
Abstract
European red deer are known to show a conspicuous phylogeographic pattern with three distinct mtDNA lineages (western, eastern and North-African/Sardinian). The western lineage, believed to be indicative of a southwestern glacial refuge in Iberia and southern France, nowadays covers large areas of the continent including the British Isles, Scandinavia and parts of central Europe, while the eastern lineage is primarily found in southeast-central Europe, the Carpathians and the Balkans. However, large parts of central Europe and the whole northeast of the continent were not covered by previous analyses. To close this gap, we produced mtDNA control region sequences from more than 500 red deer from Denmark, Germany, Poland, Lithuania, Belarus, Ukraine and western Russia and combined our data with sequences available from earlier studies to an overall sample size of almost 1,100. Our results show that the western lineage extends far into the European east and is prominent in all eastern countries except for the Polish Carpathians, Ukraine and Russia where only eastern haplotypes occurred. While the latter may actually reflect the natural northward expansion of the eastern lineage after the last ice age, the present distribution of the western lineage in eastern Europe may in large parts be artificial and a result of translocations and reintroduction of red deer into areas where the species became extinct in historical times.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The cyclic climatic changes during the Quaternary have had a profound impact on the genetic structure of European temperate species in that these have been undergoing repeated range contractions and expansions in the wake of glacial and interglacial pulses (e.g. Hewitt 2000). During glacials, most temperate European species retreated to refuge areas in the south of the continent, viz. the Iberian Peninsula and southern France, Italy and the Balkan and Carpathian regions, while in the following interglacials with their milder climate these species recolonised their previously abandoned territories (Hewitt 1996, 2000; Sommer and Nadachowski 2006). However, the refugial populations that expanded from the south carried distinct genetic lineages as a legacy of their long isolation from each other. The identification of these lineages in present populations throughout Europe and their geographic distribution (“phylogeography”) allows for the reconstruction of glacial refugia and postglacial recolonisation routes. It is important to note that for present phylogeographic patterns, it is the last glacial maximum (LGM) 25,000–18,000 BP that is decisive because in this time distribution ranges were more contracted than ever before in the history of extant species (Sommer and Zachos 2009). Therefore, it is only the refugia during the LGM that served as reservoirs for the recolonisation of central and northern Europe in the Late Pleistocene and Holocene (Sommer and Zachos 2009). In Europe, most species have been found to follow a number of recolonisation patterns, the most common of which are the so-called “grasshopper pattern” (recolonisation from a primarily Balkan refuge), “bear pattern” (main recolonisation from a western/Iberian refuge) and the “hedgehog pattern” (recolonisation from Iberia, Italy and the Balkans) (Hewitt 1999).
The European red deer Cervus elaphus has been in the focus of evolutionary and conservation biologists alike in the recent past. As one of the most widespread and most important game species it has undergone a variety of human influences such as selective hunting, translocations and habitat fragmentation which have locally and regionally resulted in high levels of genetic drift, inbreeding and the blurring of natural patterns of genetic structure (e.g. Hartl et al. 2003; Zachos et al. 2007). Human impact on genetic diversity is not specific to red deer: there are many other species where human translocations and restocking modified genetic structure, e.g. the brown hare Lepus europaeus (Andersen et al. 2009) and the wild boar Sus scrofa (Nikolov et al. 2009).
Red deer have also been studied in detail with respect to their phylogeography, both addressing more specific questions like the origin of the Sardinian/Corsican subspecies (Hmwe et al. 2006a; Hajji et al. 2008) and continent-wide patterns (Ludt et al. 2004; Skog et al. 2009, for an inclusion of the fossil record see Sommer et al. 2008). Skog et al. (2009), in by far the most comprehensive study to date, have found three distinct genetic lineages in European red deer based on mitochondrial DNA (cytochrome b and the control region): one confined to Sardinia and the north-African Barbary red deer; a western lineage spreading from Iberia through the British Isles to Scandinavia, Germany and western Poland; and an eastern lineage covering the Balkans, the Romanian Carpathians and reaching north to Austria, Hungary and the Czech Republic. There were only very few exceptions to this geographic pattern, indicating that most of the extensive translocations in red deer—at least as far as females are concerned—were carried out within these lineages rather than among them. However, large parts of central Europe (particularly where the western and eastern lineages might meet north of the Czech Republic) and the whole northern parts of eastern Europe were not covered by the study of Skog et al. (2009). It is hence unknown how far to the east the western lineage extends and how far north the eastern lineage reaches.
