
Abstract
Recent advances in our understanding of the biology of muscle, and how anabolic and catabolic stimuli interact to control muscle mass and function, have led to new interest in the pharmacological treatment of muscle wasting. Loss of muscle occurs as a consequence of several chronic diseases (cachexia) as well as normal aging (sarcopenia). Although many negative regulators [Atrogin-1, muscle ring finger-1, nuclear factor-kappaB (NF-κB), myostatin, etc.] have been proposed to enhance protein degradation during both sarcopenia and cachexia, the adaptation of mediators markedly differs among these conditions. Sarcopenic and cachectic muscles have been demonstrated to be abundant in myostatin- and apoptosis-linked molecules. The ubiquitin–proteasome system (UPS) is activated during many different types of cachexia (cancer cachexia, cardiac heart failure, chronic obstructive pulmonary disease), but not many mediators of the UPS change during sarcopenia. NF-κB signaling is activated in cachectic, but not in sarcopenic, muscle. Some studies have indicated a change of autophagic signaling during both sarcopenia and cachexia, but the adaptation remains to be elucidated. This review provides an overview of the adaptive changes in negative regulators of muscle mass in both sarcopenia and cachexia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Loss of muscle is a serious consequence of many chronic diseases and of aging itself because it leads to weakness, loss of independence, and increased risk of death. Unfortunately, the field suffers from having more definitions than therapies; muscle wasting is an inevitable part of aging, where it is known as sarcopenia [1]. Muscle loss is also common in most organ failure diseases—heart failure, liver or renal failure, chronic obstructive pulmonary disease (COPD)—and in some types of cancer. In such settings, it is known as cachexia [2]. Another term commonly used is wasting, such as AIDS wasting, known in Africa as slim disease [2]. Of course, muscle wasting is also inevitable during starvation.
In cases of simple disuse, such as when there is a cast in place or when a patient is subjected to extended bed rest, the term used is simply muscle atrophy, which is also used to describe focal muscle loss, as seen in a variety of compression syndromes or when there is motor neuron injury or disease. Also, for some inflammatory myopathies, atrophy is used to describe the loss of muscle mass. Common to all these conditions is the fact that the muscle itself is genotypically normal, in contrast to dystrophies in which a genetic mutation, usually but not always in a structural protein, causes an eventual loss of muscle mass due to the degeneration of muscle fibers.
A loss of more than 40% of body cell mass is fatal [3]. Although the treatment of muscle wasting due to starvation is simple in principle, feeding the patient, it can be complicated by metabolic derangements known as refeeding syndrome [4]. However, for muscle wasting due to sarcopenia or cachexia, the only effective treatment has been a combination of adequate dietary protein, energy, and progressive resistance exercise [5]. This approach, although clearly efficacious in clinical trials, has limited effectiveness because of difficulties with expanding exercise to the level of a public health intervention. In particular, the effect of exercise training to sarcopenia and cachexia is described clearly on the recent excellent review by Lenk et al. [6].
A major barrier to the effective management of skeletal muscle wasting is the inadequate understanding of its underlying biological mechanisms. Therefore, in this review, we will concentrate on specific alterations discussed in the current literature that are present in the skeletal muscle of both muscle wasting disorders. In addition, we will focus on the adaptive changes in negative regulators [ubiquitin–proteasome, myostatin, tumor necrosis factor-α (TNF-α), etc.] for muscle mass.
2 The characteristics of sarcopenia and cachexia
2.1 Sarcopenia
Aging is associated with a progressive decline of muscle mass, quality, and strength, a condition known as sarcopenia [7]. The term sarcopenia, coined by I. H. Rosenberg, originates from the Greek words sarx (flesh) and penia (loss). Although this term is applied clinically to denote loss of muscle mass, it is often used to describe both a set of cellular processes (denervation, mitochondrial dysfunction, inflammation, and hormonal changes) and a set of outcomes such as decreased muscle strength, decreased mobility and function [8], increased fatigue, a greater risk of falls [9], and reduced energy needs. Estimates of the prevalence of sarcopenia range from 13% to 24% in adults over 60 years of age to more than 50% in persons aged 80 years and older [8]. Von Haehling et al. [10] have also estimated the prevalence at 5–13% for elderly people aged 60–70 years and 11–50% for those aged 80 years or above. The estimated direct healthcare costs attributable to sarcopenia in the USA in 2000 were US $18.5 billion (US $10.8 billion for men, US $7.7 billion for women), which represented about 1.5% of total healthcare expenditures for that year [11].
Lean muscle mass generally contributes up to ∼50% of total body weight in young adults, but declines with aging to 25% at 75–80 years old [12, 13]. The loss of muscle mass is typically offset by gains in fat mass. The loss of muscle mass is most notable in the lower limb muscle groups, with the cross-sectional area of the vastus lateralis being reduced by as much as 40% between the age of 20 and 80 years [14]. On a muscle fiber level, sarcopenia is characterized by specific type II muscle fiber atrophy, fiber necrosis, and fiber type grouping [14–17]. In elderly men, Verdijk et al. [16] showed a reduction in type II muscle fiber satellite cell content with aging. Although various investigators support such an age-related decrease in the number of satellite cells [16–20], some reports [21, 22] indicate no such change. In contrast, most studies point to an age-dependent reduction in muscle regenerative capacity due to reduced satellite cell proliferation and differentiation.
Several possible mechanisms for age-related muscle atrophy have been described; however, the precise contribution of each is unknown. Age-related muscle loss is a result of reductions in the size and number of muscle fibers [23] possibly due to a multifactorial process that involves physical activity, nutritional intake, oxidative stress, and hormonal changes [10, 24, 25]. The specific contribution of each of these factors is unknown, but there is emerging evidence that the disruption of several positive regulators (Akt and serum response factor) of muscle hypertrophy with age is an important feature in the progression of sarcopenia [26–29]. In contrast, many investigators have failed to demonstrate an age-related enhancement in levels of common negative regulators (Atrogin-1, myostatin, and calpain) in senescent mammalian muscles. Currently available data show that resistance training, myostatin signaling inhibition, the ingestion of amino acids, treatment with testosterone or growth hormone (GH), and CR appears to counteract sarcopenia [27, 28]. Resistance training in combination with amino acid-containing nutrition would be the best candidate to attenuate age-related muscle wasting and weakness.
2.2 Cachexia
In general, cachexia is a complication of diseases such as heart failure, COPD, and cancer. A lack of agreement exists concerning a precise clinical definition of cachexia [30], but cachexia can be defined as a wasting syndrome describing the progressive loss of both adipose and skeletal muscle tissue in concert with severe injury and chronic or end-stage malignant and infectious diseases [31]. In addition to reducing the patient’s quality of life (e.g., strength, endurance, and probably susceptibility to other disease processes), progression of body wasting has poor prognostic implications. Although the loss of muscle mass clearly can be due to tumor-derived factors, their role is complicated by the associated anorexia, the decreased nutrient supply, and the resulting endocrine changes (e.g., insulin deficiency, increased glucocorticoids, TNF-α, and TWEAK) [32].
Cancer cachexia is a wasting that arises during the course of most cancers, most often in incurable patients toward the end stages of life. Although the etiopathogenesis of cancer cachexia is poorly understood, multiple biologic pathways are known to be involved, including tumor-specific proteolysis-inducing factor (PIF) [32]. Cytokines that are important in the initiation of the acute phase response have also been implicated in the genesis of cancer cachexia [33] or the result rather than the cause of cancer cachexia. In particular, TNF-α has been shown to have a direct catabolic effect on skeletal muscle and produces muscle atrophy through the ubiquitin–proteasome system (UPS) [34]. There are currently no agents approved for the treatment of cancer cachexia. However, a number of agents are used off-label to treat the condition, for example, cannabinoids, corticosteroids, and nutritional supplements [32, 35]. However, a significant unmet need prevails in the cancer cachexia market, meaning most patients do not receive any therapy.
Cardiac heart failure (CHF) is a major public health problem in Western countries. Its incident has risen steadily from 0.02 per 1,000 people per year among those aged 25–34 years to 11.6 per 1,000 among those aged 85 years or older. Using data from studies of left ventricular dysfunction, Anker et al. [36] proposed defining cardiac cachexia as a documented non-edematous weight loss of > 6% of the previous normal weight observed over a period of > 6 months. Unlike other types of cachexia, cardiac cachexia is characterized primarily by loss of type I muscle fibers. Interestingly, it can be improved with exercise treatment, which has a major clinical benefit in cardiac cachexia without any material effect on cardiac output per se [37].
Evidence has strongly suggested that hypoxia can induce muscle atrophy. It is known that chronic exposure to high-altitude hypoxia [38] can cause a reduction of muscle fiber size and loss of skeletal muscle mass in mountaineers. COPD is characterized by airflow limitation. During the past 10 years, COPD has emerged as a multi-organ system disease [39]. Besides primary effects in the lung, COPD as a chronic disease has secondary effects on other systems, including the skeletal muscle. Specifically, COPD is characterized by skeletal muscle dysfunction and atrophy [40]. One of the major problems of patients with COPD is exercise intolerance, which in turn leads to muscle wasting. Muscle wasting should be considered a serious complication of COPD that has important implications for survival. Systemic inflammation, i.e., increased muscle wasting, in particular is a serious complication in COPD. COPD patients have a more sedentary lifestyle than normal patients. In one study, COPD patients spent 64% of their time sitting or lying down compared with age-matched healthy individuals who spent more than half of their time walking or standing [39].
3 Negative regulators of skeletal muscle mass
At least four major proteolytic pathways (lysosomal, Ca2+-dependent, caspase-dependent, and ubiquitin/proteasome-dependent) operate in the skeletal muscle and may be altered during aging, thus contributing to sarcopenia. The lysosomal and proteasomal systems lead to an exhaustive degradation of cell proteins into amino acids or small peptides, whereas the Ca2+-dependent and caspase systems can perform only limited proteolysis owing to their restricted specificity.
The endosome–lysosome system is relatively nonselective and mostly involved in the degradation of long-lived proteins [41]. Lysosomal autophagic degradation has been reported to be induced in skeletal muscle by a 6-h nutrient starvation [42], whereas autophagins, a class of cysteine proteases putatively involved in the formation of autophagosomes, are particularly abundant in the skeletal muscle [43]. Recently, autophagy-related genes have been shown to be hyperexpressed during muscle atrophy induced by denervation or fasting [44–46].
The ubiquitin–proteasome system, initially described as relevant to the catabolism of regulatory or damaged proteins, is also involved in bulk protein degradation, at least in the skeletal muscle. In particular, the identification of muscle-specific components of E3 ubiquitin ligases has improved knowledge of the regulation of the ubiquitin–proteasome system in skeletal muscle [47].
The Ca2+-dependent system comprises several cystein proteases called calpains and a physiological inhibitor named calpastatin. Calpains affect only a limited proteolysis on their substrates (i.e., protein kinase C, calcineurin, and titin), resulting in irreversible modifications that lead to changes in activity or to degradation via other proteolytic pathways [48, 49].
Finally, caspases, a family of cysteine protease, are mostly known for their role in the execution of apoptosis. Whether caspases are relevant to the regulation of skeletal muscle mass still remains to be elucidated [50, 51].
3.1 Ubiquitin–proteasome system
The ATP-dependent UPS is essential for regulating protein degradation [52]. In eukaryotic cells, most proteins in the cytosol and nucleus are degraded via the UPS. The degradation of a protein via the UPS involves two steps: (1) tagging of the substrate by covalent attachment of multiple ubiquitin molecules and (2) degradation of the tagged protein by the 26S proteasome complex with the release of a free and reusable ubiquitin [53]. In a variety of conditions such as cancer, diabetes, denervation, uremia, sepsis, disuse, and fasting, skeletal muscles undergo atrophy through the degradation of myofibrillar proteins via the UPS [54]. The fact that ubiquitin and proteasomes have been implicated in many disease states suggests that these inhibitors have therapeutic potential.
