
Abstract
Cysteine-selective proteomics approaches simplify complex protein mixtures and improve the chance of detecting low abundant proteins. It is possible that cysteinyl-peptide/protein enrichment methods could be coupled to isotopic labeling and isobaric tagging methods for quantitative proteomics analyses in as few as two or up to 10 samples, respectively. Here we present two novel cysteine-selective proteomics approaches: cysteine-selective dimethyl labeling (cysDML) and cysteine-selective combined precursor isotopic labeling and isobaric tagging (cPILOT). CysDML is a duplex precursor quantification technique that couples cysteinyl-peptide enrichment with on-resin stable-isotope dimethyl labeling. Cysteine-selective cPILOT is a novel 12-plex workflow based on cysteinyl-peptide enrichment, on-resin stable-isotope dimethyl labeling, and iodoTMT tagging on cysteine residues. To demonstrate the broad applicability of the approaches, we applied cysDML and cPILOT methods to liver tissues from an Alzheimer’s disease (AD) mouse model and wild-type (WT) controls. From the cysDML experiments, an average of 850 proteins were identified and 594 were quantified, whereas from the cPILOT experiment, 330 and 151 proteins were identified and quantified, respectively. Overall, 2259 unique total proteins were detected from both cysDML and cPILOT experiments. There is tremendous overlap in the proteins identified and quantified between both experiments, and many proteins have AD/WT fold-change values that are within ~20% error. A total of 65 statistically significant proteins are differentially expressed in the liver proteome of AD mice relative to WT. The performance of cysDML and cPILOT are demonstrated and advantages and limitations of using multiple duplex experiments versus a single 12-plex experiment are highlighted.

ᅟ
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mass spectrometry (MS)-based quantitative proteomics is an important tool to measure relative and absolute protein abundances in order to discover disease biomarkers and to provide insight into biological processes. Comprehensive proteome analysis still remains challenging, however, partially because of heterogeneity associated with biological samples, the wide dynamic range of protein concentrations, the presence of protein post-translational modifications (PTMs), and proteoforms [1]. Furthermore, even with considerable advances in MS technology, there is still a demand for proteomics workflows that are all-inclusive and offer high-throughput, high efficiency, and deep proteome coverage. A widely-used strategy to reduce sample complexity and improve detection of low-abundance proteins is to isolate cysteinyl-peptides [2]. Cysteine occurs ~2.3% among the 20 amino acids in mammals [2]. According to our in-house calculations, ~14% of peptides contain cysteine, which corresponds to ~96% of proteins in the mouse proteome (Uniprot database, 05/21/2014 release, 51,344 sequences). This trend is similar for human, yeast, and other species [2] and suggests that cysteinyl-enrichment can greatly reduce sample complexity while affording high proteome coverage. Cysteine is a highly reactive nucleophilic amino acid and is implicated in biological processes, such as cell recognition and apoptotic signaling [2], cellular homeostasis, immune signaling, and redox chemistry [3]. Cysteine can be subject to a variety of covalent oxidative PTMs (e.g., sulfinic acid, disulfide formation, S-nitrosylation, and S-glutathionylation) [3, 4], and the study of these oxidation states gives insight to cellular redox status.
Cysteinyl-peptides can be enriched directly via the reactions of sulfhydryl groups, such as solid phase thiopropyl resin [5–9], superparamagnetic [10] and gold nanoparticles [11], organomercurial beads [12], and aldehyde resin [13]. Alternatively, cysteine residues may be captured indirectly [14], through derivatization [15], biotin/avidin affinity chromatography [16, 17], or with chemical tagging and antibody enrichment [18, 19]. After the enrichment of cysteinyl-peptides, the incorporation of chemical tagging steps with stable isotopes can be used to design cysteine-selective quantitative proteomics approaches. Non-cell-based isotopic labeling methods (e.g., acetylation, dimethylation) rely on “light,” “medium,” or “heavy” forms of a chemical reagent to label multiple samples that can be pooled and analyzed simultaneously with a precursor MS scan. Doublets or triplets are observed in the spectra, and peak intensities or areas provide a direct readout of relative protein abundances in the multiple samples [20, 21]. Isobaric tags, e.g., tandem mass tag (TMT), isobaric tag for relative and absolute quantification (iTRAQ), N,N-dimethyl leucine (DiLeu) [22], on the other hand, present the same mass in the MS survey scan for peptides tagged with the isobaric reagents. However, upon collision in the gas phase with MS/MS or MS3 different reporter ions are detected [23] and their intensities used to provide relative quantification across as many as 10 samples [24].
One of the first and most widely used cysteine-selective quantitative proteomics approaches is isotope-coded affinity tag (ICAT) [25]. The first version of ICAT consisted of a thiol-reactive group (iodoacetyl), an isotopically light or heavy linker, and an affinity group (biotin) for capturing tagged peptides. Since that time, new versions of ICAT reagents have been developed with optimized performance [26–32], and other quantitative approaches have been reported for cysteinyl-peptides: coupling thiopropyl resin enrichment with 16O/18O exchange [33], on-resin acetylation [34], and off-resin alkylation [35]. Several authors demonstrated the applicability of isobaric tags in either cysteine subproteome characterization or cysteine redox quantification. For instance, TMT and iTRAQ have been combined with cysteine-reactive covalent capture tag [36], biotin/avidin [37], and thiopropyl resin [38–41]. Recently, iodoTMT—a cysteine-reactive TMT reagent—was applied to map and quantify nitrosylation [18, 19, 42]. Enrichment of iodoTMT-tagged peptides is achieved by utilizing antiTMT antibody resin. Although there are attractive features to many of these approaches, few cysteinyl-based quantitative proteomics workflows provide all the following features: (1) effective cysteinyl-peptide enrichment; (2) simple and straightforward sample processing; (3) moderate sample multiplexing (at least > 2-plex and up to 8-plex or higher); and (4) cost-effective reagents.
Herein, we developed two novel cysteine-based quantitative proteomics workflows. The first method is cysteine-selective precursor dimethyl labeling (cysDML). In this workflow, cysteinyl-peptides are captured on a commercially available Thiopropyl Sepharose 6B resin and captured peptides are labeled on resin with either light (–C2H6) or heavy (–13C2 2H6) dimethyl tags [43]. CysDML appears to be a convenient, efficient, accurate, and affordable cysteine-selective quantitative proteomic technique. However, this approach is limited to a maximum of two samples in this report; thus, we sought to develop another approach that could significantly improve on sample multiplexing capabilities. Higher multiplexing capacity is useful for reducing sample preparation and analysis time, minimizing errors, and allowing a readout of differences in relative protein abundances from a variety of sample types, conditions, time points, etc. Recently, our laboratory developed combined precursor isotopic labeling and isobaric tagging (cPILOT), a method that increases multiplexing capabilities of isobaric tags to 12 and 16 samples for TMT and iTRAQ, respectively. We [44, 45] and others [46, 47] have used enhanced multiplexing to study global and PTM-specific protein abundances in complex mixtures. To date, there is no report of a cysteine-selective enhanced multiplexing method. The second approach that we present is a cysteine-selective cPILOT approach using a 12-plex experiment. This novel technique relies on cysteinyl-peptide enrichment and on-resin isotopic dimethyl labeling, in combination with iodoTMT6 reagent tagging. The combination of duplex dimethyl labeling and 6-plex iodoTMT6 tagging results in 12 channels available for sample multiplexing in a single experiment. We note that this method could be extended to 16 or 20 samples if cysteine-reactive iTRAQ or TMT10 [24] reagents were available. Both cysDML and cysteine-selective cPILOT workflows were benchmarked relative to each other and applied to liver tissues from an Alzheimer’s disease (AD) mouse model. The performance of these methods and results from the application are discussed.
Experimental
Animal Husbandry
Fourteen-month-old APP/PS-1 male mice [B6.Cg-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax, stock number 005864, genetic background C57BL/6J express the chimeric mouse/human (Mo/Hu) APP695swe (i.e., K595N and M596L) and a mutant human PS1-dE9] and the genetically heterogeneous wild type (WT) (stock number 000664, genetic background C57BL/6J) were purchased from Jackson Laboratory. Mice were housed in the Division of Laboratory Animal Resources at the University of Pittsburgh and fed standard Purina rodent laboratory chow ad libitum on a 12-hour light/dark cycle. APP/PS-1 (hereafter referred to as AD) and WT mice (n = 6 for each genotype) were euthanized using CO2. Liver tissues were harvested immediately and stored at –80°C until further experiments. Animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh.
