
Abstract
Mass spectrometric techniques and more particularly collision-induced dissociation (CID) experiments represent a powerful method for the determination of the primary sequence of (bio)molecules. However, the knowledge of the ion fragmentation patterns say the dissociation reaction mechanisms is a prerequisite to reconstitute the sequence based on fragment ions. Previous papers proposed that protonated peptoids dissociate following an oxazolone-ring mechanism starting from the O-protonation species and leading to high mass Y sequence ions. Here we revisit this backbone cleavage mechanism by performing CID and ion mobility experiments, together with computational chemistry, on tailor-made peptoids. We demonstrated that the B/Y cleavages of collisionally activated O-protonated peptoids must involve the amide nitrogen protonated structures as the dissociating species, mimicking the CID behavior of protonated peptides. Upon the nucleophilic attack of the oxygen atom of the N-terminal adjacent carbonyl group on the carbonyl carbon atom of the protonated amide, the peptoid ions directly dissociate to form an ion-neutral complex associating an oxazolone ion to the neutral truncated peptoid residue. Dissociation of the ion/neutral complex predominantly produces Y ions due to the high proton affinity of the secondary amide function characteristic of truncated peptoids. Whereas the production of Yx ions from acetylated peptoids also involves the B/Y pathway, the observation of abundant Yx ions from non-acetylated peptoid ions is shown in the present study to arise from an A1-Yx mechanism. The consecutive and competitive characters of the A1-Yx and the B/Y mechanisms are also investigated by drift time-aligned CID experiments.

Similar content being viewed by others

Introduction
Poly-N-substituted glycines, better known as peptoids, represent a class of synthetic polymers which are closely related to peptides (Scheme 1) [1]. The main difference between peptoids and peptides lies in the side chain position in the building block, located respectively on the nitrogen atom rather than on the α-carbon [1]. Efficient synthetic protocols are now available and are basically based on a stepwise incorporation of the peptoid residues [1,2,3]. Due to the large diversity of commercially available amines, the number of accessible structures is in principle infinite and can vary in terms of the nature of the side chains and their sequence. Synthetic strategies to finely tune the peptoid sequence are now abundantly exploited to master the secondary structure of peptoids to target applications, such as chiral sensors, biomimetics, or piezoelectric devices [4,5,6,7,8,9,10,11]. Shin and co-workers investigated the influence of the achiral/chiral monomer sequence on the conformation of peptoid heptamers in solution using circular dichroism [6]. They demonstrated that the helical conformation of peptoids is dictated by the specific position and number of the chiral monomer units [6]. More recently, Jin et al. studied the formation of stiff nanotubes from sequence-defined peptoids [8]. They determined that the length of the hydrophobic sequence impacts the mechanical properties and the structure of the nanotubes, including their diameter and their thickness [8].

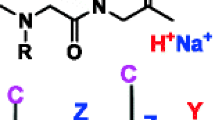
General structure of peptoid with (X = –C(=O)CH3) or without (X = H) acetylation of the terminal amine group. R stands for the side chain group
Mass spectrometry (MS) represents an elegant way to elucidate the primary structure of compounds provided that they can be transferred/ionized in the gas phase of the mass spectrometer. In the context of peptides, MS methods are widely used to determine the amino acid sequence, paving the way to the high throughput collision-induced dissociation (CID) sequencing of peptides and proteins [12, 13]. The mass differences between two successive sequence ions, i.e., ions which arise from the backbone cleavage, provide information on the amino acid sequence [13, 14]. In the late 1990s, Heerma et al. compared the fragmentation patterns of peptide ions and their peptoid analogs [15, 16]. When submitted to collisional activation, both kinds of ions dissociate at the amide bond leading to the formation of b/y and B/Y sequence fragment ions [15, 16]. Note that the b/y sequence ions are capitalized for the peptoid sequence ions [14, 17]. Interestingly, when comparing isomeric peptide/peptoid, the relative abundance of the y/Y fragment ions is most of the time higher from protonated peptoids than from peptide ions [16]. More recently, Morishetti and co-workers studied the influence of the nature of the charge (alkali vs proton) on the peptoid ion dissociation in the gas phase [17]. Their results show that protonated peptoids mainly produce high mass Y-type fragment ions contrarily to alkali peptoid ions which decompose in both B and Y-type fragment ions [17]. Shortly afterward, Ren et al. undertook a mechanistic study associating MS experiments and computational chemistry to investigate the B/Y pathway of singly and doubly protonated peptoids [18]. They propose that, upon protonation of the carbonyl oxygen atom of a given amide function, the C(=O)–N is broken after a nucleophilic attack of the oxygen atom of the adjacent amide function on the N-term side, yielding an ion/neutral dimer as intermediate (Scheme 2) [18]. Proton transfer from the oxazolone ion to the neutral C-terminal fragment, before the dissociation of the ion/neutral dimer, leads to the formation of the Y-type fragment ions [18]. The occurrence of this proton transfer is supported by density functional theory (DFT) calculations and isotope labeling experiments [18]. Such a mechanism has been also proposed to account for the formation of y ions from protonated peptides upon CID [13, 19].

The B/Y pathway proposed by Ren et al. [18]: oxazolone mechanism and intramolecular proton transfer. R stands for the side chain
We recently reported a mechanistic study on the CID dissociations of protonated peptoids with a special attention paid to the side chain loss (SCL) reaction that competes with the B/Y pathway [20]. In that study, beside the SCL fragment ions, we obviously detected B and Y ions in the CID spectra of the protonated peptoids. Interestingly and motivating the study reported hereafter, we also observed the Y ions corresponding to the dissociation of the first N–C(=O) group at the N-term side in the case of non-acetylated peptoids (Scheme 1). These peculiar fragment ions are denoted Yx (x standing for the ultimate Y ions). Based on the oxazolone mechanism [18], the observation of these Y ions from protonated non-acetylated peptoids is not explained. In the present paper, we demonstrate that the production of the Yx fragment ions from non-acetylated peptoid ions is best described as an A1-Yx mechanism, whereas the production of the other Y ions remains associated to B/Y dissociation routes. The Yx ions formally correspond to protonated truncated peptoids, i.e., the precursor peptoids shortened by a peptoid residue specifically at the N-term side. Therefore, consecutive A1-Yx reactions can be envisaged as a direct sequencing method for peptoids. The applicability of the A1-Yx sequencing will be tested in the present report.