Ubiquitin, composed of 76 amino acids, is an 8.45-kDa protein that is highly conserved in nearly all eukaryotes. The ubiquitination of proteins is regulated by at least three enzymes: ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3) [55]. Kwak et al. [56] suggested that the 14-kDa ubiquitin-conjugating enzyme E214K and the ubiquitin ligase E3 are particularly important for the degradation of muscle proteins. Indeed, recent findings assert that muscle atrophy in these conditions shares a common mechanism in the induction of the muscle-specific E3 ubiquitin ligase atrophy gene-1 (Atrogin-1) and muscle ring finger-1 (MuRF-1) [47, 57, 58]. The labeled proteins are then fed into the cells’ “waste disposers,” the so-called proteasomes, where they are chopped into small pieces and destroyed [59].
The proteasome is a highly conserved, intracellular, multi-catalytic proteinase complex [60] which can catalyze the cleavage of peptide bonds on the carboxyl side of basic, acidic, and hydrophobic amino acid residues in both natural peptides and synthetic substrates [61]. The 26S proteasome is composed of a 19S cap-shaped regulatory particle and a 20S catalytic core particle. X-ray crystallography of the 26S proteasome revealed the following: (1) the 19S proteasome is made up of 18 subunits which control the recognition, deubiquitination, and unfolding of protein substrates prior to its translocation into the 20S proteasome [62]; (2) the cylindrically shaped 20S proteasome is composed of two copies of 14 different subunits, arranged as (α1–α7, β1–β7)2 with four stacked rings.
In the skeletal muscle, anabolic signals influence protein synthesis and accumulation by activating phosphatidylinositol-3-kinase (PI3K) which is involved in the Akt-mTOR (mammalian target of rapamycin) signaling pathway leading to protein anabolism [63, 64]. Interestingly, the activation of PI3K is also associated with the phosphorylation, and therefore inactivation, of forkhead box O (FOXO) [58], a transcription factor known to participate in the transcription of Atrogin-1 and MuRF-1.
3.1.1 The UPS in aged muscle
In a variety of conditions such as cancer, diabetes, denervation, uremia, sepsis, disuse, and fasting, skeletal muscles undergo atrophy through the degradation of myofibrillar proteins via the UPS [54, 57]. Recent advances assert that muscle atrophy in these conditions shares a common mechanism in the induction of the muscle-specific E3 ubiquitin ligases Atrogin-1 and MuRF-1 [58, 65, 66]. Only very indirect measurements (small increases in mRNA levels encoding some components of the UPS [67–69] or ubiquitin–conjugate accumulation) in old muscles of rodents or humans suggested a modest activation of this pathway. Atrogin-1 and/or MuRF-1 mRNA levels in aged muscle are reportedly increased [70–72] or unchanged [73, 74] in humans and rats, or decreased in rats [73, 75, 76]. Even when the mRNA expression of these atrogenes increased in sarcopenic muscles, it was very limited (1.5- to 2.5 fold) as compared with other catabolic conditions (10-fold).
Although various findings have been made regarding the mRNA levels of both ubiquitin ligases in aged mammalian muscle, the examination of protein levels in sarcopenic muscles did not support age-related increases in the mRNA of several ubiquitin ligases. For instance, a descriptive analysis using muscle samples (n = 10) by Edström et al. [76] indicated the marked upregulation of phosphorylated Akt and FOXO4 in the gastrocnemius muscle of aged female rats, probably contributing to the downregulation of Atrogin-1 and MuRF-1 mRNA. This result is further supported by the more recent finding of Léger et al. [77] who, using human subjects 70 years old, demonstrated a decrease in nuclear FOXO1 and FOXO3a by 73% and 50%, respectively, although they did not recognize an significant age-dependent change in the expression of Atrogin-1 and MuRF-1 mRNA. Since Goldberg’s group [78] has defined the rapid and transient expression of Atrogin-1 and MuRF-1, the mRNA levels of these atrogenes would not necessarily correspond with those of proteins in various conditions. Furthermore, the major peptidase activities of the proteasome (i.e., the chymotrypsin-like, trypsin-like, and caspase-like activities) were always reduced (as reported in other tissues) [79] or unchanged with aging [70, 79, 80]. In contrast, Altun et al. [72] recently found that the hindlimb muscles of aged (30-month-old) rats contained two to threefold more 26S proteasomes than those of adult (4-month-old) controls. 26S proteasomes purified from muscles of aged and adult (control) rats showed a similar capacity to degrade peptides, proteins, and a ubiquitinated substrate, but differed in the levels of proteasome-associated proteins (e.g., the deubiquitinating enzyme USP14). Although the activities of many other deubiquitinating enzymes were greatly enhanced in the aged muscles, levels of polyubiquitinated proteins were higher than in the adult animals. Further systematic study (more frequent time points) remains to be conducted using several mammalian species because it is unclear whether the UPS contributes to the establishment of sarcopenia. Since no attempt to block sarcopenia-related atrophy and/or loss of muscle fiber in mice and humans has been successful [81], activation of the UPS would contribute little to sarcopenic symptoms.
3.1.2 UPS in skeletal muscle of cachexia
Studies with animal models of cancer cachexia, as well as in cancer patients, suggest that the UPS plays a prominent role in the degradation of myofibrillar proteins, particularly in patients with a weight loss of > 10% [82]. Increased protein degradation by the UPS is implicated in skeletal muscle wasting in cachectic conditions [83, 84]. The expression of mRNA for both α- and β-proteasome subunits was increased in the gastrocnemius muscle of weight-losing mice bearing the MAC16 adenocarcinoma [85]. Several experimental models of cancer cachexia [for example AH-130, colon 26 (C26), Lewis lung carcinoma] reported increased UPS activity as well as the overexpression of both Atrogin-1 and MuRF-1 [86, 87]. The expression of MuRF-1 in muscle after cancer cachexia has recently been shown to be regulated by TNF receptor-associated factor (TRAF) 6-dependent signaling [88]. In CHF, diaphragm weakness is associated with myosin loss and activation of the UPS [89]. In those cachectic states investigated to date, UPS activity is upregulated in the skeletal muscle; the transcript levels for UPS members are also upregulated 8-, 2-, 40-, and 8-fold for ubiquitin, E2 ubiquitin-conjugating enzymes, E3 ubiquitin ligases, and subunits of the 26S proteasome, respectively [83, 90, 91].
3.2 Autophagy-dependent signaling
Autophagy (derived from Greek and meaning “to eat oneself”) occurs in all eukaryotic cells and is evolutionarily conserved from yeast to humans [92]. Autophagy is a ubiquitous catabolic process that involves the bulk degradation of cytoplasmic components through a lysosomal pathway [93–95]. This process is characterized by the engulfment of part of the cytoplasm inside double-membrane vesicles called autophagosomes. Autophagosomes subsequently fuse with lysosomes to form autophagolysosomes in which the cytoplasmic cargo is degraded and the degradation products are recycled for the synthesis of new molecules [96]. The turnover of most long-lived proteins, macromolecules, biological membranes, and whole organelles, including the mitochondria, ribosomes, the endoplasmic reticulum and peroxisomes, is mediated by autophagy [97].
Three major mechanisms of autophagy have been described: (1) microautophagy, which destroys only a small portion of the cytoplasm, proteins, and maybe glycogen in mammals [98]; (2) chaperone-mediated autophagy, in which soluble proteins with a particular pentapeptide motif are recognized and transported across the lysosomal membrane for degradation; and (3) macroautophagy, in which a portion of the cytoplasm, including subcellular organelles, is sequestered within a double-membrane-bound vacuole that ultimately fuses with a lysosome [99].
Noglaska et al. [100] suggested the prominent accumulation of p62/SQSTM1, an autophagy-regulating protein, in the muscle fibers of patients with sporadic myositis. p62/SQSTM1 interacts with the autophagosome membrane microtubule-associated protein light chain (LC3) protein which facilitates the delivery of its polyubiquitinated cargo for lysosomal degradation [101, 102]. Interestingly, that the UPS and the lysosomal–autophagy system in the skeletal muscle are interconnected was suggested by the reports of Mammucari et al. [44] and Zhao et al. [103]. Both studies identified FOXO3 as a regulator of the lysosomal and proteasomal pathways in muscle wasting. FOXO3 is a transcriptional regulator of the ubiquitin ligases MuRF-1 and Atrogin-1. It has now been linked to the expression of autophagy-related genes in skeletal muscle in vivo and C2C12 myotubes [103]. More recently, Masiero et al. [104] found an intriguing characteristic using muscle-specific autophagy knockout mice. The atrophy, weakness, and mitochondrial abnormalities in these mice are also features of sarcopenia.
3.2.1 A possible contribution of autophagic signaling with age
A decline in autophagy during normal aging has been described for invertebrates and higher organisms [105]. Inefficient autophagy has been attributed a major role in the apparent age-related accumulation of damaged cellular components, such as undegradable lysosome-bound lipofuscin, protein aggregates, and damaged mitochondria [106]. Interestingly, the degree of age-related changes in autophagy appears to be organ-specific. While the autophagic activity in liver declines with age [107], one group suggests that autophagy is maintained in the heart of aged rats [108]. Several recent studies have dealt with the changes of autophagy with age in mammalian skeletal muscle [109–112]. Compared with those in young male Fischer 344 rats, amounts of Beclin-1, important for the formation of preautophagosome structures [113], were significantly increased in the plantaris muscles of senescent rats [112]. In contrast, aging did not influence the amounts of Atg7 and Atg9 proteins in rat plantaris muscle [112], although these autophagy-linking proteins possess a crucial role in the formation and expansion of the autophagosome [114]. Indeed, Western blot analysis by Wohlgemuth et al. [112] clearly showed a marked increase in the amount of LC3 in muscle during aging. However, they could not demonstrate an aging-related induction of the ratio of LC3-II to LC3-I, a better biochemical marker to assess ongoing autophagy [114]. In contrast, Wenz et al. [111] recognized a significant increase in both the amount of phosphorylated p65 and in the ratio of LC3-II to LC3-I during aging (3 versus 22 months) in the biceps femoris muscle of wild-type mice. Intriguingly, no significant age-related increase was observed in MCK-PGC-1α (peroxisome proliferator-activated receptor-γ coactivator-1α) mice. Therefore, PGC-1α would attenuate the autophagic process probably through increased antioxidant defense and mitochondrial biogenesis, and then preserved muscle integrity (e.g., protection from age-associated fibrosis). None of the studies determining the transcript level of autophagy-linked molecules found a significant increase with age [109, 110, 112]. Therefore, not all contributors to autophagy signaling seem to change similarly at both the mRNA and protein levels in senescent skeletal muscle.
3.2.2 A possible contribution of autophagic signaling to cachexia
Ryter et al. [115] have described increased autophagy in clinical specimens of the lung from patients with COPD relative to normal tissue, as evidenced by morphological and biochemical markers. This evidence included the electron microscopic evaluation of lung tissue morphology as well as the increased expression and activation of autophagic regulator proteins (i.e., LC3B, Beclin-1, Atg5, Atg7). Only two recent studies have dealt with the changes of autophagy during cachexia in mammalian skeletal muscle. Using biopsy samples, Plant et al. [116] demonstrated that there was no difference in the levels of Beclin-1 and LC3 transcripts in the quadriceps muscle of patients with COPD compared with control individuals. In contrast, endotoxin-induced cachexia seems to include the upregulation of autophagic signaling. A significant increase in the Atg7 and Atg12 proteins was observed in the soleus and white vastus lateralis muscles as early as 8 h after the injection of lipopolysaccharide (1 mg/kg). The amount of Beclin-1 and the ratio of LC3-II to LC3-I also increased in the soleus and white vastus lateralis muscles, respectively. A more recent study [88] demonstrated that the depletion of TRAF6 prevented tumor-induced muscle loss and the upregulation of LC3B and Beclin-1 mRNA expression. Therefore, the activation of autophagy under cachexia seems to be regulated by TRAF6-dependent signaling.
