
Abstract
A one-step enzymatic reaction for improving the collision-induced dissociation (CID)-based tandem mass spectrometry (MS/MS) analysis of phosphorylated peptides in an ion trap is presented. Carboxypeptidase-B (CBP-B) was used to selectively remove C-terminal arginine or lysine residues from phosphorylated tryptic/Lys-C peptides prior to their MS/MS analysis by CID with a Paul-type ion trap. Removal of this basic C-terminal residue served to limit the extent of gas-phase neutral loss of phosphoric acid (H3PO4), favoring the formation of diagnostic b and y ions as determined by an increase in both the number and relative intensities of the sequence-specific product ions. Such differential fragmentation is particularly valuable when the H3PO4 elimination is so predominant that localizing the phosphorylation site on the peptide sequence is hindered. Improvement in the quality of tandem mass spectral data generated by CID upon CBP-B treatment resulted in greater confidence both in assignment of the phosphopeptide primary sequence and for pinpointing the site of phosphorylation. Higher Mascot ion scores were also generated, combined with lower expectation values and higher delta scores for improved confidence in site assignment; Ascore values also improved. These results are rationalized in accordance with the accepted mechanisms for the elimination of H3PO4 upon low energy CID and insights into the factors dictating the observed dissociation pathways are presented. We anticipate this approach will be of utility in the MS analysis of phosphorylated peptides, especially when alternative electron-driven fragmentation techniques are not available.

ᅟ
Similar content being viewed by others

1 Introduction
Protein phosphorylation is a particularly prevalent post-translational modification (PTM), having critical regulatory roles in most cellular signaling systems [1]. Mass spectrometry (MS) has become the technique of choice for the qualitative [2] and quantitative [3–5] study of protein phosphorylation because of its unparalleled versatility and sensitivity. ‘Bottom-up’ phosphoproteomics analysis takes place at the peptide-level and usually requires phosphopeptide enrichment [6–8] because of the generally sub-stoichiometric levels of phosphorylated peptides relative to their non-modified counterparts [9] and their somewhat reduced ionization efficiency [10, 11], both factors that are detrimental in data-dependent analysis (DDA) strategies where the selection and fragmentation of precursor ions is dictated by their relative intensities.
The sequence and exact site(s) of phosphorylation are routinely determined by fragmenting the precursor ions using a range of techniques, with collision-induced dissociation (CID) still being the most widely used [12, 13]. During CID, unmodified peptides undergo facile cleavage of the amidic C–N bond, generating b and y ions in accordance with the mobile proton model [14–17]. The presence of a labile functional group, such as an O-phosphoester on the peptide, dramatically changes its fragmentation pathways under CID conditions. Owing to the slow heating nature of CID, as experienced particularly with ion trap instruments, the pathway characterized by the most easily accessible energetic profile will be favored, and the balance between different dissociation channels will be dictated by their relative energetic demands [13, 18–22]. Phosphorylated peptides undergo facile loss of H3PO4 during CID, to an extent which is ultimately dependent on the mobility of the ionizing proton(s), dictated by the ratio between the number of basic residues and the charge state of the observed ion [13, 20, 21]. The extent of neutral loss is generally greater with ion trap mass spectrometers compared with ‘tandem-in-space’ instruments (e.g., Q-ToFs) [23], mainly because of the difference in the amount of energy deposited in the precursor ion and the time involved in the energy transfer [24]. When the number of ionizing protons is larger than the number of basic residues, the peptide ion is believed to be under a mobile proton environment (MPE) [13, 20], where the ionizing proton(s) can be easily relocated and randomized over several amide nitrogen atoms. Relocation of these protons directs C–N bond dissociation and balances the competition between backbone cleavage of the modified peptide and the elimination of H3PO4 [20]. In a limited proton mobility environment (LPME), the neutral loss pathway is characterized by considerably smaller bond dissociation energies than those associated with cleavage of the amide bond and can therefore become predominant [20]. The C-termini of tryptic (and Lys-C) peptides can act as proton ‘sinks’, trapping the ionizing protons because of their high proton affinity, thereby resulting in a more LPME and favoring the gas-phase elimination of phosphoric acid over the C–N bond dissociation. Although several mechanisms have been suggested for the elimination of H3PO4, charge-directed intramolecular nucleophilic substitution (SN2) reactions are characterized by the lowest energetic demands and are, therefore, prevalent (Scheme 1 and Supplementary Scheme 1) [13, 20, 21].

Proposed pathways for the elimination of H3PO4 from a phosphorylated peptide in a (a) mobile proton or (b) limited proton mobility environment. (a) Intramolecular nucleophilic substitution (SN2) reactions invoked under a mobile proton environment (MPE); (b) charge directed intramolecular nucleophilic substitution (SN2) reaction involving formation of a hydrogen bond between a protonated guanidinium side chain and the phosphate group
Under MPE, one of the ionizing protons can be relocated on the sp2 oxygen of the phosphate, thus inducing the formation of a more electrophilic β-carbon atom, which can then undergo a SN2 reaction (Scheme 1a). In the LPME, it is the interaction between the protonated side chain of an arginine (or lysine) residue and the phosphate that plays a major role in dictating the outcome of the CID (Scheme 1b). This interaction, reported as a particularly strong ionic hydrogen bond [25], facilitates the transfer of a proton from the C-terminal residue to that which is phosphorylated. This results in a lower activation barrier of the process, which would otherwise be considerably higher because of the much greater proton affinity (PA) of Arg (PA = 251.2 kcal/mol) [26] and Lys (PA = 239.4 kcal/mol) [26], compared with phosphorylated Ser (PA = 224.2 kcal/mol) [27] and phosphorylated Thr (PA = 224.8 kcal/mol) [27]. The interplay between the different mechanisms is mainly dictated by the charge state and the primary and secondary structures of the gas-phase peptide ions [13, 20]. The role played by basic residues in the product ions generated following CID of a phosphorylated peptide has recently been readdressed by Laskin et al. [28] using energy-resolved surface-induced dissociation (SID) experiments on the model phosphorylated peptide (R/K)GGGpSGGG. These authors concluded that neutral loss of H3PO4 occurs via a two-step process, which involves abstraction of the phosphate to form an ion-molecule complex, whose dissociation eventually leads to the neutral loss product ion and neutral (meta)phosphoric acid.