Liver Homogenization and Protein Digestion
Liver tissues were homogenized in an ice-cold phosphate buffer saline (PBS) solution containing 8 M urea with 100 passes of a Wheaton homogenizer. Homogenate solution was collected, sonicated, and centrifuged at 13,000 rpm for 10 min (4°C). Supernatants were collected, aliquoted into ~50 μL portions, and stored at –80°C. Protein concentrations were determined using the BCA assay according to the manufacturer’s instructions (Pierce Thermo, Rockford, IL, USA). Liver proteins (100 μg and 75 μg) were digested for each sample in cysDML and cPILOT experiments, respectively. After dilution to 1 μg/μL, the liver proteins were denatured and reduced in 50 mM Tris buffer (pH = 8.2), 8 M urea, 10 mM dithiothreitol (DTT) for 1 h at 37°C. The resulting protein mixture was diluted 10-fold with 20 mM Tris buffer (pH = 8.2). TPCK-treated trypsin from bovine pancreas (Sigma, St. Louis, MO, USA) was added to each sample in a 4% w/w enzyme/protein ratio and incubated at 37°C for 18 h. Samples were acidified with 0.5% formic acid, cleaned using Waters Oasis HLB C18 cartridges, and lyophilized.
Cysteinyl-Peptide Enrichment
All solutions used in the following steps were degassed to prevent oxidation of thiols. Tryptic digests were reduced with 5 mM DTT in 20 μL of 50 mM Tris buffer (pH = 7.5) with 1 mM EDTA (coupling buffer) for 1 h at 37°C, after which the samples were diluted to 100 μL by adding coupling buffer. Thiopropyl Sepharose 6B thiol-affinity resin (35 mg each) was prepared from dried powder per the manufacturer’s instruction (Sigma, St. Louis, MO, USA). Briefly, the dried powder was rehydrated in 1 mL water for 15 min, suspended and transferred to spin columns (Pierce Thermo, Rockford, IL, USA), and washed with 0.5 mL water six times. Next, the slurry was washed with 0.5 mL coupling buffer 10 times. Reduced peptide samples were incubated with the resin for 1.5 h at room temperature with a shaking speed of ~800 rpm, and the unbound portion (non-cysteinyl peptides) was removed by centrifugation. The resin was washed in the spin column sequentially with the following solutions: 0.5 mL of 50 mM Tris buffer (pH = 8.0) with 1 mM EDTA (washing buffer), 2 M NaCl, 80% acetonitrile/0.1% TFA, and 100 mM tetraethylammonium bromide (TEAB). Each wash was repeated six times.
On-Resin Stable-Isotope Dimethyl Labeling
Washed samples were contained in spin columns and 100 μL of 100 mM TEAB was added. Then, 11.2 μL of 4% CH2O/13C2H2O (98% 2H and 99% 13C) and 11.2 μL of 0.6 M NaBH3CN/NaB2H3CN (96% 2H) were added to the sample for light and heavy labeling, respectively. In the cysDML experiments, WT samples were labeled with light (–C2H6) dimethyl tag and AD samples were labeled with heavy (–13C2 2H6) dimethyl tag. In the cPILOT experiment, randomly selected WT and AD samples (n = 3 each) were labeled with the light dimethyl tag and heavy dimethyl tags (n = 3 each). Samples were incubated for 1 h at room temperature while mixing at a speed of ~800 rpm. The reaction was terminated by adding ammonia to a final 0.2% (v/v) concentration, and then formic acid to a final 0.3% (v/v) concentration. Buffer and reagents were removed by centrifugation, and the resin was washed with 0.5 mL 100 mM TEAB (three times) and 0.5 mL washing buffer (six times). The captured and labeled cysteinyl-peptides were released by incubating the resin with 100 μL of washing buffer with freshly prepared 20 mM DTT at room temperature for 30 min while shaking. The above step was repeated two more times with shorter 10-min incubations followed by a final incubation with 80% acetonitrile. Flow-through fractions were collected and combined. In cysDML experiments, the released peptides were further alkylated with 80 mM of iodoacetamide (IAM) for 1 h at room temperature in the dark. AD and WT samples were pooled, concentrated, acidified, desalted using C18 cartridges, and lyophilized. CysDML samples were stored at –80°C for LC-MS/MS. In the cPILOT experiment, the released peptides were concentrated, acidified, desalted using C18 tips (Pierce Thermo, Rockford, IL, USA), and lyophilized.
iodoTMT Tagging
In cPILOT experiments, light and heavy labeled AD and WT samples were labeled with iodoTMT6 reagents according to the manufacturer’s protocol (Pierce Thermo) with modifications. Briefly, each peptide sample was dissolved in 10 μL of degassed washing buffer containing 5 mM DTT, reduced for 1 h at 37°C, and diluted by adding 65 μL washing buffer. Each iodoTMT6 reagent was solubilized with 10 μL of MS-grade methanol and transferred to the peptide mixture. After 1 h incubation at 37°C in the dark, the reaction was quenched by adding 20 mM DTT. All tagged samples were pooled into a single cPILOT sample, concentrated, acidified, desalted using C18 cartridges, and lyophilized.
Offline SCX Fractionation
Strong cation-exchange (SCX) fractionation of the cPILOT sample was carried out on a PolySulfoethyl A 100 mm × 2.1 mm, 5 μm, 200 Å column (The Nest Group, Inc., Southborough, MA, USA) with buffers as follows: mobile phase A was 5 mM monopotassium phosphate (25% v/v acetonitrile, pH 3.0), and mobile phase B was 5 mM monopotassium phosphate, 350 mM potassium chloride (25% v/v acetonitrile, pH 3.0). Dried sample was resuspended in 300 μL of mobile phase A and injected onto the SCX column. The gradient for SCX was 0–5 min, 0% B; 5–45 min, 0%–40% B; 45–90 min, 40%–80% B; 90–100 min, 80%–100% B; 100–110 min, 100% B; 110–121 min, 0% B. One-minute fractions were collected into a 96-well-plate and pooled into a final eight fractions, which were desalted using a C18 tip.
LC-MS/MS Analysis
Online desalting and reversed-phase chromatography was performed with a Nano-LC system equipped with an autosampler (Eksigent, Dublin, CA, USA). Mobile phases A and B were 3% (v/v) acetonitrile with 0.1% formic acid and 100% (v/v) acetonitrile with 0.1% formic acid, respectively. Sample (5 μL) was loaded onto a trapping column (100 μm i.d. × 2 cm), which was packed in-house with C18 200 Å 5 μm stationary phase material (Michrom Bioresource Inc., Auburn, CA, USA) at 3 μL/min in 3% mobile phase B for 3 min. The sample was loaded onto an analytical column (75 μm i.d. × 13.2 cm), which was packed in-house with C18 100 Å 5 μm stationary phase material (Michrom Bioresource Inc.). The following gradient was used for both cysDML and cPILOT experiments: 0–5 min, 10% mobile phase B; 5–40 min, 10%–15% B; 40–90 min, 15%–25% B; 90–115 min, 25%–30% B; 115–130 min, 30%–60% B; 130–135 min, 60%–80% B; 135–145 min, 80% B; 145–150 min, 80%–10% B; 150–180 min, 10%B. The LC eluent was analyzed with positive ion nanoflow electrospray using a LTQ-Orbitrap Velos mass spectrometer (Thermo-Fisher Scientific, Waltham, MA, USA).
CysDML samples were analyzed by employing three gas-phase fractionations (GPF). Specifically, each sample was injected seven times and subject to different MS scans: first injection) precursor scan over the m/z range 350–1700, second to fourth injections) m/z 350–800, m/z 785–975, and m/z 960–1700, respectively, and the fifth to seventh injections were repeats of the second to fourth injections. The three GPF mass ranges were determined from a preliminary analysis of the full m/z range scan and optimized to generate similar numbers of peptide spectral matches (PSMs) in each GPF. The following data-dependent acquisition (DDA) parameters were used in each injection: the MS survey scan in the Orbitrap was 60,000 resolution; the top 15 most intense peaks in the MS survey scan were isolated and fragmented with CID at an isolation width of 3 m/z; CID was performed in the ion trap with normalized collision energy 35%. The maximum fill time for MS and MS/MS is 500 ms and 50 ms, respectively. A complete duty cycle timing is ~3 s.
SCX fractions of the cPILOT sample were injected three times and subjected to various top ion acquisitions. The MS survey scan in the Orbitrap was 60,000 resolution over m/z 350–1700. The first injection included the top five ions for DDA. The second and third injections included the sixth to tenth and eleventh to fifteenth most intense peaks in the MS survey scan for DDA, respectively. DDA parameters were as follows: precursor ions were isolated with a width of 3 m/z and normalized collision energy of 35%, the most intense CID fragment ion over the m/z range 400–1300 was selected for HCD-MS3. The HCD fragment-ion isolation width was set to 4 m/z, the normalized collision energy was 60%, and HCD resolution was 7500 in the Orbitrap. The maximum fill time for MS and MS/MS is 500 ms and 50 ms, respectively. A complete Top15 CID MS/MS duty cycle timing is ~3 s. The maximum fill time for MS, MS/MS, and MS3 is 500, 50, and 250 ms, respectively. The total duty cycle timing for a Top5 CID and HCD MS3 is ~2.4 s.