Experimental
Peptoid Synthesis
All reactants and solvents are commercially available (VWR chemicals) and are used without any supplementary purification. Peptoids are synthesized on Rink amide resin using the solid-phase reaction protocol reported by Zuckermann and co-workers, which involves successive acylation and nucleophilic substitution steps [1, 3]. The acylation of the terminal amine group occurs with activation of bromoacetic acid by N,N′-diisopropylcarbodiimide, followed by the nucleophilic displacement of bromide by a primary amine selected to incorporate the desired side chain. Both steps are repeated until the targeted sequence is obtained [1, 3]. Peptoids are cleaved from the resin using a 95/5 trifluoroacetic/water solution. After filtration, the cleavage solution is subsequently diluted in 50/50 acetonitrile/water solution to reach the appropriate concentration for MS analysis (1 μg/mL). To define the peptoid structure, we use the abbreviation reported in the literature, and the sequence is always given from the N- to the C-terminal ends [21].
Mass Spectrometry Analysis
Most of the CID experiments are performed on a Waters QToF API-US mass spectrometer. The diluted peptoid cleavage solution (see peptoid synthesis section) are delivered to the electrospray ionization (ESI) source (direct injection) operating in the positive ion mode by a Harvard Apparatus syringe pump at a flow rate of 5 μL/min. The ESI parameters, i.e., capillary and cone voltage, are tuned to maximize the signal of the analyte. Typical parameters are 3.1 kV for capillary, 20 V for cone voltage, 80 °C and 120 °C for respectively source and desolvation temperature. Dry nitrogen is used as the ESI gas with a flow rate of 50 L/h for the gas cone and 500 L/h for the desolvation gas. For ESI-MS analysis, the quadrupole is set to pass ions from m/z 50 to 1000 and all ions are transmitted into the pusher region of the time-of-flight analyzer for mass-analysis with 1 s integration time (FWHM 7000 at m/z 500). Data are acquired in continuum mode until acceptable averaged data are obtained (typically 20 scans).
For the ESI-MSMS experiments, the ions of interest are mass-selected by the quadrupole mass filter. The selected ions are then submitted to collision against argon in the rf-only hexapole collision cell (pressure estimated at 0.9–1 mBar). The laboratory frame kinetic energy (ELab) is selected to afford intense enough fragment ion signals (see spectra). Fragment ions, as well as the non-dissociated precursor ions, are finally mass-measured with the orthogonal axis ToF analyzer. Data are acquired in continuum mode until acceptable averaged data are obtained.
Ion Mobility Experiments
Ion mobility measurements are performed using a hybrid quadrupole (Q)—traveling wave ion mobility spectrometry (TWIMS)—time-of-flight (ToF) mass spectrometer (Synapt G2-Si, Waters, UK) equipped with an electrospray (ESI) ionization source. Typical ion source conditions are the following: capillary voltage, 3.1 kV; sampling cone, 30 V; source offset, 80 V; source temperature, 80 °C; and desolvation temperature, 120 °C. Dry nitrogen is used as the ESI gas with a flow rate 500 L/h for the desolvation gas. The core of the instrument is constituted by the so-called TriWave® setup that is composed of three successive Traveling-wave (T-Wave) devices named the trap cell, the IMS (ion mobility spectrometry) cell, and the transfer cell, in which the wave speed and amplitude are user-tunable. The trap and transfer cells are filled with argon whereas the IMS cell is filled with nitrogen. A small rf-only cell filled with helium is fitted between the trap and the IMS cells. Collision energy can be applied to the trap cell and to the transfer cell to fragment ions respectively before and after the ion mobility separation. The typical IMS parameters are: wave height 40 V; wave velocity 1000 m.s−1; mass to charge range m/z 50 to 2000, argon trap flow 10 mL.min−1; nitrogen IMS flow 80 ml.min−1; helium cell gas flow 180 ml.min−1; and trap bias 45 V.
Nomenclature of Peptoid Fragment Ions
As previously mentioned, peptoids dissociate in a similar way to peptides giving sequence fragment ions and side chain fragment ions [15,16,17,18, 20, 22]. For the sequence fragment ions, we use the Roepstorff nomenclature, also employed in previous peptoid MS article, wherein fragment ions of peptoids are distinguished from peptides using the uppercase character. The Y ions are defined as the C-terminal fragment ions, whereas the B are the N-terminal fragment ions [14,15,16,17,18, 20, 22]. Regarding the side chain fragment ions, the loss of the charged or the neutral side chain is indicated respectively by the uppercase U or V (in superscript) letter [18]. In addition, when the number (n) and/or the position (m) of the lost side chain is clearly established, the data are expressed with the nVm symbolism. The internal fragments, i.e., fragments requiring at least two amide bond cleavages, are also named by adopting the Roepstorff nomenclature which indicates by a two-letter code both cleavages involved in the formation of the fragment ion [13, 14].
Computational Chemistry
Calculations are carried out at the density functional theory (DFT) level using the B3LYP functional and a 6-31G** double zeta basis set, as implemented in Gaussian 16 (A03 revision) [23]. Empirical Grimme’s term D3 correction is used to improve the description of dispersion interactions [24]. The functional is abbreviated B3LYP-D3 hereafter. All molecular structures are fully optimized, and minima are confirmed through a vibrational frequency analysis. Reported energies are in kJ/mol and correspond to the relative Gibbs free energy. Two starting geometries of an acetylated hexamer (helix-like and loop-like) are used as neutral inputs and fully optimized. In the case of the helix-like structure, the backbone and side chain dihedrals are set as described by Armand et al. [25]. For the loop-like structure, dihedrals were inspired from peptide ions [26]. From the optimized neutral geometries, a proton is added on each oxygen/nitrogen from each amide on the backbone, and a new geometry optimization is performed on each structure (26 in total, see SI).