In the present study we aim at closing this gap by analysing mtDNA control region sequences from more than 500 red deer from seven countries in central and eastern Europe (Denmark, Germany, Poland, Lithuania, Ukraine, Belarus and Russia). We combine our data set with that of Skog et al. (2009, n = 587) and base our conclusions on data from nearly 1,100 animals, making this study one of the most comprehensive analyses ever of European phylogeography. In addition, by means of an extensive literature search focusing on sources from eastern Europe and Russia, we try to estimate in how far the patterns observed today may be attributed to natural colonisation and to which extent translocations might have blurred the natural genetic structure in east-European red deer.
Material and methods
Sampling information
Muscle or skin samples from 507 legally culled red deer were collected in Denmark, Germany, Poland, Belarus, Ukraine, Lithuania and Russia (see Fig. 1 and Table 1 for details). It is important in the context of this study to consider human influences on red deer distribution. In the seventeenth to nineteenth centuries, red deer numbers declined in many countries of the continent (Hartl et al. 2003; Kuehn et al. 2003, 2004; Hmwe et al. 2006b; Nielsen et al. 2008), and in some places, especially in central and eastern Europe (east of the Vistula River), the species even became extinct (Mager 1941; Baleišis and Škerys 1984; Baleišis 1988). Since then, red deer have been translocated and/or migrated from places where densities were still high to areas where they were absent or occurred in low numbers (Mager 1941; Barabash-Nikiforov and Pavlovskii 1949; Frevert 1964; Kozlo 1970; Vereshchagin and Rusakov 1979; Baleišis and Škerys 1984; Baleišis 1988; Hmwe et al. 2006b; Nussey et al. 2006). In our study area, red deer were moved e.g. from Germany, Austria and Russia to north-eastern Poland, from central, northern and western Poland to eastern Poland, from Voronezkskii Zapovednik (Russia) to Lithuania and Belarus (Mager 1941; Frevert 1964; Kozlo 1970; Baleišis and Škerys 1984; Baleišis 1988; Niedziałkowska 2008). Red deer migrated from Latvia and the Kaliningrad area (Russia) to Lithuania and northern Russia (near Petersburg) (Vereshchagin and Rusakov 1979; Baleišis and Škerys 1984; Baleišis 1988). Before these migrations started, red deer were translocated to Latvia (Baleišis 1988) and Voronezskii Zapovednik (Russia) from Germany at the end of the nineteenth century (Barabash-Nikiforov and Pavlovskii 1949). Red deer were also transferred to Latvia from Poland and the Caucasian Mountains (Baleišis 1988). Moreover, there have been numerous translocations to Germany since the Middle ages from many countries including Russia, the UK, Austria, Hungary and Poland (Niethammer 1963, and unpublished material). In eastern Europe native populations survived only in some places in north-eastern Poland (Niedziałkowska 2008), the Kaliningrad area (Russia), Latvia (Vereshchagin and Rusakov 1979) and in Karpatskii Zapovednik in Ukraine (Tatarinov 1973).

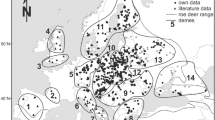
Map showing red deer distribution in Europe and sample sites of the present study and Skog et al.’s (2009). Sample sites: 1 Smolensk Region, Russia; 2 Bransk oblast, Russia; 3 Vitebsk Region, Belarus; 4 Mogilev Region, Belarus; 5 Gomel Region, Belarus; 6 Grodno Region, Belarus and Joniškis and Širvintos Districts, Lithuania; 7 Augustów, Borki and Rominta Forests, Poland; 8 Białowieża Forest (both Polish and Belarusian parts), Knyszyn and Mielnik Forests, Łomża and Sokołów Forest Districts, Poland, Brest Region, Belarus; 9 Napiwoda and Pisz Forests, Poland; 10 Bytów, Młynary, Susz, Zaporowo Forest Districts, Poland; 11 Spała and Poddębice Forest Districts, Poland; 12 Brynek, Milicz, Olesno, Prószków, Świerklaniec Forest Districts (Silesia) Poland; 13 Bieszczady Mts., Poland; 14 Tismenickii Raion, Ivano-Frankivskaya oblast, Ukraine; 15 Dolinskii Raion, Ivano-Frankivskaya oblast, Ukraine; 16 Saxony, Germany; 17 Rostock, Germany; 18 Mecklenburg, Germany; 19 Lauenburg, Germany; 20 southern Denmark; 21 Hils region, Germany; 22 Spessart and Vogelsberg, Germany; 23 Kyllwald and Gerolstein, Germany
Molecular and statistical analysis
DNA was extracted from tissue samples using the Qiagen DNeasy blood and tissue kit following the manufacturer’s protocol. A part of the mitochondrial control region was amplified by means of PCR using the following primer pairs: Pro-L/Phe-Hb and LD5/HD6 (see Nagata et al. 1998 and Zachos et al. 2003 for details). Amplicons were then sequenced on an automated sequencer ABI 3100.
