
Abstract
Pyoderma gangrenosum (PG) is an uncommon, serious, ulcerating skin disease of uncertain etiology. It manifests as a noninfectious, progressive necrosis of the skin characterized by sterile neutrophilic infiltrates. It seems to be a disorder of the immune system. PG is associated with certain underlying conditions in at least 50% of cases. Therefore, it is important to look carefully for comorbidities in every patient with PG and treat them adequately to improve the prognosis. Here, we demonstrate a 35-year-old man diagnosed with multifocal PG and hemophagocytic lymphohistiocytosis (HLH) with fatal outcome, despite combined, long-term, intensive dermatological and hematological treatment with high doses of steroids, cyclosporin, intravenous immunoglobulins (IVIG), HLH-2004 protocol with intravenously administered etoposide, and anakinra. This case is presented owing to the extremely rare coexistence of PG and HLH and the related diagnostic and therapeutic difficulties. It is also worth underlying that the diagnosis of HLH should perhaps be considered in the presence of a high percentage of double-negative T lymphocytes (DNTs) in flow cytometry, after excluding the diagnosis of lymphoma and leukemia. In this article we have also performed and present the critical literature review of local and systemic options in the management of PG lesions based on a detailed search of the PubMed database.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Pyoderma gangrenosum (PG) is an uncommon, serious, noninfectious, progressive necrosis of the skin of uncertain etiology. |
Hemophagocytic lymphohistiocytosis (HLH) is an aggressive, life-threatening disease characterized by an abnormal immune activation, which leads to excessive inflammation and tissue destruction. |
Both PG and HLH may have a potential etiology, including infection, neoplasm (especially hematological malignancy), autoimmune disease, inflammatory bowel disease, chronic hepatitis, etc. |
Once the diagnosis of HLH or PG is established, it is necessary to identify the potential etiology in order to adopt the appropriate treatment strategy. |
There are no standard therapy options for patients suffering from both PG and HLH. |
Introduction
Pyoderma gangrenosum (PG) is an uncommon, serious, ulcerating skin disease of uncertain etiology. It is a noninfectious, progressive necrosis of the skin, characterized by sterile neutrophilic infiltrates, and appears to be a disorder of the innate immune system. PG is associated with certain underlying conditions in at least 50% of cases [1, 2]. The most common comorbidities are inflammatory bowel diseases, arthritis, and hematological disorders (e.g., myelodysplastic syndrome, monoclonal gammopathy, leukemia, and others). Thus, it is important to look carefully for comorbidities in every patient with PG and treat them adequately. Otherwise, the prognosis for PG may be unfavorable [3, 4]. To date, there are no standardized treatment guidelines for PG. The therapy of PG usually combines local wound care and systemic immunosuppression based on personal experience, availability of therapy, and comorbidities [5,6,7].
Hemophagocytic lymphohistiocytosis (HLH) is an aggressive, life-threatening disease characterized by an abnormal immune activation, which leads to excessive inflammation and tissue destruction. Activated macrophages are not properly downregulated and eliminated by cytotoxic lymphocytes and/or NK cells, which are missing or insufficient [8, 9]. They secrete excessive amounts of cytokines (cytokine storm): interferon gamma, tumor necrosis factor alpha, interleukins (IL), and CD25—the soluble IL-2 receptor, leading to tissue damage, multiorgan failure, and eventually death [10]. The unique feature of HLH is hemophagocytosis of host blood cells by activated macrophages. It is diagnosed during microscopic evaluation of bone marrow, immune tissue (lymph node, spleen, liver), or cerebrospinal fluid, when erythrocytes, leukocytes, platelets, or fragments of these cells are present in the cytoplasm of macrophages. The presence of hemophagocytes in the bone marrow may indicate an infectious background [especially viral: Epstein–Barr virus (EBV) or human immunodeficiency virus (HIV), but also bacteria and parasites may play role], as well as a malignancy (lymphoma, leukemia, or solid tumor) or autoimmune disease. There is also primary HLH, which is usually observed in children and is triggered, among others, by the genetic defect in genes encoding effector proteins of cytotoxic lymphocytes and NK cells [11,12,13]. When HLH is triggered by other disease, treatment of the underlying triggering disease leads to the elimination of stimulus for immune activation. Thus, therapy of patients with secondary HLH should include the treatment of the trigger with simultaneous chemotherapy specific for HLH [14,15,16,17]. Here, we have described a patient who concomitantly developed both PG and HLH, underlining how challenging and difficult the proper treatment choice can be. We have also performed and present the critical literature review of local and systemic options in the management of PG lesions based on a detailed search of the PubMed database.