Fragmentations of non-acetylated peptoids are also investigated at the B3LYP-D3/6-31G** level of theory. Vibrational frequency calculations can also be exploited to provide thermal enthalpies which are used to compute proton affinities (PA) defined as the opposite of the protonation reaction enthalpy (−∆Hprotonation = −(Hprotonated − Hneutral)). Reported PA are obtained from thermal enthalpies calculated at 298.15 K. First, the neutral fragment is optimized, which gives access to the Hneutral. Then, the optimized structure is used as input the protonation, and a new optimization is carried out to obtain Hprotonated.
Results and Discussion
Collision-Induced Dissociation of Protonated Peptoids
When submitted to low-energy collisional activation, acetylated (S)-phenylethylamine peptoid pentamer ions, [AcNspe5 + H]+ (m/z 865.5), produce several B/Y sequence ions, i.e., Y4 and Y3 as well as B3 and B2 (see Figure 1a). First, when compared with peptide ion chemistry, we note again the predominance of Y type ions over B type ions [15,16,17,18]. The production of these Y ions may proceed via the oxazolone mechanism of Scheme 2 involving the carbonyl group of the precedent amide function at the N-term side. Proton transfer within an ion/neutral complex, here associating protonated oxazolone to the neutral truncated peptoid (Scheme 2), is under thermochemical control; in the case of low collision energy conditions, the neutral fragment with the higher proton affinity (PA) will predominantly retain the proton [18]. The difference between the PA of the primary and secondary amines, produced respectively from protonated peptides and peptoids upon CID, is often put forward to explain the highest intensity of the Y fragment ions from peptoid ions [16]. In the CID spectrum of Figure 1a, we also detect low abundance side-chain fragment ions [20] at m/z 761.4 produced from the precursor ions and at m/z 422.2 from the B3 fragment ions.

Collision-induced dissociation mass spectra (Waters QToF API-US) of (a) [AcNspe5+H] + ions and (b) [Nspe5 + H] + ions, at respectively 12 V and 22 V collision energy
For peptides, it is now well admitted that backbone fragmentations are preceded by the migration of the extra proton from the most basic site of the molecule to an amide nitrogen atom, within the so-called mobile proton model [13, 27,28,29]. In contrast to O-protonated isomers, the protonation at the nitrogen atom weakens the amide bond and increases the partial positive charge on the amide carbon making it vulnerable to the attack of a nucleophilic group [13, 30, 31]. Paizs and Suhai examined the b/y fragmentation patterns of numerous peptides starting from the O-protonated species [32]. Their theoretical investigations show that the five-membered ring intermediate produced from the O-protonated amide bond (similarly to what is proposed in Scheme 2) is energetically not favored [32]. In addition, the 1,2-elimination step from this intermediate on the way to the oxazolone ions involves a highly strained four-membered ring transition state [33]. The same authors also reported an exhaustive discussion regarding the mechanism of the cleavage of protonated amide bonds, and they concluded that, under low energy collision conditions, most of the sequence ions are formed on the b/y pathways which are energetically, kinetically, and entropically accessible. This was ascribed to the fact that the reactive protomers, i.e., the amide nitrogen-protonated species, can be easily formed during ion excitation from the O-protonated species [29]. Due to the great similarity between peptides and peptoids, we thus propose that the B/Y fragmentation pathway of protonated peptoids also involves as transient species the N-protonated isomers that are generated upon collisional activation from the O-protonated forms. Upon the nucleophilic attack of the oxygen atom of the N-terminal adjacent carbonyl group on the electrophilic carbonyl carbon of the N-protonated amide, the peptoid ions directly dissociate to form an ion-neutral complex associating an oxazolone ion to the neutral truncated peptoid residue (Scheme 3). Further dissociation of the so-produced ion/neutral complex will end up with the production of Y ions, as previously proposed in Scheme 2. In their publication related to the CID reactions of protonated peptoid-peptide hybrids [34], Ruijtenbeek et al. propose a similar mechanism to explain the backbone cleavages between peptide and peptoid residues.

The B/Y pathway proposed starting from N-protonated peptoids: direct formation of the oxazolone and intramolecular proton transfer. R stands for the side chain
[AcNspe5 + H]+ is almost exclusively dissociate to form Y4 ions, as depicted in Figure 1a. The dominant production of high mass Y-type fragment ions from collisionally activated protonated peptoids has already been observed by Ren et al. [18]. They propose that this is related to the increased proton affinities (PA) of the C-term fragments going from Y1 to Yx [18]. In their 2004 paper, using RRKM calculations, Paizs and Suhai demonstrated that the differences between the rate constants for the b/y reactions are small when considering the different amide bonds all along the peptide backbone. Therefore, the increase intensity of the b/y fragment ions from the N-term to the C-term in peptide CID was explained by the fact that the amide nitrogen protonated structures are energetically more favored if protonation occur closer to the C-term [29]. This was further confirmed by Haeffner et al. that suggested that the thermodynamic stability of the competing N-protonated isomers, created all along the peptide skeleton, dictates the sites of primary backbone cleavages. They indeed found that backbone N-protonation is most favorable near the C-term extremity for non-polar or weakly polar peptides such as polyalanine [35]. Recently, Armentrout et al. studied the decomposition of protonated oligopeptides, and they concluded that the CID reactions begin at the ground conformation for electrospray-produced peptide ions [36, 37]. They also suggest that the multitude of fragment ions does not require a large distribution of conformations formed in the ion source. Their data thus nicely correlates with the “mobile proton model” [13, 27, 28].