Electropherograms were checked by eye, and the sequences were aligned with Clustal implemented in BioEdit (Hall 1999). When combining the different data sets we had to shorten the data set used by Skog et al. (2009) and various other sequences so as to cover the largest possible geographical scale. Our final alignment comprised 249 bp and 1,094 red deer (507 from this study plus 587 from Skog et al. 2009) from virtually the whole European distribution range of the species. Sequences were collapsed into haplotypes with the FaBox package (http://www.birc.au.dk/~biopv/php/fabox/, Villesen 2007). Haplotype and nucleotide diversities were calculated with DnaSP (Rozas and Rozas 1999). While haplotype diversity is based on haplotype frequency alone, nucleotide diversity also takes into account the amount of divergence among the haplotypes found in a sample.
To address the phylogenetic and phylogeographic relationships among the haplotypes we constructed a median-joining network with Network (Bandelt et al. 1999). Networks are generally better suited to depict intraspecific phylogenies than are tree algorithms because they allow for the co-existence of ancestral and descendant alleles in a sample, whereas trees treat all sequences as terminal taxa (see Posada and Crandall 2001). We only considered different haplotypes for the construction of the network and disregarded their frequencies as the large discrepancies in sample size among localities would have introduced a severe sampling bias. To compare our data with the trees in Skog et al. (2009) we also generated phylogenetic trees. We first inferred a model of nucleotide substitution for our data set (GTR plus Gamma correction, shape parameter α = 0.189) with the Modeltest algorithm (Posada and Crandall 1998) implemented in the Findmodel software (http://www.hiv.lanl.gov/content/sequence/findmodel/findmodel.html) and then calculated a neighbour-joining (NJ) tree with the MEGA software (Tamura et al. 2007) under a TrN model (which is the closest available MEGA model to GTR). The same programme was also used to calculate a maximum parsimony tree. Node support was assessed by means of 1,000 bootstrap replicates in both approaches. As outgroups, we chose a wapiti Cervus elaphus canadensis and a sika deer Cervus nippon sequence (see Skog et al. 2009 for details).
Results
Our alignment (249 bp) of the 507 red deer from central and eastern Europe yielded 50 polymorphic sites with altogether 38 transitions, 15 transversions and one indel (four sites showed three different character states), defining 28 haplotypes. Five of them were identical to haplotypes found by Skog et al. (2009), and our combined alignment of 69 haplotypes (GenBank accession numbers HQ290005-HQ290073) included 41 further haplotypes from Skog et al. (2009) not found in the red deer from the present study. This means that, of the originally 58 haplotypes from the Skog et al. (2009) study, 12 were lost due to the reduction in sequence length from 332 to 249 bp, but structuring into the known red deer lineages was still evident (see below). Ten haplotypes were singletons, and the most frequent one (haplotype 1) occurred 108 times, but obviously these numbers are biased by the huge differences in sample size across our study area. Table 1 gives diversity values and haplotype distributions for different sampling regions. In most cases we combined different sampling sites based on geographical proximity and sample sizes. We are aware that this is in cases somewhat arbitrary, but for the main aim of our study the exact delineation of (genetic) populations is not relevant. A detailed overview of all haplotypes, their frequencies, geographical distribution and identity with those of Skog et al. (2009) is given in Table 2.
The phylogenetic analyses yielded congruent results. In the network (Fig. 2) and both trees (MP tree: Appendix) the three lineages A, B and C can easily be identified, although A is not monophyletic in the neighbour-joining tree (not shown) and C is split into two groups in the network (one of which comprises only haplotypes 9 and 15). Lineage B, typical of the North-African Barbary stag Cervus elaphus barbarus and the Corsican red deer Cervus elaphus corsicanus from Sardinia and Corsica, was monophyletic in both trees and also clearly formed a separate cluster in the network. As in Skog et al. (2009) haplogroup B was basal to A and C in the MP tree. None of our samples carried a B haplotype.

Median-joining network of the 69 red deer mtDNA control region haplotypes found in the present study. Vertical dashes along branches denote the number of mutational steps between haplotypes. Absence of dashes is equivalent to a single mutational event (irrespective of branch length). A, B and C refer to the western, North-African/Sardinian and eastern lineages, respectively
Haplotypes 16 and 41 deserve particular attention. The latter, found exclusively in Mesola in Italy, belonged to the eastern lineage C according to all approaches which is in contrast to results from Skog et al. (see “Discussion”). Haplotype 16, found in a single red deer from western Germany, was the most aberrant sequence in our data set. In the network, it forms a long branch attached to lineage A, and in the maximum parsimony tree it is the sister taxon to all other ingroup sequences, while in the NJ tree it is basal to clade B with both of them nested within lineage A. Bootstrap support, however, was below 50% in both cases.