Case Report
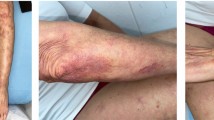
A 35-year-old man was admitted to the department of dermatology presenting plenty of ulcers with purple and undermined edges, located on the upper and lower limbs (Fig. 1a–d; Informed consent for publication, including publication of the images within the article was obtained from the patient). Five months earlier, the patient was hospitalized in another hospital because of significant neutropenia, lymphopenia, thrombocytopenia, hepatosplenomegaly, and skin lesions. At that time, PG was diagnosed clinically. Further diagnostic procedures were performed then and they included bone marrow trephine biopsy, myelogram, colonoscopy, computed tomography (CT) of head, thorax, abdomen, and pelvis, as well as many laboratory and autoimmune tests, but they remain inconclusive. The general condition of the patient was also good and no other symptoms were present. Oral glucocorticosteroid (prednisone 0.8–0.5 mg/kg/day) was administered as the first-line therapy, however, without significant clinical improvement.

Multifocal ulcerations in the course of pyoderma gangrenosum on the lower limbs (a, b) and upper limbs (c, d) on the day of admission. e–h The same lesions 2 weeks after intravenous administration of methylprednisolone followed by prednisone 30 mg and cyclosporin 150 mg twice a day. i–l The same lesions after the second administration of etoposide
At the time of admission to our department, the clinical manifestation of skin lesions was also typical for PG. Additionally, the skin biopsy was performed to exclude other conditions, e.g., cutaneous lymphoma (Fig. 2a–d). Furthermore, the patient still presented peripheral pancytopenia and hepatosplenomegaly. In addition, there were new symptoms such as daily high-spiking fevers of 39–40 °C, transaminitis, significantly elevated level of lactate dehydrogenase (LDH = 969 U/L) and β-2-microglobulin (17.5 mg/L). No lymphadenopathy nor loss of body weight was observed. The detailed screening for malignancies, autoimmune diseases, bowel inflammatory diseases, and infections was performed. CT of the head, thorax, abdomen, and pelvis was taken again, as well as ultrasonography of peripheral lymph nodes, panoramic teeth radiograph, protein electrophoresis, serum immunoglobulin levels, antinuclear antibodies, complement component (C3c and C4) serum levels, liver autoantibodies, serology for viral hepatitis, EBV (anti-VCA/EA IgG, anti-VCA IgM) and HIV, pharyngeal swab, blood culture, urine culture, and gastroenterological consultation. The background of PG and other symptoms remained unidentified.

Histopathology of the ulcer edge: subcorneal abscess formation (a); fibrinoid necrosis of the dermal vessel wall (b); pseudoepitheliomatous hyperplasia (c); infiltrate of chronic inflammatory cells and extravasation of red blood cells (d) [hematoxylin and eosin stain, original magnification: a ×40, b–d ×20]
As a result of serious lymphopenia (247 cells/µL), T cell receptor (TCR) gene rearrangement test was performed, but did not reveal any pathology. However, the immunophenotyping of peripheral blood lymphocytes showed important abnormalities: the presence of a significantly increased percentage of “double-negative” CD3+/CD4−/CD8− T lymphocytes (“double-negative” T cells; DNTs), constituting approximately 50% of the total population of T lymphocytes, and an increased percentage of the CD16+/CD3− NK cell population, constituting approximately 45% of all lymphocytes (Figs. 3 and 4). The presence of residual populations of DNTs and NK cells constituting, respectively, 0.6% and 0.8% of all nucleated cells, was confirmed about 2 months later in the bone marrow. Additionally, it has been observed that cells of granulocytic lineage in the bone marrow showed about 95% expression of CD64 and HLA-DR, which could indicate their increased activation status.