Therefore, by comparison with the chemistry of peptide ions [35], the relative stability of the corresponding O- and N-protonated peptoids must be considered when trying to decipher the product ion intensities. Unfortunately, the gas phase structures of protonated peptoids are hitherto not established making all the discussions related to the energetics of the CID reactions difficult. Based on the solution phase data on Nspe peptoids revealing the presence of helical or loop structures [38,39,40], DFT calculations give us access to the relative Gibbs free energy (Table 1) of all the protomers of AcNspe6 at all the oxygen and nitrogen atoms for two different conformations, i.e., helix and loop structures, see Figure SI1–4 for all the optimized structures. Note that no helix-to-loop or loop-to-helix isomerization has been observed during the DFT optimizations. Table 1 suggests that protonation occurs most favorably at the carbonyl oxygen atom of the acetyl group at the N-terminus and that the protonated ions adopt a loop structure stabilized by the creation of H-bonds with the C-term amide atoms. The occurrence of these H-bonds also explains the trend observed in the relative energies for the O-protonated loop structures with the less stable structure corresponding to the protonation at the central positions. This evolution is no longer observed for the O-protonated helical peptoids that show a monotonic decrease in thermodynamic stability from N-term to C-term. Whatever the conformation, the O-protonated ions are always calculated more stable than their N-protonated counterparts (Table 1). However, no direct correlation between the fragment ion abundances and the relative stability of the different protomers is detected. A complete examination of dissociation energetics and kinetics for the peptoid ions, including computed transition state energies for the B/Y pathways, is required to rationalize the product ion intensities. However, the prerequisite for such a work will be the identification of the more stable protomers and conformers and their interconversion upon activation. We are currently conducting a joint ion mobility/molecular dynamics investigation to get further insight in the conformations of protonated peptoids. For the present work, we will conclude that, upon CID, the O-protonated most stable peptoids will first isomerize to the N-protomers before B-Y dissociation as presented in Scheme 3.
In Figure 1, the CID spectrum of the [AcNspe5 + H]+ ions (1a) is also compared with the CID spectrum recorded for their non-acetylated counterparts (1b), namely [Nspe5 + H]+. The [Nspe5 + H]+ ions also dissociate into B/Y sequence ions, i.e., Y4 (= Yx), Y3 and Y2 and B4, B3 and B2. Whereas the Y3 ions (= Yx-1) are here the most abundant fragment ions, the fragmentation of the [Nspe5 + H]+ ions also produces Y4 fragment ions at m/z 662.4 in good yield (about 15% of the base peak). However, if the production of Y4 ions from [AcNspe5 + H]+ ions is easily explained based on the oxazolone mechanism [18], the absence of an acetyl group at the N-term amine in the [Nspe5 + H]+ ions precludes this participation, thus questioning the origin of the Y4 ions from the non-acetylated peptoid ions. This also implies that the highest-mass Y ions produced via the oxazolone B/Y mechanism are the most abundant Y3 (Yx-1) ions. Before entering into mechanistic considerations, we selected a second peptoid with alkyl chain as the side group, i.e. [Namy2 + H]+ ions with n-pentyl side chains. As shown in Figure 2, [Namy2 + H]+ ions mostly dissociate upon CID into Y1 fragment ions. [Namy2 + H]+ ions cannot produce Y1 fragment ions following the oxazolone-ring mechanism [18] due to the absence of the carbonyl group at the N-term extremity. Among the fragment ions, m/z 100 ions are detected and correspond to iminium ions bearing the n-pentyl side chain, C5H11–NH+=CH2, see Figure 2. The dissociation of peptoid ions upon CID into iminium ions has previously been reported by Heerma et al. [15]. In the peptide ion chemistry, iminium ions and high-mass y-type fragment ions are often associated in the so-called a/y pathway (Scheme 4a) [13, 41, 42]. This dissociation route begins by the protonation of the nitrogen atom of an amide group followed by an extensive suite of bond cleavages ultimately yielding an iminium ion (a1), carbon monoxide, and the neutral truncated peptide [13, 41, 42]. Quantum-chemical and RRKM studies from Paizs et al. demonstrate that, after the loss of CO, an ion/neutral complex, associating the iminium ion to the neutral peptide, is produced. The lifetime of this complex is sufficient for a proton transfer from the iminium ion to the neutral peptide to take place [41], generating either the a1 and yx fragment ions, depending on the relative PA (See Scheme 4a).

Collision-induced dissociation mass spectrum (Waters QToF API-US) of [Namy2 + H]+ ions at 15 V collision energy

As an alternative, Cordero et al. proposed that the formation of the y1 ions from dipeptide ions rather occurs following the aziridinone mechanism [43]. Harrison et al. subsequently suggested that the unstable aziridinium ions can dissociate into a1 ions by loss of CO [44]. However, Paizs et al. demonstrated for protonated glycylglycine that the aziridinone pathway has a relatively high threshold energy and is kinetically disfavored compared with the iminium pathway [41]. Consequently, it is reasonable to assume that protonated peptoids without N-terminal acetylation also dissociate into iminium ions (A1 ions) and protonated truncated peptoids (Yx ions) following the A1-Yx pathway presented in Scheme 4b for the [Namy2 + H+] ions. As it is the case for the B/Y fragment notation used for peptoids [15,16,17,18], the uppercase character A/Y is also employed hereafter to distinguish peptide–peptoid fragments. From [Namy2 + H+] ions, Y1 (= Yx) ions at m/z 145 are the most abundant fragment ions whereas A1 iminium ions at m/z 100 are by far less abundant (Figure 2). The PA of the imine – N–pentylformimine – and pentyl peptoid monomer have been calculated using DFT at the B3LYP-D3/6-31G(d,p) level of theory and respectively amount to 941 and 988 kJ/mol, making the protonation of the peptoid residue thermochemically more favorable (Scheme 4b).