Of the 28 haplotypes found in our 507 red deer, 17 belonged to the western lineage A and ten to the eastern lineage C. The remaining one is the aberrant haplotype 16 which shows closest affinities to lineage A (in line with its occurrence in western Germany), but is separated by 26 mutational steps from the closest A haplotype (see Fig. 2). GenBank comparisons have confirmed its red deer origin. We sequenced this sample twice to confirm the haplotype; the quality of the electropherograms was the same as for the other sequences. The geographical distribution of the 28 haplotypes showed that most of east-central and eastern Europe is inhabited by western-type red deer (Table 1 and Fig. 3). Indeed, the only sample sites with exclusively eastern lineage haplotypes were Bieszczady Mountains in the Polish Carpathians, western Russia and Ukraine. Apart from these regions, C haplotypes were also found together with A haplotypes in eastern Belarus (four C sequences in 13 individuals, i.e. 31%), Lithuania (11%), and central/western Poland (8%). A haplotypes dominate in these populations. Haplotype 9 from the eastern lineage C was exclusively found in eastern Belarus, Lithuania and western Russia. Some haplotypes showed wide and often disjunct distributions, e.g. haplotypes 2, 8 and 10 (found in western Germany and Poland) or, even more striking, haplotype 4 which was found in western Germany and some of our easternmost sample sites including north-eastern Poland, Belarus and Lithuania.

Geographical distribution of mtDNA control region haplotypes from the three major genetic lineages in Europe; asterisk at site 23 one individual with the most divergent haplotype 16 was found; double asterisks Mesola haplotype (41) was intermediate between lineages A and C in Skog et al. (2009).
Discussion
We analysed mitochondrial control region sequences from almost 1,100 red deer sampled throughout their entire European distribution range. Some 500 of these sequences were new, and the vast majority was sampled in areas not covered by the previous large phylogeographic study by Skog et al. (2009). Based on these new data, we found that the western lineage A extends far into the east of Europe before being replaced by the eastern lineage in western Russia. In central Europe, both lineages occur in close proximity.
While the geographic coverage of our data set is nearly complete with respect to Europe, our alignment is rather short with 249 bp. This may account for the poor bootstrap support for both neighbour-joining and maximum parsimony trees and is also the likely explanation why in the former haplogroup A is not monophyletic (in contrast to results by Skog et al. 2009 and our maximum parsimony tree, although bootstrap support for clade A in Skog et al. was also low). Nevertheless, the length of our sequences was sufficient to clearly discriminate the three haplogroups known to occur in red deer. When differentiation between major genetic lineages is large, short alignments have been shown to yield good results in phylogeographic studies, e.g. in lions (130 bp, Barnett et al. 2006) and wolves (257 bp, Pilot et al. 2006).
Our results are in line with those of Skog et al. (2009) in that we found the two predominant European lineages A and C with the western A much more diverse and structured than the eastern C. Both Skog et al. (2009) and the present study revealed 10 singletons, but the geographical scale and the overall number of haplotypes in Skog et al.’s study was much larger, which may be taken as evidence that there are more rare haplotypes in central and eastern Europe than in the continent as a whole. We also found high nucleotide diversity and a high number of haplotypes in the Polish Carpathians two of which (13 and 19) are identical with haplotypes found in the Romanian part of the mountain range. One of these (19) is exclusively found in the Carpathians. This is in line with Carpathian red deer being genetically special (cf. Feulner et al. 2004), although introduction of Austrian red deer into the Carpathians has been recorded (Micu et al. 2010). The present data set is clearly insufficient to settle this question. In general, genetic diversity was within the range known for red deer with areas where both the A and C lineages were found and the German sample with the aberrant haplotype 16 expectedly showing higher values.
The phylogenetic position of haplotype 41 is interesting. It is exclusively found in Mesola, in the Po delta region in Italy. The Mesola red deer are the only surviving native Italian red deer (see Zachos et al. 2009), and since red deer are known to have occurred in Italy during the Last Glacial Maximum (Sommer et al. 2008), they are important from a phylogeographic point of view. The study of Skog et al. (2009) yielded unequivocal results as to whether the Mesola haplotype was more closely related to lineage A or C. In our analyses, it is consistently grouped with lineage C, but it remains unclear if it really belongs to one of them or perhaps rather represents a remnant of an otherwise extinct Italian refugial lineage. Perhaps both Skog et al.’s alignment (332 bp) and ours (249 bp) are too short to reliably infer the phylogenetic position of the Mesola haplotype.
