
Abstract
The red deer Cervus elephus has been a common species in Italy until the Middle Ages and the Renaissance, when its distribution range started to considerably decrease, due to gradual deforestation and hunting pressure. Afterwards, the red deer has been reintroduced to many regions of the world, including Italy. In the Italian Apennines, the Acquerino-Cantagallo Natural Reserve (ACQUERINO) hosts one of the largest peninsular red deer populations, originated from a series of successful reintroductions. In this study, we meant to detect the level of genetic variability of Acquerino-Cantagallo Natural Reserve deer population and to investigate the genetic relationships with the other Italian and European populations. We identified five mitochondrial DNA control region (D-loop) haplotypes, four falling in lineage A and one falling in lineage C, derived from at least two maternal lineages, confirming that ACQUERINO population should be the result of multiple reintroductions. Haplotype diversity (H = 0.50) and nucleotide (π = 0.004) diversity were low, but included into the deer range values. ACQUERINO population showed low levels of genetic diversity when compared to other European and Mediterranean populations, confirming that this expanding population may have been generated from a low number of founders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ungulates (Artiodactyla) are a group of mammals which importance in terrestrial zoocenoses is paramount (Putman 1996; Reimoser 2003; Murray et al. 2013). They are often the largest species in size, thus arousing a strong hunting interest and becoming a substantial management commitment, especially in the last decades (Hobbs 1996; Reimoser 2003). Among ungulates, the European red deer Cervus elaphus Linnaeus, 1758 is not an endangered species, but its populations have suffered from increased isolation processes and frequent reintroductions by allochthonous stocks (Hartl et al. 2003; Frantz et al. 2017). Accordingly, introgression of genes of non-native or captive origin red deer into native populations is relatively frequent throughout all of Europe, as a consequence of translocations (Iacolina et al. 2019).
Until the tenth/eleventh centuries, the red deer was distributed over most of Europe, and it was still a common species in almost all the Italian mainland (Mattioli 1990). During the late Middle Ages and the Renaissance, its distribution range decreased considerably, due to gradual deforestation and hunting pressure (Lovari 1993). With the further improvement of firearms, between the eighteenth and nineteenth centuries, red deer populations suffered a rapid decrease, so that, at the beginning of the 1900s, the red deer was nearly extinct over almost the entire Italian territory, except for the Mesola Natural Reserve (Northern-Central Italy, Ferrara District) and Sardinia (Mattioli 1990; Mattioli et al. 2003; Zachos et al. 2009, 2014; Gippoliti et al. 2020). In peninsular Italy, the only native population is currently represented by the Mesola population, on the eastern coastline. At present, in Italy, several populations are the result of a series of reintroductions between 1950 and 2003, followed by a natural expansion in the Alps from neighbouring European areas (Switzerland, Austria, and Slovenia: Mattioli et al. 2001; Carnevali et al. 2009) (Fig. S1). Other small nuclei in the Apennines are indeed the results of local unmanaged escapes (e.g. Monte Penna in Castell’Azzara-Grosseto and Merse river valley in the province of Siena). In 2010, the Italian territory included over 70,000 individuals divided as follows: Central-Western Alps 20,000, Eastern Alps 32,000, Northern Apennines 8,000, Central-Southern Apennines 2,000, Islands 8,000 (cf. Apollonio et al. 2010). Since then, no reliable estimation of red deer population has been conducted in Italy. For a species with a potential annual increase of up to 30%, current numbers could be quite different compared to those collected 12 years ago, especially for populations of Apennine protected areas (see Chirichella et al. 2017). In the Italian Alps, the deer shows a favourable conservation status and has occupied a large part of the potential range again, so much as in some geographical regions, the populations must be controlled in order to avoid excessive forest damages. Red deer populations in Northern Apennines are currently distributed in three main continuous areas (Becciolini et al. 2016; Gippoliti et al. 2020): in the north-west, with a population estimated to be nearly 3,100 individuals; in the south-eastern area, with a population of 2,500–3,000 deer; in the central area, with about 3,000–3,500 individuals. The progressive expansion of distribution area of the deer population presents in the Tosco-Emilian Apennines led in 1999 to the creation of a protocol of agreement for the management of the species at the inter-regional level, named ACATER (Areale del Cervo dell'Appennino Tosco-Emiliano e Romagnolo), which aim is to guarantee a correct management of the red deer in the long term, taking into account the biological and behavioural characteristics of the species. In Central and Southern Apennines, the perspective for a natural expansion of populations seems to be highly probable, given the extent of protected areas in the Abruzzo and Lazio regions. In addition, several areas of the Southern Apennines have environmental conditions suitable for hosting this species and may be interested by future reintroductions.