Regarding the [Nspe5 + H]+ ions, as depicted in Figure 1b, the A1 fragment ions at m/z 134.1 are also sparsely observed compared with the Y4 (= Yx) fragment ions at m/z 662.4. A difference of ~ 40 kJ/mol is calculated between the PA of the N- and the C-terminal fragments (995 vs 952 kJ/mol respectively) again in nice agreement with the major formation of the truncated peptoid ions. The observation of both ions, i.e., the iminium (A1) and the protonated peptoid (Yx), together with the nice agreement between the experimental data and the calculated PA, gives support to the proposed A1-Yx pathway. Globally, we can now conclude that the production of the Yx ions from protonated acetylated peptoid proceeds via the B/Y mechanism proposed by Ren et al. [18], but considering a transient N-protonation, whereas the observation of the Yx ions from the non-acetylated peptoid ions is associated to an A1-YX mechanism. Of course, all primary Yx-1 to Y1 fragment ions from non-acetylated peptoid precursor ions remain generated via the oxazolone B/Y mechanism (Scheme 2).
A sequence-defined peptoid with four different side chains and presenting the Nste-Namy-Nspe-Nipr sequence (with Nste, Namy, Nspe, and Nipr corresponding to N-(S)-tolylethyl, N-pentyl, N-(S)-phenylethyl, and N-isopropyl—Figure 3b) has been prepared and will be named P1 hereafter. Upon CID of the [P1 + H]+ ions, all Y sequence ions are detected, namely Y3 (Yx − m/z 405.3), Y2 (m/z 278.2), and Y1 (m/z 117.1). As previously observed in Figure 1b, the Yx-1 ions, here Y2 ions at m/z 278.2, are the most abundant Y sequence ions for non-acetylated peptoid ions and arise from a B/Y mechanism, together with the abundant B2 ions at m/z 303.2. Fragment ions arising from the losses of one or several neutral side chains are also detected from the precursor ions as well as from the sequence ions. In our recent paper dealing with the SCL reactions, we observed that Nspe and Nste side chains are prone to be expelled as neutral Ph–CH=CH2 and CH3–Ph–CH=CH2, even from low internal energy ions [20]. As observed from the CID spectrum in Figure 3a, the detected side chain losses concern these two side chains. For instance, from the precursor [P1 + H]+ ions, the Nspe and Nste side chain losses generate fragment ions at m/z 476.4 and 462.3, respectively, labeled [P1 + H]+ V2 and [P1 + H]+ V4. As far as the Y3 ions are concerned, the occurrence of the A1-Yx (x = 3) mechanism is confirmed by the detection of the A1 ions at m/z 148.1.

Collision-induced dissociation mass spectra (Waters QToF API-US) of (a) [P1 + H] + ions, (b) in-source Y3 fragment ions, and (c) in-source Y2 fragment ions. All spectra are recorded at 30 V collision energy. The detected fragment ions (b), (c) in the result from consecutive dissociations from the precursor [P1 + H]+ ions; they must be considered as internal fragment ions of the [P1 + H] + precursor ions, and they are assigned accordingly (see experimental section for the nomenclature). Note that, in Fig. 3a, the intense signal at m/z 156.1 remains unattributed for the time being
Consecutive A1-Yx Reactions Highlighted by MSn Reactions
The CID fragment ions in Figure 3a are thus created following competitive and consecutive dissociation reactions that can be classified into B/Y, A1-Yx and SCL processes. The A1-Yx pathway is specific to protonated non-acetylated peptoids and was demonstrated above to rationalize the detection of abundant Yx fragment ions. These so-produced Yx ions are then expected to consecutively undergo the A1-Yx mechanism upon CID, generating a second generation of Yx-type ions, and so on. This also reveals that, in the CID of a given non acetylated peptoid ions, Yx-1 to Y1 ions can be generated following competitive pathways, namely direct B/Y mechanism from the precursor ions and consecutive formal A1-Yx pathway from fragment ions. The second process can be seen as cutting up the peptoid backbone, specifically from the N-terminal extremity.
In order to shed light on this competition, we first built a MSn strategy to monitor the [P1 + H]+ → Y3 → Y2 → Y1 sequence of decomposition. On a QToF instrument, MS3 is prohibited, except if we consider the source region as a collision cell by applying higher voltages on the sample cone. We thus increase the collision voltage to induce the in-source fragmentation of [P1 + H]+ precursor ions to promote the production of Y3 sequence ions (Yx − m/z 405.3) following the A1-Yx mechanism. The so-produced Y3 fragment ions are then mass-selected and subjected to CID in the hexapole cell of the QToF mass spectrometer (Figure 3b). Among the fragment ions, Y2 ions are also detected at m/z 278.2 and arise from an A1-Yx type reaction from Y3. To further probe the CID behavior of these Y2 ions, we must first produce them in the ion source by increasing the cone voltage and record their CID in the hexapole cell after mass selection with the quadrupole analyzer. We then recorded the CID spectrum featured in Figure 3c that again highlights the Y2 to Y1 dissociation following the A1-Yx mechanism. The occurrence of the residual mass-to-charge ratio at m/z 117 (Y1) confirms that the C-type fragment corresponds to the Nipr residue, thus demonstrating the consecutive nature of the [P1 + H]+ → Y3 → Y2 → Y1 process. Basically, such an MSn suite of experiments can be useful for sequencing (non-acetylated) peptoids using the highly specific A1-Yx reaction in a kind of back-biting mechanism.
Competition between A1-Yx and B/Y Mechanisms Highlighted by Ion Mobility Experiments
By means of the TriWave device of the Waters Synapt G2-Si mass spectrometer, it is possible to record the arrival time distributions (ATD) of fragment ions produced from mass-selected precursor ions in the trap cell upstream the ion mobility cell [45]. In Figure 4, the CID spectrum of m/z 580.4 precursor ions is presented. We also measured the arrival times (tA) of all other ions, and all data are presented in Table 2. In the CID spectrum, we mostly focused on the Y and B sequence ions under interest. The tA of m/z 580.4 (precursor ions), m/z 405.3 (Y3), m/z 303.3 (B2), m/z 278.2 (Y2), and m/z 117.1 (Y1) are respectively 8.8 ms, 6.1 ms, 4.5 ms, 4.2 ms, and 2.1 ms (see Figure 4). The ATD of the Y2, B2, and Y3 ions are also gathered in Figure 5(a-c-e).

