Lewandowski A, Litkowiec M (2016) Genetic structure of the old black poplar population along the bank of the Vistula River in Poland. Acta Soc Bot Pol 86:3524. https://doi.org/10.5586/asbp.3524
Liro A, Głowacka I, Jakubowski W, Keftan J, Matuszkiewicz AJ, Szacki J (1994) Koncepcja krajowej sieci ekologicznej EECONET-POLSKA [The concept of the national ecological network EECONET-POLSKA]. Fundacja IUCN Polnad, Warszawa
Lsebrands JG, Aronsson P, Carlson M, Ceulemans R, Coleman M, Dickinson N, Dimitriou J, Doty S, Gardiner E, Heinsoo K, Johnson JD, Koo YB, Kort JJ, Kuzovkina J, Licht L, McCracken AR, Mcivor I, Mertens P, Perttu K, Riddeii-Biack D, Robinson BB, Scarascia-Mugnozza G, Schroeder WR, Stanturf J, Volk TA, Weih M (2014) Environmental applications of poplars and willows. In: Isebrands JG, Richardson J (eds) Poplars and willows-trees for society and the environment. Oxfordshire, England, pp 258–336
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Research 27:209–220
Migoń P (2006) Geomorfologia [Geomorphology]. Wyd. Nauk, PWN, Warszawa
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L, Estoup A (2004) Geneclass2: a software for genetic assignment and first generation migrants detection. J Hered 95:536–539. https://doi.org/10.1093/jhered/esh074
Pospíšková M, Bartáková I (2004) Genetic diversity of a black poplar population in the Morava river basin assessed by microsatellite analysis. For Genet 11:257–262
Pospíšková M, Šálková I (2006) Population structure and parentage analysis of black poplar along the Morava River. Can J For Res 36:1067–1076. https://doi.org/10.1139/x06-003
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Ramasamy RK, Ramasamy S, Bindroo BB, Naik VG (2014) STRUCTURE PLOT: a program for drawing elegant STRUCTURE bar plots in user friendly interface. Springer Plus 3:431. https://doi.org/10.1186/2193-1801-3-431
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. PNAS 94:9197–9201
Rathmacher G, Niggemann M, Köhnen M, Ziegenhagen B, Bialozyt R (2010) Short distance gene flow in Populus nigra L. accounts for small-scale spatial genetic structures: implications for in situ conservation measures. Conserv Genet 11:1327–1338. https://doi.org/10.1007/s10592-009-9961-6
Rousset F (2008) GENEPOP’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Res 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Rozenfeld AF, Arnaud-Haond S, Hernandez-Garcia E, Eguiluz VM, Serrao EA, Duarte CM (2007) Spectrum of genetic diversity and networks of clonal populations. J R Soc Interface 4:1093–1102. https://doi.org/10.1098/rsif.2007.0230
Šiler B, Skorić M, Mišić D (2014) General considerations of the European black poplar biology, significance and conservation prospects. In: Tomović Z, Vasić M (eds) Variability of European Black Poplar (Populus nigra L.) in the Danube Basin. PE Vojevodinašume, pp 9–51
Slatkin M (1993) Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47:264–279. https://doi.org/10.2307/2410134
Smulders MJM, Van Der Schoot J, Arens P, Vosman B (2001) Trinucleotide repeat microsatellite markers for black poplar (Populus nigra L.). Mol Ecol Notes 1:188–190. https://doi.org/10.1046/j.1471-8278.2001.00071.x
Smulders MJM, van der Schoot J, Ivens B, Strome V, Castiglione S, Grassi F, Bovenschen J, van Dam BC, Vosman B (2002) Clonal propagation in black poplar (Populus nigra). In: Van Dam BC, Bordács S (eds) Proceedings of an International Symposium: Genetic Diversity in River Populations of European Black Poplar. Csiszár Nyomda Ltd Ltd, pp 39–52
