
Abstract
Many adult and most childhood neurological diseases have a genetic basis. CRISPR/Cas9 biotechnology holds great promise in neurological therapy, pending the clearance of major delivery, efficiency, and specificity hurdles. We applied CRISPR/Cas9 genome editing in its simplest modality, namely inducing gene sequence disruption, to one adult and one pediatric disease. Adult polyglucosan body disease is a neurodegenerative disease resembling amyotrophic lateral sclerosis. Lafora disease is a severe late childhood onset progressive myoclonus epilepsy. The pathogenic insult in both is formation in the brain of glycogen with overlong branches, which precipitates and accumulates into polyglucosan bodies that drive neuroinflammation and neurodegeneration. We packaged Staphylococcus aureus Cas9 and a guide RNA targeting the glycogen synthase gene, Gys1, responsible for brain glycogen branch elongation in AAV9 virus, which we delivered by neonatal intracerebroventricular injection to one mouse model of adult polyglucosan body disease and two mouse models of Lafora disease. This resulted, in all three models, in editing of approximately 17% of Gys1 alleles and a similar extent of reduction of Gys1 mRNA across the brain. The latter led to approximately 50% reductions of GYS1 protein, abnormal glycogen accumulation, and polyglucosan bodies, as well as ameliorations of neuroinflammatory markers in all three models. Our work represents proof of principle for virally delivered CRISPR/Cas9 neurotherapeutics in an adult-onset (adult polyglucosan body) and a childhood-onset (Lafora) neurological diseases.
Similar content being viewed by others


Introduction
Glucose chains as short as 12 units form double helices and can precipitate, yet glycogen with up to 55,000 units is soluble. Glycogen is the product principally of two enzymes acting in concert, glycogen synthase and glycogen branching enzyme (GBE1). Glycogen synthase links glucose units through α-1,4 bonds until a certain chain length is reached, when GBE1 cleaves the terminal hexamer and reattaches it upstream through an α-1,6 bond, thus converting the chain to a fork and doubling the sites for glycogen synthase to extend. This doubling occurring with every extension generates structures that are regularly and highly branched, allowing water to permeate and retain the large molecules in solution [1]. GBE1 deficiency in mammals results in glycogen with overlong branches, which precipitates and over time accumulates into polyglucosan bodies (PBs). The precipitation is thought to be driven, like in plant starches, by the long chains winding round each other and extruding water [2, 3].
In humans, profound GBE1 deficiency leads to massive polyglucosan accumulations and early childhood death from multi-organ failure (Andersen disease) [4]. Adult polyglucosan body disease (APBD) is an allelic form of Andersen disease with lesser GBE1 deficiency (15% residual activity with the most common GBE1 mutation p.Y239S). APBD patients have lesser systemic polyglucosan accumulations, and clinical disease limited to the central and peripheral nervous systems, namely a motor neuron disease with onset after age 50 and course resembling amyotrophic lateral sclerosis (ALS) [5, 6].
Two other enzymes known to regulate glycogen structure, though not understood how, are the glycogen phosphatase laforin (EPM2A) and its interacting ubiquitin E3 ligase malin (EPM2B) [7, 8]. In the absence of either, at any one time, a portion of glycogen molecules appears to acquire overlong branches. This leads these particular molecules to precipitate and accumulate, as above, into PBs (in this context also called Lafora bodies) [3, 9]. The associated disease, Lafora disease (LD), is also neurological, characterized by teenage-onset progressive myoclonus, epilepsy, and dementia, and death within 10 years of onset [9,10,11].
Why APBD and LD have different neurological presentations is unknown. Notwithstanding, in both the PBs drive an immune response, which at least in part underlies neurodegeneration and the neurological disease [12,13,14,15,16,17,18,19,20]. Since long glycogen branches are at the root of PB formation in both conditions, it was reasoned that targeting the glycogen synthase isoform expressed in the brain, Gys1, for downregulation may represent a therapeutic modality for both diseases. APBD (Gbe1Y239S) and LD (Epm2a−/− and Epm2b−/−) mouse models replicate their corresponding diseases [21,22,23]. Crossing these mice with mice deficient of GYS1 activity dramatically reduced PBs and neuroimmune markers, and rescued behavioural phenotypes [12, 13, 17, 19, 20, 24,25,26]. This confirmed the role of PB accumulation in the pathogeneses of these diseases and supported the targeting of GYS1 as a potential, shared, therapeutic approach.
CRISPR/Cas9 genome editing is widely expected to revolutionize neurology [27,28,29]. Among the hurdles to be overcome are achieving wide distribution of the enzyme/guide complex across the brain, and accurate target editing. The latter is not a concern when the goal is not actually to edit but to delete a gene function. In the present work, we show that AAV9-delivered Gys1-directed CRISPR/Cas9 significantly reduces PB accumulations and immune markers in APBD and LD mouse models. Our results represent proof of principle for GYS1-targeting CRISPR/Cas9 as a potential therapy for both these fatal diseases.