Collision-induced dissociation mass spectrum of the [P1 + H] + ions in the trap cell (trap CE = 20 V) of the Waters Synapt G2-Si mass spectrometer. The data in red correspond to the arrival times (tA) of the fragment ions generated in the trap cell (trap CID)

Collision-induced dissociation and drift time-aligned CID of the [P1 + H]+ ions (mass selected by the quadrupole analyzer) in the trap cell (trap CE = 20 V) of the Waters Synapt G2-Si mass spectrometer: Arrival time distributions (ATD) of the (a), (b) m/z 405.3 (Y3), (c), (d) m/z 278.2 (Y2), and (e), (f) m/z 303.2 (B2) ions. For the (b), (d), (f) lower ATDs, the transfer cell is also activated for CID to generate fragment ions from the mobility-separated trap cell fragment ions (transfer CE 20 V and 15 V for B2 ions)
In a second set of experiments, we used the transfer cell to perform CID experiments on all ion mobility separated ions (i.e., all ions from the trap cell - Figure 4) in a CID-ion mobility-CID sequence of events. Such an experiment will allow identifying the precursor ions of all fragment ions since fragment ions produced upon CID in the transfer cell will appear at the tA of their precursor ions. Fragment and parent ions are drift time-aligned and these experiments are often referred to as drift time-aligned CID experiments [45]. Doing so, we confirmed that the Y3 ions are direct fragment (A/Y mechanism) from the [P1 + H]+ ions since, in Figure 5b, a second tA is detected at 8.8 ms for m/z 405.3 ions, namely the tA of the [P1 + H]+ ions. The situation is more complicated for the Y2 ions (m/z 278.2), as observed when comparing Figure 5c and d. Indeed, beside the tA at 4.2 ms that is characteristic of the Y2 ions, three additional ATDs are detected in the mobilogram of the m/z 278.2 ions at 6.1 ms, 6.8 ms, and 8.8 ms. From Table 2, it is possible to assign these tA to the Y3 ions, [P1 + H+]V4 ions and the [P1 + H]+ ions, respectively. In other words, the Y2 ions are produced from three different routes: (i) direct B/Y mechanism from the precursor [P1 + H]+ ions (signal at 8.8 ms), (ii) consecutive A1-Yx mechanism from the Y3 ions (signal at 6.1 ms), and (iii) consecutive B/Y mechanism from the [P1 + H+]V4 ions (signal at 6.8 ms). On the other hand, as depicted in Figure 5e, f, B2 ions (m/z 303.2) are only produced from the [P1 + H]+ ions (signal at 8.8 ms) via a first generation B/Y process and by a consecutive B/Y mechanism from the [P1 + H+]V2 ions (signal at 7.1 ms).
Conclusion
Low energy CID reactions of protonated peptoids were investigated by combining the CID experiments with ion mobility experiments as well as theoretical calculations. We demonstrated that collisionally activated protonated peptoids competitively dissociate following three different reactions, namely the B/Y, the A1-Yx, and the side chain loss (SCL) [20] processes, with all dissociation pathways being best described based on the intermediacy of the amide nitrogen–protonated structures.
We also compared the CID spectra of protonated Nspe peptoids with free or acetylated terminal amino function. We observed that both dissociate by producing mostly high-mass Y-type (say Yx or Yx-1) fragment ions [18], oppositely to what is often observed for peptide ions. For peptides ions, literature results reveal that (i) the multitude of exit channels upon CID is best explained based on the mobile proton model rather than to the sampling of ESI-produced multiple protomers and conformers [36, 37] and (ii) the b/y fragment ion intensities are best correlated to the stability of the various N-protonated structures [29] rather than being under kinetic control. Unfortunately, we did not identify any direct correlation between the B/Y fragment ion abundances and the relative stability of the different O- and N-protomers. A complete examination of dissociation energetics and kinetics for the peptoid ions, including computed transition state energies for the B/Y pathways, will be required in the future to rationalize the product ion intensities. The production of abundant Yx ions from non-acetylated peptoid ions is proposed in the present study to arise from an A1-Yx mechanism, whereas the production of the other Y ions remains associated to B/Y dissociation routes starting from N-amide protonated species. By considering that the Yx ions formally correspond to protonated truncated peptoids, i.e., the precursor peptoids shortened by a peptoid residue specifically at the N-term side, consecutive A1-Yx reactions are envisaged as a direct sequencing method for peptoids in a kind of back-biting process. The applicability of the peptoid sequencing using the consecutive A1-Yx reactions has been demonstrated with MSn experiments. Using the combination of CID and ion mobility experiments, we also demonstrated that the production of Yx-1 to Y1 ions upon CID of the precursor protonated peptoids is the combination of competitive and consecutive A1-Yx and B/Y reactions.