Mitochondrial markers (for a review see Zachos and Hartl 2011) identified in European red deer modern populations three major haplogroups including the several subspecies described for this taxon (Skog et al. 2009): C. e. hippelaphus (synonim of C. e. germanicus), C. e. elaphus, C. e. atlanticus, C. e. scoticus, and C. e. hispanicus belong to the Western European haplogroup A (corresponding to haplogroup WERD: Skog et al. 2009), occurring in Western and Northern Europe. Haplogroup B includes C. e. corsicanus and C. e. barbarus and it is confined to Corsica, Sardinia, and Northern Africa. The third haplogroup, i.e. the South-Eastern lineage, named C, is represented by C. e. montanus and it characterizes the eastern European populations. Furthermore, haplogroup D occurs in Turkey, Iran, the Republic of Dagestan (Russian Federation) in the Northern Caucasus — i.e. the distribution range of C. elaphus maral — and the Mesola deer in the Po delta area in Italy. C. e. italicus (formerly in haplogroup C) (Ludt et al. 2004; Skog et al. 2009) and haplogroup E occur in Armenia, Crimea, and Southern Russia (Doan et al. 2022). The above distinct mitochondrial DNA (mtDNA) lineages would reflect the main refugia during the Last Glacial Maximum (LGM). From Iberia, lineage A colonized the British Isles and parts of Central Europe; in the Balkan region, lineage C migrated to South-East Central Europe and the Carpathians. The Mediterranean (lineage B) could be originated from the Italian refugial lineage, and lineage D has been proposed for the Middle East and South-Eastern Europe (Zachos and Hartl 2011; Meiri et al. 2013; Doan et al. 2017; Schnitzler et al. 2018; Queiros et al. 2019). These hypotheses have been recently reconsidered and new data suggested that the species survived the LGM not only in the well-known southern European refugia, but also in more northern areas of Western and Eastern Europe and in the Urals (Niedziałkowska et al. 2021).
The genetic structure of the red deer in Europe has been widely investigated (Ludt et al. 2004; Skog et al. 2009; Perez-Espona et al. 2009; Niedziałkowska et al. 2011; Fernández-García et al. 2014; Zachos et al. 2016; Rey-Iglesia et al. 2017; Schnitzler et al. 2018; Doan et al. 2022) even though a small percentage of data involved peninsular Italian populations (Lorenzini et al. 2005; Hmwe et al. 2006A, BA; Zachos et al. 2016; Doan et al. 2017, 2022). Doan et al. (2022) also highlighted Italy as one of the countries with the lowest genetic amount of data as to red deer populations. The ACQUERINO population is one of the largest populations in Central Italy, and its morphometry is well studied (Becciolini et al. 2016). Even if originated by reintroductions, we considered of interest a genetic characterization of this population that has experienced strong demographic expansion. The ACQUERINO population was originated by seven red deer individuals reintroduced in 1958 and 1965 from the State Forest of Tarvisio, North-Eastern Italy (Mazzarone and Mattioli 1996) in the Acquerino-Cantagallo Natural Reserve followed by some not documented introductions.
In this paper, we aimed at evaluating the level of genetic variability of Acquerino-Cantagallo Natural Reserve deer population and to investigate the genetic relationships with the other Italian and European populations.