“Double-negative” CD4−/CD8− T lymphocytes* (red), CD4+ helper T lymphocytes (green), CD8+ cytotoxic T lymphocytes (yellow), CD3−/CD16+ NK cells (purple). *Full immunophenotypic profile of “double-negative” T lymphocytes: CD3+ strong/CD4−/CD8−/CD2+/CD5+ dim/CD7+/CD56±/CD57−/CD45RA−/CD45RO−/TCRαβ−/TCRγδ+

CD3−/CD16+ NK cells* (purple), “double-negative” CD4−/CD8− T lymphocytes (red), CD8+ cytotoxic T lymphocytes (yellow), CD4+ helper T lymphocytes (green), B lymphocytes (orange). *Full immunophenotypic profile of NK cells: CD3−/CD16+/CD56+/CD57−/CD45RA+/CD45RO−/CD2+/CD5−/CD7+
In a meantime, the treatment with intravenous methylprednisolone pulses every 4 weeks was started, followed by prednisone 0.5 mg/kg daily and cyclosporin dosed depending on serum drug level. With each pulse, clinical improvement was achieved and lasted no longer than 2 weeks from administration of methylprednisolone (Fig. 1e–h), and then PG progressed slowly again. Topical treatment of PG lesions included hydrofiber silver-impregnated antimicrobial dressing (Aquacel® Ag), hydrogel wound dressing (Purilon® Gel), lipid fat-impregnated sterile dressing (Grassolind® Neutral), and 0.05% betamethasone with 0.1% gentamicin ointment applied on the ulcer edges. Because of the lack of full response, in December 2019 IVIG (2 g/kg) were added to the therapy, again, without therapeutic effect. In addition, the hematological disorder continued to progress. Thus, bone marrow was evaluated both cytomorphologically in aspirate and histopathologically in biopsy. The aspirate showed of coarse toxic granules in granulocytes and numerous hemophagocytes (Fig. 5), present also in the histopathological sample. HLH was suggested and diagnosed quickly in January 2020, according to HLH-2004 diagnostic guidelines [17]. Five out of eight HLH-2004 criteria were confirmed (Fig. 6). HLH-2004 protocol therapy was initiated with etoposide 150 mg/m2 i.v. twice weekly, dexamethasone 10 mg/m2 p.o. daily, cyclosporin depending on its blood levels, and supportive therapy indicated in the aforementioned protocol [12]. The treatment resulted in grade 4 neutropenia (according to CTCAE v.3.0) and serious bacteremia quickly after the second administration of etoposide, and subsequently the patient withdrew his consent to continue etoposide, despite some improvement of skin lesions (Fig. 1i–l).

Hemophagocytes in the patient’s bone marrow. Fragments of phagocytosed cells of the erythroid lineage are present within the macrophage cytoplasm

HLH-2004 diagnostic criteria for hemophagocytic lymphohistiocytosis fulfilled by the patient [17]
Two weeks after discontinuation of etoposide therapy, the progression of both HLH and PG was noted. In March 2020, the rescue treatment with anakinra (100 mg subcutaneously once a day) was started, in addition to previous treatment with steroids and cyclosporin, as its effectiveness was reported in both PG and HLH [18,19,20,21,22,23,24,25]. However, no clinical improvement was observed and the patient’s condition gradually deteriorated. Recurrent thrombocytopenia, neutropenia, and lymphopenia progressed to grade 4 (according to CTCAE v.3.0), resulting in the patient’s death from sepsis with multiorgan failure. The timeline of therapeutic management and outcomes is presented in Table 1.
Discussion and Literature Review
The management of PG remains a challenge as therapeutic decisions need to be personalized, focused on the presence of concomitant diseases, and the specific quantity, location, diameter, and depth of lesions. The topical therapy of PG often does not allow proper control of disease activity. Therefore, the additional use of systemic therapeutic agents is necessary in more severe PG cases.
For the purposes of this article, a detailed search of the PubMed database was carried out, using the following search terms: “pyoderma gangrenosum”, “hemophagocytic lymphohistiocytosis”, “HLH”, “double negative T lymphocytes”, “treatment of pyoderma gangrenosum”, “topical treatment of pyoderma gangrenosum”, and “systemic treatment of pyoderma gangrenosum”. Articles published till January 2021 were selected on the basis of their relevance. The list of suitable articles was amended by manual search of references of identified papers. All studies published in languages other than English were excluded. The most relevant papers on PG treatment involving more than 15 patients are listed in Table 2.
Topical Therapy
The most common topical treatments for PG include calcineurin inhibitors and potent or ultra-potent corticosteroids. According to Marzano et al., monotherapy with tacrolimus could be the first-line treatment of idiopathic, early, localized PG [29]. In a case series of five patients treated with 0.1% tacrolimus ointment, complete clinical remission was achieved within a mean time of 6 weeks [37]. Moreover, in another clinical trial conducted by Lyon et al., 11 patients with peristomal PG were treated with topical tacrolimus 0.3%, whereas 17 patients received topical clobetasol propionate 0.05% [30]. This study suggested that 0.3% tacrolimus might be more effective than clobetasol in managing PG lesions larger than 2 cm in diameter. Importantly, in most case reports, topical tacrolimus was used together with other systemic therapies.
Systemic Therapy
Systemic corticosteroids and cyclosporin, either in monotherapy or combined, are considered first-line agents for PG. The results from a randomized controlled trial comparing oral cyclosporin (4 mg/kg per day) with prednisolone (0.75 mg/kg per day) showed no significant difference between these two regimens [26]. Despite similar adverse reactions, infections were more common in the prednisolone group, while renal impairment and hypertension were frequent in the cohort receiving cyclosporin. Hence, the specific side effect profiles of both drugs should be crucial in making treatment decisions.