The formation of a strong hydrogen bond between a protonated arginine residue and the phosphate has also been reported to be responsible for the gas-phase rearrangement of phosphate observed to different extents in the CID of tryptic peptides, especially under LPME [25].
A considerable amount of research has been devoted to improving the tandem MS analysis of phosphorylated peptides, mainly looking at derivatization reactions aimed at manipulating the physical chemical properties phosphorylated peptides and thereby improving their ionization efficiency and fragmentation [29]. Gas-phase ion–molecule reactions using trivalent boron species have also been exploited to modulate the collisional dissociation behavior of simple model phosphorylated peptides in a quadrupole ion trap [30], showing a partial reduction of H3PO4 elimination of the derivatized peptides.
Owing to their purportedly nonergodic nature [31], electron-driven strategies such as electron transfer dissociation (ETD) and electron capture dissociation (ECD) are generally believed to outperform CID for determining sites of covalent modifications [32], but they are not always implemented on commercially available instrumental platforms. Moreover, fragmentation efficiencies are largely dependent on the charge state or, more appropriately, charge density of the precursor ion [33, 34]. MS analysis of modified peptides therefore still largely relies on collisional dissociation, and a strategy able to modify the dissociation features of these species to favor the formation of more sequence diagnostic ions would be of significant benefit.
Here we describe the enzymatic removal of the C-terminal basic residue of tryptic and/or Lys-C phosphorylated peptides with carboxypeptidase-B (CBP-B), which effectively removes the C-terminal proton ‘sink’, releasing the ionizing proton(s). CBP-B treatment thereby overcomes the problems associated with predominant loss of H3PO4 and improves the quality of CID product ion mass spectra for phosphopeptide identification. This procedure can be performed in the same buffer conditions as most standard proteases and, therefore, does not require additional clean-up steps. The use of alternative or complementary proteases in the bottom-up MS analysis of peptides is not unprecedented [35, 36], and we ourselves have shown utility of CBP-B in manipulating peptide dissociation by both CID and ETD [37]. Ultimately, this study demonstrates significant benefits of CBP-B treatment for enhanced analysis of phosphopeptides by CID-MS/MS.
2 Experimental
2.1 Sample Preparation
Synthetic phosphorylated peptides were obtained from Intavis AG (Köln, Germany) and diluted to a final concentration of 1 pmol/μL in 3 % (vol/vol) MeCN, 0.1 % (vol/vol) TFA.
An equimolar mixture of α-casein (comprising α-S1-casein, α-S2-casein), β-casein (Sigma Aldrich, Gillingham, Dorset, UK), and Aurora A [38] was diluted to a final concentration of 1 μg/μL in 50 mM NH4HCO3. Disulphide bonds were reduced with 4.2 mM dithiothreitol (DTT) for 10 min at 60 °C. Samples were cooled to room temperature and alkylated with 16 mM iodoacetamide (IAA) for 45 min in the dark at room temperature. Excess IAA was quenched by addition of DTT to a final concentration of 8 mM. Proteins were digested with either trypsin or Lys-C (Roche, Hertfordshire, UK) (1:50 enzyme:protein) at 37 °C overnight.
Peptide mixtures (trypsin or Lys-C generated) were incubated with CBP-B (Worthington Biochemicals, Lakewood Township, NJ, USA) (12 units/mg protein) at room temperature for 2 h. An aliquot of the digestion solutions (before or after CBP-B treatment) was diluted in 0.1 % (vol/vol) TFA to a final concentration of 100 fmol/μL and analyzed by nanoflow-ultra high performance liquid chromatography-electrospray-tandem mass spectrometry (nUHPLC-ESI-MS/MS).
2.2 Liquid Chromatography and Mass Spectrometry
Data-dependent n-UHPLC-ESI-MS/MS analyses were performed using a Bruker amaZon ETD ion trap MS arranged in-line with a Thermo Scientific Ultimate 3000 nanoflow n-UHPLC system™, using CID as the activation technique (see Supplementary Method for details). Peaklists from the CID MS/MS spectra were extracted using Data Analysis software 4.0 (Bruker, Bremen, Germany) and converted to Mascot generic files (.mgf).
2.3 Database Searching and Data Analysis
.mgf files were searched against the concatenated SwissProt database (2011.05.03) using the Mascot search engine (2.2.06), specifying either H. sapiens or other mammalia for taxonomy as appropriate. Precursor mass tolerance was set to 0.4 Da and fragment ion tolerance to 0.6 Da. Up to 1 missed cleavage was permitted. Trypsin, Lys-C, trypsin/CBP-B, and Lys-C/CBP-B were selected as the enzyme(s) depending on the combination of proteases used. CBP-B cleavage was defined as cleavage N-terminal to K/R. Carbamidomethylation of Cys was set as a fixed modification. Deamidation of Asn and Gln, oxidation of Met, and phosphorylation of Ser, Thr, and Tyr were set as variable modifications. The completeness of the CBP-B treatment as well as the effect of the treatment on the ionization efficiency of the phosphorylated peptides were assessed by comparing the peak areas of extracted ion chromatograms (XIC) of the corresponding protonated peptides before and after the treatment (Supplementary Tables 1 and 2). As the purpose of the Mascot search was to show an increased performance of the search engine before and after the treatment, all of the peptides identified as phosphorylated were retained, and the associated CID tandem mass spectra were manually inspected. As for the non-modified peptides, a minimum Mascot ion score of 25 was required for them to be included in Supplementary Table 1.