References
Zuckermann, R.N., Kerr, J.M., Kent, S.B.H., Moos, W.H.: Efficient method for the preparation of peptoids [oligo(N- substituted glycines)] by submonomer solid-phase synthesis. J. Am. Chem. Soc. 114, 10646–10647 (1992)
Seo, J., Lee, B.-C., Zuckermann, R.N.: Peptoids: synthesis, characterization, and nanostructures. Compr. Biomater. 2, 53–76 (2011)
Tran, H., Gael, S.L., Connolly, M.D., Zuckermann, R.N.: Solid-phase submonomer synthesis of peptoid polymers and their self-assembly into highly-ordered nanosheets. J. Vis. Exp. (2011). https://doi.org/10.3791/3373
Gorske, B.C., Blackwell, H.E.: Tuning peptoid secondary structure with pentafluoroaromatic functionality: a new design paradigm for the construction of discretely folded peptoid structures. J. Am. Chem. Soc. 128, 14378–14387 (2006)
Shin, S.B.Y., Kirshenbaum, K.: Conformational rearrangements by water-soluble peptoid foldamers. Org. Lett. 9, 5003–5006 (2007)
Shin, H.-M., Kang, C.-M., Yoon, M.-H., Seo, J.: Peptoid helicity modulation: precise control of peptoid secondary structures via position-specific placement of chiral monomers. Chem. Commun. 50, 4465–4468 (2014)
Rudzińska-Szostak, E., Berlicki, Ł.: Sequence engineering to control the helix handedness of peptide foldamers. Chem. - A Eur. J. 23, 14980–14986 (2017)
Jin, H., Ding, Y.H., Wang, M., Song, Y., Liao, Z., Newcomb, C.J., Wu, X., Tang, X.Q., Li, Z., Lin, Y., Yan, F., Jian, T., Mu, P., Chen, C.L.: Designable and dynamic single-walled stiff nanotubes assembled from sequence-defined peptoids. Nat. Commun. 9, 1–11 (2018)
Maayan, G., Ward, M.D., Kirshenbaum, K.: Folded biomimetic oligomers for enantioselective catalysis. Proc. Natl. Acad. Sci. U. S. A. 106, 13679–13684 (2009)
Wu, H., Su, X., Li, K., Yu, H., Ke, Y., Liang, X.: Improvement of peptoid chiral stationary phases by modifying the terminal group of selector. J. Chromatogr. A. 1265, 181–185 (2012)
Marvin, C.W., Grimm, H.M., Miller, N.C., Horne, W.S., Hutchison, G.R.: Interplay among sequence, folding propensity, and bio-piezoelectric response in short peptides and peptoids. J. Phys. Chem. B. 121, 10269–10275 (2017)
Biemann, K.: Contributions of mass spectrometry to peptide and protein structure. Biomed. Environ. Mass Spectrom. 16, 99–111 (1988)
Paizs, B., Suhai, S.: Fragmentation pathways of protonated peptides. Mass Spectrom. Rev. 24, 508–548 (2005)
Roepstorff, P.: Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 11, 601 (1984)
Heerma, W., Versluis, C., De Kostert, C.G., Kruijtzer, J.A.W., Zigrovic, I., Liskamp, R.M.J.: Comparing mass spectrometric characteristics of peptides and peptoids. Rapid Commun. Mass Spectrom. 10, 459–464 (1996)
Heerma, W., Boon, J.P.J.L., Versluis, C., Kruijtzer, J.A.W., Hofmeyer, L.J.F., Liskamp, R.M.J.: Comparing mass spectrometric characteristics of peptides and peptoids - 2. J. Mass Spectrom. 32, 697–704 (1997)
Morishetti, K.K., Russell, S.C., Zhao, X., Robinson, D.B., Ren, J.: Tandem mass spectrometry studies of protonated and alkali metalated peptoids: enhanced sequence coverage by metal cation addition. Int. J. Mass Spectrom. 308, 98–108 (2011)
Ren, J., Tian, Y., Hossain, E., Connolly, M.D.: Fragmentation patterns and mechanisms of singly and doubly protonated peptoids studied by collision induced dissociation. J. Am. Soc. Mass Spectrom. 27, 646–661 (2016)
Paizs, B., Suhai, S.: Combined quantum chemical and RRKM modeling of the main fragmentation pathways of protonated GGG. II. Formation of b2, y1, and y2 ions. Rapid Commun. Mass Spectrom. 16, 375–389 (2002)
Halin, E., Hoyas, S., Lemaur, V., De Winter, J., Laurent, S., Cornil, J., Roithová, J., Gerbaux, P.: Side-chain loss reactions of collisionally activated protonated peptoids : a mechanistic insight. Int. J. Mass Spectrom. 435, 217–226 (2019)
Lear, S., Cobb, S.L.: Pep-Calc.com: a set of web utilities for the calculation of peptide and peptoid properties and automatic mass spectral peak assignment. J. Comput. Aided Mol. Des. 30, 271–277 (2016)
Ren, J., Mann, Y.S., Zhang, Y., Browne, M.D.: Synthesis and mass spectrometry analysis of oligo-peptoids. J. Vis. Exp. (2018). https://doi.org/10.3791/56652
Gaussian 16, Revision A.03, Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G. A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A. V., Bloino, J., Janesko, B. G., Gomperts, R., Mennucci, B., Hratchian, H. P., Ortiz, J. V., Izmaylov, A. F., Sonnenberg, J. L., Williams-Young, D., Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V. G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery, J. A., Jr., Peralta, J. E., Ogliaro, F., Bearpark, M. J., Heyd, J. J., Brothers, E. N., Kudin, K. N., Staroverov, V. N., Keith, T. A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A. P., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Millam, J. M., Klene, M., Adamo, C., Cammi, R., Ochterski, J. W., Martin, R. L., Morokuma, K., Farkas, O., Foresman, J. B., Fox, D. J. Gaussian, Inc., Wallingford CT, (2016)
Grimme, S., Antony, J., Ehrlich, S., Krieg, H.: A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010)
Armand, P., Kirshenbaum, K., Falicov, A., Dunbrack, R.L.J., Dill, K.A., Zuckermann, R.N., Cohen, F.E.: Chiral N-substituted glycines can form stable helical conformations. Fold. Des. 2, 369–375 (1997)