Database Searching and Data Analysis
RAW files were analyzed using the SEQUEST HT search engine with Proteome Discoverer 1.4 software (Thermo-Fisher Scientific, Waltham, MA, USA) and searched against the Uniprot mouse database (05/21/2014, 51,344 sequences). SEQUEST HT search parameters of cysDML data are as follows: precursor mass tolerance 15 ppm; fragment mass tolerance 1 Da; static modifications light dimethyl/ + 28.031 Da (Lys) or heavy dimethyl/ + 36.076 Da (Lys), carbamidomethyl modification/ + 57.021 Da (Cys); dynamic modifications light dimethyl/ + 28.031 Da (N-terminal) or heavy dimethyl/ + 36.076 Da (N-terminal), oxidation/ + 15.995 Da (Met). Decoy database searching was employed to calculate false discovery rate (FDR). Only peptides with at least medium confidence (<5% FDR) were used for further analysis [48]. Proteome Discoverer 1.4 provided peak area information for light and heavy labeled peptides and protein ratio calculations. Protein ratios were normalized based on the protein median ratio in each biological replicate experiment for cysDML. SEQUEST HT search parameters of cPILOT data are the same as cysDML data except the static modification on cysteine is iodoTMT6/ + 329.226 Da. The reporter ions (i.e., m/z 126–131) were identified with the following parameters: centroid with smallest delta mass, 30 ppm for reporter ion mass tolerance. The isotope correction was employed according to the manufacturer’s data sheet (Pierce Thermo, Rockford, IL). The median reporter ion intensity of each channel was calculated across all PSMs. The median of all reporter ion channels (from light and heavy) was used to normalize reporter ion intensities. Peptide ratios were calculated and, finally, protein ratios were determined from peptide median ratios. Noncysteinyl-peptides were excluded from quantification.
Statistics
Normalized AD/WT ratios were transformed to log2 scale and subjected to permutation. Permutation testing calculates P-values by randomly enumerating all possible permutations. The null hypothesis is H 0 : µ = 0 with alternative of H 1 : µ ≠ 0. The P-value was calculated as P = (1 + b)/(1 + m), where b is the number of times in the 10,000 permuting counts, m, that t permuted (test statistics in permutation test) is larger than t observed (observed test statistic) [49–51]. Calculations were performed in MATLAB R2014a. A P < 0.05 was considered statistically significant. Stringent filter criteria were applied to generate a list of statistically significant differentially expressed proteins as follows: (1) protein must be quantified in n = 6 biological replicates; (2) for cysDML, AD/WT ratios < 0.78 or > 1.20 and for cPILOT, AD/WT ratios < 0.72 or > 1.40 (manuscript under review), and (4) standard deviation < 0.5 for protein AD/WT ratios across all biological replicates.
Results and Discussion
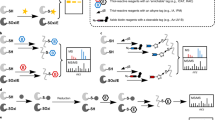
Here we present two novel multiplexing approaches based on the enrichment of cysteinyl-peptides termed cysDML and cPILOT. Both strategies are depicted in Figure 1 and were used to compare differences in the liver proteomes of AD and WT mice. First, twelve liver protein samples (i.e., six WT and six AD) were serially digested by trypsin. Next, cysteinyl-peptides were enriched using a Thiopropyl Sepharose 6B resin. On-resin captured peptides were labeled with either light (–C2H6) or heavy (–13C2 2H6) dimethyl tags on primary amines such as the N-termini and lysine residues. The cysDML approach relies on precursor labeling to quantify relative protein abundances between WT and AD samples. Because cysDML is a duplex experiment, it was necessary to repeat six independent times to accommodate all biological replicates. On the other hand, the cPILOT approach is a 12-plex experiment and dimethylation is used to double the number of channels accessible with the TMT isobaric tagging method. Here, three WT and three AD samples were labeled with the light dimethyl group, whereas the remaining samples in each group were labeled with the heavy dimethyl group. After precursor labeling steps, peptides were released from the resin using DTT. CysDML samples were alkylated, six WT and AD pairs were pooled independently, and analyzed using GPF [52] and LC-MS/MS. cPILOT samples were cleaned, tagged with iodoTMT6 reagents, and the 12 samples were pooled into a single mixture that was analyzed using LC-MS/MS and HCD-MS3.

Schematic representation of cysteine-selective proteomics workflow. Mouse liver peptides are enriched by a thiol-affinity resin. Samples are labeled with either light (–C2H6) or heavy dimethyl (–13C2 2H6) tags on resin. In the cysDML experiment: WT and AD samples are tagged with light and heavy dimethyl groups, respectively (middle center); peptides are eluted from the resin with 20 mM dithiothreitol (DTT) and free cysteines are alkylated by iodoacetamide (middle left); WT and AD samples are combined, desalted and analyzed by LC-MS/MS (bottom left). In the cPILOT experiment: WT and AD samples are tagged with light or heavy dimethyl groups on resin (middle center); after elution with DTT, iodoTMT6 reagents are added to each sample (middle right); all 12 samples are combined, cleaned, fractionated and analyzed by LC-MS3 (bottom right)
Optimization of On-Resin Dimethylation Reaction Conditions
Stable-isotope dimethylation is an attractive precursor isotopic labeling technique because (1) the tag is inexpensive [20], (2) it offers up to five sample channels [53], (3) the reaction is versatile and can be performed in solution or on resin [54], and (4) the reaction is pH-dependent and site-selective [55]. In order to minimize sample loss, we performed dimethylation on the thiolpropyl Sepharose 6B resin. Initially, we achieved an ~90% labeling efficiency (Supplemental Figure S1a) using starting conditions that mimicked in-solution labeling conditions (i.e., 25 mM NaBH3CN, 55 mM CH2O, and 1 h incubation). Significant improvement of the labeling efficiency to > 98% was achieved with a longer incubation time (i.e., 24 h). Because we are interested in maximizing the overall throughput of multiplexing experiments, we sought to reduce the reaction time while maintaining high efficiency. This was made possible by increasing the reagent concentrations ~2.5-fold (60 mM NaBH3CN, 145 mM CH2O) with a 1-h incubation period (Supplemental Figure S1b). These conditions are consistent with dimethyl labeling performance on solid-phase hydrazide beads [56] and were used for the remaining cysDML and cPILOT experiments. Because NaBH3CN is a much weaker reducing regent than NaBH4, it will not affect aldehydes, ketones as well as disulfides between peptides and resin [57]. We also did not observe physical property changes of resin after dimethyl labeling, indicating that the disulfide bonds stay intact after dimethyl labeling [58].
Evaluation of Quantification Accuracy and Resin Loading Range for cysDML
CysDML is a novel precursor dimethylation technique. Thus, we assessed the quantitative accuracy and linear dynamic range using tryptic peptides from WT mouse liver. The first experiment evaluated quantitative accuracy of a mixture of 1:1 light:heavy labeled tryptic peptides that were separated using a 3-h LC gradient. A total of 689 proteins were identified and 424 of these were quantified (i.e., proteins had reported ratios for light and heavy peptides from Proteome Discoverer report). The average heavy/light ratio for the quantified proteins is 0.98 ± 0.21 (mean ± standard deviation) as shown in Supplemental Figure S2a; this error is consistent with other reports [59]. More than 95% of the proteins have ratios falling within two standard deviations of the mean and thus fits a normal distribution. To understand the effects of resin loading amount on quantitative accuracy, we varied the sample loading on resin as follows: six cysDML samples contained a fixed amount (100 μg) of peptides prior to resin loading, whereas the heavy channel varied from 12.5, 25, 50, 100, 200 to 400 μg. When the sample loading amount was between 25 and 200 μg, accurate heavy/light ratios were obtained (Supplemental Figure S2b). However, on the low and high ends, the ratios were skewed. We attribute this to dilute samples on the low end that result in an overall minimal capture of cysteinyl-peptides. On the high end, inefficient capture on the resin occurred as the amount of DTT concentration was not increased to accommodate higher concentrations of peptide thiols. Excessive DTT concentrations are damaging to the thiolpropyl Sepharose 6B resin. The measured dynamic range is 8-fold, which is comparable to other reports [9, 60], and the maximum standard deviation is ~0.5. Results of these experiments were used to establish appropriate criteria for determining differential expression of proteins.