Materials and methods
Study area
We conducted our study in the Acquerino-Cantagallo Natural Reserve (provinces of Prato and Pistoia, Central Italy, 44.003°N–11.015°E: 100–1100 m a.s.l., hereafter ACQUERINO), an historical reproductive area of the Northern Apennine red deer population (Becciolini et al. 2016). The local climate shows sub-montane characteristics. Average monthly temperatures range from 0 to 6 °C in the coldest months (October–March) and from 17 to 24 °C in the warmest ones (April–September). Annual precipitations are generally over 1,000 mm, with seasonal maximum in autumn and spring, and the minimum in summer (Becciolini et al. 2016). Forests cover the 84% of the study area, with broad-leaved forests being the most abundant type (73%), followed by mixed broad-leaved and conifer (7%) and conifer woodlands (4%). Farmlands and olive groves represent 3% and 2% of the territory, respectively (Becciolini et al. 2016).
Sampling and DNA extraction
We collected 26 blood red deer samples (13 males and 13 females) in 2008–2009 as part of a selective hunting program. Samples were stored in EDTA 0.25 M and kept in a freezer at − 80 °C. DNA was extracted using the Qiagen Qiamp Tissue and Blood kit (Qiagen, Inc, Tokyo, Japan). The analysis of genetic variability was carried out using a portion (738 bp) of the mitochondrial DNA control region (mt CR, D-loop). PCR amplification was obtained using two pairs of primers already available in the literature: CE-CR-FOR (5′-CAATACACTGGTCTTGTAACC-3′) and CE-CR-REV (5′-TAATAGGAAGGGGGGCC-3′; McDevitt et al. 2009). The PCR was carried out with an Applied Biosystems 2700 machine in 25 μl comprising buffer 10 × , 12 mM MgCl2, 200 μM dNTPs, 0 2 μM of each primer, and 1 unit Taq polymerase (Life Technologies). The amplification conditions included initial denaturing at 94 °C for 2 min, followed by 35 cycles of 94 °C for 45″, 55 °C (T annealing) for 30″, 72 °C for 1 min, and a final extension to 72 °C for 10 min PCR products were run by electrophoresis on 15% agarose gel, containing 05 mg/ml of SYBR green gel staining, purified (Sure Clean method, Bioline), and sequenced with the ABI Big Dye™ Terminator Cycle Sequencing version 20-ABI PRISM (Applied Biosystems, Foster City, USA) kit. The sequence analyses were carried out using an automatic ABI 310 sequencer (Applied Biosystems). Electropherograms have been displayed with the CHROMAS 145 program (http://www.technelysium.com/au). The sequences have been manually corrected and analyzed with the MEGA 6 program (Tamura et al. 2013).
Genetic and statistical analyses
We used DNA-sp version 5 (Librado and Rozas 2009) to calculate the number of haplotypes (Nh), haplotypic diversity (H), and nucleotide diversity (π). We used DNA-sp also to investigate the demographic history, calculating deviations from neutrality with Tajima’s D (Tajima 1989), Fu and Li’s F (Fu and Li 1993), and Fu’s Fs (Fu 1997) tests. Significant D, F, and Fs negative values indicated an excess of low-frequency polymorphisms relative to expectation, indicating population size expansion (following a bottleneck or a selective sweep) or purifying selection. Fu’s Fs is considered a more powerful tool than Tajima’s D. Its power comes from the ability to differentiate between population growth and genetic hitchhiking and in rejecting the hypothesis of neutral mutations. JModelTEST 304 (Posada and Crandall 1998) was used to test the most accurate model of substitution using the Bayesian information criterion (BIC; Schwarz 1978) and Akaike’s information criterion (AIC; Akaike 1974), corrected for the heterogeneity between sites (gamma [G]). Red deer sequences obtained in this study were aligned with previously published D-loop sequences available on GenBank (http://www.ncbi.nlm.nih.gov/) (datasets 1 and 2, Table 1). A first dataset (dataset 1) consisted of longer sequences of about 700 bp and a second dataset (dataset 2) consisted of shorter sequences (about 320 bp) but included more European samples (Table 1). A median-joining network was constructed to determine the genealogical relationships and haplotype relative frequencies for ACQ population (Bandelt et al. 1999) by software PopART (Leigh and Bryant 2015). We applied a Bayesian approach performed by MrBayes v 3.2.6. (http://mrbayes.sourceforge.net/download.php). We ran four Markov chains in two independent analyses for 10 million generations, sampling every 1,000 generations. The first 25% of samples were discarded as burn-in. The sequences of C. canadensis and C. nippon (Table 1) were used as outgroups. Support node values were given as posterior probability values (pp).