2.4 Quantification of Product Ion Currents
To quantify relative product ion currents, an in-house R script was developed. Peak lists corresponding to the peptide spectral matches were extracted using the following characteristic identifiers: file name, elution time, m/z ratio, and charge state of precursor ion, and annotated according to the highest-scoring sequences identified by Mascot and validated by manual inspection of the corresponding CID product ion mass spectra. Theoretical product ions considered included neutral loss of H3PO4 and H2O from the precursor ion, b and y ions ± loss of H3PO4, and b and y ions ± loss of H2O or NH3 as appropriate, with charge state ≤ +2. The relative product ion current (defined here as the summation of ion current within a given category against the total ion current) was then computed.
2.5 Computational Phosphorylation Site Localization
Mascot Delta score (MD) [39] and Ascore [40] were employed as scoring measures to assess confidence in phosphosite localization. MD values were computed manually from the Mascot output. Ascore values were determined post-database search, using the identified phosphopeptides from Mascot and extracting their corresponding MS/MS as input for the Ascore webserver (http://ascore.med.harvard.edu/ascore.html).
3 Results and Discussion
3.1 Reduced Elimination of H3PO4 with CBP-B Treatment and Enhanced Formation of Sequence-Specific Product Ions
It was our hypothesis that removal of the C-terminal basic residue, which plays a fundamental role in the gas-phase dissociation of phosphorylated peptides by facilitating the H3PO4 elimination pathway [20, 21, 25], should result in an increase in the number and relative intensity of diagnostic b and y ions and thereby improve the quality of the CID product ion mass spectra. In an attempt to develop a fast, one-step protocol to alter the gas-phase behavior of protonated phosphopeptide ions, the C-terminal basic residue (arginine or lysine) was removed from tryptic/Lys-C generated phosphorylated peptides with a simple enzymatic reaction using CBP-B, which catalyzes the hydrolysis of basic amino acids from the C-terminal position of a polypeptide. Based on current understanding of the mechanisms of dissociation of phosphorylated peptides, removal of this residue should balance the competition between elimination of phosphoric acid and cleavage of the amidic C–N bond.
To test our hypothesis, we assessed the effect of CBP-B treatment on a mixture of phosphorylated proteins after digestion with either trypsin or Lys-C. Twenty-four peptides from the digested mixture of proteins (66 % of the total peptide number) were observed as their cognate CBP-B analogues, with the C-terminal arginine or lysine residue having been removed (Supplementary Table 1). All of the phosphopeptides identified prior to CBP-B treatment were converted into their CBP-B analogues. Four further phosphorylated peptides (DIGSEpSTEDQAMEDI, DIGpSEpSTEDQAMEDI, QLQApTSVPHPVS, and RpTpTLCGTLDYLPPEMIEG) were also identified after treatment (Table 1 and Supplementary Table 1), in addition to a further nine non-phosphorylated peptides. The efficiency of the enzymatic reaction with CBP-B, monitored by measuring the peak areas of the extracted ion chromatograms (XICs) associated with the native and CBP-B treated peptides, was close to 100 % for most of the identified peptides, with limited residual tryptic or Lys-C peptides found in the treated samples. Three of the phosphorylated species showed incomplete conversion, varying from 40 % (FQpSEEQQQTEDELQDK) to 62 % (TVDMEpSTEVFTK) and 82 % (VPQLEIVPNpSAEER) (Supplementary Table 1). Interestingly, the same rate of incomplete CBP-B conversion was also observed for the non-phosphorylated form of TVDMEpSTEVFTK, suggesting that proteolytic efficiency may be sequence-dependent.
The charge states in which the untreated and treated peptides were observed were also comparable for the two sample sets, although unsurprisingly there was a reduction in the number of treated peptides that were further observed as triply charged species: 25 % untreated peptides were observed as triply charged species, which reduced to 8 % for the CBP-B peptides (Supplementary Table 1). No singly charged peptides could be identified under our instrumental conditions before or after the treatment, even when singly charged ions were included in the precursor ion selection.
The ionization efficiency of the phosphorylated peptides showed a sequence-dependent response to the treatment, ranging from increased relative signal intensity of 2.7-fold for VLVpTQQFPCQNPLPVNSGQAQ(R), to 0.4 for LVpSSHKPVQNQ(K) (Supplementary Table 2), suggesting a more complex scenario than one would envisage for the relationship between the presence of a C-terminal basic residue and the efficiency of ESI.
Comparison of the CID product ion mass spectra of the nine phosphorylated peptide pairs listed in Table 2A and B, before and after CBP-B treatment, indicated that the effects of removal of the C-terminal basic residue can be divided into three main groups: (1) absence of neutral loss of H3PO4 and the concurrent enhanced formation of b and y ions; (2) substantial reduction in the extent of neutral loss; (3) no change in the predominance of neutral loss attributable to the presence of internal basic residues, examples of which will be presented.
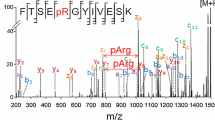
The CID product ion mass spectrum of the untreated peptide VLVpTQQFPCQNPLPVNSGQAQR (Figure 1a) is dominated by the peaks corresponding to loss of H3PO4 (m/z 1231.7) and (H3PO4 + H2O) (m/z 1222.7).