Wyttenbach, T., Bushnell, J.E., Bowers, M.T.: Salt bridge structures in the absence of solvent ? The case for the oligoglycines. J. Am. Chem. Soc. 120, 5098–5103 (1998)
Boyd, R., Somogyi, Á.: The mobile proton hypothesis in fragmentation of protonated peptides: a perspective. J. Am. Soc. Mass Spectrom. 21, 1275–1278 (2010)
Dongré, A.R., Jones, J.L., Somogyi, Á., Wysocki, V.H.: Influence of peptide composition, gas-phase basicity, and chemical modification on fragmentation efficiency: evidence for the mobile proton model. J. Am. Chem. Soc. 118, 8365–8374 (1996)
Paizs, B., Suhai, S.: Towards understanding the tandem mass spectra of protonated oligopeptides. 1: mechanism of amide bond cleavage. J. Am. Soc. Mass Spectrom. 15, 103–113 (2004)
McCormack, A.L., Somogyi, A., Dongré, A.R., Wysocki, V.H.: Fragmentation of protonated peptides: surface-induced dissociation in conjunction with a quantum mechanical approach. Anal. Chem. 65, 2859–2872 (1993)
Somogyi, A., Wysocki, V.H., Mayer, I.: The effect of protonation site on bond strengths in simple peptides: application of ab initio and modified neglect of differential overlap bond orders and modified neglect of differential overlap energy partitioning. Am. Soc. Mass Spectrom. 5, 704–717 (1994)
Paizs, B., Suhai, S.: Combined quantum chemical and RRKM modeling of the main fragmentation pathways of protonated GGG. I. Cis - trans isomerization around protonated amide bonds. Rapid Commun. Mass Spectrom. 15, 2307–2323 (2001)
Paizs, B., Csonka, I.P., Lendvay, G., Suhai, S.: Proton mobility in protonated glycylglycine and N -formylglycylglycinamide : a combined quantum chemical and RKKM study. Rapid Commun. Mass Spectrom. 15, 637–650 (2001)
Ruijtenbeek, R., Versluis, C., Heck, A.J.R., Redegeld, F.A.M., Nijkamp, P., Liskamp, R.M.J.: Characterization of a phosphorylated peptide and peptoid and peptoid – peptide hybrids by mass spectrometry. J. Mass Spectrom. 37, 47–55 (2002)
Haeffner, F., Merle, J.K., Irikura, K.K.: N-protonated isomers as gateways to peptide ion fragmentation. Am. Soc. Mass Spectrom. 22, 2222–2231 (2011)
Armentrout, P.B., Heaton, A.L.: Thermodynamics and mechanisms of protonated diglycine decomposition : a guided ion beam study. J. Am. Soc. Mass Spectrom. 23, 632–643 (2012)
Mookherjee, A., Van Stipdonk, M.J., Armentrout, P.B.: Thermodynamics and reaction mechanisms of decomposition of the simplest protonated tripeptide, triglycine: a guided ion beam and computational study. J. Am. Soc. Mass Spectrom. 28, 739–757 (2017)
Armand, P., Kirshenbaum, K., Goldsmith, R.A., Farr-Jones, S., Barron, A.E., Truong, K.T.V., Dill, K.A., Mierke, D.F., Cohen, F.E., Zuckermann, R.N., Bradley, E.K.: NMR determination of the major solution conformation of a peptoid pentamer with chiral side chains. Proc. Natl. Acad. Sci. 95, 4309–4314 (1998)
Wu, C.W., Sanborn, T.J., Zuckermann, R.N., Barron, A.E.: Peptoid oligomers with alpha-chiral, aromatic side chains: effects of chain length on secondary structure. J. Am. Chem. Soc. 123, 2958–2963 (2001)
Huang, K., Wu, C.W., Sanborn, T.J., Patch, J.A., Kirshenbaum, K., Zuckermann, R.N., Barron, A.E., Radhakrishnan, I.: A threaded loop conformation adopted by a family of peptoid nonamers. J. Am. Chem. Soc. 128, 1733–1738 (2006)
Paizs, B., Suhai, S.: Theoretical study of the main fragmentation pathways for protonated glycylglycine. Rapid Commun. Mass Spectrom. 15, 651–663 (2001)
Paizs, B., Schnölzer, M., Warnken, U., Suhai, S., Harrison, A.G.: Cleavage of the amide bond of protonated dipeptides. Phys. Chem. Chem. Phys. 6, 2691–2699 (2004)
Cordero, M.M., Houser, J.J., Wesdemiotis, C.: The neutral products formed during backbone fragmentations of protonated peptides in tandem mass spectrometry. Anal. Chem. 65, 1594–1601 (1993)
Harrison, A.G., Csizmadia, I.G., Tang, T.-H., Tu, Y.: Reaction competition in the fragmentation of protonated dipeptides. J. Mass Spectrom. 35, 683–688 (2000)
Rathore, D., Dodds, E.D.: Collision-induced release, ion mobility separation, and amino acid sequence analysis of subunits from mass-selected noncovalent protein complexes. Am. Soc. Mass Spectrom. 25, 1600–1609 (2014)
Acknowledgements
The UMONS MS laboratory acknowledges the “Fonds National de la Recherche Scientifique (FRS-FNRS)” for continuing support. EH and SH thank the “Fonds pour la Recherche Industrielle et Agricole” for their PhD grant. The work in the Laboratory for Chemistry of Novel Materials was supported by the European Commission/Région Wallonne (FEDER–BIORGEL project), the Consortium des Équipements de Calcul Intensif (CÉCI), funded by the Fonds National de la Recherche Scientifique de Belgique (F.R.S.-FNRS) under Grant No. 2.5020.11. J.C. is FNRS research fellow. EH thanks the FRS-FNRS and the UMONS (Fonds Franeau) for financial support granted for the stay in the Lawrence Berkeley National Laboratory (LBNL–Molecular Foundry). Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 1736 kb)
Rights and permissions
About this article

Cite this article
Halin, E., Hoyas, S., Lemaur, V. et al. Backbone Cleavages of Protonated Peptoids upon Collision-Induced Dissociation: Competitive and Consecutive B-Y and A1-YX Reactions. J. Am. Soc. Mass Spectrom. 30, 2726–2740 (2019). https://doi.org/10.1007/s13361-019-02342-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-019-02342-z