A likelihood-ratio test (LRT) was performed to test the molecular clock hypothesis based on D-loop gene sequences. The likelihoods with the molecular clock assumption (L0) and without the molecular clock assumption (L1) were calculated using MEGA software. Since the clock-like behaviour of the data was rejected for the complete dataset (the equal evolutionary rate throughout the tree was rejected at a 5% significance level (P = 1.181E-030)), a relaxed clock (uncorrelated lognormal) was applied, as implemented in BEAST 1.5.3 using a Bayesian MCMC approach (Drummond and Rambaut 2007). We used a CR gene substitution rate of 3,849 × 10–7 (per site per year, Queirós et al. 2019). Two separate MCMC analyses were run for 20 million generations for each dataset with parameters sampled every 1,000 generations. Independent runs were combined using LogCombiner, and the first 20% of the generations from each run were discarded as “burnin”. The convergence of the chains was checked using TRACER. The searches achieved adequate mixing as assessed by the high effective sampling size (ESS) values for all parameters of 100 or greater. Node ages and upper and lower bounds of the 95% highest posterior density interval for divergence times were calculated using TreeAnnotator and visualized using FigTree 1.31 (Rambaut 2009).
Results
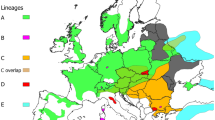
We detected five new haplotypes among the 26 examined samples: HAP1_ACQUERINO, HAP2_ACQUERINO, HAP3_ACQUERINO, HAP4_ACQUERINO, HAP5_ACQUERINO (GenBank Acc. No: MZ997342-MZ997346): the HAP1_ACQUERINO is the most frequent one (18 individuals), followed by HAP2_ACQUERINO (5 individuals). The other three haplotypes were found in one individual each (Fig. 1). In the median-joining network, the HAP5_ACQUERINO haplotype is included within haplogroup C, whereas the other 4 haplotypes are inserted in haplogroup A. The ACQUERINO population showed levels of genetic diversity (H = 0.50; π = 0.004) low when compared to those of various European and Mediterranean populations (H = 0.93 ± 0.14; π = 0.009 Meiri et al. 2013; H = 0.93 ± 0.12; π = 0.007 Doan et al. 2017), but they are similar to average values obtained by Queiros et al. (2019) (H = 0.51 ± 0.12; π = 0.006) for Iberia.

Median-joining network of red deer haplotypes. Haplogroup A: red, orange, and yellow tones; haplogroup B: green tones; haplogroup C: blue tones; haplogroup D: pink tones; ACQUERINO haplotypes are indicated in black. Haplotypes downloaded from NCBI web site are indicated as geographical area only; details about GenBank numbers are in Table 1. Size of each circle corresponds to the number of haplotypes; white circles represent mutation steps
Both trees (obtained with dataset 1 and dataset 2; tree with dataset 2 not shown) gave very similar results (Fig. 2): all ACQUERINO haplotypes belong to the clade Cervus elaphus, with HAP1_ ACQUERINO1-HAP4_ ACQUERINO part of haplogroup A and HAP5_ ACQUERINO of haplogroup C (lineages sensu Skog et al. 2009). The red deer haplogroup A, including most ACQUERINO haplotypes, was well supported statistically (Fig. 2, P = 0.98), unlike haplogroup C that showed a low support value (P = 0.62). All neutrality tests showed a demographic expansion with negative and significant values (Tajima D: − 243, P-value < 0.001; Fu and Li’s D: − 401, P-value < 0.002; Fu’s Fs: − 413 P-value < 0.002). Times of divergence between the three haplogroups reported in the chronograms (Fig. S2) showed as the European red deer population diverged during the last 3–1 million years.