CID product ion mass spectra of (a) the doubly protonated peptide before (VLVpTQQFPCQNPLPVNSGQAQR; m/z 1280.7) and (b) triply protonated peptide after (VLVpTQQFPCQNPLPVNSGQAQ, m/z 802.2) CBP-B treatment, demonstrating complete absence of neutral loss of phosphoric acid from the precursor and product ions following CBP-B treatment
However, the corresponding CBP-B treated peptide VLVpTQQFPCQNPLPVNSGQAQ (Figure 1b) clearly shows how removal of the C-terminal basic residue results in complete absence of neutral loss product ions; a dramatic improvement in the formation of diagnostic b and y ions was observed, leading to an almost complete generation of sequence determining ions. Some relatively intense ions were also present in both the treated and untreated spectra, which could not be assigned based on the known sequence of the peptide. Of particular interest are the diagnostic ions y17 2+ (at low intensity) and y18 2+, which were observed following CBP-B treatment, allowing unambiguous assignment of the phosphorylation site at position Thr4. Assignment of the modification site in the CID product ion mass spectrum of the untreated peptide is indirect, relying primarily on observation of loss of H3PO4 from a limited number of b ions (b6Δ, b7Δ, b11Δ, b15Δ). Pinpointing phosphorylation sites in phosphopeptides on the observation of species corresponding to the elimination of H3PO4 from product ions has been recently questioned [23, 25], as a net dissociation transition of 98 Da could be the result of elimination of metaphosphoric acid (HPO3) from the phosphorylation site and H2O from a non-modified serine or threonine residue. In this particular example, CID of the untreated phosphopeptide failed to generate product ions for most of the N-terminal portion of the peptide, which serves to decrease confidence in the phosphopeptide identification compared with the treated peptide.
Very similar effects were seen for the peptide VPQLEIVPNpSAEER, which undergoes extensive loss of H3PO4 (m/z 781.8) as a result of CID (Figure 2a). The analogous neutral loss species is almost completely eliminated in the product ion mass spectrum of the CBP-B-treated peptide (Figure 2b), thus confirming the anticipated effect of the removal of the C-terminal basic residue on the decreased prevalence of the neutral loss pathway.

CID product ion mass spectra of doubly protonated peptide: (a) before (VPQLEIVPNpSAEER, m/z 830.7) and (b) after (VPQLEIVPNpSAEE, m/z 752.7) CBP-B treatment, demonstrating a significant reduction in the extent of neutral loss of phosphoric acid following CBP-B treatment
An increased ion current towards diagnostic b and y ions is of particular value in all those circumstances where unambiguous identification of the peptide relies on the detection of sequence specific product ions, as is the case for SRM-based assays for phosphopeptide quantification [41]. The synthetic peptide ILSDVTHpSAVFGVPAS(K) was chosen to investigate whether an internal basic residue such as histidine, with a proton affinity (PA = 228.3 kcal/mol) [26] lower than that of arginine and lysine, can limit the benefits of the CBP-B treatment. This peptide demonstrated very similar behavior indicating that the internal histidine residue in this peptide does not have sufficient gas-phase basicity to trap the proton and hinder the formation of b and y ions, and confirms that the basicity of the proton ‘sink’ plays a major role in dictating the outcome of CID (Supplementary Figure 1).
Of particular interest, therefore, are those peptides characterized by the presence of a non C-terminal Lys or Arg residue, generated either as a consequence of a missed cleavage site or an internal [K/R]P, which does not undergo trypsin proteolysis. The Aurora A phosphopeptide RpTpTTLCGTLDYLPPEMIEG, which has an N-terminal basic residue arising because of incomplete tryptic cleavage, was observed as a doubly charged ion at m/z 1113.2 (Supplementary Figure 2) and generated an almost complete series of b and y ions upon CID, allowing unambiguous localization of the two phosphorylation sites at positions 2 and 3. The base peak in the product ion spectrum is the y7 ion, and no neutral loss of H3PO4 or (H3PO4 + H2O) could be observed. Despite the presence of a basic residue (Arg at position 1), the doubly charged ion is still under a MPE as the extra proton can potentially relocate to any of the amidic nitrogen atoms upon CID due to the lack of any other proton ‘sink’ within the peptide sequence, therefore balancing the competition between potential dissociation pathways towards dissociation of the amidic bond. An internal basic side chain (Figure 3 and Supplementary Figure 3) can also serve as a proton ‘sink’, meaning that even after CBP-B treatment, doubly charged peptides are under a LPME, as the number of basic residues (three) is greater than the number of ionizing protons. Figure 3 compares the CID product ion spectra of LVpSSHKPVQNQK before (Figure 3a) and after (Figure 3b) treatment with CBP-B. In both cases, the product ion mass spectrum is dominated by a base peak corresponding to the neutral loss of H3PO4 from the precursor ion (m/z 673.7 and 609.7, respectively) with limited formation of product ions. The internal lysine at position 6 acts as a proton sink, potentially along with the histidine residue at position 5, which is the second most thermodynamically favored protonation site [26]. CBP-B treatment of this peptide therefore has little effect on the extent of loss of H3PO4 as a result of the limited mobility of the ionizing protons and the occurrence of a hydrogen bond between the internal lysine and the phosphate on Ser at position 3, which is likely to facilitate the elimination of H3PO4 in the treated phosphorylated peptide.