3.3 Myostatin
Growth and differentiation factor 8, otherwise known as myostatin, was first discovered during screening for novel members of the transforming growth factor-β (TGF-β) superfamily and was shown to be a potent negative regulator of muscle growth [117]. Studies indicate that myostatin inhibits cell cycle progression and reduces levels of myogenic regulatory factors, thereby controlling myoblastic proliferation and differentiation during developmental myogenesis [118–120]. Mutations in myostatin can lead to massive hypertrophy and/or hyperplasia in developing animals, as evidenced by knockout experiments in mice. Moreover, mouse skeletal muscle engineered to overexpress the myostatin propeptide, the naturally occurring myostatin inhibitor follistatin, or a dominant-negative form of the Activin IIB receptor (ActRIIB—the main myostatin receptor) [121] all display similar, if not greater, increases in size [121]. Myostatin binds to and signals through a combination of ActRIIA/B receptors on the cell membrane, but has higher affinity for ActRIIB. On binding to ActRIIB, myostatin forms a complex with a second surface type I receptor, either activin receptor-like kinase (ALK) 4 or ALK5, to stimulate the phosphorylation of receptor Smad and the Smad2/3 transcription factors in the cytoplasm. Then, Smad2/3 are translocated and modulate the nuclear transcription of genes such as MyoD [122] via a TGF-β-like mechanism. In contrast, FOXO1 and Smad2 appear to control the differentiation of C2C12 myoblasts by regulating myostatin mRNA and its promoters [122]. More recently, the insulin-like growth factor I (IGF-I)-Akt-mTOR pathway, which mediates both differentiation in myoblasts and hypertrophy in myotubes, has been shown to inhibit myostatin-dependent signaling. Blockade of the Akt-mTOR pathway using siRNA to RAPTOR, a component of TORC1 (TOR signaling complex 1), facilitates myostatin’s inhibition of muscle differentiation because of an increase in Smad2 phosphorylation [123]. In contrast, Smad2/3 inhibition promotes muscle hypertrophy partially dependent on mTOR signaling [124].
Taking notice of the functional role in muscle mass by myostatin, many researchers have conducted experiments on inhibiting myostatin for treating various muscle disorders. The use of neutralizing antibodies to myostatin improved muscle disorders in rodent models of Duchenne muscular dystrophy (mdx), limb girdle muscular dystrophy 2F (Sgcg−/−), and amyotrophic lateral sclerosis (SOD1G93A transgenic mouse) [125–127]. Indeed, myostatin inhibition using MYO-029 was tested in a prospective, randomized, placebo-controlled US phase I/II trial in 116 adults with muscular dystrophy such as Becker muscular dystrophy, facioscapulohumeral muscular dystrophy, and limb girdle muscular dystrophy [128]. Long-term administration of MYO-029 at 1-, 3-, 10-, or 30-mg/kg doses had good safety and tolerability, except for cutaneous hypersensitivity at 10 and 30 mg/kg. No improvements in muscle function were noted, but some subjects had increased muscle fiber size. The study concluded that the systemic administration of myostatin inhibitors was relatively safe and that more effective inhibitor of myostatin signaling should be considered to attenuate muscular dystrophy.
3.3.1 Myostatin signaling in sarcopenia
Myostatin levels increase with muscle atrophy due to neuromuscular inactivity in mice and humans [129–131], and with severe muscle wasting in HIV patients [132]. For example, our recent study [130], using healthy university students, showed significantly increased levels of myostatin mRNA in muscle after the unilateral suspension of a hindlimb for 2 weeks. Together, these studies suggest that increased levels of myostatin lead to muscle wasting. Unfortunately, there are only few studies that have examined the changes of the myostatin level in sarcopenia. These studies have yielded conflicting results such as marked increases in humans at the mRNA and protein levels [77], no change in mice at the protein level [133], and a decrease in rats at the mRNA level [134]. Intriguingly, Carlson et al. [133] showed the enhanced levels of Smad3, but not myostatin, TGF-β2, or follistatin, in sarcopenic muscles of mice. The functional role of myostatin in aged mammalian muscle seems to be revealed by further descriptive analysis using other methods (e.g., immunofluorescence) and examining the adaptive changes in downstream modulators (e.g., ActRIIB, Smad3) of myostatin signaling.
Although it is ambiguous for the functional role in myostatin to sarcopenia, many researchers have investigated the effect of inhibiting myostatin to counteract sarcopenia using animals. A lack of myostatin caused by gene manipulation increased the number of satellite cells and enlarged the cross-sectional area of predominant type IIB/X fibers in tibialis anterior muscles of mice [135]. In addition, these myostatin-null mice showed prominent regenerative potential, including accelerated fiber remodeling after an injection of notexin [135]. Lebrasseur et al. [136] reported several positive effects of 4 weeks of treatment with PF-354 (24 mg/kg), a drug for inhibiting myostatin, in aged mice. They found that PF-354-treated mice exhibited significantly greater muscle mass (by 12%) and increased performance such as treadmill time and distance to exhaustion. Furthermore, PF-354-treated mice exhibited decreased levels of phosphorylated Smad3 and MuRF-1 in aged muscle. More recently, Murphy et al. [137] showed, by way of once-weekly injections, that a lower dose of PF-354 (10 mg/kg) significantly increased the fiber cross-sectional area (by 12%) and in situ muscle force (by 35%) of aged mice (21 months old). In addition, this manner of PF-354 treatment reduced the markers of apoptosis by 56% and reduced caspase3 mRNA by 65%. Myostatin blockade enhances muscle protein synthesis [138] by inhibiting the UPS which is controlled, in part, by Akt [139, 140], although the mechanism has not been apparently demonstrated. In contrast, microarray analysis of the skeletal muscle of Mstn−/− mice showed an increased expression of anti-apoptotic genes compared with control mice [140]. These findings clearly highlight the therapeutic potential of antibody-directed myostatin inhibition for sarcopenia by inhibiting protein degradation and/or apoptosis.
3.3.2 Myostatin signaling in cachexia
Increased signaling by the ActRIIB pathway has been implicated in many cancers [141, 142]. The administration of myostatin in vivo to adult mice induces profound muscle loss analogous to that seen in human cachexia syndromes [143]. In vitro studies showed that myostatin-induced cachexia caused the activation of the UPS through a FOXO1-dependent signaling [139]. Myostatin induction was also recognized in an animal model of cardiac cachexia such as the peri-infact zone in sheep [144] and a rat model of volume overload induced by an aortocaval shunt [145].
As support to these findings, Heineke et al. [146] demonstrated that cardiomyocyte-specific overexpression of myostatin significantly reduced both cardiac and skeletal muscle mass. Furthermore, several recent reports suggested that the upregulation of myostatin is involved in the development of muscle atrophy associated with COPD [116, 147]. For example, Hayot et al. [148] found an increased skeletal muscle myostatin expression in hypoxemic COPD patients (PaO2 = 64 ± 2 mm Hg) compared with that in healthy controls. Therefore, abundant myostatin protein induces muscle atrophy in various different cachexias.
The attenuation of myostatin-dependent signaling markedly improves the muscle wasting of cachexia. Skeletal muscle atrophy in heart failure was markedly prevented by myostatin deletion from the heart but not the skeletal muscle [148]. More recently, Zhou et al. [149] indicated the therapeutic potential of blocking the myostatin receptor, ActRIIB, on cancer cachexia. They utilized various models of cancer cachexia models, including C26 tumor-bearing mice and inhibin-deficient mice, as well as nude mice bearing human G361 melanoma and TOV-21G ovarian carcinoma xenografts. Blockade of the ActRIIB pathway using the decoy receptor (sActRIIB) completely reversed the pathology and muscle atrophy caused by cancer cachexia. Treatment with sActRIIB abolished the activation of the UPS and the induction of atrophy-specific ubiquitin ligases, probably due to the decreased phospho-Smad2 and the increased inactivated (phospho)-FOXO3 and phospho-Akt proteins in cachectic muscles. Klimek et al. [150] also showed that the use of a soluble ActRIIB receptor increased mass by between 23% and 40%. ActRIIB-Fc is not specific for myostatin and known ActRIIB ligands include growth and differentiation factor-11 and the activins [151]. Thus, the protective effect of ActRIIB-Fc may be due to the inhibition of these other targets, alone or with myostatin. Furthermore, Heineke et al. [146] observed a therapeutic effect of an anti-myostatin antibody (JA-16) to the atrophy of several skeletal muscles occurring after cardiac cachexia caused by transverse aortic constriction.
3.4 TNF-α and NF-κB signaling in sarcopenia
Inflammation may negatively influence skeletal muscle through direct catabolic effects or through indirect mechanisms (i.e., decreases in GH and IGF-I concentrations, induction of anorexia, etc.) [152]. There is growing evidence that higher levels of inflammatory markers are associated with physical decline in older persons, possibly through the catabolic effects of inflammatory markers on muscle. In an observational study of more than 2,000 men and women, TNF-α showed a consistent association with declines in muscle mass and strength [153]. The impact of inflammation on the development of sarcopenia is further supported by a recently published animal study showing that a reduction of low-grade inflammation by ibuprofen in old (20 months) animals resulted in a significant decrease in muscle mass loss [154]. An age-related disruption in the intracellular redox balance appears to be a primary causal factor of a chronic state of low-grade inflammation. More recently, Chung et al. [155] hypothesized that abundant NF-κB protein induced age-related increases in interleukin (IL)-6 and TNF-α. Moreover, reactive oxygen species (ROS) also appear to function as second messengers for TNF-α in skeletal muscle, activating NF-κB either directly or indirectly [156]. Indeed, a marked production of ROS has been documented in muscle of the elderly [157, 158]. However, it is not clear whether NF-κB signaling is enhanced with age. Despite some evidence supporting enhanced NF-κB signaling in type I fibers of aged skeletal muscle, direct evidence for an increased activation and DNA binding of NF-κB is lacking [159, 160]. For example, Phillips and Leeuwenburgh [160] found that neither p65 protein expression nor the binding activity of NF-κB was significantly altered in the vastus lateralis muscles of 26-month-old rats despite the marked upregulation of TNF-α expression in both blood and muscle. Upregulated TNF-α expression in serum and muscle seems to enhance apoptosis in the mitochondria, resulting in the loss of muscle fibers [160–162]. It has been shown that TNF-α is one of the primary signals inducing apoptosis in muscle.
3.4.1 PIF, TNF-α, and NF-κB in cachectic muscle
TNF-α, IL-1, IL-6, and interferon (IFN)-γ have been implicated in cancer-related muscle wasting [163]. For example, the transplantation of Chinese hamster ovary cells transfected with the human TNF-α gene produced a syndrome resembling cachexia, with progressive wasting, anorexia, and early death [164]. Treatment with TNF-α disrupted the regeneration of skeletal muscle because of a marked delay in the expression of muscle differentiation markers such as myogenin and neonatal myosin heavy chain [165]. TNF-α, either on its own [166] or in combination with IFN-r [167], causes the reduced expression of myosin heavy chain in cultured myotubes or whole muscle possibly due to the MyoD-stabilizing, RNA-binding protein HuR [168]. The enhanced production of nitric oxide (NO) synthase by TNF-α/IFN-r led to HuR moving away from the MyoD message, thereby causing MyoD mRNA decay. In fact, the atrophic muscles in a rodent model of cancer cachexia possessed low levels of MyoD [169]. The NO induction by TNF-α/IFN-α in cachectic muscles seems to be mediated by NF-κB-dependent signaling [168]. Several studies of cachexia have indicated enhanced NF-κB signaling in skeletal muscle [170, 171], different from sarcopenic muscle. For example, NF-κB DNA binding and transactivation potential have been demonstrated in atrophic muscle bearing C26 tumors [172, 173]. In addition, a study using an injected decoy oligonucleotide to NF-κB further supports the role of this transcription factor in tumor-regulated muscle wasting [173]. Furthermore, MuRF-1, a downstream mediator of the IKKβ/NF-κB pathway, is more abundant in cachectic muscle [65, 174, 175]. In contrast, TNF-α, IFN-r, and IL-1β, known to be increased in cachectic patients, are potent activators of inducible NO synthase expression [176], which in turn produces toxic levels of NO high enough to inhibit the key enzymes of oxidative phosphorylation. More recent findings showed cachexia-induced selective atrophy of glycolytic muscles probably due to small amounts of catalase and superoxide dismutase to inhibit oxidative stress [177].