The two major mitochondrial lineages in European red deer indicate a western refuge in Iberia and southern France and an eastern refuge in the Balkans and perhaps the Carpathian region (Sommer et al. 2008; Skog et al. 2009). Together with the North-African/Sardinian lineage B they probably diverged between roughly 200,000 and 400,000 BP (Skog et al. 2009), i.e. before the last glaciation as often found for mitochondrial lineages in European species (Taberlet et al. 1998). Our results show a wide expansion of the western haplotype lineage A into eastern Europe. The eastern lineage C occurs in our southernmost and easternmost sample sites, the Polish Carpathians, Ukraine and western Russia. In addition, it is found together with A haplotypes in parts of Poland, Lithuania and Belarus. In eastern Belarus, the proportion of C haplotypes is higher than in the other regions, and it may not be a coincidence that these sample sites are close to regions where only the C lineage is found (although our all-C western Russian sample was very small with n = 4, so A haplotypes might have been missed there). It is well documented that the red deer in Ukraine at least partly originated from the autochthonous Carpathian red deer population (Tatarinov 1973), so the exclusive occurrence of C haplotypes is probably natural there. It is also possible that the red deer from Ukraine migrated to the north to Belarus and western Russia which would explain why C haplotypes dominate in these areas. However, in Belarus, Lithuania and Russia most of the present red deer populations originated from animals that were either translocated (mainly from Germany) or migrated there from Latvia, the Kaliningrad area or Poland (Sztolcman 1918; Barabash-Nikiforov and Pavlovskii 1949; Vereshchagin and Rusakov 1979; Baleišis and Škerys 1984; Baleišis 1988). This origin of red deer in Belarus and Lithuania is in accordance with the finding of the western lineage A in that region of Europe.
The geographical distribution of lineages A and C in our sample regions suggests that the western lineage A has expanded far more to the east than hitherto assumed. Lineage C may not have successfully recolonised the northern parts of eastern Europe (Poland north of the Carpathians, Belarus and Lithuania) but, as suggested by our Russian sample, became confined to the east of the distribution range of lineage A. This is in line with the fact that the proportion of C haplotypes increases towards the south and the east. However, since we are dealing with red deer, arguably the most important and most highly held European game species, translocations are the crucial unknown entering the equation. These translocations may have contributed to high genetic diversity in some areas through the introduction of divergent haplotypes. This may particularly hold for western Germany with the aberrant haplotype 16. The very high nucleotide diversity found in eastern Belarus is due to the presence of both most divergent haplotypes from the C lineage (9 and 15), but whether this is due to translocations or natural migration remains unclear. Haplotype 1 is found in eastern Germany and (particularly north-eastern) Poland, haplotype 20 in eastern Germany and Lithuania, and an even more disjunct distribution is shown by haplotypes 2, 8 and 10 which are found in western Germany and Poland (including the northeast of the country). However, the most unlikely candidate for a natural distribution is haplotype 4: it is found in western Germany and then again in northeastern Poland, Belarus and Lithuania. Such a pattern is almost certainly a result of translocations. There are several documented records of translocations of red deer at the end of the nineteenth and beginning of the twentieth centuries from Germany to north-eastern Poland, e.g. to Pisz, Białowieża and Rominta Forests (Genko 1902–1903; Karcov 1903; Mager 1941, 1960; Frevert 1964; Gautschi and Winsmann-Steins 1992; Serafin 1995; Niedziałkowska 2008), and to Voronezkskii Zapovednik (Russia) and Latvia (Barabash-Nikiforov and Pavlovskii 1949; Baleišis 1988) so it is possible that the haplotypes of the introduced individuals have survived there and in places where they were translocated or migrated from them. After the translocations, the number of red deer in north-eastern Poland increased, and in the twentieth century they were translocated or migrated to neighbouring countries, e.g. Lithuania and Belarus where the red deer was absent or occurred in lower numbers (Sztolcman 1918; Baleišis and Škerys 1984; Baleišis 1988; Niedziałkowska 2008). From Latvia and Voronezkskii Zapovednik (Russia) red deer were moved or migrated to Lithuania and Belarus (Barabash-Nikiforov and Pavlovskii 1949; Kozlo 1970; Vereshchagin and Rusakov 1979; Baleišis and Škerys 1984; Baleišis 1988).