Methods
Study Design
We hypothesized that CRISPR/Cas9–mediated disruption of the Gys1 gene in Gbe1Y239S, Epm2a−/−, and Epm2b−/− mice would improve the neuropathology of these models. Following packaging in adeno-associated virus 9 (AAV9), we administered Gys1-targeting CRISPR/Cas9 at postnatal day 2 by intracerebroventricular (ICV) injection, sacrificed the mice at 3 months of age, and studied the effect of the treatment on PB quantity and neuroinflammation markers.
Plasmid Construction and Viral Packaging
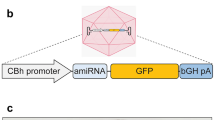
We selected multiple Gys1 targeting candidate guide RNAs (sgRNAs) using the CRISPR Design Tool software [30], which we custom-synthesized to include 5′-end guanines for efficient expression from the U6 promoter. These were cloned into a published Staphylococcus aureus Cas9 (SaCas9)-expressing AAV vector [31], replacing that vector’s SaCas9 CMV promoter with the small synthetic ubiquitously expressing JeTI promoter for more efficient viral packaging [32], with final arrangement as in Fig. 1. Candidate plasmids were transfected into mouse N2A cells, with genomic DNA extracted 48 h post-transfection for the Inference of CRISPR Edits assay (ICE), as published [https://tinyurl.com/y43gtzux], to identify the sgRNA causing the most indel formation (final plasmid ICE primer and sgRNA sequence in Table 1). The final plasmid was packaged into AAV9 at the University of North Carolina (UNC) Vector Core facility, as described [33].

AAV-SaCas9 targeting murine Gys1. Schematic representation of AAV vector, guide RNA sequence, and Gys1-Exon1 target site. ITR inverted terminal repeats, Myc c-myc, NLS nuclear localization signal sequence, bGH-pA bovine growth hormone polyadenylation signal, PAM protospacer adjacent motif
Mice
APBD (Gbe1Y239S) and LD (Epm2a−/− and Epm2b−/−) mouse models were described previously [21,22,23]. Of relevance to the interpretation of some of the results (see penultimate paragraph in the ‘Discussion’ section) that while APBD is ‘milder’ than LD (later age of onset and slower progression) in humans, the reverse is true in mouse where APBD mice die by approximately 10 months of age, while LD mice only begin to have mild behavioural phenotypes at that age [21,22,23]. Both sexes were used in approximately equal proportion in all experiments. All procedures were carried out according to NIH guidelines and the animal care committee regulations at the University of Texas Southwestern Medical Center. About 7 × 1011 vector genomes (as 5-μL solution) or 5 μL PBS were injected ICV, as described [34]. Mice were sacrificed by cervical dislocation, and brain harvested and cut into two hemispheres, one fixed in formalin for paraffin embedding and histo- and immunohisto-chemistry, the other snap-frozen in liquid nitrogen, ground into powder on the same by mortar and pestle, and aliquoted into 30–40 mg powder in screwcap tubes for the genetic and biochemical experiments below.
Cas9 Immunohistochemistry
Sections (5 µm) were mounted on glass slides, de-paraffinized and rehydrated by processing through xylenes and decreasing concentrations of ethanol in water, and subjected to antigen retrieval using citrate buffer pH 6.0 (Sigma). Endogenous peroxidase activity was blocked for 10 min with BLOXALL solution (Vector labs). Sections were incubated with rabbit anti-CRISPR-Cas9 antibody (1:50, Abcam) diluted in normal horse serum overnight at 4 °C, then successively incubated with Amplifier Antibody and ImmPRESS Polymer Reagent (Vector labs) and the ImmPACT DAB EqV working solution (Vector labs) until desired stain intensity.
Gys1 Indel and Expression Quantifications by Droplet Digital PCR
Drop-off assay (QX200 Droplet Digital PCR system, Bio-Rad Laboratories) was used to quantify the indels generated by AAV-SaCas9 per manufacturer’s instructions. Genomic DNA was extracted using PureLink Genomic DNA Mini Kit (Invitrogen). 40× Taqman SNP genotyping assay and custom-designed primer–probe set (Life Technologies; Table 1) were used to detect indels. Briefly, the primer set amplifies the sequence that includes the predicted cut site, a FAM-labelled reference probe distant from the site of potential non-homologous end joining site counts all copies of the amplicon, and a VIC-labelled drop-off probe binds the predicted cut site. The reaction mix consisted of 10 ml of 2× ddPCR SuperMix for Probes (Bio-Rad Laboratories), 0.5 ml of the 40× assay, 9.5 ml nuclease-free water, and 1.0 ml gDNA. Cycling conditions were 95 °C for 10 min, 45 cycles of 94 °C for 30 s and 58 °C for 1 min, 98 °C for 10 min and finally a 10 °C hold on a Life Technologies Veriti thermal cycler. The loss of VIC signal (while maintaining FAM signal) was used as an indel indicator. The resulting data was quantified by QuantaSoft v1.4 (Bio-Rad).