PIF is a 24-kDa sulfated glycoprotein originally isolated from cachexia-inducing MAC16 tumor cells using an antibody cloned from splenocytes of mice bearing the same tumor, but with a delayed cachexia [178]. This substance was evident in the urine of weight-losing patients with a variety of tumor types, including pancreatic, lung, breast, ovary, rectum, colon, and liver cancer, but absent in urine from patients without weight loss [179]. Unlike TNF-α, it occurred without a depression of either food or water intake and was a result of the specific depletion of lean body mass [178]. There were specific reductions in the weight of several skeletal muscles, but not the heart or kidney. Aside from TNF-α and IL-1β, recent developments indicate that NF-κB is also under the control of PIF [180]. Wyke and Tisdale [181] showed that the activation of NF-κB in C2C12 myotubes occurred through the phosphorylation and degradation of IκB (inhibitor of NFκB)α, leading to the nuclear translocation of the p50/p65 heterodimer. Although this activation is relatively modest in comparison with cytokine stimulation, it is sufficient to induce an ATP-dependent proteasome activity because addition of the IκBSR transdominant inhibitor of NF-κB inhibits the proteasome in response to PIF treatment.
3.5 Apoptotic signaling in sarcopenic muscle
Several reports indicate that apoptosis is enhanced in aged skeletal muscle, likely contributing to the development of sarcopenia [182]. Apoptosis is an evolutionary conserved process of programmed cell death, which is performed via a systematic set of morphological and biochemical events, resulting in cellular self-destruction without inflammation or damage to the surrounding tissue. The execution of apoptosis in skeletal muscle displays unique features due to the multinucleated nature of myofibers. Therefore, apoptosis in myocytes may result in the elimination of individual myonuclei and the surrounding portion of sarcoplasm, without the dismantling of the entire fiber [183].
The mitochondria play an important part in apoptosis by activating caspases (cystein-dependent, aspartate-specific proteases) [184, 185]. Mitochondrial dysfunction can lead to mitochondrial cytochrome c release [185]. In the cytoplasm, cytochrome c, apoptotic protease activating factor-1 , caspase-9, and dATP form an apoptosome which can activate caspase-3, a key cell death protease. Importantly, in the white gastrocnemius and soleus muscles of Fischer 344 rats, it was recently demonstrated that aging significantly increased DNA fragmentation, and the amounts of cleaved caspase-3 and pro-apoptotic Bax, and decreased anti-apoptotic Bcl-2 protein content [186]. In addition, several researchers [186, 187] have suggested that the mitochondrial permeability transition pore could induce the release of cytochrome c. Mitochondria are essential for proper cellular functioning and viability, being the main site for energy production and playing an essential role in the maintenance of redox homeostasis [188]. MtDNA is especially prone to oxidative damage [189], particularly in aged mammalian muscles [190, 191], due to its proximity to the electron transport chain, the lack of protective histones, and a less efficient repair system compared with nuclear DNA [192].
Aside from mitochondria-mediated apoptosis, other pathways of myonuclear apoptosis may be involved at an advanced age. In particular, the death receptor-mediated pathway, triggered by TNF-α, appears to be upregulated in aged rodent muscles [160, 182]. Recent findings suggest that apoptotic signaling is required for and precedes protein degradation during muscle atrophy [193]. Although a clear mechanistic link between myonuclear apoptosis and sarcopenia is yet to be established, data from animal models suggest that this link exists [161, 194]. Moreover, other rodent studies indicate an association between myocyte apoptosis and declines in muscle mass and strength [184]. Despite that most studies with animal models appear to support a key role for apoptosis in age-related muscle atrophy, evidence in humans is still scarce. A recent report indicated increases in the expression of the mitochondrial caspase-independent mediator apoptosis-inducing factor in the semitendinousus muscle of middle-aged men relative to younger controls [195]. This finding is consistent with previously described data from animal experiments and supports the involvement of mitochondria-driven apoptosis in the development of sarcopenia.
3.5.1 Apoptotic signaling in cachectic muscle
Busquets et al. [196] have shown that, together with myofibrillar protein loss, skeletal muscle of cachectic tumor-bearing animals is subject to an important activation of DNA fragmentation and, therefore, apoptosis. Interestingly, they have found that different anti-cachectic treatments resulted not only in a restoration of skeletal muscle protein content but also in an amelioration of apoptosis [197]. For instance, formoterol (a β2-adrenergic agent) treatment in tumor-bearing animals resulted in a dramatic reduction of DNA fragmentation in skeletal muscle.
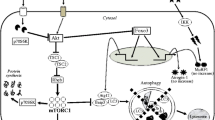
Several reports suggest that certain molecules involved in signaling pathways that result in apoptosis can promote or inhibit NF-κB activation in particular cell lines. Caspase-8, Fas-associated death domain, caspase-8-like inhibitory protein (cFLIP), TNF-α-related apoptosis-inducing ligand receptors, and TNF-R1 activate NF-κB in a variety of cells using a TRAF2/NF-κB-inducing kinase/IKK-dependent pathway [197–199] that can be blocked by their inhibitors or dominant-negative mutants [198, 200]. Cellular cFLIP is a caspase-8 homologue devoid of protease activity and a catalytically inactive caspase-like domain. cFLIP inhibits procaspase-8 processing at the DISC in most cells, thus inhibiting also the apoptosis induced by cancer cells [201]. Jiang and Clemens [201] studied the effect of secreted factors (e.g., TNF-α) from different cancer cell lines on myoblast differentiation in vitro. In their study, cFLIP overexpression inhibited IL-1β or PC-3 or Mel media-induced NF-κB activation in myoblast cells, thus promoting the myogenic differentiation of myoblasts. The stable expression of cFLIP yielded results similar to the stable expression of IκBSR, a mutated form of IκBα that prevents the activation of NF-κB. In contrast, the overexpression of Bcl-xL enhanced NF-κB activation in myoblast cells, with a further increase upon exposure to conditioned medium from PC-3 cells. Bcl-xL is another anti-apoptotic protein and is localized to the endoplasmic reticulum so as to prevent the release of cytochrome c from the mitochondria [202]. Although several studies have shown that both cFLIP and Bcl-xL influence NF-κB activation and cell survival in a variety of cell lines [197, 200], the two may play a role in apoptosis via a different pathway, at least in muscle cell lines [201]. Figure 1 provides an overview of the adaptive changes in negative regulators for muscle mass in sarcopenia and cachexia.

Myostatin signals through the activin receptor IIB (Actr2b) ALK4/5 heterodimer activate Smad2/3 with the consequent nuclear translocation of Smad4 and blocking of MyoD transactivation in an autoregulatory feedback loop. In both sarcopenic and cachectic muscles, abundant activated Smad2/3 inhibit protein synthesis, probably due to blocking of the functional role of Akt. In sarcopenic muscle, Akt can block the nuclear translocation of FOXO to inhibit the expression of Atrogin-1. In contrast, the impaired regulation of FOXO by Akt results in the abundant expression of Atrogin-1 and the consequent protein degradation in cachectic muscle. Although the blood and muscle levels of TNF-α were increased in both muscle wasting, the elevation of NF-κB occurs in cachectic but not sarcopenic muscles. Both muscle wasting include the enhancement of apoptosis signaling. IGF-I insulin-like growth factor I, IGF-IR IGF-I receptor, PI3-K phosphatidylinositol 3-kinase, TORC1 component of TOR signaling complex 1, Rheb Ras homolog enriched in brain, mTOR mammalian target of rapamycin, eIF4E eukaryotic initiation factor 4E, FOXO forkhead box O, IKK inhibitor of κB kinase, NF-κB nuclear factor of kappa-B, MuRF1 muscle ring finger protein 1, TNF tumor necrosis factor
3.6 Anabolic hormones in sarcopenic muscle
Among hormonal changes that might be related to aging, a primary role is likely had by the aging-related deficit of anabolic hormones, promoting a milieu that favors catabolism. This aging-related deficiency in the anabolic hormone milieu takes several different forms, being relatively sudden and dramatic in the case of estrogen in women, but more gradual and steady for testosterone in men, and GH in both genders [203–207]. Indeed, circulating GH levels decline progressively after 30 years of age at a rate of ∼1% per year [203]. In addition, there is evidence that the age-associated decline in GH levels in combination with lower IGF-I levels contributes to the development of sarcopenia [208].
IGF-I is perhaps the most important mediator of muscle growth and repair [209] possibly by utilizing Akt-mTOR-p70S6K signaling. Although the transgenic approach of upregulating IGF-I expression in skeletal muscle would be appropriate for inhibiting sarcopenia, the administration of IGF-I to the elderly has resulted in controversial findings on muscle strength and function [210]. GH treatment in elderly subjects has also yielded conflicting results [27, 211]. The ineffectiveness may be attributable to age-related insulin resistance to amino acid transport and protein synthesis [212] or a marked decrease in IGF-I receptors [213, 214] and receptor affinity for IGF-I [215] in muscle with age. In addition, reduced mRNA levels of the GH receptor in skeletal muscle have been observed in older versus younger healthy men, exhibiting a significant negative relationship with myostatin levels [216]. Wilkes et al. [217] demonstrated a reduced effect of insulin on protein breakdown in the legs in older versus younger subjects, probably due to the blunted activation of Akt by insulin. More comprehensive reviews on insulin resistance in sarcopenia can be found elsewhere [212].
Numerous studies of treatment with testosterone in the elderly have been performed over the past few years [218–221]. Systemic reviews of the literature [222] have concluded that testosterone supplementation attenuates several sarcopenic symptoms in humans, including the decrease in muscle mass [219–221] and grip strength [218]. For instance, a recent study of 6 months of testosterone supplementation in a randomized placebo-controlled trial reported increased leg lean body mass and leg and arm strength [223]. Although the mechanisms by which testosterone increases skeletal muscle mass are poorly understood, several modulators such as Notch-1, the androgen receptor, and IGF-I acting via testosterone-mediated signaling have been identified [224].
3.6.1 Anabolic hormones in cachectic muscle
Low concentrations of testosterone and anabolic hormones are major contributors to cachexia-related wasting of skeletal muscle. Up to 50% of men with metastatic cancer prior to chemotherapy present with low testosterone levels [225]. A reduction in testosterone might lead to a reduced muscle strength and sexual function in both men and women [226]. In addition, reduced levels of anabolic hormones, such as testosterone and IGF-I, have been observed in COPD patients [227–229]. Furthermore, studies in patients with advanced heart failure and cardiac cachexia have revealed a possible involvement of GH and IGF-I in muscle catabolism. Elevated levels of GH with inappropriate serum levels of IGF-I have been described in cardiac cachexia [230]. Indeed, low levels of systematic IGF-I have been associated with decreased leg muscle cross-sectional area and strength in CHF patients [231]. In contrast, catabolic syndromes involving chronic inflammation, sepsis, or cancer show an altered GH/IGF-I axis most probably due to a peripheral IGF-I deficiency because of an impaired IGF-I response to GH [232]. Pro-inflammatory cytokines such as TNF-α uncouple the GH/IGF-I axis through suppression of the GH receptor [233].