CID product ion mass spectra of doubly protonated peptide: (a) before (LVpSSHKPVQNQK, m/z 722.7) and (b) after (LVpSSHKPVQNQ, m/z 658.7) CBP-B treatment. The presence of an internal lysine residue (PA = 239.4 kcal/mol) overcomes the benefits of the removal of the C-terminal lysine, forcing the system towards a reduced proton mobility and therefore favoring the pathway leading to the neutral loss of H3PO4 over the dissociation of the amidic C–N bond
Gas-phase elimination of H3PO4 upon CBP-B treatment can still be observed for some peptides that do not contain an internal K/R residue, albeit at significantly reduced levels, as for the case of QLQApTSVPHPVS (Supplementary Figure 4). However, even under these circumstances, the ion current associated with b and y ion formation (no neutral loss) remains enhanced. Supplementary Figure 5 shows CID product ion mass spectra for the peptides TVDMEpSTEVFTK (Supplementary Figure 5a), TVDMEpSTEVFTKK (Supplementary Figure 5b) and the CBP-B treated TVDMEpSTEVFT (Supp. Figure 5c) respectively. The CID product ion mass spectra of the native phosphopeptides both show base peaks corresponding to the neutral loss product ions (m/z 733.6 and m/z 614.1, respectively) which are markedly reduced in the CID product ion mass spectrum of the corresponding CBP-B treated peptide. Unambiguous assignment of the phosphorylation site was not possible for the shorter native peptide, due to the lack of sufficient diagnostic ions to safely discriminate between phosphorylation of Ser6 and Thr7 (Supplementary Figure 5a), although a minor product ion defined as y7 in the longer peptide appears to localize the site of modification to Thr7. Removal of the C-terminal lysine residue(s) (Supplementary Figure 5c) resulted in the generation of b6, b7, y5 and y6 product ions, enabling confident localization of the phosphorylation site to position 6. These data therefore appear to imply a heterogeneous population of Ser6 and Thr7 phosphopeptides in this sample. The prominence of the different dissociation pathways is particularly striking when considering the product ions arising from the mis-cleaved peptide TVDMEpSTEVFTKK, containing a dibasic C-terminus (Supplementary Figure 5b). CID of this peptide generates a product ion mass spectrum that is dominated by the neutral loss fragment ion at m/z 748.8, with most of the sequence-specific product ions being observed at very low levels.
Peptide VLCPSNpSSQR yielded an interesting CID product ion mass spectrum; although the peptide lacks an obvious proton sink, the spectrum was dominated by neutral loss of the precursor ion both before and after CBP-B treatment (Supplementary Figure 6a and b). However, the treated peptide showed additional b and y ions (y2 and y3) which enabled unambiguous identification of the phosphorylation site at position 7. For this peptide sequence, the phosphorylated serine residue at position 7 is characterized by the highest proton affinity (224.2 kcal/mol) [27]. For the doubly charged precursor ion, the oxygen of the phosphor-ester is likely to be the preferred protonation site, meaning that the system contains a -CH2-O-PO3H2 moiety where protonation of one of the oxygens (which normally requires the energy imparted to the ion upon CID) is likely pre-existent to the fragmentation process. Consequently, the species are much more labile towards H3PO4 elimination, as the leaving group is already preformed in the structure of the precursor ion.
Finally, it is worth noting one particular case where a ‘chimeric’ CID product ion mass spectrum was generated for a pair of isomeric, co-eluting phosphorylated peptides after CBP-B treatment: DIGSEpSTEDQAMEDI and DIGpSESTEDQAMEDI at m/z 860.1 (Figure 4). The tandem mass spectrum contains all of the product ions required for confident localization of the phosphorylation site on the serine residue in position 6. However, two product ions, namely y10* and y11* at m/z 1138.4 and 1267.5, respectively, account for a phosphorylated serine at position 4. The computational algorithms Mascot and Ascore identified Ser6 and Ser4 as the phosphorylation site, respectively. This contradiction in site localization highlights a largely unaddressed issue of most of the automated search algorithms which face considerable limitations when dealing with chimeric tandem mass spectra generated from co-isolation of isobaric species, and demonstrates how non-rigorous interpretation of the output from automated search engines might lead to incorrect inferences in site localization.

CID product ion spectra of the doubly protonated peptide DIGSEpSTEDQAMEDI m/z 860.1 after CBP-B treatment. Product ions specific to the co-eluting peptide DIGpSESTEDQAMEDI (y10 and y11) are highlighted in bold and italic
3.2 CBP-B Treatment Increases the Number and Intensity of Sequence-Specific b and y ions
To quantify the effects of CBP-B treatment on the appearance of useful, diagnostic product ions, an in-house R script was used to extrapolate ion currents associated with the different product ion types, namely: precursor ion-H3PO4; precursor ion-(H3PO4 + H2O); b ions (+1, +2); y ions (+1, +2); (b ions-H3PO4) (+1, +2); (y ions-H3PO4) (+1, +2); (b ions-H2O) (+1, +2); (b ions-NH3) (+1, +2); (y ions-H2O) (+1, +2); (y ions-NH3) (+1, +2). Table 2 list the intensities of these ions as percentages of the total product ion current for each of the identified phosphorylated peptides before and after removal of the C-terminal residue, respectively.
The effects of the CBP-B treatment are particularly noticeable when comparing the relative amounts of b ions for the treated and untreated peptides. The percentage ion current associated with intact b ions ([M + H]+ and [M + 2H]2+) is consistently higher for the treated peptides: up to 13 times greater in the case of TVDMEpSTEVFT(K). Chawner et al. [37] already described how removal of the C-terminal basic residue from unmodified tryptic peptides has a marked influence on the distribution of CID and ETD product ions, with an increased ion current of the b and c ion series observed after the treatment, due to the mobilization of the ionizing protons. Appearance of certain b ions was only observed upon removal of the basic C-terminus, leading the authors to hypothesize that not only does the removal of C-terminus release the ionizing protons, but it can also modify the gas-phase reactivity of the corresponding ionic species. This allows them to enter otherwise less accessible or inaccessible energetic pathways, resulting in pronounced increase in the intensity of defined b ions. A similar effect could account for the appearance of the b7 and y6 ions in Figure 2a.
The signal due to neutral loss elimination of H3PO4 (with or without concomitant elimination of H20) from the precursor ion also decreases by an average of 26.3 %, without taking into account those phosphorylated peptides which could only be identified after CBP-B treatment. Furthermore, it is interesting to note how even when the extent of gas-phase neutral loss of H3PO4 from the precursor ion is still the main channel of dissociation in the treated peptide, there is an increased number of diagnostic b and y product ions; for example, CID of peptide EQLSTpSEENS(K) generated 21 sequence specific b/y ions after treatment, but only 9 ions before treatment (Supplementary Figure 7), again allowing precise localization of the phosphorylation site due to the appearance specifically of the b6 and y5 ions. This confirms what was presciently anticipated by Rožman [20] and demonstrated by Chawner et al. [37]: by sequestering the ionizing proton, the basic C-terminal residue effectively controls the kinetic thermodynamic accessibility of several dissociation pathways, ultimately dictating the appearance of defined b and y ions.