Bayesian phylogenetic tree (MCMC = 10,000,000) of D-loop dataset 2 sequences of Cervus elaphus. Posterior probability values are reported at nodes. Lineage A: haplogroup A or western European lineage; lineage B: haplogroup B or Mediterranean lineage; lineage C: haplogroup C or eastern European lineage
Discussion
The low level of genetic variability found in ACQUERINO population is related to the small number of source individuals (Tarvisio Forest, NE Italy), along with the unknown multiple reintroductions in the 1980s by some individuals from farms located in the Bologna District (Mazzarone and Mattioli 1996). The ACQUERINO population showed low levels of genetic diversity when compared to those of various European and Mediterranean populations (Doan et al. 2017), whereas being similar to values obtained by Queiros et al. (2019). Besides, also active migrations may have occurred from close Alpine/Apennine populations since the 1960s throughout the Italian peninsula (Apollonio et al. 2010). However, the increased population size of ACQUERINO population, along with a constant increasing trend of Italian red deer population, is in agreement with the demographic tests militating for a population expansion. Another outcome of mitochondrial analysis, although preliminary, suggested a mixed composition of the ACQUERINO population with the most part of haplotypes grouped in lineage A, identified by Skog et al. (2009). The only one exception is haplotype 5 that grouped in lineage C including Tarvisio, Bulgaria, Hungary, and Poland, the last one representing the closest sequence to HAP5_ACQUERINO. Therefore, although the original population in ACQUERINO derived from free-ranging individuals captured in North-Eastern Italy (Mazzarone and Mattioli 1996), several unofficial releases from local caged areas may have occurred in the last 50 years, thus contributing to this genetic diversity.
Genetic studies on the red deer have involved mitochondrial DNA sequences (Ludt et al. 2004; Skog et al. 2009; Perez-Espona et al. 2009; Niedziałkowska et al. 2011; Fernández-García et al. 2014; Borowski et al. 2016; Schnitzler et al. 2018; Queiròs et al. 2019; Doan et al. 2022) and microsatellites (Hwme et al. 2006B; Zachos et al. 2016). These authors have shown that the large-scale genetic structure of European red deer has been shaped by the Late Pleistocene and Holocene glacial-interglacial cycles. The role of Italy as a possible refugium and the origin of North African and Sardinian red deer is likely. During the Quaternary ice ages, most of Northern Europe was covered by ice. Conversely, Southern Europe, including Italy, provided ice-free glacial refugia during the Late Pleistocene for many temperate species (Meiri et al. 2013; Carranza et al. 2016). Main European glacial refugia corresponded to the southern peninsulas of Iberia, Italy, and the Balkans. These three main refugia acted as bases for the postglacial recolonization of the continent for different taxa, carrying distinct genetic lineages as an inheritance of their long isolation from each other (Meiri et al. 2013; Carranza et al. 2016). Niedziałkowska et al. (2021) have recently suggested that the species survived the Last Glacial Maximum not only in the abovementioned southern European refugia, but also in more northern areas of Western and Eastern Europe and in the Urals. The history of haplogroups took place in a period of 3–1 mya, i.e. dating older than in previous data (Doan et al. 2022), but in agreement with other studies using the same molecular marker (Randi et al. 2001).
Even if our results are based on only 26 samples and cannot yet be considered conclusive, our data represent the first outcomes regarding the genetics of a peninsular Italian red deer population, except restriction fragment analysis (Lorenzini et al. 2005) and the Mesola Forest Reserve population (Zachos et al. 2014). The phylogenetic analysis provided information on the composition and genetic variability of the ACQUERINO deer population, confirming that the current distribution of red deer in Italy would be the result of a series of successful (re)introductions. Accordingly, it should be emphasized that carrying out initial genetic monitoring of the ACQUERINO deer represented a significant opportunity to predict any negative effects on the population viability (Borowski et al. 2016). Future phylogeographic studies involving the application of nuclear markers, as microsatellites, should be conducted to test whether this population is subjected to inbreeding depression. The results obtained in this paper integrated with eco-ethological data will be essential for the management of the species and to avoid choices that may have repercussions on the well-being of the population. In other words, the best way to avoid any future inbreeding depression is to promote the range expansion of this population (e.g. Kenney et al. 2014). Based on these results, it would be useful to examine a greater number of samples and other molecular markers, to understand what are the gene flows between the various Apennine nuclei and the actual genetic structure of the species in this region.
Data availability
All data are available by the corresponding author. Tissue samples are stored at the Institute of Biosciences and Bioresources — CNR, Sesto Fiorentino (FI) I-50019.
Code availability
Not applicable.