Application of cysDML to the Liver Proteome of an AD Mouse Model
A tradeoff that must be considered in any proteomics experiment is proteome depth or coverage versus sample preparation, acquisition, and analysis time. We wanted to minimize the number of sample handling steps (and potential sample loss) while maintaining adequate proteome coverage because each cysDML sample is only ~40 μg. Thus, GPF was used as a fractionation step for cysDML samples [52]. Supplemental Figure S3 provides example base peak chromatograms of seven GPFs for one of the pooled AD/WT sample pairs. The first injection was analyzed with a full m/z range of 350–1700. Six subsequent injections were collected over the m/z ranges of 350–800, 785–975, and 960–1700, such that each fraction was not analyzed back-to-back. An overlapping window of 15 m/z was used between adjacent GPFs to ensure that light and heavy pairs were detected within the same spectrum. Comparisons of GPF analyses in these data to the injections with the full m/z range of 350–1700 indicate that GPF increases protein and peptide identifications by 79% and 75%, respectively (data not shown). Furthermore, the replicate injections are highly reproducible.
Figure 2a displays several example spectra containing light (m/z = 974.03) and heavy (m/z = 982.07) pairs of the doubly charged peptide [V(dimethyl)AVVAGYGDVGK(dimethyl)GC(IAM)AQALR + 2H]2+ from protein adenosylhomocysteinase. The observed spacing (Δm = 16 Da) between the peaks is consistent with two dimethyl groups being present on the peptide. Also, the diversity in peptide levels across the six biological replicates is apparent. An M + 7 Da species, which has a relative abundance of ~10%, is observed for heavy dimethylated peaks, consistent with other reports [54, 61]. The presence of this peak could be from use of a less isotopically pure reducing reagent, however, does not have significant influence on quantitative accuracy and precision (Supplemental Figure S2). Overall, the average numbers of spectral counts, peptides, proteins identified, and proteins quantified across the replicates are 14005 ± 2125, 1823 ± 238, 850 ± 92, and 594 ± 65, respectively (Table 1). In total, 2085 unique proteins were identified from cysDML experiments. A large number of the spectral counts (~98%) and peptides (~91%) identified in each cysDML experiment can be attributed to cysteinyl-peptides. Thus, the cysDML approach is very efficient at enrichment and detection of cysteinyl-peptides. When assessing the AD/WT ratios for proteins quantified in each of the six cysDML experiments, we find that they are very similar across biological replicates, Figure 2b. Many proteins have ratios that fall outside of an AD/WT ratio of one. We used permutation testing and conservative filtering criteria (see the Experimental section) and identified 54 proteins that are differentially-expressed in the AD mice from cysDML experiments (Table 2). Twenty-three of these proteins have higher levels in AD mice, whereas 31 proteins have lower levels in AD mice relative to WT. Differentially-expressed proteins are involved in various biological processes, which will be briefly discussed below.

Example cysDML MS spectra for: (a) pair of light (m/z = 974.03) and heavy (m/z = 982.07) peaks assigned to the doubly charged peptide V(dimethyl)AVVAGYGDVGK(dimethyl)GC(IAM)AQALR of adenosylhomocysteinase in each biological replicate (BR); (b) scatter plot of normalized protein ratios (AD/WT) measured in cysDML experiment for each BR
Application of cPILOT to the Liver Proteome of an AD Mouse Model
Previously, our laboratory has demonstrated enhanced multiplexing using global [44] and 3-nitrotyrosine [45] specific cPILOT approaches. The combination of precursor isotopic labeling with isobaric tagging methods can increase the number of sample multiplexing channels by a factor of two to three times. Capabilities afforded by enhanced sample multiplexing include increasing biological replication, the ability to examine many tissues, sample types, environmental stimuli, longitudinal studies, etc. in a single analysis, and minimizing biases caused by multiple sample preparation steps and LC and MS acquisitions. We note that because cPILOT involves post-digestion chemical labeling, errors introduced prior to sample pooling are still inherent in the final ratios reported. In order to increase sample multiplexing capabilities, simplify the protein mixture, and potentially maintain breadth of proteome coverage, we developed a cysteine-selective cPILOT assay (Figure 1) and benchmarked its performance against the cysDML method. Compared with the cysDML sample preparation, iodoTMT was used to tag thiols after resin capture. Before iodoTMT tagging, excess DTT (~20 mM) was removed using C18 cleanup [62]. In order to reduce thiols that may have been oxidized during the cleanup, a low level of DTT (5 mM) was applied to the samples (according to the manufacturer’s protocol). IodoTMT reagent is in such excess that there is enough (~4 mM) remaining to label peptides in addition to any reagent used for thiols on DTT. Efficient labeling of peptides was tested using iodoTMT0 before application to AD and WT tissues. Tris(2-carboxyethyl)phosphine (TCEP) may be a suitable alternative for reducing agents, and the removal of the additional DTT step could be explored in future cPILOT experiments.
Data-dependent acquisition was employed on a LTQ-Orbitrap Velos mass spectrometer such that the top five most intense ions were subject to CID MS/MS and the most intense fragment ion (over the m/z range 400–1300) was further subjected to HCD-MS3. MS3 has been demonstrated to address co-isolation and ratio suppression issues of isobarically-tagged peptides [63]. Figure 3 provides example MS spectra for a tryptic peptide detected in the cysteine-specific cPILOT experiment. The precursor MS scan (Figure 3a) displays a light (m/z = 693.02) and a heavy (m/z = 698.39) pair of peaks that arise from a triply-charged ion. In independent CID MS/MS scans, both the light and heavy peaks were isolated and fragmented to provide the MS/MS spectra shown in Figure 3b. The fragmentation patterns for the light and heavy labeled precursor ions are very similar and the fragment peaks only differ by the masses of the heavy isotope atoms from the dimethyl tag. Based on the MS/MS spectral information, the peptide sequence has been assigned to the peptide [T(dimethyl)SAC(iodoTMT6)FEPSLDYMVTK(dimethyl) + 3H]3+ that belongs to the protein carbamoyl-phosphate synthase. We applied an isolation width of 3 m/z for precursor selection of fragment ions. This isolation window is large enough to give the best sensitivity for MS3 analysis. Although 35.4% of PSMs have a charge state greater than three, only 5.5% of PSMs have m/z spacing less than 2.7 between light and heavy dimethylated pairs (Supplemental Figure S4). Despite potential co-isolation of precursor pairs, MS3 isolation and fragmentation improves quantitation. Isolation and HCD fragmentation of the most intense peaks in the CID spectra (i.e., the b 6 2+ ion at m/z = 498.97 for light and m/z = 503.04 for heavy), result in the MS3 spectra shown in Figure 3c. The low m/z region of the spectra are shown and two sets of reporter ions (m/z 126–131) are detected for the light and heavy labeled fragment ions. Relative abundances of the reporter ion peaks for WT and AD samples indicate that this peptide has an overall lower level in AD liver relative to WT. When considering the average reporter ion AD/WT ratio (i.e., AD/WT = 0.81, P = 0.015) for this protein, it is excluded according to filter criteria (see the Experimental section) for differential expression.

Example cPILOT MS spectra for: (a) pair of peaks assigned to the peptide T(dimethyl)SAC(iodoTMT6)FEPSLDYMVTK(dimethyl) of carbamoyl-phosphate synthase; (b) CID MS/MS spectra of the peaks with m/z = 693.024 and m/z = 698.387 from (a). The most intense peaks (*) within the m/z range of 400–1300 were further selected and fragmented to give the HCD MS3 spectra shown in (c), which are zoomed-in over the reporter ion region
There was a total of 3318 spectral counts and 245 peptides that are specific to cysteinyl-peptides in the cPILOT experiment. Overall, this total number results in 330 identified proteins in which 151 proteins were quantified. It is clear that the performance of the 12-plex experiment compared with the cysDML duplex experiment is lower with regards to total proteins identified and quantified. These differences could be due to sample loading amounts (i.e., 75 ug for cPILOT and 100 ug for cysDML) and the additional processing steps in the cPILOT experiment that can lead to sample loss and lower recovery. In lieu of GPF with the cPILOT experiment, we performed offline SCX separations. We believe that with the additional condensed-phase separation, sample loss occurred as another sample clean-up step is necessary between SCX fractionation and final LC-MS3 analysis. Furthermore, the detection of reporter ions relies on the generation of intense fragments that contain the iodoTMT tag. Based on the location of the cysteine residue relative to the N-terminus, we observe that only half of HCD-MS3 spectra result in reporter ions when the cysteine is within three positions relative to the N-terminus. Also, it appears that the enrichment efficiency of cysteinyl-peptides is lower for cPILOT (88.5% PSMs) compared with cysDML (98.3%). However, because we are reporting efficiency after derivatization of thiols with iodoTMT or IAM, it is possible that the labeling efficiency of cysteines with iodoTMT is also less. Detection of lower numbers of cysteinyl-peptides with cPILOT could be attributed to sample loss and lower MS3 duty cycle. The latter occurred because excess iodoTMT reagent eluted throughout the course of the reversed-phase LC run, and these contaminant ions were selected and fragmented numerous times. Additionally, we noticed many instances where non-iodoTMT-tagged fragment ions were further selected for HCD-MS3. As the most intense ions are selected for HCD-MS3, this suggests that the instrument spent a great deal of time on ions that could not generate reporter ions. In the future, we plan to remove these excess reagents as well as include these ions on a reject list and incorporate potentially more selective ion [45] or multi-notch MS3 [64] approaches. After application of string criteria, eleven proteins have statistically significant differential expression in liver from AD mice relative to WT from cysteine-selective cPILOT (Table 3).