Smulders MJM, Beringen R, Volosyanchuk R, Vanden Broeck A, van der Schoot J, Arens P, Vosman B (2008a) Natural hybridisation between Populus nigra L. and P. x canadensis Moench. Hybrid offspring competes for niches along the Rhine river in the Netherlands. Tree Genet Genom 4:663–675. https://doi.org/10.1007/s11295-008-0141-5
Smulders MJM, Cottrell JE, Lefèvre F, van der Schoot J, Arens P, Vosman B, Tabbener HE, Grassi F, Fossati T, Castiglione S, Krystufek V, Fluch S, Burg K, Vornam B, Pohl A, Gebhardt K, Alba N, Agúndez D, Maestro C, Notivol E, Volosyanchuk R, Pospíšková M, Bordács S, Bovenschen J, van Dam BC, Koelewijn HP, Halfmaerten D, Ivens B, van Slycken J, Vanden Broeck A, Storme V, Boerjan W (2008b) Structure of the genetic diversity in black poplar (Populus nigra L.) populations across European river systems: consequences for conservation and restoration. For Ecol Manag 255:1388–1399. https://doi.org/10.1016/j.foreco.2007.10.063
Tylkowski T (2010) Transformation of dendroflora composition in the middle valley of the Warta River. Acta Sci Pol 9:117–124
Van der Schoot J, Pospíšková M, Vosman B, Smulders MJM (2000) Development and characterization of microsatellite markers in black poplar (Populus nigra L.). Theor Appl Genet 101:317–322. https://doi.org/10.1007/s001220051485
Vanden Broeck A, Storme A, Cottrell JE, Boerjan W, Van Bockstaele E, Quataert P, Van Slycken J (2004) Gene flow between cultivated poplars and native black poplar (Populus nigra L.): a case study along the river Meuse on the Dutch-Belgian border. For Ecol Manage 197:307–310. https://doi.org/10.1016/j.foreco.2004.05.021
Vekemans X, Hardy OJ (2004) New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13:921–935. https://doi.org/10.1046/j.1365-294X.2004.02076.x
Waples RS (2005) Genetic estimates of contemporary effective population size: to what time periods do the estimates apply? Mol Ecol 14:3335–3352. https://doi.org/10.1111/j.1365-294X.2005.02673.x
Waples RS, Do C (2008) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Res 8:753–756. https://doi.org/10.1111/j.1755-0998.2007.02061.x
Wójkiewicz B (2020) The genotypic data of black poplar populations from the Oder valley. [dataset]. V1. RepOD repository. https://doi.org/10.18150/EIGSVA.UNF:6:0NHN62QvcpaFQjbRe8mJFQ==%5bfileUNF]
Wójkiewicz B, Żukowska WB, Wachowiak W, Lewandowski A (2019) The genetic assessment of the natural regeneration capacities of black poplar populations in the modern river valley landscapes. For Ecol Manage 448:150–159. https://doi.org/10.1016/j.foreco.2019.06.003
Wojtkowiak B, Wrońska-Pilarek D, Pilarek Z (2013) Dendroflora of the projected reserve "Bełczańskie Starorzecza" of the Odra valley (Dolnośląskie Voivodeship). Acta Sci Pol Silv Colendar Rat Ind Lignar 12:37–50
Ziegenhagen B, Gneuss S, Rathmacher G, Leyer I, Bialozyt R, Heinze B, Liepelt S (2008) A fast and simple genetic survey reveals the spread of poplar hybrids at a natural Elbe river site. Conserv Genet 9:373–379. https://doi.org/10.1007/s10592-007-93494
Acknowledgments
We thank Maria Ratajczak from the Institute of Dendrology, Polish Academy of Sciences for help in DNA isolation.
Funding
This work was financed by the Polish National Science Centre (Grant No. 2016/21/N/NZ9/01515).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Handling Editor: Bruno Fady
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributions of the co-authors Conceptualization: BW and AL; methodology: BW and WW; software: BW; investigation: BW and AL; resources: BW and AL; data curation: BW, WBŻ, and ML; writing—original draft: BW; writing—review and editing: BW, AL, WW, WBŻ, and ML; supervision: WW; project administration: BW; funding acquisition: BW.
Appendix
Appendix