In addition to introductions from Germany and other European countries (Niedziałkowska 2008) red deer were transferred to the eastern part of Poland from central (Spała Forest), western (Silesia) and northern (Pomerania) Poland. This might be an explanation for the presence of haplotype 2 in only Germany and western and north-eastern Poland. The distribution of haplotypes 8 and 10 could also be the result of human impacts; alternatively, these haplotypes may be widespread in Poland through natural migration. Since obviously on this geographical scale human management has blurred the natural genetic structure of red deer in eastern Europe, it is impossible to fully disentangle natural and anthropogenic patterns. Interestingly, though, the amount of mismatch between genetic lineages and geography is much higher in our study area than in the rest of Europe where Skog et al. (2009) only very rarely found animals carrying a genetic signature not matching their geographical origin. This may suggest that inter-lineage translocations were more common in eastern Europe, or alternatively, it may simply be the result of the two lineages occurring parapatrically in the east which will make admixture more likely if translocations are carried out or natural migration occurs. Ultimately, only ancient DNA analyses of red deer remains or trophies dating back to before translocations were carried out (ideally early or middle Holocene samples), perhaps combined with detailed local population genetic analyses and admixture studies, can answer the question if the present domination of western haplotypes in northeastern Europe is natural or caused by human interference.
References
Andersen LW, Fredsted T, Wincentz T, Pertoldi C (2009) Brown hares on the edge: genetic population structure of the Danish brown hares. Acta Theriol 54: 97–110
Baleišis R (1988) Razlichiya v stroenii rogov blagorodnykh olenei Litvy. Trudy Akademii nauk Litovskoi SSR, Seria B: 87–94 [In Russian]
Baleišis R, Škerys J (1984) Introdukciya, pereselenie i mechenie blagorodnogo oleniya v Litve. Trudy Akademii nauk Litovskoi SSR, Seria B: 89-96 [In Russian]
Bandelt H-J, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48
Barabash-Nikiforov II, Pavlovskii NK (1949) Fauna nazemnykh pozvonochnykh Voronezhskogo gosudarstvennogo zapovednika [Fauna of terrestrial vertebrates of Voronezh State Reserve]. Trudy VGZ, Voronezh, vol. II. [In Russian]
Barnett R, Yamaguchi N, Barnes I, Cooper A (2006) The origin, current diversity and future conservation of the modern lion (Panthera leo). Proceedings of the Royal Society London B 273: 2119–2125 doi:10.1098%2Frspb.2006.3555
Feulner PGD, Bielfeldt W, Zachos FE, Bradvarovic J, Eckert I, Hartl GB (2004) Mitochondrial DNA and microsatellite analyses of the genetic status of the presumed subspecies Cervus elaphus montanus (Carpathian red deer). Heredity 93: 299–306 doi:10.1038%2Fsj.hdy.6800504
Frevert W (1964) Rominten. Bayerischer Landwirtschaftsverlag, München
Gautschi A, Winsmann-Steins B (1992) Rominten—Gestern und Heute. Nimrod-Verlag, Bothel
Genko N (1902–1903) Kharakteristika Belovezhskoi Pushchi i istoricheskiya o nei dannya. Lesnoi Zhurnal 22(5): 1014–1156, 22(6): 1269–1302, 23(1): 22–56 [In Russian]
Hajji GM, Charfi-Cheikrouha F, Lorenzini R, Vigne JD, Hartl GB and Zachos FE (2008) Phylogeography and founder effect of the endangered Corsican red deer (Cervus elaphus corsicanus). Biodivers Conserv 17: 659–673 doi:10.1007%2Fs10531-007-9297-9
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41: 95–98
Hartl GB, Zachos F and Nadlinger K (2003) Genetic diversity in European red deer (Cervus elaphus L.): anthropogenic influences on natural populations. Comptes Rendus Biologies 326: S37–S42 doi:10.1016%2FS1631-0691%2803%2900025-8
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58: 247–276 doi:10.1006%2Fbijl.1996.0035
Hewitt GM (1999) Post-glacial recolonization of European biota. Biol J Linn Soc 68: 87–112 doi:10.1006%2Fbijl.1999.0332
Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature 405: 907–913