Comparison of cysDML and cPILOT
Both the cysDML and cPILOT approaches described herein are novel methods to quantify cysteinyl-proteins in multiple samples simultaneously. The cysDML, duplex experiment, resulted in 2.5 times more identified and quantified proteins in comparison with the cPILOT, 12-plex experiment. Although similar amounts of starting material where used for each experiment, the number of sample handling and sample cleanup and wash steps is substantially greater in the cPILOT approach. From the proteins identified with each method, 156 overlap, and 1929 and 174 are unique to the cysDML and cPILOT experiments, respectively. Thus, there is good agreement in the proteins identified from both methods; however, each approach can give new information not reported in the other method. Also, cysDML is more advantageous for deeper proteome coverage compared with cPILOT. Six cysDML experiments were completed and compared with a single cPILOT experiment. However, if one is interested in generating a short list of starting candidates in a quick analysis, the cPILOT approach would be more beneficial.
Next, we compared the correlation in AD/WT ratios from cysDML and cPILOT experiments for all proteins quantified in six biological replicates regardless of P-values from statistical testing (Table 4) to better assess the performance of each method. In a majority of the cases, the AD/WT ratios are in good agreement (e.g., within ~20% error) between cysDML and cPILOT experiments, however, based on the results of statistical testing may not be considered as differentially-expressed in one or both methods. Furthermore, there exist a handful of proteins in which the AD/WT ratios are different between the cysDML and cPILOT experiments. In these cases, there are high standard deviation (>0.5) values across peptide ratios, differences in peptides detected, number of PSMs used for quantitation, and errors associated with variations in selection of peaks for MS/MS and HCD-MS3 during data-dependent acquisition.
There are other considerations for sample multiplexing with cysDML or cPILOT. First, the number of necessary sample channels is important for determining if it is appropriate for a researcher to perform multiple duplex experiments or a single 12-plex (or higher) experiment. It could become rather cumbersome and time-consuming to perform multiple combinatorial experiments to compare differences from more than two sample types with the cysDML approach, whereas with the cPILOT experiment every sample can be analyzed simultaneously with the noted tradeoff in breadth of proteome coverage. Incorporation of additional separation steps and improvements to the cPILOT workflow to reduce sample handling steps and minimize sample loss could significantly improve the proteome breadth of this approach. CysDML is a fairly inexpensive approach compared with cPILOT, which involves the purchase of commercial isobaric tagging reagents. We maximized commercial reagents by using each iodoTMT6 reagent vial to label two samples (75 μg for light and 75 μg for heavy dimethyl peptides). The use of isobaric reagents that could be synthesized in-house such as DiLEU tags [22, 65] could help to reduce the cost of a cPILOT experiment while maintaining enhanced sample multiplexing capability. The cysDML sample preparation steps can be carried out in less than 24 h whereas the cPILOT approach can take up to 2 or 3 d. Overall, the cPILOT approach ends up taking less total experiment time as there is only one sample used for fractionation and smaller numbers of samples for MS acquisition. A major drawback to the cysDML approach is the limited amount of multiplexing capability that it has even with recent reports of five sample multiplexing with dimethyl labeling [53]. Currently, we have demonstrated 12-plex analyses with cPILOT, however note that further multiplexing is possible with the use of iTRAQ8 reagents or TMT10 [24] reagents and additional stable-isotope precursors. The errors that arise from independent LC-MS/MS experiments in cysDML experiments are not present in cPILOT experiments, where all 12 samples are subject to the same exact MS conditions.
It must be noted that the enrichment of cysteine-containing peptides introduces additional sample handling steps that can increase variation in the workflow. In control cysDML experiments (see Supplemental Figure S2), accurate quantitation was obtained across a limited dynamic range. Care was taken to ensure that samples were treated similarly prior to the pooling steps. Normalization [46] of reporter ion signals was performed to help account for errors introduced from sample handling.
In both experiments, analysis of only cysteinyl-peptides dramatically simplifies precursor MS spectra relative to global dimethylation and cPILOT experiments. This simplification affords less spectral interference from closely-spaced precursors that are likely to be co-isolated and fragmented in global assays.
Shi et al. have identified 1000 proteins from mouse liver proteome in a single-run LC Orbitrap MS analysis [66]. Our analyses of liver tissue without enrichment generate similar results (data not shown). Although enrichment of cysteine-containing peptides should allow for the same depth of proteome coverage, lower numbers of proteins are identified [5, 6]. This could be attributed to several factors, including minimal number of cysteine-containing peptides after enrichment for a given protein, peptides not being selected during a DDA experiment, and sample loss that can occur during the sample preparation steps as additional clean-up is necessary.
Differentially-Expressed Proteins in the Liver Proteome of an AD Mouse Model
Herein, cysDML and cPILOT methods identified 65 differentially-expressed proteins in liver tissue from an AD mouse model relative to WT controls. AD is a progressive neurodegenerative disorder and the most common form of dementia. Little is reported about changes in the liver proteome of AD patients or animal models. However, it is suggested that the liver may be a major contributor to amyloid-β accumulation in the brain [67].
The liver has a wide range of functions, including metabolism, biosynthesis of proteins and small molecules, as well as detoxification; however, the most interesting changes we have observed revolve around metabolism.
Several proteins have similar trends in differential expression in AD liver compared with previously reported studies in AD brain and plasma: alpha-2-macroglobulin [68] and hydroxymethylglutyaryl-CoA synthase [69] are higher in AD, whereas ATP synthase subunit gamma [70, 71], 14-3-3 zeta/delta [71, 72], sodium/potassium-transporting ATPase subunit beta-1 (Na+/K+-ATPase) [70, 71, 73], phosphoglycerate mutase 1, enolase, and fructose-bisphosphate aldolase B [70, 72, 73] are lower in AD. One protein, superoxide dismutase [Cu-Zn] changes differently in liver tissue compared with the brain for AD subjects. In the liver, superoxide dismutase is lower in AD whereas in the brain it has higher levels in AD [70, 74]. Superoxide dismutase is a major protein targeted under oxidative stress in AD, and the Cys146 residue is irreversibly oxidized to cysteic acid [75]. In cysDML experiments, we detected a tryptic peptide containing Cys146, however, it was unmodified. Oxidized cysteine residues are likely to be lost during the enrichment steps. Thus, although we observe lower levels of the unmodified peptide in AD mice, it is possible that our ratios would be different with detection of the oxidized version of the peptide.
Metabolism
The reactome pathway database [76] was used to provide biological processes related to the differentially-expressed proteins and here we focus on a few key aspects of metabolism.
First, carbohydrate metabolism appears to be altered in AD mouse liver. Phosphorylase, a protein involved in glycogenolysis, is higher in AD mice relative to WT. Enzymes involved in glycolysis: fructose biphosphate aldolase, phosphoglycerate mutase, and enolase are lower in AD mice relative to WT. In the liver, glycogen synthesis and degradation regulate blood glucose levels. Higher phosphorylase suggests that high levels of glucose are generated in the liver; however, altered glycolysis implies that glucose is not being utilized efficiently in this tissue. Hyperglycemia is a major risk factor for vascular injury associated with AD [77], and diabetes is also a risk factor for AD [78], and it is well known that lower glucose metabolism occurs in the brains of AD patients [79].
Second, our data suggest that lipid metabolism is augmented in AD mice. For instance, methylmalonyl-CoA mutase and acyl-CoA oxidase, enzymes involved degradation of long-chain fatty acids, are higher in AD mice. Changes in this pathway are consistent with other studies in our laboratory that have utilized global cPILOT methods to compare liver tissues in AD mice (Evans, A. R., Gu, L., Guerrero, R. J., Robinson, R. A. S.: Altered liver metabolism in an APP/PS-1 mouse model revealed by proteomics. Submitted). Another interesting finding is the increased level of ketogenesis. In AD brain, higher levels of ketone bodies were observed with the decrease of brain glucose uptake [80]. We observed higher levels of hydroxymethylglutaryl-CoA synthase in AD mice. This enzyme catalyzes the synthesis of acetoacetate, major ketone bodies produced in ketogenesis. Ketone bodies migrate from the liver and enter the circulatory system. Ketone bodies have been suggested as alternative fuel for AD brain [79] and as a possible therapeutic approach of AD [81].