Studies on the use of testosterone and testosterone derivatives in cachexia patients have been limited largely to cases of COPD and HIV/AIDS, where positive effects on body weight, lean body mass, and some functional parameters have been documented [234–236]. The long experience with testosterone use makes this hormone a therapeutic candidate for use in cachectic patients. The limited number of trials that have investigated the anabolic effect of testosterone in cachectic subjects demonstrates a generally favorable effect on body weight, lean body mass, and muscle strength with an acceptable safety profile [234–236].
In a pilot study of malnourished patients with COPD by Pape et al. [237], subcutaneous GH injections (0.05 mg/kg daily) caused substantial weight gain and improved maximal inspiratory pressure. Although Burdet et al. [238] also found a significant increase in free fatty mass after 3 weeks of treatment with GH, none of the other parameters (handgrip strength, etc.) improved. Furthermore, in several trials, GH treatment failed to improve the clinical status of CHF patients [239, 240]. A recombinant GH has been approved by the U.S. Food and Drug Administration for the treatment of HIV/AIDS wasting or pediatric chronic kidney disease. Indeed, in several studies in cachectic patients with HIV, recombinant GH has significantly increased lean body mass, physical endurance, and quality of life [241]. However, the effect is not very large and treatment of the underlying disease with highly effective antiretroviral agents has largely eliminated AIDS wasting as a clinical problem in developed countries.
An indirect way in which GH secretion increases is via GH secretagogues [242]. The prototype, ghrelin, is a 28-amino acid peptide mainly produced by cells in the stomach, intestines, and hypothalamus [243]. When released by neuroendocrine cells of the hypothalamus, it binds to receptors on somatotropes and causes the release of GH into the circulation. In contrast, ghrelin inhibits the production of anorectic pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α [244]. In addition, ghrelin increases hunger by acting on hypothalamic feeding centers and stimulating gastric emptying, which facilitates increased food intake [245]. Because of their combined anabolic effects on skeletal muscle and appetite, ghrelin and low-molecular-weight agonists of the ghrelin receptor are considered attractive candidates for the treatment of cachexia [242].
Although glucocorticoids are useful adjuvants in the treatment of cachexia because of their beneficial effects on symptoms such as appetite, food intake, and a sensation of well-being, their use should be confined to the end stage of the disease, and limited to a few weeks, because of their ability to induce atrophy of the skeletal muscle, which primarily affects type II muscle fibers. Glucocorticoids may play a role in the development of cancer-related cachexia [246], although adrenalectomy has been shown not to alter the course of cachexia in other animal models [247]. The effect of glucocorticoids on muscle atrophy is mediated by upregulation of the UPS [248] and intramuscular myostatin expression. Intriguingly, deletion of the myostatin gene prevented glucocorticoid-induced muscle atrophy [249].
Abbreviations
- ActRIIB:
-
Activin type IIB receptor
- ALK:
-
Activin receptor-like kinase
- cFLIP:
-
Caspase-8-like inhibitory protein
- CHF:
-
Chronic heart failure
- COPD:
-
Chronic obstructive pulmonary disease
- CR:
-
Caloric restriction
- FOXO:
-
Forkhead box O
- GH:
-
Growth hormone
- IFN:
-
Interferon
- IGF-I:
-
Insulin-like growth factor I
- IκB:
-
Inhibitor of NFκB
- IL:
-
Interleukin
- LC3:
-
Microtubule-associated protein light chain
- mTOR:
-
Mammalian target of rapamycin
- MuRF-1:
-
Muscle ring finger-1
- NF-κB:
-
Nuclear factor-kappaB
- NO:
-
Nitric oxide
- PGC-1α:
-
Peroxisome proliferator-activated receptor-γ coactivator 1α
- PIF:
-
Proteolysis-inducing factor
- PI3K:
-
Phosphatidylinositol-3-kinase
- ROS:
-
Reactive oxygen species
- TGF-β:
-
Transforming growth factor-β
- TNF-α:
-
Tumor necrosis factor-α
- TORC1:
-
TOR signaling complex 1
- TWEAK:
-
TNF-like weak promoter of apoptosis
- TRAF:
-
TNF receptor-associated factor
- UPS:
-
Ubiquitin–proteasome system
- Atrogin-1:
-
Atrophy gene-1
References
Rosenberg I. Summary comments. Am J Clin Nutr. 1989;50:1231–3.
Roubenoff R, Heymsfield SB, Kehayias JJ, Cannon JG, Rosenberg IH. Standardization of nomenclature of body composition in weight loss. Am J Clin Nutr. 1997;66:192–6.
Roubenoff R, Kehayias J. The meaning and measurement of lean body mass. Nutr Rev. 1991;46:163–75.
Keys A, Brozek J, Henschel A, Mickelsen O, Taylor HL. The biology of human starvation. Minneapolis: University of Minnesota Press; 1950.
Hughes V, Roubenoff R. Sarcopenia: current concepts. J Gerontol Med Sci. 2000;55A:M716–24.
Lenk K, Schuler G, Adams V. Skeletal muscle wasting in cachexia and sarcopenia: molecular pathophysiology and impact of exercise training. J Cachexia Sarcopenia Muscle. 2010;1:9–21.
Candow DG, Chilibeck PD. Differences in size, strength, and power of upper and lower body muscle groups in young and older men. J Gerontol A Biol Sci Med Sci. 2005;60:148–56.
Melton 3rd LJ, Khosla S, Crowson CS, O’Fallon WM, Riggs BL. Epidemiology of sarcopenia. J Am Geriat Soc. 2000;48:625–30.
von Haehling S, Morley JE, Anker SD. An overview of sarcopenia: facts and numbers on prevalence and clinical impact. J Cachexia Sarcopenia Muscle. 2010;1:129–33.
Baumgartner RN, Waters DL, Gallagher D, Morley JE, Garry PJ. Predictors of skeletal muscle mass in elderly men and women. Mech Ageing Dev. 1999;107:123–36.
Janssen I, Shepard DS, Katzmarzyk PT, Roubenoff R. The healthcare costs of sarcopenia in the United States. J Am Geriatr Soc. 2004;52:80–5.
Short KR, Nair KS. The effect of age on protein metabolism. Curr Opin Clin Nutr Metab Care. 2000;3:39–44.
Short KR, Vittone JL, Bigelow JL, Proctor DN, Nair KS. Age and aerobic exercise training effects on whole body and muscle protein metabolism. Am J Physiol Endocrinol Metab. 2004;286:E92–E101.
Lexell J. Human aging, muscle mass, and fiber type composition. J Gerontol A Biol Sci Med Sci. 1995;50:11–6.
Larsson L. Morphological and functional characteristics of the ageing skeletal muscle in man. A cross-sectional study. Acta Physiol Scand Suppl. 1978;457:1–36.
Verdijk LB, Koopman R, Schaart G, Meijer K, Savelberg HH, van Loon LJ. Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am J Physiol Endocrinol Metab. 2007;292:E151–7.
Verdijk LB, Gleeson BG, Jonkers RAM, Meijer K, Savelberg HHCM, Dendale P, et al. Skeletal muscle hypertrophy following resistance training is accompanied by a fiber type-specific increase in satellite cell content in elderly men. J Gerontol A Biol Sci Med Sci. 2009;64:332–9.
Brack AS, Bildsoe H, Hughes SM. Evidence that satellite cell decrement contributes to preferential decline in nuclear number from large fibres during murine age-related muscle atrophy. J Cell Sci. 2005;118:4813–21.
Collins CA, Zammit PS, Ruiz AP, Morgan JE, Partridge TA. A population of myogenic stem cells that survives skeletal muscle aging. Stem Cells. 2007;25:885–94.
Day K, Shefer G, Shearer A, Yablonka-Reuveni Z. The depletion of skeletal muscle satellite cells with age is concomitant with reduced capacity of single progenitors to produce reserve progeny. Dev Biol. 2010;340:330–43.
Conboy IM, Conboy MJ, Smythe GM, Rando TA. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302:1575–7.
Wagners AJ, Conboy IM. Cellular and molecular signatures of muscle regeneration: current concepts and controversies in adult myogenesis. Cell. 2005;122:659–67.
Lexell J. Ageing and human muscle: observations from Sweden. Can J Appl Physiol. 1993;18:2–18.
Roubenoff R, Hughes VA. Sarcopenia: current concepts. J Gerontol A Biol Sci Med Sci. 2000;55:M716–24.
Scott D, Blizzard L, Fell J, Jones G. The epidemiology of sarcopenia in community living older adults: what role does lifestyle play? J Cachexia Sarcopenia Muscle. 2010;2:125–34.
Sakuma K, Akiho M, Nakashima H, Akima H, Yasuhara M. Age-related reductions in expression of serum response factor and myocardin-related transcription factor A in mouse skeletal muscles. Biochim Biophys Acta Mol Basis Dis. 2008;1782:453–61.
Sakuma K, Yamaguchi A. Molecular mechanisms in aging and current strategies to counteract sarcopenia. Curr Aging Sci. 2010;3:90–101.
Sakuma K, Yamaguchi A (2011) Sarcopenia: molecular mechanisms and current therapeutic strategy. In: Perloft JW, Wong AH (eds) Cell aging. Nova Science, New York (in press)
Thomson DM, Gordon SE. Impaired overload-induced muscle growth is associated with diminished translational signaling in aged rat fast-twitch skeletal muscle. J Physiol. 2006;574:291–305.
Dahele M, Fearon KC. Research methodology: cancer cachexia syndrome. Palliat Med. 2004;84:209–38.
Anker SD, Sharma R. The syndrome of cardiac cachexia. Int J Cardiol. 2002;85:51–66.
Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev. 2009;89:381–410.
Deans C, Wigmore SJ. Systemic inflammation, cachexia and prognosis in patients with cancer. Curr Opin Clin Nurs Metal Care. 2005;8:265–9.
Mitch WE, Price SR. Transcription factors and muscle cachexia: is there a therapeutic target? Lancet. 2001;357:734–5.
Kumar NB, Kazi A, Smith T, Crocker T, Yu D, Reich RR, et al. Cancer cachexia: traditional therapies and novel molecular mechanism-based approaches to treatment. Curr Treat Options Oncol. 2010;11:107–17.
Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, et al. Wasting as independent risk factor for mortality in chronic heart failure. Lancet. 1997;349:1050–3.
Pu CT, Johnson MT, Forman DE, Hausdorff JM, Roubenoff R, Foldvari M, et al. Randomized trial of progressive resistance training to counteract the myopathy of chronic heart failure. J Appl Physiol. 2001;90:2341–50.
MacDougall JD, Green HJ, Sutton JR, Coates G, Cymerman A, Yung P, et al. Operation Everest II: structural adaptations in skeletal muscle in response to extreme stimulated altitude. Acta Physiol Scand. 1991;142:421–7.
Wouters EF. Management of severe COPD. Lancet. 2004;364:883–95.
Wouters EF. Local and systemic inflammation in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:26–33.
Kadowaki M, Kanazawa T. Amino acids as regulators of proteolysis. J Nutr. 2003;133:2052S–6S.
Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, et al. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med. 2010;16:1313–20.
Marino G, Uria JA, Puente XS, Quesada V, Bordallo J, Lopez-Otin C. Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. J Biol Chem. 2003;278:3671–8.
Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–71.
Ogata T, Oishi Y, Higuchi M, Muraoka I. Fasting-related autophagic response in slow- and fast-twitch skeletal muscles. Biochem Biophys Res Commun. 2010;394:136–40.
O’Leary MFN, Hood DA. Denervation-induced oxidative stress and autophagy signaling in muscle. Autophagy. 2009;5:230–1.
Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–8.
Costelli P, Reffo P, Penna F, Autelli R, Bonelli G, Baccino FM. Ca2+-dependent proteolysis in muscle wasting. Int J Biochem Cell Biol. 2005;37:2134–46.