Hmwe SS, Zachos FE, Eckert I, Lorenzini R, Fico R and Hartl GB (2006a). Conservation genetics of the endangered red deer from Sardinia and Mesola with further remarks on the phylogeography of Cervus elaphus corsicanus. Biol J Linn Soc 88: 691–701 doi:10.1111%2Fj.1095-8312.2006.00653.x
Hmwe SS, Zachos FE, Sale JB, Rose HR and Hartl GB (2006b) Genetic variability and differentiation in red deer (Cervus elaphus) from Scotland and England. Biol J Zool 270: 479–487 doi:10.1111%2Fj.1469-7998.2006.00123.x
Karcov G (1903) Belovezhskaya Pushcha. Ee istoricheskii ocherk, sovremennoe okhotniche khozaistvo i vysochaishe okhoty v Pushche. A. Marks, Sankt Petersburg: 1–413 [In Russian]
Kozlo PG (1970) Chislennost i razmeshchenie kopytnykh v Berezinskom zapovednike [Numbers and distribution of ungulates in Berezinskii Reserve]. Berezinskii Zapovednik, Issledovaniya 1: 122–128 [In Russian]
Kuehn R, Schroeder W, Pirchner F, Rottmann O (2003) Genetic diversity, gene flow and drift in Bavarian red deer populations (Cervus elaphus). Conserv Genet 4: 157–166. doi:10.1023/A:1023394707884
Kuehn R, Haller H, Schroeder W, Rottmann O (2004) Genetic roots of the red deer (Cervus elaphus) population in Eastern Switzerland. J Heredity 95: 136–143 http://dx.doi.org/10.1093%2Fjhered%2Fesh019
Ludt CJ, Schroeder W, Rottmann O and Kuehn R (2004) Mitochondrial DNA phylogeography of red deer (Cervus elaphus). Molecular Phylogenetics and Evolution 31: 1064–1083 doi:10.1016%2Fj.ympev.2003.10.003
Mager F (1941) Wildbahn und Jagd Altpreussens im Wandel der geschichtlichen Jahrhunderte. Verlag J, Neumann, Neudamm und Berlin
Mager F (1960) Der Wald in Altpreussen als Wirtschaftsraum. Bohlau-Verlag, Köln
Micu I, Nahlik A, Negus S, Mihalache I, Szabo I (2010) Ungulates and their management in Romania. In: Apollonio M, Andersen R, Putman R (eds) European Ungulates and their Management in the 21st Century. Cambridge University Press, Cambridge
Nagata J, Masuda R, Kaji K, Kaneko M and Yoshida MC (1998) Genetic variation and population structure of the Japanese sika deer (Cervus nippon) in Hokkaido Island, based on mitochondria D-loop sequences. Mol Ecol 7: 871–877 doi:10.1046%2Fj.1365-294x.1998.00404.x
Niedziałkowska M (2008) Struktura genetyczna populacji jelenia Cervus elaphus w puszczach północno-wschodniej Polski. Dissertation, University of Warsaw [In Polish]
Nielsen K, Olesen E, Pertoldi C, Gravlund P, Barker JSF, Mucci N, Randi E, Loeschcke V (2008) Genetic structure of the Danish red deer (Cervus elaphus). Biol J Linn Soc 95: 688–701. doi:10.1111/j.1095-8312.2008.01115.x
Niethammer G (1963) Die Einbürgerung von Säugetieren und Vögeln in Europa. Paul Parey, Hamburg, Berlin
Nikolov IS, Gum B, Markov G and Kuehn R (2009) Population genetic structure of wild boar Sus scrofa in Bulgaria as revealed by microsatellite analysis. Acta Theriol 54: 193–205 doi:10.4098%2Fj.at.0001-7051.049.2008
Nussey DH, Pemberton J, Donald A, Kruuk LEB (2006) Genetic consequences of human management in an introduced island population of red deer (Cervus elaphus). Heredity 97: 56–65 doi:10.1038%2Fsj.hdy.6800838
Pilot M, Jędrzejewski W, Branicki W, Sidorovich VE, Jędrzejewska B, Stachura K and Funk SM (2006) Ecological factors influence population genetic structure of European Grey wolves. Molecular Ecology 15: 4533–4553 doi:10.1111%2Fj.1365-294X.2006.03110.x
Posada D and Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14: 817–818 doi:10.1093%2Fbioinformatics%2F14.9.817
Posada D and Crandall KA (2001) Intraspecific gene genealogies: trees grafting into networks. Trends Ecol Evol 16: 37–45 doi:10.1016%2FS0169-5347%2800%2902026-7
Rozas J and Rozas R (1999) DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics 15: 174–175 doi:10.1093%2Fbioinformatics%2F15.2.174
Serafin D (1995) Jeleń w Puszczy Piskiej. Znad Pisy, Czasopismo poświęcone Ziemi Piskiej 1: 51–55 [In Polish]
Skog A, Zachos FE, Rueness EK, Feulner PGD, Mysterud A, Langvatn R, Lorenzini R, Hmwe SS, Lehoczky I, Hartl GB, Stenseth NC, Jakobsen KS (2009) Phylogeography of red deer (Cervus elaphus) in Europe. J Biogeogr 36: 66–77. doi:10.1111/j.1365-2699.2008.01986.x
Sommer RS and Nadachowski A (2006) Glacial refugia of mammals in Europe: evidence from fossil records. Mamm Rev 36: 251–265 doi:10.1111%2Fj.1365-2907.2006.00093.x