Finally, it appears that higher levels of ammonia that occur in the blood and brain of AD patients [82, 83] may be linked to our observation of decreased consumption of ammonia by key enzymes in the liver. Aspartate aminotransferase and glutamine synthetase, involved in ammonia regulation, have lower levels in AD mice relative to WT. These lower levels suggest that ammonia is not being consumed by the liver and thus correlates well with reported higher levels of ammonia in blood and brain of AD subjects [82, 83]. Hyperammonemia in the liver links to cognitive impairment in a model animal study [84].
Conclusions
Two novel cysteine-selective quantitative proteomics approaches were presented in this work: cysDML and cPILOT. These are two approaches that allow moderate and high levels of sample multiplexing in proteomics workflows. Based on our results, cysDML allows higher proteome coverage compared with cysteine-selective cPILOT. However, cysteine-selective cPILOT offers a more high-throughput approach to study many samples simultaneously. There are direct advantages and limitations to perform multiple duplex experiments or single 12-plex experiments, as we have thoroughly discussed. It is up to the researchers to design which approach is most suitable for their given research questions. Our application of both cysDML and cPILOT to the liver proteome from an AD mouse model resulted in identification of more than 2200 proteins, in which 65 were differentially expressed in the AD model relative to WT controls. These are amongst the first studies to report on changes in the liver proteome for this AD mouse model and AD in general. Many interesting findings, especially involved in metabolism, occur in the liver of AD mice.
Our laboratory is working to improve upon the cysteine-selective cPILOT approach by minimizing sample preparation and loss of steps, improving proteome coverage and breadth, optimizing instrument data acquisition parameters, increasing sample channels available for multiplexing, and automating aspects of the entire process.
References
Wu, L., Han, D.K.: Overcoming the dynamic range problem in mass spectrometry-based shotgun proteomics. Expert. Rev. Proteomics 3, 611–619 (2006)
Giron, P., Dayon, L., Sanchez, J.C.: Cysteine tagging for MS-based proteomics. Mass Spectrom. Rev. 30, 366–395 (2011)
Bachi, A., Dalle-Donne, I., Scaloni, A.: Redox proteomics: chemical principles, methodological approaches, and biological/biomedical promises. Chem. Rev. 113, 596–698 (2013)
Held, J.M., Gibson, B.W.: Regulatory control or oxidative damage? Proteomic approaches to interrogate the role of cysteine oxidation status in biological processes. Mol. Cell. Proteomics 11, R111 013037 (2012)
Wang, H., Qian, W.J., Chin, M.H., Petyuk, V.A., Barry, R.C., Liu, T., Gritsenko, M.A.,Mottaz, H.M., Moore, R.J.,Camp Ii, D.G.,Khan, A.H.,Smith, D.J.,Smith, R.D.: Characterization of the mouse brain proteome using global proteomic analysis complemented with cysteinyl-peptide enrichment. J. Proteome Res. 5, 361–369 (2006)
Liu, T., Qian, W.J., Chen, W.N., Jacobs, J.M., Moore, R.J., Anderson, D.J., Gritsenko, M.A., Monroe, M.E., Thrall, B.D.,Camp, D.G.,2nd, Smith, R.D.: Improved proteome coverage by using high efficiency cysteinyl peptide enrichment: the human mammary epithelial cell proteome. Proteomics 5, 1263–1273 (2005)
Forrester, M.T., Hess, D.T., Thompson, J.W., Hultman, R., Moseley, M.A., Stamler, J.S., Casey, P.J.: Site-specific analysis of protein S-acylation by resin-assisted capture. J. Lipid Res. 52, 393–398 (2011)
Liu, M., Hou, J., Huang, L., Huang, X., Heibeck, T.H., Zhao, R., Pasa-Tolic, L., Smith, R.D., Li, Y., Fu, K., Zhang, Z., Hinrichs, S.H., Ding, S.J.: Site-specific proteomics approach for study protein S-nitrosylation. Anal. Chem. 82, 7160–7168 (2010)
Paulech, J., Solis, N., Edwards, A.V., Puckeridge, M., White, M.Y., Cordwell, S.J.: Large-scale capture of peptides containing reversibly oxidized cysteines by thiol-disulfide exchange applied to the myocardial redox proteome. Anal. Chem. 85, 3774–3780 (2013)
Palani, A., Lee, J.S., Huh, J., Kim, M., Lee, Y.J., Chang, J.H., Lee, K., Lee, S.W.: Selective enrichment of cysteine-containing peptides using SPDP-functionalized superparamagnetic Fe(3)O(4)@SiO(2) nanoparticles: application to comprehensive proteomic profiling. J. Proteome Res. 7, 3591–3596 (2008)
Xu, Y., Cao, Q., Svec, F., Frechet, J.M.: Porous polymer monolithic column with surface-bound gold nanoparticles for the capture and separation of cysteine-containing peptides. Anal. Chem. 82, 3352–3358 (2010)
Raftery, M.J.: Enrichment by organomercurial agarose and identification of cys-containing peptides from yeast cell lysates. Anal. Chem. 80, 3334–3341 (2008)
Giron, P., Dayon, L., David, F., Sanchez, J.C., Rose, K.: Enrichment of N-terminal cysteinyl-peptides by covalent capture. J. Proteome 71, 647–661 (2009)
Dai, J., Wang, J., Zhang, Y., Lu, Z., Yang, B., Li, X., Cai, Y., Qian, X.: Enrichment and identification of cysteine-containing peptides from tryptic digests of performic oxidized proteins by strong cation exchange LC and MALDI-TOF/TOF MS. Anal. Chem. 77, 7594–7604 (2005)
Giron, P., Dayon, L., Mihala, N., Sanchez, J.C., Rose, K.: Cysteine-reactive covalent capture tags for enrichment of cysteine-containing peptides. Rapid Commun. Mass Spectrom. 23, 3377–3386 (2009)
Jaffrey, S.R., Snyder, S.H.: The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE. 2001, pl1. (2001). doi:10.1126/stke.2001.86.pl1
Lin, D., Li, J., Slebos, R.J., Liebler, D.C.: Cysteinyl peptide capture for shotgun proteomics: global assessment of chemoselective fractionation. J. Proteome Res. 9, 5461–5472 (2010)
Qu, Z., Meng, F., Bomgarden, R.D., Viner, R.I., Li, J., Rogers, J.C., Cheng, J., Greenlief, C.M., Cui, J., Lubahn, D.B., Sun, G.Y., Gu, Z.: Proteomic quantification and site-mapping of S-nitrosylated proteins using isobaric iodoTMT reagents. J. Proteome Res. 13, 3200–3211 (2014)
Pan, K.T., Chen, Y.Y., Pu, T.H., Chao, Y.S., Yang, C.Y., Bomgarden, R.D., Rogers, J.C., Meng, T.C., Khoo, K.H.: Mass spectrometry-based quantitative proteomics for dissecting multiplexed redox cysteine modifications in nitric oxide-protected cardiomyocyte under hypoxia. Antioxid. Redox Signal. 20, 1365–1381 (2014)
Bantscheff, M., Lemeer, S., Savitski, M.M., Kuster, B.: Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal. Bioanal. Chem. 404, 939–965 (2012)
Bantscheff, M., Schirle, M., Sweetman, G., Rick, J., Kuster, B.: Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 (2007)
Xiang, F., Ye, H., Chen, R., Fu, Q., Li, L.: N, N-dimethyl leucines as novel isobaric tandem mass tags for quantitative proteomics and peptidomics. Anal. Chem. 82, 2817–2825 (2010)
Thompson, A., Schafer, J., Kuhn, K., Kienle, S., Schwarz, J., Schmidt, G., Neumann, T., Johnstone, R., Mohammed, A.K., Hamon, C.: Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904 (2003)
McAlister, G.C., Huttlin, E.L., Haas, W., Ting, L., Jedrychowski, M.P., Rogers, J.C., Kuhn, K., Pike, I., Grothe, R.A., Blethrow, J.D., Gygi, S.P.: Increasing the multiplexing capacity of TMTs Using reporter ion isotopologues with isobaric masses. Anal. Chem. 84, 7469–7478 (2012)