In cases of refractory PG, intravenous immunoglobulin therapy (IVIG) can be used simultaneously with systemic steroids [36, 38,39,40]. In a systematic review of 49 patients, 43 (87.8%) achieved a complete or partial response, whereas complete response was observed in 26 (53.1%) of cases. Most patients received a dose of 2 g/kg and the average time to initial response was 3–5 weeks. These findings suggest that clinicians may consider IVIG as adjuvant therapy or a good alternative to cyclosporin and corticosteroids.
The use of other immunosuppressive agents, such as azathioprine, methotrexate, cyclophosphamide, mycophenolate mofetil, or thalidomide have been described in the literature; however, more data are required to evaluate their effectiveness in the management of PG, as currently only individual case series are available [41,42,43,44].
In recent years, many new treatment approaches have been introduced for a subset of patients who only partially respond to conventional immunosuppressive therapy [45]. Although the role of cytokines in pathogenesis of PG is not fully understood, overexpression of tumor necrosis factor alpha (TNFα) is associated with the neutrophil infiltration characteristic of PG; therefore, this group of drugs may be useful. A multicenter study evaluated the efficacy and safety of adalimumab in active ulcers of PG [46]. In this clinical trial, 22 enrolled patients received adalimumab 160 mg at week 0, followed by 80 mg at week 2, and then 40 mg every week starting at week 4. Of this group, seven patients (32%) who achieved Physician Global Assessment (PGA) 0 (all ulcers completely healed) completed the study at week 26, while nine patients (41%) who reached improvement of ulcers with a PGA score of 1–3 at week 26 entered the extension period to receive adalimumab 40 mg every week until week 52. Eighteen patients experienced adverse events, most commonly infections (n = 11), whereas serious adverse events, such as anemia, cataract, bacterial arthritis, and pain due to PG, were reported by four patients. Nevertheless, the results of this study suggest that adalimumab is an effective and generally well-tolerated treatment for patients with PG.
The efficacy of infliximab for the treatment of PG was evaluated in a randomized, double-blind, placebo-controlled trial conducted by Brooklyn et al. [27]. In this study, 30 patients were randomized to receive an infusion of infliximab at 5 mg/kg or placebo at week 0. Thereafter, 13 patients received infliximab, and 17 patients received a placebo. At week 2, the infliximab group achieved better improvement (46%; 6/13) compared to placebo group (6%; 1/17). Subsequently, patients were reassessed, and those who did not respond were offered open-label infliximab at the same dose. At the end of the study, of the 29 patients who received infliximab, 69% (20/29) demonstrated a favorable clinical response.
Although data for the effectiveness of etanercept are limited to case reports and small case series, it appears to be useful in the treatment of recalcitrant and widespread PG [47,48,49,50,51,52,53]. The majority of studies showed clinical improvement or complete resolution with no serious adverse reactions. However, compared to infliximab, etanercept was less effective in the treatment of PG coexisting with active Crohn’s disease. Another novel TNFα inhibitor golimumab could be an interesting treatment option, especially for patients who did not respond to infliximab and adalimumab therapies [54]. In one case report, 24 weeks after the start of the treatment with golimumab, the patient showed complete remission of PG. It is worth noting, that TNFα inhibitors, especially adalimumab and etanercept, may elicit potential paradoxical effects and induce PG lesions. In such cases, the IL-12/23 inhibitor ustekinumab has proved to be beneficial [55], as several reports have demonstrated the complete resolution of resistant PG lesions with this drug. However, further research is required to confirm these results.
In some genetic syndromes such as PAPA (pyogenic arthritis, PG, and acne) mutation in the PSTPIP1 gene is common. In these cases, increased IL-1 production was noted. Therefore, IL-1 inhibitors could be the treatment of choice for this group of patients. Several studies have described the rapid and lasting response to anakinra in patients with PAPA syndrome [20]. Nevertheless, data suggesting its superiority over other biological drugs are still limited. Another monoclonal antibody targeting IL-1β canakinumab was found to be effective in the case of steroid-refractory PG [56]. In this prospective open-label study, five patients received canakinumab subcutaneously in a single dose of 150 mg at weeks 0 and 2, and a dose of 150–300 mg at week 4. Four out of five patients achieved reduction in wound size, as well as improvement of PGA score and the Dermatology Life Quality Index (DLQI). In addition, three out of five patients showed complete remission at week 16.