3.3 CBP-B Improves Search Engine-Based Data Analysis of CID Product Ion Mass Spectra
Having shown that removal of the C-basic residue results in enhanced formation of sequence and phosphorylation site-specific b and y ions, we investigated whether the better quality of the CID product ion mass spectra could be reflected in improved performance of the search engine Mascot, by comparing the ion scores and expectation values associated to the identified phosphorylated peptides before and after the CBP-B treatment. Mascot delta (MD) score [39] and Ascore [40] were further implemented to assess whether our protocol could help with discriminating between two potential candidate phosphorylated isoforms for those peptides containing more than one potential site of phosphorylation.
Mascot Delta is a search-engine difference score, which relies on the difference in Mascot Ion score between the top-and second-ranked peptide spectrum matches to assess the reliability of the sequence identification and site location. In contrast, Ascore is a probability-based localizer which does not rely on scoring metrics but rather applies its own scoring algorithm to the product ion spectrum using the inferred phosphopeptide identification generated by the search engine. The Ascore algorithm only annotates intact phosphorylated product ions [40] (i.e. product ions carrying the intact phosphoester moiety) and makes use of an intensity-based peak depth filter to maximize the annotated-to-unannotated ion ratio.
Table 1 lists all of the phosphorylated peptides identified in the untreated and treated peptide sets, correlating Mascot ion scores, expectation values, MD scores and Ascore values for the two sets of peptides. Under circumstances where the same peptide was identified more than once, the CID product ion mass spectrum associated with the highest score was selected. Averaged ion scores, resulting from eight technical replicates, are also provided. Apart from VLVpTQQFPCQNPLPVNSGQAQ which appeared as a triply, rather than a doubly charged ion after CBP-B treatment, all comparisons were performed between precursor ions of the same charge state. With the exception of peptide FQpSEEQQQTEDELQDK (the score of which decreased from 97 to 83), the highest ion scores of each phosphopeptide were improved after removal of the C-terminal residue. The average ion scores confirmed this trend, with the CBP-B treated peptides all having scores equal to or greater than their unmodified counterparts. The average increase in ion score was 14.7, without taking into account the four phosphopeptides which could only be identified after CBP-B treatment. Tandem mass spectra generated from these four phosphopeptides all yielded ion scores above 30, with a maximum value of 74, demonstrating the beneficial effects of enhanced formation of product ions on the performance of the search engine. Moreover, improved expectation values (sometimes by two orders of magnitude) are associated with the treated set of phosphorylated peptides, confirming that sequence assignment of the modified peptides can be achieved with greater confidence.
MD scores were also improved, especially for peptides containing more than one potential site of modification, as in the case of peptides EQLSTpSEENSK, TVDMEpSTEVFTK(K), VLCPpSNSSQR and VLVpTQQFPCQNPLPVNSGQAQR, whose delta scores were increased by 6.1, 6.3, 16.4 and 15.6 respectively. For the first three phosphopeptides above, CBP-B treatment increased the MD score above the cutoff value of 10, which approximates to a 5 % false localization rate on an ion-trap instrument with CID fragmentation [39].
As the CBP-B treatment increases the number and relative intensity of sequence specific b and y ions with no neutral loss, the CID product ion mass spectra typically contain more peaks likely to survive the filtering process required for Ascore. With the exception of FQpSEEQQQTEDELQDK, (Supplementary Figure 8), for which the Ascore value dropped from 137.1 to 84.9, the Ascore values for all of the other phosphorylated peptides increased upon CBP-B treatment (Table 1) by an average of 20.2. Unsurprisingly, the Ascore value of LVpSSHKPVQNQK, whose CID product ion mass spectrum was unaffected by the treatment, was unchanged. A remarkable increase in the Ascore value was obtained for peptide EQLSTpSEENSK, which went from 0 to 20.4, confirming that the algorithm greatly benefits from the improved quality of the CID product ion mass spectra.
Overall, computational phosphopeptide sequence identification and phosphosite localization for this relatively small dataset both benefitted (or were largely unchanged) from this sample preparation step. However, it will be useful to assess the effect of our protocol on the performance of search engines with a much larger dataset prior to global implementation of this strategy for high-throughput phosphoproteomics studies. Irrespective, this procedure seems to be of particular benefit when the appearance of one or few diagnostic product ions is the limiting factor in the possibility of distinguishing between potential phosphorylated isomeric peptides with internal, sequential Ser and Thr residues.
4 Conclusions
Enzymatic removal of the C-terminal basic residue from tryptic and Lys-C phosphorylated peptides using CBP-B resulted in the generation of species whose CID, as performed with a Paul-type ion trap, showed reduced elimination of phosphoric acid and enhanced formation of sequence specific, diagnostic product ions. Removing the C-terminal proton ‘sink’ from such phosphopeptides eliminates the possibility for the charged C-terminus to promote the H3PO4 loss, resulting in transition towards a more mobile proton environment in which the competition between neutral loss of phosphoric acid and cleavage of the C-N amidic bond is thermodynamically more balanced. The result is improved confidence in both sequence determination and site localization. This strategy is particularly beneficial in those cases where defining the exact location of the modification on the peptide sequence is problematic (and oftentimes incorrect) due to the presence of multiple potential phosphorylation sites which may be clustered in a short region of the sequence. Considering the role described for the C-terminal basic residue in the gas-phase scrambling of the phosphorylation site [20, 25], removal of this residue may also limit the occurrence of this phenomenon, which has been reported to affect the CID-based analysis of phosphorylated peptides to differing extents [25, 42].