Smith IJ, Dodd SL. Calpain activation causes a proteasome-dependent increase in protein degradation and inhibits the Akt signaling pathway in rat diaphragm muscle. Exp Physiol. 2007;92:561–73.
Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest. 2004;113:115–23.
Moresi V, Presterá A, Scicchitano BM, Molinaro M, Teodori L, Sassoon D, et al. Tumor necrosis factor-alpha inhibition of skeletal muscle regeneration is mediated by a caspase-dependent stem cell response. Stem Cells. 2008;26:997–1008.
Hershko A, Ciechanover A. Mechanisms of intracellular protein breakdown. Annu Rev Biochem. 1982;51:335–64.
Glickman MH, Ciechanover A. The ubiquitin–proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428.
Attaix D, Aurousseau E, Combaret L, Kee A, Larbaud D, Ralliére C, et al. Ubiquitin–proteasome-dependent proteolysis in skeletal muscle. Reprod Nutr Dev. 1998;38:153–65.
Tisdale MJ. The ubiquitin–proteasome pathway as a therapeutic target for muscle wasting. J Support Oncol. 2005;3:209–17.
Kwak KS, Zhou X, Solomon V, Baracos VE, Davis J, Bannon AW, et al. Regulation of protein catabolism by muscle-specific and cytokine-inducible ubiquitin ligase E3alpha-II during cancer cachexia. Cancer Res. 2004;64:8193–8.
Cai D, Frantz JD, Tawa Jr NE, Melendez PA, Oh BC, Lidov HG, et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119:285–9.
Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412.
Deshaies RJ. SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol. 1999;15:435–67.
Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–47.
Rivett A. Proteasomes: multicatalytic proteinase complexes. Biochem J. 1993;291:1–10.
Baumeister W, Walz J, Zühl F, Seemüller E. The proteasome: paradigm of a self-compartmentalizing protease. Cell. 1998;92:367–80.
Glass DJ. Signaling pathways perturbing muscle mass. Curr Opin Clin Nutr Metabol Care. 2010;13:225–9.
Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, et al. Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem. 2005;280:2737–44.
Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51.
Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, et al. The IGF-I/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell. 2004;14:395–403.
Bossola M, Pacelli F, Costelli P, Tortorelli A, Rosa F, Doglietto GB. Proteasome activities in the rectus abdominis muscle of young and older individuals. Biogerontology. 2008;9:261–8.
Cai D, Lee KK, Li M, Tang MK, Chan KM. Ubiquitin expression is up-regulated in human and rat skeletal muscles during aging. Arch Biochem Biophys. 2004;425:42–50.
Pattison JS, Folk LC, Madsen RW, Childs TE, Booth FW. Transcriptional profiling identifies extensive downregulation of extracellular matrix gene expression in sarcopenic rat soleus muscle. Physiol Genomics. 2003;15:34–43.
Combaret L, Dardevet D, Béchet D, Taillandier D, Mosoni L, Attaix D. Skeletal muscle proteolysis in aging. Curr Opin Clin Nutr Metab Care. 2009;12:37–41.
DeRuisseau KC, Kavazis AN, Powers SK. Selective downregulation of ubiquitin conjugation cascade mRNA occurs in the senescent rat soleus muscle. Exp Gerontol. 2005;40:526–31.
Altun M, Besche HC, Overkleeft HS, Piccirillo R, Edelmann MJ, Kessler BM, et al. Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. J Biol Chem. 2010;285:39597–608.
Welle S, Brooks AL, Delehany JM, Needler N, Thornton CA. Gene expression profile of aging in human muscle. Physiol Genomics. 2003;14:149–59.
Whitman SA, Wacker MJ, Richmond SR, Godard MP. Contributions of the ubiquitin–proteasome pathway and apoptosis to human skeletal muscle wasting with age. Pflugers Arch. 2005;450:437–46.
Clavel S, Coldefy AS, Kurkdjian E, Salles J, Margaritis I, Derijard B. Atrophy-related ubiquitin ligases, atrogin-1 and MuRF1 are up-regulated in aged rat tibialis anterior muscle. Mech Ageing Dev. 2006;127:794–801.
Edström E, Altun M, Hägglund M, Ulfhake B. Atrogin-1/MAFbx and MuRF1 are downregulated in ageing-related loss of skeletal muscle. J Gerontol A Biol Sci Med Sci. 2006;61:663–74.
Léger B, Derave W, De Bock K, Hespel P, Russell AP. Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res. 2008;11:163–75.
Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR, et al. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 2007;21:140–55.
Attaix D, Mosoni L, Dardevet D, Combaret L, Mirand PP, Grizard J. Altered responses in skeletal muscle protein turnover during aging in anabolic and catabolic periods. Int J Biochem Cell Biol. 2005;37:1962–73.
Husom AD, Peters EA, Kolling EA, Fugere NA, Thompson LV, Ferrington DA. Altered proteasome function and subunit composition in aged muscle. Arch Biochem Biophys. 2004;421:67–76.
Sakuma K, Yamaguchi A. Inhibitors of myostatin- and proteasome-dependent signaling for attenuating muscle wasting. Recent Pat Regen Med. 2011;1:284–98.
Khal J, Hine AV, Fearon KCH, Dejong CHC, Tisdale MJ. Increased expression of proteasome subunits in skeletal muscle of cancer patients with weight loss. Int J Biochem Cell Biol. 2005;37:2196–206.
Filippatos GS, Anker SD, Kremastinos DT. Pathophysiology of peripheral muscle wasting in cardiac cachexia. Curr Opin Clin Nutr Metab Care. 2005;8:249–54.
Temparis S, Asensi M, Taillandier D, Aurousseau E, Larbaud D, Obled A, et al. Increased ATP-ubiquitin-dependent proteolysis in skeletal muscle of tumor-bearing rats. Cancer Res. 1994;54:5568–73.
Khal J, Wyke SM, Russell ST, Hine AV, Tisdale MJ. Expression of the ubiquitin-proteasome pathway and muscle loss in experimental cancer cachexia. Br J Cancer. 2005;93:774–80.
Llovera M, Carbó N, García-Martínez C, Costelli P, Tessitore L, Baccino FM, et al. Anti-TNF treatment reverts increased muscle ubiquitin gene expression in tumor-bearing rats. Biochem Biophys Res Commun. 1996;221:653–5.
Costelli P, Muscaritoli M, Bossola M, Penna F, Reffo P, Bonetto A, et al. IGF-I is downregulated in experimental cancer cachexia. Am J Physiol Regul Integr Comp Physiol. 2006;291:R674–83.
Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, et al. Targeting ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol. 2010;191:1395–411.
Murton AJ, Constantin D, Greenhalf PL. The involvement of the ubiquitin proteasome system in human skeletal muscle remodeling and atrophy. Biochim Biophys Acta Mol Basis Dis. 2008;1782:730–43.
Baracos VE, DeVivo C, Hoyle DH, Goldberg AL. Activation of the ATP–ubiquitin–proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am J Physiol Endocrinol Metab. 1995;268:E996–E1006.
Chai J, Wu Y, Sheng ZZ. Role of ubiquitin–proteasome pathway in skeletal muscle wasting in rats with endotoxemia. Crit Care Med. 2003;31:1802–7.
Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–77.
Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cell. Int J Biochem Cell Biol. 2004;36:2445–62.
Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–9.
Sandri M. Autophagy in health and disease. 3. Involvement of autophagy in muscle atrophy. Am J Physiol Cell Physiol. 2010;298:C1291–7.
Lee HK, Marzella L. Regulation of intracellular protein degradation with special reference to lysosomes: role in cell physiology and pathology. Int Rev Exp Pathol. 1994;35:39–147.
Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263:55–72.
Raben N, Schreiner C, Baum R, Takikita S, Xu S, Xie T, et al. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder—murine Pompe disease. Autophagy. 2010;6:1078–89.
Wang CW, Klionsky DJ. The molecular mechanism of autophagy. Mol Med. 2003;9:65–76.
Noglaska A, Terracciano C, D’Agostino C, Engel WK, Askanas V. p62/SQSTM1 is overexpressed and prominently accumulated in inclusion of sporadic inclusion-body myositis muscle fibers, and can help differentiating it from polymyositis and dermatomyositis. Acta Neuropath. 2009;118:407–13.
Bjorkoy G, Lamak T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–14.
Pankiv S, Clausen TH, Lamak T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45.
Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–83.
Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009;10:507–15.
Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy. 2005;1:131–40.
Terman A, Brunk UT. Oxidative stress, accumulation of biological ‘garbage’, and aging. Antioxid Redox Signal. 2006;8:197–204.
Donati A, Cavallini G, Paradiso C, Vittorini S, Pollera M, Gori Z, et al. Age-related changes in the regulation of autophagic proteolysis in rat isolated hepatocytes. J Gerontol A Biol Sci Med Sci. 2001;56:B288–93.
Wohlgemuth SE, Julian D, Akin DE, Fried J, Toscano K, Leeuwenburgh C, et al. Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res. 2007;10:281–92.
Gaugler M, Brown A, Merrell E, Disanto-Rose M, Rathmacher JA, Reynolds 4th TH. PKB signaling and atrogene expression in skeletal muscle of aged mice. J Appl Physiol. 2011;111:192–9.
McMullen CA, Ferry AL, Gamboa JL, Andrade FH, Dupont-Versteegden EE. Age-related changes of cell death pathways in rat extraocular muscle. Exp Gerontol. 2009;44:420–5.
Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci USA. 2009;106:20405–10.
Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, Leeuwenburgh C. Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp Gerontol. 2010;45:138–48.
Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152:519–30.
Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34.
Ryter SW, Chen Z-H, Kim HP, Choi AMK. Autophagy in chronic obstructive pulmonary disease. Autophagy. 2009;5:235–7.
Plant PJ, Brooks D, Faughnan M, Bayley T, Bain J, Singer L, et al. Cellular markers of muscle atrophy in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2010;42:461–71.
Lee SJ. Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol. 2004;20:61–86.
Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem. 2002;277:49831–40.
Thomas M, Langley B, Berry C, Sharma M, Kirk S, Bass J, et al. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast differentiation. J Biol Chem. 2000;275:40235–43.
Yang W, Zhang Y, Li Y, Wu Z, Zhu D. Myostatin induces cyclin D1 degradation to cause cell cycle arrest through a phosphatidylinositol 3-kinase/AKT/GSK-3β pathway and is antagonized by insulin-like growth factor 1. J Biol Chem. 2007;282:3799–808.
Lee SJ, McPherron AM. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA. 2001;98:9306–11.
Allen DL, Unterman TG. Regulation of myostatin expression and myoblast differentiation by FoxO and SMAD transcription factors. Am J Physiol Cell Physiol. 2007;292:C188–99.
Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296:C1258–70.
Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, et al. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol. 2009;296:C1248–57.
Bradley L, Yaworsky PJ, Walsh FS. Myostatin as a therapeutic target for musculoskeletal disease. Cell Mol Life Sci. 2008;65:2119–24.
Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–21.
Holzbaur EL, Howland DS, Weber N, Wallace K, She Y, Kwak S, et al. Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;23:697–707.
Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, et al. A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol. 2008;63:561–71.
Sakuma K, Watanabe K, Sano M, Uramoto I, Totsuka T. Differential adaptation of growth and differentiation factor 8/myostatin, fibroblast growth factor 6 and leukemia inhibitory factor in overloaded, regenerating, and denervated rat muscles. Biochim Biophys Acta, Mol Cell Res. 2000;1497:77–88.
Sakuma K, Watanabe K, Hotta N, Koike T, Ishida K, Katayama K, et al. The atrophy of skeletal muscle after lower limb unloading in humans. Acta Physiol (Oxf). 2009;197:151–9.
Wehling M, Cai B, Tidball JG. Modulation of myostatin expression during modified muscle use. FASEB J. 2000;14:103–10.
Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S, et al. Organization of the human myostatin gene and expression in health men and HIV-infected men with muscle wasting. Proc Natl Acad Sci USA. 1998;95:14938–43.
Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors is old muscle stem cells. Nature. 2008;454:528–32.
Haddad F, Adams GR. Aging-sensitive cellular and molecular mechanisms associated with skeletal muscle hypertrophy. J Appl Physiol. 2006;100:1188–203.
Siriett V, Platt L, Salerno MS, Ling N, Kambadur R, Sharma M. Prolonged absence of myostatin reduces sarcopenia. J Cell Physiol. 2006;209:866–73.
Lebrasseur NK, Schelhorn TM, Bernardo BL, Cosgrove PG, Loria PM, Brown TA. Myostatin inhibition enhances the effects on performance and metabolic outcomes in aged mice. J Gerontol A Biol Sci Med Sci. 2009;64:940–8.
Murphy KT, Koopman R, Naim T, Léger B, Trieu J, Ibebunjo C, et al. Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB J. 2010;24:4433–42.
Amirouche A, Durieux AC, Banzet S, Koulmann N, Bonnefoy R, Mouret C, et al. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology. 2009;150:286–94.
McFarlane C, Plummer E, Thomas M, Hennebry A, Ashby M, Ling N, et al. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-κB-independent, FoxO1- dependent mechanism. J Cell Physiol. 2006;209:501–14.
Chelh I, Meunier B, Picard B, Reecy JM, Chevalier C, Hocquette JF, et al. Molecular profile of quadriceps muscle in myostatin-null mice reveal PI3K and apoptotic pathways as myostatin targets. BMC Genomics. 2009;10:196.
Costelli P, Muscaritoli M, Bonetto A, Penna F, Reffo P, Bossola M, et al. Muscle myostatin signaling is enhanced in experimental cancer cachexia. Eur J Clin Invest. 2008;38:531–8.
Petraglia F, Florio P, Luisi S, Gallo R, Gadducci A, Viganó P, et al. Expression and secretion of inhibin and activin A in normal and neoplastic uterine tissues. High levels of serum activin A in women with endometrial and cervical carcinoma. J Clin Endocrinol Metab. 1998;83:1194–200.
Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, et al. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296:1486–8.
Sharma M, Kambadur R, Matthews KG, Somers WG, Devlin GP, Conaglen JV, et al. Myostatin, a transforming growth factor-beta superfamily member, is expressed in heart muscle and is upregulated in cardiomyocytes after infarct. J Cell Physiol. 1999;180:1–9.
Shyu KG, Lu MJ, Wang BW, Sun HY, Chang H. Myostatin expression in ventricular myocardium in a rat model of volume-overload heart failure. Eur J Clin Invest. 2006;36:713–9.
Heineke J, Auger-Messier M, Xu J, Sargent M, York A, Welle S, et al. Genetic deletion of myostatin from the heart prevents skeletal muscle atrophy in heart failure. Circulation. 2010;121:419–25.
Testelmans D, Crul T, Maes K, Agten A, Comach M, Decramer M, et al. Atrophy and hypertrophy signaling in the diaphragm of patients with COPD. Eur Respir J. 2010;35:549–56.
Hayot M, Rodriguez J, Vernus B, Carnac G, Jean E, Allen D, et al. Myostatin up-regulation is associated with the skeletal muscle response to hypoxic stimuli. Mol Cell Endocrinol. 2011;332:38–47.
Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142:531–43.
Klimek MEB, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun. 2010;391:1548–54.
Lee SJ, Reed LA, Davies MV, Girgenrath S, Goad ME, Tomkinson KN, et al. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci USA. 2005;102:18117–22.
Roubenoff R. Catabolism of aging: is it an inflammatory process? Curr Opin Clin Nutr Metab Care. 2003;6:295–9.
Schaap LA, Pluijim SMF, Deeg DJH, Harris TB, Kritchevsky SB, Newman, et al. Higher inflammatory marker levels in older persons: associations with 5-year change in muscle mass and muscle strength. J Gerontol A Biol Sci Med Sci. 2009;64A:1183–9.
Rieu I, Magne H, Savary-Auzeloux I, Averous J, Bos C, Peyron MA, et al. Reduction of low grade inflammation restores blunting of postprandial muscle anabolism and limits sarcopenia in old rats. J Physiol. 2009;587:5483–92.
Chung HY, Cesari M, Anton S, Marzetti E, Giovannini S, Seo AY, et al. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res Rev. 2009;8:18–30.
Reid MB, Li YP. Tumor necrosis factor-α and muscle wasting: a cellular perspective. Respir Res. 2001;2:269–72.
Aoi W, Sakuma K. Oxidative stress and skeletal muscle dysfunction with aging. Curr Aging Sci. 2011;4:101–9.
Meng S-J, Yu L-J. Oxidative stress, molecular inflammation and sarcopenia. Int J Mol Sci. 2010;11:1509–26.
Bar-Shai M, Carmeli E, Coleman R, Rozen N, Perek S, Euchs D, et al. The effect of hindlimb immobilization on acid phosphatase, metalloproteinase and nuclear factor-kappaB in muscles of young and old rats. Mech Ageing Dev. 2005;126:289–97.
Phillips T, Leeuwenburgh C. Muscle fiber-specific apoptosis and TNF-α signaling in sarcopenia are attenuated by life-long calorie restriction. FASEB J. 2005;19:668–70.
Marzetti E, Carter CS, Wohlgemuth SE, Lees HA, Giovannini S, Anderson B, et al. Changes in IL-15 expression and death-receptor apoptotic signaling in rat gastrocnemius muscle with aging and life-long calorie restriction. Mech Ageing Dev. 2009;130:272–80.
Pistilli E, Jackson JR, Always SE. Death receptor-associated pro-apoptotic signaling in aged skeletal muscle. Apoptosis. 2006;11:2115–26.
Argilés JM, Busquets S, Toledo M, López-Soriano FJ. The role of cytokines in cancer cachexia. Curr Opin Support Palliat Care. 2009;3:263–8.
Malik ST, Naylor MS, East N, Oliff A, Balkwill FR. Cells secreting tumour necrosis factor show enhanced metastasis in nude mice. Eur J Cancer. 1990;26:1031–4.
Moresi V, Pristera A, Scicchitano BM, Molinaro M, Teodori L, Sassoon D, et al. Tumor necrosis factor-α inhibition of skeletal muscle regeneration is mediated by a caspase-dependent stem cell response. Stem Cells. 2008;26:997–1008.
Coletti D, Moresi V, Adamo S, Molinaro M, Sassoon D. Tumor necrosis factor-alpha gene transfer induces cachexia and inhibits muscle regeneration. Genesis. 2005;43:120–8.
Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin Jr AS. NF-κB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science. 2000;289:2363–6.
Di Marco S, Mazroui R, Dallaire P, Chittur S, Tenenbaum SA, Radzioch D, et al. NF-kappa B-mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR protein, and nitric oxide release. Mol Cell Biol. 2005;25:6533–45.
Costelli P, Muscaritoli M, Bossola M, Moore-Carrasco R, Crepaldi S, Grieco G, et al. Skeletal muscle wasting in tumor-bearing rats is associated with MyoD down-regulation. Int J Oncol. 2005;26:1663–8.
Remels AH, Gosker HR, Schrauwen P, Hommelberg PP, Sliwinski P, Polkey M, et al. TNF-alpha impairs regulation of muscle oxidative phenotype: implication for cachexia? FASEB J. 2010;24:5052–62.
Wysong A, Couch M, Shadfar S, Li L, Rodriguez JE, Asher S, et al. NF-κB inhibition protects against tumor-induced cardiac atrophy in vivo. Am J Pathol. 2011;178:1059–68.
Acharyya S, Butchbach ME, Sahenk Z, Wang H, Saji M, Carathers M, et al. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell. 2005;8:421–32.
Kawamura I, Morishita R, Tomita N, Lacey E, Aketa M, Tsujimoto S, et al. Intratumoral injection of oligonucleotides to the NF-κB binding site inhibits cachexia in a mouse tumor model. Gene Ther. 1999;6:91–7.
Mastrocola R, Reffo P, Penna F, Tomasinelli CE, Boccuzzi G, Baccino FM, et al. Muscle wasting in diabetic and in tumor-bearing rats: role of oxidative stress. Free Radical Biol Med. 2008;44:584–93.
Doucet M, Russell AP, Léger B, Debigaré R, Joanisse DR, Caron MA, et al. Muscle atrophy and hypertrophy signaling in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;176:261–9.
Adams V, Nehrhoff B, Späte U, Linke A, Schulze PC, Baur A, et al. Induction of iNOS-expression in skeletal muscle by IL-1β and NFκB activation: an in vitro and in vivo study. Cardiovasc Res. 2002;54:95–104.
Yu Z, Li P, Zhang M, Hannink M, Stamler JS, Yan Z. Fiber type-specific nitric oxide protects oxidative myofibers against cachectic stimuli. PLoS One. 2008;3:e2086.
Todorov PT, Cariuk P, McDevitt T, Coles B, Fearon K, Tisdale M. Characterization of a cancer cachectic factor. Nature. 1996;379:739–42.
Cariuk P, Lorite MJ, Todorov PT, Field WN, Wigmore SJ, Tisdale MJ. Induction of cachexia in mice by a product isolated from the uterine of cachectic cancer patients. Br J Cancer. 1997;76:606–13.
Tisdale MJ. Molecular pathways leading to cancer cachexia. Physiology (Bethesda). 2005;20:340–8.
Wyke SM, Tisdale MJ. NF-κB mediates proteolysis-inducing factor induced protein degradation and expression of the ubiquitin–proteasome system in skeletal muscle. Br J Cancer. 2005;92:711–21.
Marzetti E, Hwang JC, Lees HA, Wohlgemuth SE, Dupont-Versteegden EE, Carter CS, et al. Mitochondrial death effectors: relevance to sarcopenia and disuse muscle atrophy. Biochim Biophys Acta, Gen Subj. 2009;1800:235–44.
Dupont-Versteegden EE. Apoptosis in muscle atrophy: relevance to sarcopenia. Exp Gerontol. 2005;40:473–81.
Marzetti E, Lawler JM, Hiona A, Manini T, Seo AY, Leeuwenburgh C. Modulation of age-induced apoptotic signaling and cellular remodeling by exercise and calorie restriction in skeletal muscle. Free Radic Biol Med. 2008;44:160–8.
Wang C, Youle RJ. The role of mitochondria in apoptosis. Ann Rev Genet. 2009;43:95–118.
Song W, Kwak HB, Lawler JM. Exercise training attenuates age-induced changes in apoptotic signaling in rat skeletal muscle. Antioxid Redox Signal. 2006;8:517–28.
Pollack M, Leeuwenburgh C. Apoptosis and aging: role of the mitochondria. J Gerontol A Biol Sci Med Sci. 2001;56:B475–82.
Jezek P, Hlavata L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int J Biochem Cell Biol. 2005;37:2478–503.
Yakes FM, Van HB. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA. 1997;94:514–9.
Bua EA, McKiernan SH, Eanagat J, McKenzie D, Aiken JM. Mitochondrial abnormalities are more frequent in muscles undergoing sarcopenia. J Appl Physiol. 2002;92:2617–24.
Wanagat J, Cao Z, Pathare P, Aiken JM. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 2001;15:322–32.
Wei YH, Lee HC. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp Biol Med (Maywood). 2002;227:671–82.
Argiles JM, Lopez-Soriano FJ, Busquets S. Apoptosis signaling is essential and precedes protein degradation in wasting skeletal muscle during catabolic conditions. Int J Biochem Cell Biol. 2008;40:1674–8.
Baker DJ, Hepple RT. Elevated caspase and AIDF gene expression correlate with progression of sarcopenia during aging in male F344BN rats. Exp Gerontol. 2006;41:1149–56.