Peculiarities of Current Case
Among possible therapeutic options for PG, some molecules, such as corticosteroids, cyclosporin, and IVIG, may also be used in treatment of HLH. Simultaneously, the pathogenesis of these diseases is still not fully understood, which makes choosing the optimal treatment modality difficult. Moreover, both PG and HLH can coexist with various systemic disorders, and the requirement for successful therapy is to treat these comorbidities effectively, as well. Here, we demonstrate a case of the concomitance of these two rare diseases, where, despite intensive cooperative treatment, no successful outcome was obtained. A long thorough analysis of the interview and medical documentation also did not lead us to determine which of these conditions developed primarily and which was secondary.
The detection and further research of abnormalities within cell populations will possibly help us to understand mechanisms responsible for occurrence of these two conditions and their symptoms. In our patient we observed an increased NK cell population, constituting approximately 45% of all lymphocytes, while normally in adults they only represent about 10–15% of the total peripheral blood lymphocytes. In HLH, cytotoxic T lymphocytes and NK cells are dysfunctional, which leads to pathological immune stimulation resulting in a significant increase in proinflammatory cytokine concentrations and in reduced or absent NK cell activity. The functioning of T lymphocytes and NK cells may be affected also by genetic disorders, which results in abnormal synthesis of their intracellular cytotoxic granules [57, 58].
Additionally, an increased CD4−/CD8− (TCRγ/δ) DNT population was also revealed, constituting approximately 50% of all peripheral blood T lymphocytes, instead of the normally observed less than 5% [59]. The exact role and function of DNTs within the immune system remains still unclear. A high percentage of these cells is described in infections (both bacterial and viral), inflammatory diseases, post-splenectomy, cellular immunity deficiencies, autoimmune disorders, or lymphoproliferative malignancy [60,61,62]. Reports on immunophenotypic changes accompanying HLH focus mainly on the population of cytotoxic T lymphocytes, while reports on the presence of DNTs in the course of HLH are rare [9]. An increased, similarly to our observation, number of DNTs was reported by Dalal et al. [63], who studied various immunophenotypic abnormalities in adults with HLH. Splenic infiltration of atypical CD4−/CD8− α/β T cells in a 14-year-old boy diagnosed with HLH was reported by Hossny et al. [64].
PG and HLH appear to be disorders of the immune system and share common mechanisms consisting of increased production of cytokines, among others the IL-1 family. It is already well recognized that blocking of IL-1-mediated inflammation by anakinra (human IL-1 receptor antagonist) reduces consequences of tissue damage and organ failure. Anakinra is registered for treatment of rheumatoid arthritis, Still disease, and cryopyrin-associated periodic syndromes; however, it is also frequently used off-label. There are reports about successful anakinra therapy of PG in the context of PAPA syndrome [20], PG in association with hidradenitis suppurativa and acne (PASH syndrome) [21], and PG in association with rheumatoid and psoriatic arthritis [22, 23]. Anakinra was also reported as a useful therapeutic for patients suffering from severe HLH [24, 25]. Therefore, in the absence of standard therapy options, we decided to use anakinra in our patient. However, the ineffectiveness of this treatment may suggest the need to look for another common immunological checkpoint in patients with co-morbid HLH and PG. So far, there are no descriptions of such clinical cases; thus the aim of this case report is to draw attention to the need to search for new therapeutic options for these patients. Recently, monoclonal antibodies (such as emapalumab, a human IgG1 monoclonal antibody against interferon-gamma) and JAK pathway inhibitors (such as ruxolitinib) have been approved and reported to be effective in the treatment of HLH [65,66,67,68,69]. Further clinical trials are ongoing to confirm the effectiveness and safety of these and other biological and small molecule therapies in different groups of patients suffering from HLH. Currently, it seems that the use of biological drugs may be considered in the treatment of HLH patients, especially in patients refractory or intolerant to conventional HLH therapy. However, further controlled trials are warranted.
References
Brooklyn T, Dunnill G, Probert C. Diagnosis and treatment of pyoderma gangrenosum. Br Med J. 2006;333:181–4.
Braswell SF, Kostopoulos TC, Ortega-Loayza AG. Pathophysiology of pyoderma gangrenosum (PG): an updated review. J Am Acad Dermatol. 2015;73:691–8.
DeFilippis EM, Feldman SR, Huang WW. The genetics of pyoderma gangrenosum and implications for treatment: a systematic review. Br J Dermatol. 2015;172:1487–97.
Montagnon CM, Fracica EA, Patel AA, et al. Pyoderma gangrenosum in hematologic malignancies: a systematic review. J Am Acad Dermatol. 2020;82:1346–59.