The efficiency of CBP-B treatment for improving high-throughput global phosphoproteomic analysis on a whole cell lysate is hard to predict based on this necessarily limited dataset for comprehensive analysis, and consideration will need to be given to the database search strategies employed, due in part to issues associated with defining the proteolytic rules for CBP-B treatment on the database. However, our current data indicates that this approach could indeed be useful for more complex phosphoproteomics datasets given firstly that the majority of peptides were converted to their CBP-B analogues, and secondly that both the number of phosphopeptides identified and the confidence in phosphopeptide identification increased following CBP-B treatment. Consequently, we envisage that this approach, based on a simple enzymatic reaction performed immediately after digestion of the target proteins that does not require any additional sample clean-up, will be of notable benefit in phosphopeptide analysis, particularly when alternative, complementary dissociation strategies, i.e. ETD/ECD, are not available. Ultimately, this study provides experimental proof of the role played by basic residues and charge distribution in the low-energy CID of protonated, phosphorylated peptides, confirming the theory which describes their gas-phase dissociation pathways.
References
Hunter, T.: Philos Trans. Soc. Lond. B Biol. Sci. 367, 2513–2516 (2012)
Zhou, H., Di Palma, S., Preisinger, C., Peng, M., Polat, A., Heck, A.J., Mohammed, S.: Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 12, 260–271 (2013)
Jia, W., Andaya, A., Leary, J.A.: Novel mass spectrometric method for phosphorylation quantification using cerium oxide nanoparticles and tandem mass tags. Anal. Chem. 84, 2466–2473 (2012)
Johnson, H., Eyers, C.E., Eyers, P.A., Beynon, R.J., Gaskell, S.J.: Rigorous determination of the stoichiometry of phosphorylation using mass spectrometry. J. Am. Soc. Mass Spectrom. 20, 2211–2220 (2009)
Engholm-Keller, K., Birck, P., Storling, J., Pociot, F., Mandrup-Poulsen, T., Larsen, M.R.: TiSH-a robust and sensitive global phosphoproteomics strategy employing a combination of TiO2, SIMAC, and HILIC. J. Proteomics 75, 5749–5761 (2012)
Graauw, M., Thingholm, T., Jensen, O., Larsen, M.: In: Phospho Proteomics. Humana Press: Vol. 527, pp 67–78 (2009)
Gates, M.B., Tomer, K.B., Deterding, L.J.: Comparison of metal and metal oxide media for phosphopeptide enrichment prior to mass spectrometric analysis. J. Am. Soc. Mass Spectrom. 21, 1649–1659 (2010)
Haydon, C.E., Eyers, P.A., Aveline-Wolf, L.D., Resing, K.A., Maller, J.L., Ahn, N.G.: Identification of novel phosphorylation sites on Xenopus laevis Aurora A and analysis of phosphopeptide enrichment by immobilized metal-affinity chromatography. Mol. Cell Proteomics 2, 1055–1067 (2003)
Steen, H., Jebanathirahjah, J.A., Rush, J., Morrice, N., Kirschner, M.W.: Phosphorylation analysis by mass spectrometry. Mol. Cell Proteomics 5, 172–181 (2006)
Marcantonio, M., Trost, M., Courcelles, M., Desjardins, M., Thibault, P.: Combined enzymatic and data mining approaches for comprehensive phosphoproteome analyses. Mol. Cell Proteomics 7, 645–660 (2008)
Ishihana, Y., Wei, F.Y., Aoshiama, K., Sato, T., Kuromitsu, J., Oda, Y.: Enhancement of the efficiency of phosphoproteome identification by removing phosphates after phosphopeptide enrichment. J. Proteome Res. 6, 1139–1144 (2007)
Hoffert, J.D., Knepper, M.A.: Taking aim at shotgun phosphoproteomics. Anal. Biochem. 375, 1–10 (2008)
Boersema, P.J., Mohammed, S., Heck, A.J.: Phosphopeptide fragmentation and analysis by mass spectrometry. J. Mass Spectrom. 44, 861–878 (2009)
Burlet, O., Yang, C.-Y., Gaskell, S.: Influence of cysteine to cysteic acid oxidation on the collision-activated decomposition of protonated peptide: evidences for intraionic interactions. J. Am. Soc. Mass Spectrom. 3, 337–344 (1992)
Harrison, A.G., Yalcin, T.: Proton mobility in protonated amino acids and peptides. Int. J. Mass Spectrom. Ion Process. 166, 339–347 (1997)
Wysocki, V.H., Tsaprailis, G., Smith, L.L., Breci, L.A.: Mobile and localized protons: a framework for understanding peptide dissociation. J. Mass Spectrom. 35, 1399–1406 (2000)
Paizs, B., Suhai, S.: Fragmentation pathways of protonated peptides. Mass Spectrom. Rev. 24, 508–548 (2005)
Tholey, A., Reed, J., Lehmann, W.D.: Electrospray tandem mass spectrometric studies of phosphopeptides and phosphopeptide analogues. J. Mass Spectrom. 34, 117–123 (1999)
Medzihradsky, K., Trinidad, J.: Unusual fragmentation of pro-ser/thr-containing peptides detected in collision-induced dissociation spectra. J. Am. Soc. Mass Spectrom. 23, 602–607 (2012)
Rožman, M.: Modeling of the gas-phase phosphate group loss and rearrangement in phosphorylated peptides. J. Mass Spectrom. 46, 949–955 (2011)
Palumbo, A.M., Tepe, J.J., Reid, G.E.: Mechanistic insights into the multistage gas-phase fragmentation behavior of phosphoserine- and phosphothreonine-containing peptides. J. Proteome Res. 7, 771–779 (2008)
Degnore, J., Qin, J.: Fragmentation of phosphopeptides in an ion trap mass spectrometer. J. Am. Soc. Mass Spectrom. 9, 1175–1188 (1998)