Sommer RS and Zachos FE (2009) Fossil evidence and phylogeography of temperate species: ‘glacial refugia’ and post-glacial recolonization. J Biogeogr 36: 2013–2020 doi:10.1111%2Fj.1365-2699.2009.02187.x
Sommer RS, Zachos FE, Street M, Jöris O, Skog A and Benecke N (2008) Late Quaternary distribution dynamics and phylogeography of the red deer (Cervus elaphus) in Europe. Quat Sci Rev 27: 714–733 doi:10.1016%2Fj.quascirev.2007.11.016
Sztolcman J (1918) Łowiectwo dla szkół leśnych i rolnych. Wydanie Warszawskiego Towarzystwa Prawidłowego Myśliwstwa. Druk Gazety Rolniczej (W. Musielewicza), Warszawa [In Polish]
Taberlet P, Fumagalli L, Wust-Saucy A-G and Cosson J-F (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7: 453–464 doi:10.1046%2Fj.1365-294x.1998.00289.x
Tamura K, Dudley J, Nei M and Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24: 1596–1599 doi:10.1093%2Fmolbev%2Fmsm092
Tatarinov KA (1973) Fauna khrebetnikh zakhodu Ukraini [Vertebrate fauna of western Ukraine]. Lviv, Ukraine [In Ukrainian]
Vereshchagin NK, Rusakov OS (1979) Kopytnye severo-zapada SSSR [Ungulates of north-western USSR]. Nauka, Leningrad [In Russian]
Villesen P (2007) FaBox: an online toolbox for fasta sequences. Molecular Ecology Notes 7: 965–968 doi:10.1111%2Fj.1471-8286.2007.01821.x
Zachos FE, Althoff C, v Steynitz Y, Eckert I and Hartl GB (2007) Genetic analysis of an isolated red deer (Cervus elaphus) population showing signs of inbreeding depression. European Journal of Wildlife Research 53: 61–67 doi:10.1007%2Fs10344-006-0065-z
Zachos FE, Hajji GM, Hmwe SS, Hartl GB, Lorenzini R, Mattioli S (2009) Population viability analysis and genetic diversity of the endangered red deer population from Mesola, Italy. Wildlife Biol 15: 175–186. doi:10.2981/07-075
Zachos F, Hartl GB, Apollonio M and Reutershan T (2003) On the phylogeographic origin of the Corsican red deer (Cervus elaphus corsicanus): evidence from microsatellites and mitochondrial DNA. Mamm biol 68: 284–298 doi:10.1078%2F1616-5047-00097
Acknowledgements
We would like to thank the personnel of the Polish State Forests, hunters, Rimvydas Juškaitis, Hermann Ansorge, students and colleagues from MRI PAS for their help in collecting samples. We are grateful to Barbara Marczuk for her help with the laboratory work. The project was financed by the Polish Ministry of Sciences and Higher Education (grants nr 2 P06L 006 29 and N303 006 32/0379), the budget of the Mammal Research Institute PAS in Białowieża, and by a Marie Curie Transfer of Knowledge project BIORESC within the European Commission’s 6th Framework Programme (contract no. MTKD-CT-2005-029957). The Danish and some of the German red deer sequences were generated as part of the model and demonstration project “Sicherung genetischer Diversität beim Rothirsch in der Kulturlandschaft” (514–06.01.-07BM014) funded via the Bundesanstalt für Landwirtschaft und Ernährung (BLE) by the Bundesministerium für Ernährung, Landwirtschaft und Verbraucherschutz (BMELV) of the Federal Republic of Germany (grant to FEZ and GBH).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited. The open access option was financed by the Polish Ministry of Sciences and Higher Education within the Springer Open Choice/Open Access Programme.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Jan M. Wójcik
Appendix
Appendix
Maximum parsimony tree of the 69 red deer mtDNA control region haplotypes found in the present study. A, B and C refer to the western, North-African/Sardinian and eastern lineages, respectively. All three lineages form monophyletic clades, albeit with low bootstrap support (numbers at branches give percentage values based on 1,000 replicates). The two encircled haplotypes are the one from Mesola (41) and the aberrant one found in western Germany (16).

Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Niedziałkowska, M., Jędrzejewska, B., Honnen, AC. et al. Molecular biogeography of red deer Cervus elaphus from eastern Europe: insights from mitochondrial DNA sequences. Acta Theriol 56, 1–12 (2011). https://doi.org/10.1007/s13364-010-0002-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13364-010-0002-0