Goldust M, Hagstrom EL, Rathod D, et al. Diagnosis and novel clinical treatment strategies for pyoderma gangrenosum. Expert Rev Clin Pharmacol. 2020;13:157–61.
Reichrath J, Bens G, Bonowitz A, Tilgen W. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005;53:273–83.
Teagle A, Hargest R. Management of pyoderma gangrenosum. J R Soc Med. 2014;107:228–36.
Jordan MB, Allen CE, Weitzman S, et al. How I treat hemophagocytic lymphohistiocytosis. Blood. 2011;118:4041.
Dalal BI, Vakil AP, Khare NS, et al. Abnormalities of the lymphocyte subsets and their immunophenotype, and their prognostic significance in adult patients with hemophagocytic lymphohistiocytosis. Ann Hematol. 2015;94:1111–7.
Filipovich A, McClain K, Grom A. Histiocytic disorders: recent insights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. 2010;16:S82.
Saxena R, Pati HP, Mahapatra M. Atlas of hematology. New Delhi: Jaypee Brothers Medical Publishers; 2012. 110 p. ISBN 978-93-5025-508-7.
Naeim F, Rao NP, Song SX, Grody WW. Atlas of hematopathology: morphology, immunophenotype, cytogenetics, and molecular approaches, 1st ed. London: Academic; 2013. ISBN 13: 9780123851833, ISBN 10: 0123851831.
Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Program. 2009;2009:127–31.
Lehmberg K, Nichols KE, Henter JI, et al. Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica. 2015;100:997–1004.
Arca M, Fardet L, Galicier L, et al. Prognostic factors of early death in a cohort of 162 adult haemophagocytic syndrome: impact of triggering disease and early treatment with etoposide. Br J Haematol. 2015;168:63–8.
Kleynberg RL, Schiller GJ. Secondary hemophagocytic lymphohistiocytosis in adults: an update on diagnosis and therapy. Clin Adv Hematol Oncol. 2012;10:726–32.
Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31.
Pazyar N, Feily A, Yaghoobi R. An overview of interleukin-1 receptor antagonist, anakinra, in the treatment of cutaneous diseases. Curr Clin Pharmacol. 2012;7:271–5.
Wollina U, Tchernev G. Pyoderma gangrenosum: pathogenetic oriented treatment approaches. Wien Med Wochenschr. 2014;164:263–73.
Brenner M, Ruzicka T, Plewig G, et al. Targeted treatment of pyoderma gangrenosum in PAPA (pyogenic arthritis, pyoderma gangrenosum and acne) syndrome with the recombinant human interleukin-1 receptor antagonist anakinra. Br J Dermatol. 2009;161:1199–201.
Braun-Falco M, Kovnerystyy O, Lohse P, et al. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)—a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. 2012;66:409–15.
Beynon C, Chin MF, Hunasehally P, et al. Successful treatment of autoimmune disease-associated pyoderma gangrenosum with the IL-1 receptor antagonist anakinra: a case series of 3 patients. J Clin Rheumatol. 2017;23:181–3.
Acquitter M, Plantin P, Kupfer I, et al. Anakinra improves pyoderma gangrenosum in psoriatic arthritis: a case report. Ann Intern Med. 2015;163:70–1.
Wohlfarth P, Agis H, Gualdoni GA, et al. Interleukin 1 receptor antagonist anakinra, intravenous immunoglobulin, and corticosteroids in the management of critically ill adult patients with hemophagocytic lymphohistiocytosis. J Intensive Care Med. 2019;34:723–31.
Eloseily EM, Weiser P, Crayne CB, et al. Benefit of anakinra in treating pediatric secondary hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. 2020;72:326–34.
Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958.
Brooklyn TN, Dunnill MGS, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo-controlled trial. Gut. 2006;55:505–9.
Bhat RM, Nandakishore B, Sequeira FF, et al. Pyoderma gangrenosum: An Indian perspective. Clin Exp Dermatol. 2011;36:242–7.
Marzano AV, Trevisan V, Lazzari R, et al. Pyoderna gangrenosum: a study of 21 patients and proposal of a ‘clinicotherapeutic’ classification. J Dermatolog Treat. 2011;22:254–60.
Lyon CC, Stapleton M, Smith AJ, et al. Topical tacrolimus in the management of peristomal pyoderma gangrenosum. J Dermatolog Treat. 2001;12:13–7.
Argüelles-Arias F, Castro-Laria L, Lobatón T, et al. Characteristics and treatment of pyoderma gangrenosum in inflammatory bowel disease. Dig Dis Sci. 2013;58:2949–54.