References
Akaike H (1974) A new look at the statistical model identification. IEEE Trans Automat Contr 19:716–723
Apollonio M, Andersen R, Putman R (2010) European ungulates and their management in the XXI Century. Cambridge University Press Editions, Cambridge, UK pp: 1–578.
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Becciolini V, Bozzi R, Viliani M, Biffani S, Ponzetta MP (2016) Body measurements from selective hunting: biometric features of red deer (Cervus elaphus) from Northern Apennine, Italy. Ital J Anim Sci 15:461–472
Boitani L, Lovari S, Vigna Taglianti A (2003) Fauna of Italy Mammalia III Carnivora-Arctiodactyla. Calderini Editins, Bologna, Italy: pp 1–412.
Borowski Z, Świsłocka M, Matosiuk M, Mirski P, Krysiuk K, Czajkowska M, Borkowska A, Ratkiewicz M (2016) Purifying selection, density blocking and unnoticed mitochondrial DNA diversity in the red deer. Cervus Elaphus Plos One 11:e0163191
Carnevali L, Pedrotti L, Riga F, Toso S (2009) Ungulates in Italy: status, distribution, abundance, management and hunting of ungulate populations in Italy—Report 2001–2005. Biol Cons Fauna 117:1–168
Carranza J, Salinas M, de Andrés D, Pérez-González J (2016) Iberian red deer: paraphyletic nature at mt DNA but nuclear markers support its genetic identity. Ecol Evol 6:905–922
Chirichella R, Mustoni A, Apollonio M (2017) Alpine landscape and canopy cover from 1973 to 2011: are roe and red deer population dynamics linked? Wildl Res 44:504–513
Doan K, Zachos FE, Wilkens B, Vigne JD, Piotrowska N, Stankovic A, Jędrzejewska B, Stefaniak B, Niedziałkowska M (2017) Phylogeography of the Tyrrhenian red deer (Cervus elaphus corsicanus) resolved using ancient DNA of radiocarbon-dated subfossils. Sci Rep 7:2331
Doan K, Niedziałkowska M, Stefaniak K, Sykut M, Jędrzejewska B, Ratajczak-Skrzatek U, Piotrowska N, Ridush B, Zachos FE, Popovic D, Baca M, Mackiewics P, Kosintsev P, Makowiecki D, Charniauski M, Boeskorov G, Anatolievich Bondarev A, Danila G, Kusak J, Manaseryan N, Krasnodebski D, Titov V, Hulva P, Balasescu A, Trantalidou K, Dimitrijevic V, Shpansky A, Kovalchuk O, Klementiev AM, Foronova I, Malikov DG, Juras A, Nikolskiy P, Egorovich Grigoriev S, Cheprasov MY, Novgorodov GV, Sorokin AD, Wilczinski J, Vasilievich Protopopov A, Lipecki G, Stankovic A (2022) Phylogenetics and phylogeography of red deer mtDNA lineages during the last 50,000 years in Eurasia. Zool J Linn Soc 194:431–456
Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:1–8
Fernández-García JL, Carranza J, Martínez JG, Randi E (2014) Mitochondrial D-loop phylogeny signals two native Iberian red deer (Cervus elaphus) lineages genetically different to Western and Eastern European red deer and infers human-mediated translocations. Biodivers Conserv 23:537–554
Frantz AC, Zachos FE, Bertouille S, Eloy MC, Colyn M, Flamand MC (2017) Using genetic tools to estimate the prevalence of non-native red deer (Cervus elaphus) in a Western European population. Ecol Evol 7:7650–7660
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Fu YX, Li WH (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Gippoliti S, Lai M, Milana G (2020) A roadmap to an evolutionarily significant conservation strategy for Cervus corsicanus. J Anim Div 2:8–15
Hartl GB, Zachos F, Nadlinger K (2003) Genetic diversity in European red deer (Cervus elaphus L.): anthropogenic influences on natural populations. C R Biol 326:37–42
Hmwe SS, Zachos FE, Eckert I, Lorenzini R, Fico R, Hartl GB (2006A) Conservation genetics of the endangered red deer from Sardinia and Mesola with further remarks on the phylogeography of Cervus elaphus corsicanus. Biol J Linn Soc 88:691–701
Hmwe SS, Zachos FE, Sale JB, Rose HR, Hartl GB (2006B) Genetic variability and differentiation in red deer (Cervus elaphus) from Scotland and England. J Zool 270(3):479–487