Gygi, S.P., Rist, B., Gerber, S.A., Turecek, F., Gelb, M.H., Aebersold, R.: Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 17, 994–999 (1999)
Shi, Y., Xiang, R., Crawford, J.K., Colangelo, C.M., Horvath, C., Wilkins, J.A.: A simple solid-phase mass tagging approach for quantitative proteomics. J. Proteome Res. 3, 104–111 (2004)
Zhou, H., Ranish, J.A., Watts, J.D., Aebersold, R.: Quantitative proteome analysis by solid-phase isotope tagging and mass spectrometry. Nat. Biotechnol. 20, 512–515 (2002)
Guo, M., Galan, J., Tao, W.A.: A novel quantitative proteomics reagent based on soluble nanopolymers. Chem. Commun. (Camb). 1251–1253 (2007)
Ahrends, R., Pieper, S., Kuhn, A., Weisshoff, H., Hamester, M., Lindemann, T., Scheler, C., Lehmann, K., Taubner, K., Linscheid, M.W.: A metal-coded affinity tag approach to quantitative proteomics. Mol. Cell. Proteomics 6, 1907–1916 (2007)
Zhang, L., Guo, Y.L., Liu, H.Q.: A novel class of chemically modified iodo-containing resins: design, synthesis, and application to mass spectrometry-based proteome analysis. J. Mass Spectrom. 39, 447–457 (2004)
Olsen, J.V., Andersen, J.R., Nielsen, P.A., Nielsen, M.L., Figeys, D., Mann, M., Wisniewski, J.R.: HysTag—a novel proteomic quantification tool applied to differential display analysis of membrane proteins from distinct areas of mouse brain. Mol. Cell. Proteomics 3, 82–92 (2004)
Qiu, Y., Sousa, E.A., Hewick, R.M., Wang, J.H.: Acid-labile isotope-coded extractants: a class of reagents for quantitative mass spectrometric analysis of complex protein mixtures. Anal. Chem. 74, 4969–4979 (2002)
Liu, T., Qian, W.J., Strittmatter, E.F., Camp II, D.G., Anderson, G.A., Thrall, B.D., Smith, R.D.: High-throughput comparative proteome analysis using a quantitative cysteinyl-peptide enrichment technology. Anal. Chem. 76, 5345–5353 (2004)
Forrester, M.T., Thompson, J.W., Foster, M.W., Nogueira, L., Moseley, M.A., Stamler, J.S.: Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol. 27, 557–559 (2009)
Shen, M., Guo, L., Wallace, A., Fitzner, J., Eisenman, J., Jacobson, E., Johnson, R.S.: Isolation and isotope labeling of cysteine- and methionine-containing tryptic peptides: application to the study of cell surface proteolysis. Mol. Cell. Proteomics 2, 315–324 (2003)
Giron, P., Dayon, L., Turck, N., Hoogland, C., Sanchez, J.C.: Quantitative analysis of human cerebrospinal fluid proteins using a combination of cysteine tagging and amine-reactive isobaric labeling. J. Proteome Res. 10, 249–258 (2011)
Liu, P., Zhang, H., Wang, H., Xia, Y.: Identification of redox-sensitive cysteines in the Arabidopsis proteome using OxiTRAQ, a quantitative redox proteomics method. Proteomics 14, 750–762 (2014)
Guo, J., Gaffrey, M.J., Su, D., Liu, T., Camp II, D.G., Smith, R.D., Qian, W.J.: Resin-assisted enrichment of thiols as a general strategy for proteomic profiling of cysteine-based reversible modifications. Nat. Protoc. 9, 64–75 (2014)
Su, D., Shukla, A.K., Chen, B., Kim, J.S., Nakayasu, E., Qu, Y., Aryal, U., Weitz, K., Clauss, T.R., Monroe, M.E., Camp II, D.G., Bigelow, D.J., Smith, R.D., Kulkarni, R.N., Qian, W.J.: Quantitative site-specific reactivity profiling of S-nitrosylation in mouse skeletal muscle using cysteinyl peptide enrichment coupled with mass spectrometry. Free Radic. Biol. Med. 57, 68–78 (2013)
Su, D., Gaffrey, M.J., Guo, J., Hatchell, K.E., Chu, R.K., Clauss, T.R.W., Aldrich, J.T., Wu, S., Purvine, S., Camp, D.G., Smith, R.D., Thrall, B.D., Qian, W.J.: Proteomic identification and quantification of S-glutathionylation in mouse macrophages using resin-assisted enrichment and isobaric labeling. Free Radic. Biol. Med. 67, 460–470 (2014)
Guo, J., Nguyen, A., Dai, Z., Su, D., Gaffrey, M.J., Moore, R.J., Jacobs, J.M., Monroe, M.E., Smith, R.D., Koppenaal, D.W., Pakrasi, H.B., Qian, W.J.: Proteome-wide light/dark modulation of thiol oxidation in cyanobacteria revealed by quantitative site-specific redox proteomics. Mol. Cell. Proteomics 13, 3270-3285 (2014)
Murray, C.I., Uhrigshardt, H., O’Meally, R.N., Cole, R.N., Van Eyk, J.E.: Identification and quantification of S-nitrosylation by cysteine reactive tandem mass tag switch assay. Mol. Cell. Proteomics 11, M111 013441 (2012)
Hsu, J.L., Huang, S.Y., Chow, N.H., Chen, S.H.: Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 75, 6843–6852 (2003)
Evans, A.R., Robinson, R.A.: Global combined precursor isotopic labeling and isobaric tagging (cPILOT) approach with selective MS(3) acquisition. Proteomics 13, 3267–3272 (2013)
Robinson, R.A., Evans, A.R.: Enhanced sample multiplexing for nitrotyrosine-modified proteins using combined precursor isotopic labeling and isobaric tagging. Anal. Chem. 84, 4677–4686 (2012)
Dephoure, N., Gygi, S.P.: Hyperplexing: a method for higher-order multiplexed quantitative proteomics provides a map of the dynamic response to rapamycin in yeast. Sci. Signal. 5, rs2 (2012)
Everley, R.A., Kunz, R.C., McAllister, F.E., Gygi, S.P.: Increasing throughput in targeted proteomics assays: 54-plex quantitation in a single mass spectrometry run. Anal. Chem. 85, 5340–5346 (2013)
Qian, W.J., Liu, T., Monroe, M.E., Strittmatter, E.F., Jacobs, J.M., Kangas, L. J., Petritis, K., Camp II, D.G., Smith, R.D.: Probability-based evaluation of peptide and protein identifications from tandem mass spectrometry and SEQUEST analysis: the human proteome. J. Proteome Res. 4, 53–62 (2005)
Chen, D., Shah, A., Nguyen, H., Loo, D., Inder, K.L., Hill, M.M.: Online quantitative proteomics p-value calculator for permutation-based statistical testing of peptide ratios. J. Proteome Res. 13, 4184-4191 (2014)
Ludbrook, J., Dudley, H.: Why permutation tests are superior to t- and F tests in biomedical research. Am. Stat. 52, 127–132 (1998)
Musunuri, S., Wetterhall, M., Ingelsson, M., Lannfelt, L., Artemenko, K., Bergquist, J., Kultima, K., Shevchenko, G.: Quantification of the brain proteome in Alzheimer’s disease using multiplexed mass spectrometry. J. Proteome Res. 13, 2056–2068 (2014)
Dayon, L., Sonderegger, B., Kussmann, M.: Combination of gas-phase fractionation and MS(3) acquisition modes for relative protein quantification with isobaric tagging. J. Proteome Res. 11, 5081–5089 (2012)
Wu, Y., Wang, F., Liu, Z., Qin, H., Song, C., Huang, J., Bian, Y., Wei, X., Dong, J., Zou, H.: Five-plex isotope dimethyl labeling for quantitative proteomics. Chem. Commun. (Camb.) 50, 1708–1710 (2014)
Boersema, P.J., Raijmakers, R., Lemeer, S., Mohammed, S., Heck, A.J.: Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 4, 484–494 (2009)
Qin, H., Wang, F., Zhang, Y., Hu, Z., Song, C., Wu, R.A.,Ye, M.L.,Zou, H.F.: Isobaric cross-sequence labeling of peptides by using site-selective N-terminus dimethylation. Chem. Commun. (Camb.) 48, 6265–6267 (2012)
Sun, Z., Qin, H., Wang, F., Cheng, K., Dong, M., Ye, M., Zou, H.: Capture and dimethyl labeling of glycopeptides on hydrazide beads for quantitative glycoproteomics analysis. Anal. Chem. 84, 8452–8456 (2012)
Jentoft, N., Dearborn, D.G.: Labeling of Proteins by reductive methylation using sodium cyanoborohydride. J. Biol. Chem. 254, 4359–4365 (1979)