Park SY, Kim HY, Lee JH, Yoon KH, Change MS, Park SK. The age-dependent induction of apoptosis-inducing factor (AIF) in the human semitendinosus skeletal muscle. Cell Mol Biol Lett. 2010;15:1–12.
Busquets S, Figueras M, Fuster G, Almendro V, Moore-Carrasco R, Ametller E, et al. Anticachectic effects of formoterol: a drug for potential treatment of muscle wasting. Cancer Res. 2004;64:6725–31.
Kataoka T, Budd RC, Holler N, Thome M, Martinon F, Irmler M, et al. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr Biol. 2000;10:640–8.
Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21:5299–305.
Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, et al. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7:715–25.
Wajant H, Haas E, Schwenzer R, Muhlenbeck F, Kreuz S, Schubert G, et al. Inhibition of death receptor-mediated gene induction by a cycloheximide-sensitive factor occurs at the level of or upstream of Fas-associated death domain protein (FADD). J Biol Chem. 2000;275:24357–66.
Jiang Z, Clemens PR. Cellular caspase-8-like inhibitory protein (cFLIP) prevents inhibition of muscle cell differentiation induced by cancer cells. FASEB J. 2006;20:E1979–89.
Daniel NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–19.
Hermann M, Berger P. Hormonal changes in aging men: a therapeutic indication? Exp Gerontol. 2001;36:1075–82.
Kamel H, Maas D, Duthie E. Role of hormones in the pathogenesis and management of sarcopenia. Drugs Aging. 2002;19:869–77.
Ryall JG, Schertzer JD, Lynch GS. Cellular and molecular mechanisms underlying age-related skeletal muscle wasting and weakness. Biogerontology. 2008;9:213–28.
Szulc P, Duboeuf F, Marchand F, Delmas PD. Hormonal and lifestyle determinants of appendicular skeletal muscle mass in men: the MINOS Study. Am J Clin Nutr. 2004;80:496–503.
Tenover JL. Testosterone and the aging male. J Androl. 1997;18:103–6.
Ferrucci L, Penninx BW, Volpato S, Harris TB, Bandeen-Roche K, Balfour J, et al. Change in muscle strength explains accelerated decline of physical function in older women with high interleukin-6 serum levels. J Am Geriatr Soc. 2002;50:1947–54.
Philippou A, Maridaki M, Halapas A, Koutsilieris M. The role of the insulin-like growth factor-I (IGF-I) in skeletal muscle physiology. In Vivo. 2007;21:45–54.
Butterfield GE, Thompson J, Rennie MJ, Marcus R, Hintz RL, Hoffman AR. Effect of thGH and rhIGF-I treatment on protein utilization in elderly women. Am J Physiol Endocrinol Metab. 1997;272:E94–9.
Giovannini S, Marzetti E, Borst SE, Leeuwenburgh C. Modulation of GH/IGF-I axis: potential strategies to counteract sarcopenia in older adults. Mech Ageing Dev. 2008;129:593–601.
Evans WJ, Paolisso G, Abbatecola AM, Corsonello A, Bustacchini S, Strollo F, et al. Frailty and muscle metabolism dysregulation in the elderly. Biogerontology. 2010;11:527–36.
Dardevet D, Sornet C, Attaix D, Baracos VE, Grizard J. Insulin-like growth factor-I and insulin resistance in skeletal muscles of adults. Endocrinology. 1994;134:1475–84.
Martineau LC, Chadan SG, Parkhouse WS. Age-associated alterations in cardiac and skeletal muscle glucose transporters, insulin and IGF-I receptors, and PI3-kinase protein contents in the C57BL/6 mouse. Mech Ageing Dev. 1999;106:217–32.
Arvat E, Broglio F, Ghigo E. Insulin-like growth factor I: implication in aging. Drugs Aging. 2000;16:29–40.
Marcell TJ, Harman SM, Urban RJ, Metz DD, Rodgers BD, Blaskman MR. Comparison of GH, IGF-I, and testosterone with mRNA of receptors and myostatin in skeletal muscle in older men. Am J Physiol Endocrinol Metab. 2001;281:E1159–64.
Wilkes EA, Selby AL, Atherton PJ, Patel R, Rankin D, Smith K, et al. Blunting of insulin inhibition of proteolysis in legs of older subjects may contribute to age-related sarcopenia. Am J Clin Nutr. 2009;90:1343–50.
Bakhshi V, Elliott M, Gentili A, Godschalk M, Mulligan T. Testosterone improves rehabilitation outcomes in ill older men. J Am Geriatr Soc. 2000;48:550–3.
Ferrando AA, Sheffield-Moore M, Yeckel CW, Gilkison C, Jiang J, Achacosa A, et al. Testosterone administration to older men improves muscle function: molecular and physiological mechanisms. Am J Physiol Endocrinol Metab. 2002;282:E601–7.
Morley JE, Perry 3rd HM. Androgen deficiency in aging men: role of testosterone replacement therapy. J Lab Clin Med. 2000;135:370–8.
Snyder PJ, Peachey H, Hannoush P, Berlin JA, Loh L, Lenrow DA, et al. Effect of testosterone treatment on body composition and muscle strength in men over 65 years of age. J Clin Endocrinol Metab. 1999;84:2647–53.
Bhasin S, Calof O, Storer TW, Lee M, Mazer N, Jasuja R, et al. Drug insight: testosterone and selective androgen receptor modulators as anabolic therapies for physical dysfunction in chronic illness and ageing. Nat Clin Pract Endocrinol Metab. 2006;2:146–59.
Sinha-Hikim I, Cornford M, Gaytan H, Lee ML, Bhasin S. Effects of testosterone supplementation on skeletal muscle fiber hypertrophy and satellite cells in community-dwelling older men. J Clin Endocrinol Metab. 2006;91:3024–33.
Kovacheva EL, Hikim AP, Shen R, Sinha I, Sinha-Hikim I. Testosterone supplementation reverses sarcopenia in aging through regulation of myostatin, c-Jun NH2-terminal kinase, Notch, and Akt signaling pathways. Endocrinology. 2010;151:628–38.
Chebowski RT, Heber D. Hypogonadism in male patients with metastatic cancer prior to chemotherapy. Cancer Res. 1982;42:2495–8.
Zitzmann M, Nieschlag E. Hormone substitution in male hypogonadism. Mol Cell Endocrinol. 2000;161:73–88.
Crul T, Spruit MA, Gayan-Ramirez G, Quarck R, Gosselink R, Troosters T, et al. Markers of inflammation and disuse in vastus lateralis of chronic obstructive pulmonary disease patients. Eur J Clin Invest. 2007;37:897–904.
Debigare R, Marquis K, Cote CH, Tremblay RR, Michaud A, LeBlanc P, et al. Catabolic/anabolic balance and muscle wasting in patients with COPD. Chest. 2003;124:83–9.
Kamischke A, Kemper DE, Castel MA, Luthke M, Rolf C, Behre HM, et al. Testosterone levels in men with chronic obstructive pulmonary disease with or without glucocorticoid therapy. Eur Respir J. 1998;11:41–5.
Anker SD, Volterrani M, Pflaum CD, Strasburger CJ, Osterziel KJ, Doehner W, et al. Acquired growth hormone resistance in patients with chronic heart failure: implications for therapy with growth hormone. J Am Coll Cardiol. 2001;38:443–52.
Niebauer J, Pflaum CD, Clark AL, Strasburger CJ, Hooper J, Poole-Wilson PA, et al. Deficient insulin-like growth factor I in chronic heart failure predicts altered body composition, anabolic deficiency, cytokine and neurohormonal activation. J Am Coll Cardiol. 1998;32:393–7.
Ng EH, Rock CS, Lazarus DD, Stiaino-Coico L, Moldawer LL, Lowry SF. Insulin-like growth factor I preserves host lean tissue mass in cancer cachexia. Am J Physiol. 1992;262:R426–31.
Denson LA, Menon RK, Shaufl A, Bajwa HS, Williams CR, Karpen SJ. TNF-alpha downregulates murine hepatic growth hormone receptor expression by inhibiting Sp1 and Sp3 binding. J Clin Invest. 2001;107:1451–8.
Casaburi R, Bhasin S, Cosentino L, Porszasz J, Somfay A, Lewis MI, et al. Effects of testosterone and resistance training in men with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;170:870–8.
Knapp PE, Storer TW, Herbst KL, Singh AB, Dzekov C, Dzekov J, et al. Effects of a supraphysiological dose of testosterone on physical function, muscle performance, mood, and fatigue in men with HIV-associated weight loss. Am J Physiol Endocrinol Metab. 2008;294:E1135–43.
Mulligan K, Zackin R, Von Roenn JH, Chesney MA, Egorin MJ, Sattler FR, et al. Testosterone supplementation of megestrol therapy does not enhance lean tissue accrual in men with human immunodeficiency virus associated weight loss: a randomized, double-blind, placebo-controlled, multicenter trial. J Clin Endocrinol Metab. 2007;92:563–70.
Pape GS, Friedman M, Underwood LE, Clemmons DR. The effect of growth hormone on weight gain and pulmonary function in patients with chronic obstructive lung disease. Chest. 1991;99:1495–500.
Burdet L, de Muralt B, Schutz Y, Pichard C, Fitting JW. Administration of growth hormone to underweight patients with chronic obstructive pulmonary disease. A prospective, randomized, controlled study. Am J Respir Crit Care Med. 1997;156:1800–6.
Frustaci A, Gentiloni N, Russo MA. Growth hormone in the treatment of dilated cardiomyopathy. N Engl J Med. 1996;335:672–3.
Osterziel KJ, Strohm O, Schuler J, Friedrich M, Hänlein D, Willenbrock R, et al. Randomised, double blind, placebo-controlled trial of human recombinant growth hormone in patients with chronic heart failure due to dilated cardiomyopathy. Lancet. 1998;351:1233–7.
Gullett NP, Hebbar G, Ziegler TR. Update on clinical trials of growth factors and anabolic steroids in cachexia and wasting. Am J Clin Nutr. 2010;91:1143S–7S.
Akamizu T, Kangawa K. Ghrelin for cachexia. J Cachexia Sarcopenia Muscle. 2010;1:169–76.
Kojima M, Kangawa K. Ghrelin: structure and function. Physiol Rev. 2005;85:495–522.
Dixit VD, Schaffer EM, Pyle RS, Collins GD, Sakthivel SK, Palaniappan R, et al. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J Clin Invest. 2004;114:57–66.
Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. 2001;86:5992.
Russell ST, Tisdale MJ. The role of glucocorticoids in the induction of zinc-α2-glycoprotein expression in adipose tissue in cancer cachexia. Br J Cancer. 2005;92:876–81.
Tessitore L, Costelli P, Baccino FM. Pharmacological interference with tissue hypercatabolism in tumour-bearing rats. Biochem J. 1994;299:71–8.
Hasselgren PO. Glucocorticoids and muscle metabolism. Curr Opin Clin Nutr Metab Care. 1999;2:201–5.
Gilson H, Schakman O, Combaret L, Lause P, Grobet L, Attaix D, et al. Myostatin gene deletion prevents glucocorticoid-induced muscle atrophy. Endocrinology. 2007;148:452–60.
von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. J Cachexia Sarcopenia Muscle. 2010;1:7–8.
Acknowledgments
All authors of this manuscript comply with the guidelines of ethical authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle [250]. This work was supported by a research Grant-in-Aid for Scientific Research C (no. 23500778) from the Ministry of Education, Science, Culture, Sports, Science and Technology of Japan.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Sakuma, K., Yamaguchi, A. Sarcopenia and cachexia: the adaptations of negative regulators of skeletal muscle mass. J Cachexia Sarcopenia Muscle 3, 77–94 (2012). https://doi.org/10.1007/s13539-011-0052-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13539-011-0052-4