Lehmann, W.D., Krüger, R., Salek, M., Hung, C.-W., Wolschin, F., Weckwerth, W.: Neutral-loss based phosphopeptide recognition: a collection of caveats. J. Proteome Res. 6, 2866–2873 (2006)
Johnson, J.V., Yost, R.A., Kelly, P.E., Bradford, D.C.: Tandem-in-space and tandem-in-time mass spectrometry: triple quadrupoles and quadrupole ion traps. Anal. Chem. 626, 21662–22172 (1990)
Palumbo, A.M., Reid, G.E.: Evaluation of gas-phase transfer and competing fragmentation reactions on protein phosphorylation site assignment using collision induced dissociation-MS/MS and MS3. Anal. Chem. 80(24), 9735–9747 (2008)
Hunter, E.P.L., Lias, S.G.: Evaluated gas phase basicities and proton affinities of molecules: an update. J. Phys. Chem. Ref. Data 27, 413–656 (1998)
Paladini, A., Scuderi, D., Laganá, A., Giardini, A., Filippi, A., Speranza, M.: Gas-phase basicity and enantio-discrimination of some phosphorous-containing α-amino acid mimics. Int. J. Mass Spectrom. 228, 349–358 (2003)
Laskin, J., Kong, R.P.W., Song, T., Chu, I.K.: Effects of the basic residue on the energetic and dynamics of dissociation of phosphopeptides. Int. J. Mass Spectrom. 330, 295–301 (2012)
Knight, Z.A., Schilling, B., Row, R.H., Kenski, D.M., Gibson, B.W., Shokat, K.M.: Phosphospecific proteolysis for mapping sites of protein phosphorylation. Nat. Biotechnol. 21, 1047–1054 (2003)
Gronert, S., Li, K.H., Horiuchi, M.: Manipulating the fragmentation patterns of phosphopeptides via gas-phase boron derivatization: determining phosphorylation sites in peptides with multiple serines. J. Am. Soc. Mass Spectrom. 16(12), 1905–1914 (2005)
Tureček, F.: N–Cα bond dissociation energies and kinetics in amide and peptide radicals. Is the dissociation a non-ergodic process? J. Am. Chem. Soc. 125(19), 5954–5963 (2003)
Zubarev, R.A., Horn, D.M., Fridriksson, E.K., Kelleher, N.L., Kruger, N.A., Lewis, M.A., Carpenter, B.K., McLafferty, F.W.: Electron capture dissociation for structural characterization of multiply charged protein cations. Anal. Chem. 72, 563–573 (2000)
Molina, H., Horn, D.M., Tang, N., Mathivanan, S., Pandey, A.: Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 104, 2199–2204 (2007)
Xia, Y., Gunawardena, H.P., Erickson, D.E., McLuckey, S.A.: Effect of cation charge-site identity and position on electron-transfer dissociation of polypeptide cations. J. Am. Soc. Mass Spectrom. 129, 12232–12243 (2007)
Gilmore, J., Kettenbach, A., Gerber, S.: Increasing phosphoproteomic coverage through sequential digestion by complementary proteases. Anal. Bioanal. Chem. 402, 711–720 (2012)
Taouatas, N., Altellar, A.F.M., Drugan, M.M., Helbig, A.O., Mohammed, S., Heck, A.J.R.: Strong cation exchange-based fractionation of Lys-N-generated peptides facilitates the targeted analysis of post-translational modifications. Mol. Cell. Proteomics 8, 190–200 (2009)
Chawner, R., Eyers, C.E., Gaskell, S.J.: The influence of a C-terminal basic residue on peptide fragmentation pathways. Int. J. Mass Spectrom. 316, 284–291 (2012)
Sloane, D.A., Trikic, M.Z., Chu, M.L.H., Lamers, M.B.A.C., Mason, C.S., Mueller, I., Savory, W.J., Williams, D.H., Eyers, P.A.: Drug-resistant Aurora A mutants for cellular target validation of the small molecule kinase inhibitors MLN8054 and MLN8237. ACS Chem. Biol. 5, 563–576 (2010)
Savitski, M.M., Lemeer, S., Boesche, M., Lang, M., Mathieson, T., Bantscheff, M., Kuster, B.: Confident phosphorylation site localization using the Mascot delta score. Mol. Cell Proteomics 10, 1–12 (2011)
Beausoleil, S.A., Villen, J., Gerber, S.A., Rush, J., Gygi, S.P.: A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol 24, 1285–1292 (2006)
Unwin, R.D., Griggiths, J.R., Whetton, A.D.: A sensitive mass spectrometric method for hypothesis-driven detection of peptide post-translational modifications: multiple reaction monitoring-initiated detection and sequencing (MIDAS). Nat. Protoc. 4, 870–877 (2009)
Mischerikow, N., Maarten Altelaar, A.F., Navarro, J.D., Mohammed, S., Heck, A.J.R.: Comparative assessment of site assignments in CID and ETD spectra of phosphopeptides discloses limited relocation of phosphate groups. Mol. Cell Proteomics 9, 2140–2148 (2010)
Acknowledgments
This work was supported by funding from Biotechnology and Biological Sciences Research Council (BBSRC) grants BB/F00561X/1 (Systems Approach to Biological Research) and BB/H007113/1 awarded to C.E. D.L. is supported by a BBSRC/EPSRC funded Systems Biology studentship.
The authors are grateful to Dr. Stephen W. Holman and Dr. Simon Hubbard for helpful discussions during the preparation of this manuscript, Dr. Craig Lawless for his assistance with Ascore and Dr. Patrick Eyers for provision of purified recombinant autophosphorylated Aurora A.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 3397 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Lanucara, F., Chi Hoo Lee, D. & Eyers, C.E. Unblocking the Sink: Improved CID-Based Analysis of Phosphorylated Peptides by Enzymatic Removal of the Basic C-Terminal Residue. J. Am. Soc. Mass Spectrom. 25, 214–225 (2014). https://doi.org/10.1007/s13361-013-0770-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-013-0770-2