Liu, T., Qian, W.J., Camp II, D.G., Smith, R.D.: The use of a quantitative cysteinyl-peptide enrichment technology for high-throughput quantitative proteomics. Methods Mol. Biol. 359, 107–124 (2007)
Carr, S.A., Abbatiello, S.E., Ackermann, B.L., Borchers, C., Domon, B., Deutsch, E. W., Grant, R. P., Hoofnagle, A. N., Huttenhain, R., Koomen, J. M., Liebler, D. C., Liu, T., MacLean, B., Mani, D., Mansfield, E., Neubert, H., Paulovich, A. G., Reiter, L., Vitek, O., Aebersold, R., Anderson, L., Bethem, R., Blonder, J., Boja, E., Botelho, J., Boyne, M., Bradshaw, R. A., Burlingame, A. L., Chan, D., Keshishian, H., Kuhn, E., Kinsinger, C., Lee, J. S. H., Lee, S. W., Moritz, R., Oses-Prieto, J., Rifai, N., Ritchie, J., Rodriguez, H., Srinivas, P. R., Townsend, R. R., Van Eyk, J., Whiteley, G., Wiita, A., Weintraub, S.: Targeted peptide measurements in biology and medicine: best practices for mass spectrometry- based assay development using a fit-for-purpose approach. Mol. Cell. Proteomics 13, 907–917 (2014)
Lau, H.T., Suh, H.W., Golkowski, M., Ong, S.E.: Comparing SILAC- and stable isotope dimethyl-labeling approaches for quantitative proteomics. J. Proteome Res. 13, 4164-4174 (2014)
Boersema, P.J., Aye, T.T., van Veen, T.A., Heck, A.J., Mohammed, S.: Triplex protein quantification based on stable isotope labeling by peptide dimethylation applied to cell and tissue lysates. Proteomics 8, 4624–4632 (2008)
El-Khatib, A.H., Esteban-Fernandez, D., Linscheid, M.W.: Dual labeling of biomolecules using MeCAT and DOTA derivatives: application to quantitative proteomics. Anal. Bioanal. Chem. 403, 2255–2267 (2012)
Ting, L., Rad, R., Gygi, S.P., Haas, W.: MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940 (2011)
McAlister, G.C., Nusinow, D.P., Jedrychowski, M.P., Wuhr, M., Huttlin, E.L., Erickson, B.K., Rad, R., Haas, W., Gygi, S.P.: MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150–7158 (2014)
Sturm, R.M., Lietz, C.B., Li, L.: Improved isobaric tandem mass tag quantification by ion mobility mass spectrometry. Rapid Commun. Mass Spectrom. 28, 1051–1060 (2014)
Shi, R., Kumar, C., Zougman, A., Zhang, Y., Podtelejnikov, A., Cox, J., Wisniewski, J.R., Mann, M.: Analysis of the mouse liver proteome using advanced mass spectrometry. J. Proteome Res. 6, 2963–2972 (2007)
Sutcliffe, J.G., Hedlund, P.B., Thomas, E.A., Bloom, F.E., Hilbush, B.S.: Peripheral reduction of beta-amyloid is sufficient to reduce brain beta-amyloid: implications for Alzheimer’s disease. J. Neurosci. Res. 89, 808–814 (2011)
Hye, A., Lynham, S., Thambisetty, M., Causevic, M., Campbell, J., Byers, H.L., Hooper, C., Rijsdijk, F., Tabrizi, S.J.,Banner, S., Shaw, C.E., Foy, C., Poppe, M., Archer, N., Hamilton, G., Powell, J., Brown, R.G., Sham, P., Ward, M., Lovestone, S.: Proteome-based plasma biomarkers for Alzheimer’s disease. Brain 129, 3042–3050 (2006)
VanItallie, T.B., Nufert, T.H.: Ketones: metabolism’s ugly duckling. Nutr. Rev. 61, 327–341 (2003)
Martin, B., Brenneman, R., Becker, K.G., Gucek, M., Cole, R.N., Maudsley, S.: iTRAQ analysis of complex proteome alterations in 3xTgAD Alzheimer’s mice: understanding the interface between physiology and disease. PLoS One 3, e2750 (2008)
Shevchenko, G., Wetterhall, M., Bergquist, J., Hoglund, K., Andersson, L.I., Kultima, K.: Longitudinal characterization of the brain proteomes for the tg2576 amyloid mouse model using shotgun based mass spectrometry. J. Proteome Res. 11, 6159–6174 (2012)
Musunuri, S., Wetterhall, M., Ingelsson, M., Lannfelt, L., Artemenko, K., Bergquist, J., Kultima, K., Shevchenko, G.: Quantification of the brain proteome in Alzheimer’s disease using multiplexed mass spectrometry. J. Proteome Res. 13, 2056-2068 (2014)
Andreev, V.P., Petyuk, V.A., Brewer, H.M., Karpievitch, Y.V., Xie, F., Clarke, J. Camp, D., Smith, R.D., Lieberman, A.P., Albin, R.L., Nawaz, Z., El Hokayem, J., Myers, A.J.: Label-free quantitative LC-MS proteomics of Alzheimer’s disease and normally aged human brains. J. Proteome Res. 11, 3053–3067 (2012)
Schonberger, S.J., Edgar, P.F., Kydd, R., Faull, R.L., Cooper, G.J.: Proteomic analysis of the brain in Alzheimer’s disease: molecular phenotype of a complex disease process. Proteomics 1, 1519–1528 (2001)
Choi, J., Rees, H.D., Weintraub, S.T., Levey, A.I., Chin, L.S., Li, L.: Oxidative modifications and aggregation of Cu,Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J. Biol. Chem. 280, 11648–11655 (2005)
Croft, D., Mundo, A.F., Haw, R., Milacic, M., Weiser, J., Wu, G. M., Caudy, M., Garapati, P., Gillespie, M., Kamdar, M. R., Jassal, B., Jupe, S., Matthews, L., May, B., Palatnik, S., Rothfels, K., Shamovsky, V., Song, H., Williams, M., Birney, E., Hermjakob, H., Stein, L., D'Eustachio, P.: The reactome pathway knowledgebase. Nucleic Acids Res. 42, D472–D477 (2014)
Carvalho, C., Katz, P.S., Dutta, S., Katakam, P.V., Moreira, P.I., Busija, D.W.: Increased susceptibility to amyloid-beta toxicity in rat brain microvascular endothelial cells under hyperglycemic conditions. J. Alzheimers Dis. 38, 75–83 (2014)
Vignini, A., Giulietti, A., Nanetti, L., Raffaelli, F., Giusti, L., Mazzanti, L., Provinciali, L.: Alzheimer’s disease and diabetes: new insights and unifying therapies. Curr. Diabetes Rev. 9, 218–227 (2013)
Cunnane, S., Nugent, S., Roy, M., Courchesne-Loyer, A., Croteau, E., Tremblay, S., Castellano, A., Pifferi, F., Bocti, C., Paquet, N., Begdouri, H., Bentourkia, M., Turcotte, E., Allard, M., Barberger-Gateau, P., Fulop, T., Rapoport, S.I.: Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 27, 3–20 (2011)
Ding, F., Yao, J., Rettberg, J.R., Chen, S., Brinton, R.D.: Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PLoS One 8, e79977 (2013)
Henderson, S.T.: Ketone bodies as a therapeutic for Alzheimer’s disease. Neurotherapeutics 5, 470–480 (2008)
Branconnier, R.J., Dessain, E.C., Mcniff, M.E., Cole, J.O.: Blood ammonia and Alzheimers-disease. Am. J. Psychiatry 143, 1313 (1986)
Hoyer, S., Nitsch, R., Oesterreich, K.: Ammonia is endogenously generated in the brain in the presence of presumed and verified dementia of Alzheimer type. Neurosci. Lett. 117, 358–362 (1990)
Rodrigo, R., Cauli, O., Gomez-Pinedo, U., Agusti, A., Hernandez-Rabaza, V., Garcia-Verdugo, J.M., Felipo, V.: Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy. Gastroenterology 139, 675–684 (2010)
Acknowledgments
This research was supported by the University of Pittsburgh Start-up Funds. The authors acknowledge Dr. Ryan D. Bomgarden, Dr. John C. Rogers, and Mr. Brian Hulsebus of Thermo Fisher Scientific for discussions about iodoTMT tagging, and Xi Wang for assistance with statistical testing.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplemental Table S1
(XLSX 171 kb)
Supplemental Table S2
(XLSX 487 kb)
Supplemental Table S3
(XLSX 36 kb)
Supplemental Table S4
(XLSX 60 kb)
ESM 1
(DOCX 178 kb)
Rights and permissions
About this article

Cite this article
Gu, L., Evans, A.R. & Robinson, R.A.S. Sample Multiplexing with Cysteine-Selective Approaches: cysDML and cPILOT. J. Am. Soc. Mass Spectrom. 26, 615–630 (2015). https://doi.org/10.1007/s13361-014-1059-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-014-1059-9