Adişen E, Erduran F, Gürer MA. Pyoderma gangrenosum: a report of 27 patients. Int J Low Extrem Wounds. 2016;15:148–54.
Funayama Y, Kumagai E, Takahashi KI, Fukushima K, Sasaki I. Early diagnosis and early corticosteroid administration improves healing of peristomal pyoderma gangrenosum in inflammatory bowel disease. Dis Colon Rectum. 2009;52:311–4.
Li J, Kelly R. Treatment of pyoderma gangrenosum with mycophenolate mofetil as a steroid-sparing agent. J Am Acad Dermatol. 2013;69:565–9.
Pereira N, Brites MM, Gonçalo M, Tellechea Ó, Figueiredo A. Pyoderma gangrenosum—a review of 24 cases observed over 10years. Int J Dermatol. 2013;52:938–45.
Song H, Lahood N, Mostaghimi A. Intravenous immunoglobulin as adjunct therapy for refractory pyoderma gangrenosum: systematic review of cases and case series. Br J Dermatol. 2018;178:363–8.
Marzano AV, Trevisan V, Lazzari R, et al. Topical tacrolimus for the treatment of localized, idiopathic, newly diagnosed pyoderma gangrenosum. J Dermatolog Treat. 2010;21:140–3.
Kreuter A, Reich-Schupke S, Stucker M, et al. Intravenous immunoglobulin for pyoderma gangrenosum. Br J Dermatol. 2008;158:856–7.
Cummins DL, Anhalt GJ, Monahan T, Meyerle JH. Treatment of pyoderma gangrenosum with intravenous immunoglobulin. Br J Dermatol. 2007;157:1235–9.
Gupta AK, Shear NH, Sauder DN. Efficacy of human intravenous immune globulin in pyoderma gangrenosum. J Am Acad Dermatol. 1995;32:140–2.
Herberger K, Dissemond J, Hohaus K, et al. Treatment of pyoderma gangrenosum: retrospective multicentre analysis of 121 patients. Br J Dermatol. 2016;175:1070–2.
Alavi A, French LE, Davis MD, et al. Pyoderma gangrenosum: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol. 2017;18:355–72.
Fletcher J, Raed A, Afsaneh A. Recent advances in managing and understanding pyoderma gangrenosum. F1000Res. 2019;8:2092.
Quist SR, Kraas L. Treatment options for pyoderma gangrenosum. J Dtsch Dermatol Ges. 2017;5:34–40.
McKenzie F, Cash D, Gupta A. Biologic and small-molecule medications in the management of pyoderma gangrenosum. J Dermatolog Treat. 2019;30:264–76.
Yamasaki K, Yamanaka K, Zhao Y, et al. Adalimumab in Japanese patients with active ulcers of pyoderma gangrenosum: twenty-six-week phase 3 open-label study. J Dermatol. 2000;47:1383–90.
Charles CA, Leon A, Banta MR, Kirsner RS. Etanercept for the treatment of refractory pyoderma gangrenosum: a brief series. Int J Dermatol. 2007;46:1095–9.
McGowan JW, Johnson CA, Lynn A. Treatment of pyoderma gangrenosum with etanercept. J Drugs Dermatol. 2004;3:441–4.
Pastor N, Betlloch I, Pascual JC, Blanes M, Banuls J, Silvestre JF. Pyoderma gangrenosum treated with anti-TNF alpha therapy (etanercept). Clin Exp Dermatol. 2006;31:152–3.
Roy DB, Conte ET, Cohen DJ. The treatment of pyoderma gangrenosum using etanercept. J Am Acad Dermatol. 2006;54(Suppl 2):S128–34.
Goldenberg G, Jorizzo JL. Use of etanercept in treatment of pyoderma gangrenosum in a patient with autoimmune hepatitis. J Dermatolog Treat. 2005;16:347–9.
Rogge FJ, Pacifico M, Kang N. Treatment of pyoderma gangrenosum with the anti-TNF alpha drug—etanercept. J Plast Reconstr Aesthet Surg. 2008;61:431–3.
Disla E, Quayum B, Cuppari GG, Pancorbo R. Successful use of etanercept in a patient with pyoderma gangrenosum complicating rheumatoid arthritis. J Clin Rheumatol. 2004;10:50–2.
Benzaquen M, Monnier J, Beaussault Y, et al. Pyoderma gangrenosum arising during treatment of psoriasis with adalimumab: effectiveness of ustekinumab. Australas J Dermatol. 2017;58:e270–1.
Diotallevi F, Campanati A, Radi G, et al. Pyoderma gangrenosum successfully treated with golimumab: case report and review of the literature. Dermatol Ther. 2019;32:e12928.