Hybrid identification across the studied populations with the use of STRUCTURE software. All samples are represented by the vertical bars partitioned into segments of length proportional to the likelihood of clustering to one of two genetic clusters. The eight reference individuals included to the analysis are pointed as the population R. Based on the proportion of the membership coefficient of each individual to the inferred clusters (K = 2), among the analyzed black poplar trees 34 hybrids were detected (QI > 90%)

The distribution map of black poplar trees at the studied area located near Kędzierzyn Koźle city in Poland. The red points indicate the location of black poplar trees. The green and yellow squares indicate the location of black poplar trees which share the same genotype. The blue cross indicates the location of the hybrid individual recognized as the cultivar Populus ‘Marilandica’. The location data of each studied tree are available online: https://mygeodata.cloud/map/#9678297-Black-poplar-population-KędzierzynKozle

The distribution map of black poplar trees at the studied area located near Brzeg city in Poland. The red points indicate the location of black poplar trees. Different color squares indicate the location of black poplar trees which share the same genotype. The location data of each studied tree are available online: https://mygeodata.cloud/map/#9679027-Black-poplar-population-Brzeg

The distribution map of black poplar trees at the studied area located in Wrocław city in Poland. The red points indicate the location of black poplar trees. Different color squares indicate the location of black poplar trees which share the same genotype. The location data of each studied tree are available online: https://mygeodata.cloud/map/#9679625-Black-poplar-population-Wrocław

The distribution map of black poplar trees at the studied area located near Brzeg Dolny city in Poland. The red points indicate the location of black poplar trees. Different color squares indicate the location of black poplar trees which share the same genotype. The location data of each studied tree are available online: https://mygeodata.cloud/map/#9680702-Black-poplar-population-BrzegDolny

The distribution map of black poplar trees at the studied area located near Ciechanów city in Poland. The red points indicate the location of black poplar trees. Different color squares indicate the location of black poplar trees which share the same genotype. The location data of each studied tree are available online: https://mygeodata.cloud/map/#2742885-Black-poplar-population-Ciechan%C3%B3w

The distribution map of black poplar trees at the studied area located near Bytom Odrzański city in Poland. The red points indicate the location of black poplar trees. Different color squares indicate the location of black poplar trees which share the same genotype. The blue cross indicates the locations of the hybrid individuals recognized as the cultivar Populus ‘Marilandica’. The location data of each studied tree are available online: https://mygeodata.cloud/map/#9722590-Black-poplar-population-BytomOdrzanski

The distribution map of black poplar trees at the studied area located near Cigacice city in Poland. The red points indicate the location of black poplar trees. Different color squares indicate the location of black poplar trees which share the same genotype. The blue crosses indicate the location of the hybrid individuals recognized as the cultivar Populus ‘Marilandica’, whereas the yellow crosses indicate the location of the hybrid individuals recognized as the cultivar P. ‘Robusta’. The location data of each studied tree are available online: https://mygeodata.cloud/map/#9723423-Black-poplar-population-Cigacice

The distribution map of black poplar trees at the studied area located near Kostrzyn nad Odrą city in Poland. The red points indicate the location of black poplar trees. Different color squares indicate the location of black poplar trees which share the same genotype. The blue crosses indicate the locations of the hybrid individuals recognized as the cultivar Populus ‘Serotina’. The location data of each studied tree are available online: https://mygeodata.cloud/map/#9743889-Black-poplar-population-Kostrzyn-nad-Odr%C4%85

The distribution map of black poplar trees at the studied area located near Gozdowice city in Poland. The red points indicate the location of black poplar trees. Different color squares indicate the location of black poplar trees which share the same genotype. The blue crosses indicate the locations of the hybrid individuals. The location data of each studied tree are available online: https://mygeodata.cloud/map/#9759575-Black-poplar-population-Gazdowice

Genotype accumulation curve describing the genotypic resolution of microsatellites in a data set of studied black poplar trees containing 548 sampling units genotyped using 12 microsatellites. The edges of the boxes show the minimum and maximum number of genotypes and the central line shows the average number of genotypes identified in the sample using X microsatellites

Correlograms of the Spatial Genetic Structure (SGS) analysis. Loiselle’s kinship coefficient (Fij) is plotted for discrete distance classes with respective 95% confidence intervals (dotted lines).

The regression analysis between the genetic distance (FST/1-FST) and the geographic distance (km) performed with the use of the Mantel test
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wójkiewicz, B., Lewandowski, A., Żukowska, W.B. et al. Low effective population size and high spatial genetic structure of black poplar populations from the Oder valley in Poland. Annals of Forest Science 78, 37 (2021). https://doi.org/10.1007/s13595-021-01055-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13595-021-01055-2