Hobbs NT (1996) Modification of ecosystems by ungulates. J Wildl Manage 60:695–713
Iacolina L, Corlatti L, Buzan E, Safner T, Šprem N (2019) Hybridisation in European ungulates: an overview of the current status, causes, and consequences. Mammal Rev 49:45–59
Kenney J, Allendorf FW, McDougal C, Smith JL (2014) How much gene flow is needed to avoid inbreeding depression in wild tiger populations? Proc R Soc B 281:20133337
Leigh JW, Bryant D (2015) Popart: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452
Lorenzini R, Fico R, Mattioli S (2005) Mitochondrial DNA evidence for a genetic distinction of the native red deer of Mesola, northern Italy, from the Alpine populations and the Sardinian subspecies. Mamm Biol 70(3): 187–198
Lovari S (1993) Recent development of the populations of large mammals of the fauna of Italy. In: XIX Seminar on Biological Evolution and the Great Problems of Biology Rome, Italy: 21–37
Ludt CJ, Schroeder W, Rottmann O, Kuehn R (2004) Mitochondrial DNA phylogeography of red deer (Cervus elaphus). Mol Phylogen Evol 31:1064–1083
Mattioli S (1990) Red deer in the Italian peninsula with particular reference to the Po delta population. Deer 8:95–98
Mattioli S, Meneguz PG, Brugnoli A, Nicoloso S (2001) Red deer in Italy: recent changes in range and numbers. Hystrix 12:27–35
Mattioli S, Fico R, Lorenzini R, Nobili G (2003) Mesola red deer: physical characteristics, population dynamics and conservation perspectives. Hystrix 14:1–2
Mazzarone V, Mattioli S (1996) Survey of the deer population of Aquerino: final report 1993–1995 Tuscany Region, Florence, Italy.
Mcdevitt AD, Edwards CJ, O’Toole P, O’sullivan P, O’Reilly C, Carten RF (2009) Genetic structure of, and hybridisation between, red (Cervus elaphus) and sika (Cervus nippon) deer in Ireland. Mamm Biol 74:263–273
Meiri M, Lister AM, Higham TF, Stewart JR, Straus LG, Obermaier H, González Morales MR, Marín-Arroyo AB, Barnes I (2013) Late-glacial recolonization and phylogeography of European red deer (Cervus elaphus L). Mol Ecol 22:4711–4722
Murray BD, Webster CR, Bump JK (2013) Broadening the ecological context of ungulate–ecosystem interactions: the importance of space, seasonality, and nitrogen. Ecology 94:1317–1326
Niedziałkowska M, Jędrzejewska B, Honnen AC, Otto T, Sidorovich VE, Perzanowski K, Zachos FE (2011) Molecular biogeography of red deer Cervus elaphus from eastern Europe: insights from mitochondrial DNA sequences. Acta Theriol 56:1–12
Niedziałkowska M, Doan K, Gorny M, Sykut M, Stefaniak K, Piotrowska N, Jędrzejewska B, Ridush B, Pawełczyk S, Mackiewicz P, Schmölcke U, Kosintsev P, Makowiecki D, Charniauski M, Krasnodębski D, Rannamäe E, Saarma U, Arakelyan M, Manaseryan N, Titov VV, Hulva P, Bălășescu A, Fyfe R, Woodbridge J, Trantalidou K, Dimitrijević V, Kovalchuk O, Wilczyński J, Obadă T, Lipecki G, Arabey A, Stanković A (2021) Winter temperature and forest cover have shaped red deer distribution in Europe and the Ural Mountains since the Late Pleistocene. J Biogeogr 48:147–159
Pérez-Espona S, Pérez-Barbería FJ, Goodall-Copestake WP, Jiggins CD, Gordon IJ, Pemberton JM (2009) Genetic diversity and population structure of Scottish Highland red deer (Cervus elaphus) populations: a mitochondrial survey. Heredity 102:199–210
Posada D, Crandall KA (1998) MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818
Putman RJ (1996) Ungulates in temperate forest ecosystems: perspectives and recommendations for future research. Forest Ecol Manage 88:205–214
Queirós J, Acevedo P, Santos JPV, Barasona J, Beltran-Beck B, González-Barrio D, Armenteros JA, Diez-Delgado I, Boadella M, Fernandéz de Mera I, Ruiz-Fons JF, Vicente J, de la Fuente J, Gortázar C, Searle JB, Alves PC (2019) Red deer in Iberia: molecular ecological studies in a southern refugium and inferences on European postglacial colonization history. PLoS One. 8;14(1):e0210282.