Kolios AG, Maul JT, Meier B, et al. Canakinumab in adults with steroid-refractory pyoderma gangrenosum. Br J Dermatol. 2015;173:1216–23.
McCall CM, Mudali S, Arceci RJ, et al. Flow cytometric findings in hemophagocytic lymphohistiocytosis. Am J Clin Pathol. 2012;137:786–94.
Park MS, Yoo IY, Kim HJ, et al. Flow cytometric analysis of T cells in hemophagocytic lymphohistiocytosis. Ann Lab Med. 2019;39:430–7.
De Rosa SC, Andrus JP, Perfetto SP, et al. Ontogeny of γδ T cells in humans. J Immunol. 2004;172:1637–45.
Pituch-Noworolska A. Immunophenotype of mature hematopoietic cells—primary and secondary immunodeficiences. Post Biol Kom. 2008;35(suppl.24):45–54.
Roden AC, Morice WG, Hanson CA, et al. Immunophenotypic attributes of benign peripheral blood γδ T Cells and conditions associated with their increase. Arch Pathol Lab Med. 2008;132:1774–80.
Prochorec-Sobieszek M. Characteristics of T-cell large granular lymphocyte proliferaitons. J Transf Med. 2009;2:81–135.
Dalal BI, Khare NS, Wang SY, et al. Diverse immunophenotypic abnormalities in adult patients with hemophagocytic lymphohistiocytosis. Blood. 2012;120:3267.
Hossny EM, El-Owaidy RH, Afifi HM, Ahmed HR. Double negative alpha beta T cells in pediatric hemophagocytic syndromes. Egypt J Pediatr Allergy Immunol. 2019;17:13–9.
Vallurupalli M, Berliner N. Emapalumab for the treatment of relapsed/refractory hemophagocytic lymphohistiocytosis. Blood. 2019;134:1783–6.
Cheloff AZ, Al-Samkari H. Emapalumab for the treatment of hemophagocytic lymphohistiocytosis. Drugs Today (Barc). 2020;56:439–46.
Yildiz H, Van Den Neste E, Defour JP, et al. Adult haemophagocytic lymphohistiocytosis: a review. QJM. 2020. https://doi.org/10.1093/qjmed/hcaa011.
Ahmed A, Merrill SA, Alsawah F, et al. Ruxolitinib in adult patients with secondary haemophagocytic lymphohistiocytosis: an open-label, single-centre, pilot trial. Lancet Haematol. 2019;6:e630–7.
Goldsmith SR, Rehman SSU, Shirai CL, et al. Resolution of secondary hemophagocytic lymphohistiocytosis after treatment with the JAK1/2 inhibitor ruxolitinib. Blood Adv. 2019;3:4131–5.
Acknowledgements
Compliance with Ethics Guidelines
Informed consent for publication, including publication of the images within the article, was obtained from the patient.
Funding
No funding or sponsorship was received for this study or publication of this article. No Rapid Service Fee was received by the journal for the publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authors’ Contributions
Aleksandra Opalińska drafted the dermatological side of the whole manuscript regarding pyoderma gangrenosum and the case report. Dominika Kwiatkowska prepared the literature review. Adrian Burdacki performed the flow cytometry analysis, wrote the part about the double negative T lymphocytes, and prepared Figs. 3–5. Mirosław Markiewicz drafted the part of the hematological side of the manuscript regarding hemophagocytic lymphohistiocytosis (HLH), consulted and controlled the therapy of HLH. Dominik Samotij was responsible for the photographs of the skin lesions and supervision of the patient treatment. Marek Dudziński and Jadwiga Niemiec-Dudek played roles in making the diagnosis of HLH and choosing the HLH therapy. Elżbieta Ostańska made a histopathological evaluation and figures. Adam Reich made a concept and design, validated, supervised and edited the whole manuscript, tables and figures. All authors read the entire manuscript and made the appropriate corrections when necessary.
Disclosures
Aleksandra Opalińska, Dominika Kwiatkowska, Adrian Burdacki, Mirosław Markiewicz, Dominik Samotij, Marek Dudziński, Jadwiga Niemiec-Dudek, Elżbieta Ostańska have nothing to disclose. Adam Reich is a member of the journal’s Editorial Board.
Data Availability
All data are reported in the manuscript. No additional data were generated while preparing this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Opalińska, A., Kwiatkowska, D., Burdacki, A. et al. Multifocal Pyoderma Gangrenosum with an Underlying Hemophagocytic Lymphohistiocytosis: Case Report and the Review of the Literature. Dermatol Ther (Heidelb) 11, 1217–1237 (2021). https://doi.org/10.1007/s13555-021-00571-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-021-00571-3