Rambaut A (2009) FigTree, ver. 1.3. 1. Document available online at: https://www.tree.bio.ed.ac.uk/software/figtree. Accessed 21 Aug 2021.
Randi E, Mucci N, Claro-Hergueta F, Bonnet A, Douzery EJ (2001). A mitochondrial DNA control region phylogeny of the Cervinae: speciation in Cervus and implications for conservation. In: AA.VV. (Eds) Animal Conservation forum 4. Cambridge University Press Editions, Cambridge, UK: 1-11.
Reimoser F (2003) Steering the impacts of ungulates on temperate forests. J Nat Cons 10:243–252
Rey-Iglesia A, Grandal-d’Anglade A, Campos PF, Hansen AJ (2017) Mitochondrial DNA of pre-last glacial maximum red deer from NW Spain suggests a more complex phylogeographical history for the species. Ecol Evol 7(24):10690–10700
Schnitzler A, Granado J, Putelat O, Arbogas RM, Drucker D, Eberhard A, Schmutz A, Klaefiger Y, Salzburger W, Schibler J, Schlumbaum A, Bocherens H (2018) Genetic diversity, genetic structure and diet of ancient and contemporary red deer (Cervus elaphus L.) from north-eastern France. PloS One 13:e0189278
Schwarz G (1978) Estimating the dimension of a model. Ann Stat 6:461–464
Skog A, Zachos FE, Rueness EK, Feulner PGD, Mysterud A, Langvatn R, Lorenzini R, Hmwe SS, Lehoczky I, Hartl GB, Stenseth NC, Jakobsen KS (2009) Phylogeography of red deer (Cervus elaphus). J Biogeogr 36:66–77
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729
Zachos FE, Hartl GB (2011) Phylogeography, population genetics and conservation of the European red deer Cervus elaphus. Mamm Rev 41:138–150
Zachos FE, Hajji GM, Hartl GB, Lorenzini R, Mattioli S (2009) Population viability analysis and genetic diversity of the endangered red deer Cervus elaphus population from Mesola, Italy. Wild Biol 15:175–186
Zachos FE, Mattioli S, Ferretti F, Lorenzini R (2014) The unique Mesola red deer of Italy: taxonomic recognition (Cervus elaphus italicus nova ssp., Cervidae) would endorse conservation. Ital J Zool 81:136–143
Zachos FE, Frantz AC, Kuehn R, Bertouille S, Colyn M, Niedziałkowska M, Flamand MC (2016) Genetic structure and effective population sizes in European red deer (Cervus elaphus) at a continental scale: insights from microsatellite DNA. J Hered 107:318–326
Acknowledgements
Professor Frank Zachos and Stefano Mattioli kindly provided us with useful comments on an early draft. Bruno Esattore and Andrea Viviano helped us with subspecies identification. Dr. E. Basset (University of Birmingham) revised our manuscript and improved the English grammar and syntax. Two anonymous reviewers and the Editor Magdalena Niedzialkowska kindly improved our MS with their useful comments.
Funding
Tuscany Region, Hunting and Fishing Department.
Author information
Authors and Affiliations
Contributions
FD conceived the idea; FG collected data; MB conducted the genetic analyses and organized the dataset; all authors wrote the MS. All authors approved the final draft before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Communicated by: Karol Zub
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Giovannelli, F., Mori, E., Zaccaroni, M. et al. Genetic insights into an Apennine population of the Italian red deer. Mamm Res 67, 399–406 (2022). https://doi.org/10.1007/s13364-022-00637-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13364-022-00637-3