RNA was extracted using TriZol (Invitrogen) and purified using PureLink RNA Mini Kit (Invitrogen) following manufacturer instruction. cDNA was generated using the iScript Reverse Transcription SuperMix kit (Bio-Rad Laboratories). Gys1 RNA was quantified using the Bio-Rad QX200 Droplet Digital PCR (ddPCR) system as instructed by the manufacturer. Tfrc was used as reference gene. Custom-designed TaqMan primers and probes (Thermo Fisher Scientific) were used for both Gys1 and Tfrc. ddPCR reactions were assembled using standard protocols. The 20-μL reaction mix consisted of 10-μL 2× ddPCR SuperMix for Probes (Bio-Rad Laboratories), 1-μL Gys1 assay mix (FAM-labelled), 1-μL reference Tfrc assay mix (VIC-labelled), 3 μL of cDNA, and 5-μL nuclease-free water. Cycling conditions were 95 °C for 10 min, 45 cycles of 94 °C for 30 s and 60 °C for 1 min, and 98 °C for 10 min. PCR and data analysis equipment were the same as above. Results were expressed as ratio of Gys1/Tfrc. No-template controls were run in parallel with study samples. Primer/probe sets are in Table 1.
SaCas9 and GYS1 Western Blots
Tissue lysate was obtained by homogenizing frozen ground brain tissue using ice-cold RIPA lysis buffer (NaCl 150 mM, NP-40 1%, sodium deoxycholate 0.5%, SDS 0.1%, Tris HCl 50 mM pH 8.0) containing protease inhibitors (1 mM PMSF, 5 µg/mL Leupetin, 10 µg/mLPepstatin, 20KIU/mL Aprotinin, 50 mM NaF). Lysates were centrifuged at 15,000×g for 5 min at 4 °C and supernatants collected. Protein concentration was measured using the Bradford assay reagent (ThermoScientific). Serial dilution of albumin standard (ThermoScientific) generated the standard curve for protein quantification. Equal amounts of whole protein from each sample were subjected to SDS-PAGE using the TGX Stain-Free FastCast Acrylamide kit (BioRad). Protein bands were transferred to polyvinylidene difluoride (PVDF) membrane (Millipore) overnight at 4 °C. Primary antibodies for SaCas9 (1:1000, abcam) and for GYS1 (1:1000, Cell Signaling) were used. Protein dansitometry was performed using Image Lab software (Bio-Rad Laboratories). Intensity of each protein band was normalized to the intensity of its corresponding whole protein lane image obtained on the same membrane.
PASD Staining and PB Quantitation
Paraffin-embedded brain tissue was sectioned and stained using the periodic acid-Schiff diastase (PASD) method as described previously [13, 20]. Stained slides were scanned using the Hamamatsu Nanozoomer 2.0 HT digital slide scanner (40× objective), and the % area of hippocampus covered by PBs was quantified using ImageJ [35] and iLastik (v. 1.3.3) [36].
Quantification of Degradation-Resistant Glycogen
Aliquot of frozen ground brain was left out to thaw at room temperature for 1 h to allow soluble glycogen to degrade, leaving behind degradation-resistant, i.e. PB, glycogen [37]. The sample was then boiled in 30% KOH and glycogen precipitated in 70% ethanol with 57 mM sodium sulfate. Three further rounds of centrifugation and suspension in 67% ethanol with 15 mM LiCl followed after which the pellet was resuspended in 100 mM sodium acetate pH 4.5 and digested with amyloglucosidase (Megazyme) in a 55 °C oven for 1 h along with digestion of blank controls. Glucose amount was determined in triplicate for each sample using a modified version of Lowry and Passonneau [38]. Briefly, samples and glucose standards were mixed with 170 µL of G6PDH reaction mix (200 mM tricine buffer/10 mM MgCl2 pH 8, 0.66 mM NADP; 1 mM of ATP; 0.5 units of G6PDH). Absorbance at 340 nm was acquired before (Abs1) and after (Abs2) addition of hexokinase (0.6 units in 4 μL of 200 mM Tricine/KOH, pH 8, 10 mM MgCl2). (Abs2–Abs1) of glucose standards was used to derive glucose quantity in samples. Following normalization to fresh weight, glucose quantity represents degradation-resistant glycogen [37].
Quantitative Real-Time PCR
Immune system–related genes Cxcl10, Ccl5, Lcn2, and C3 were quantified by quantitative real-time PCR (qRT-PCR) using the QuantStudio 7 Pro System thermo-cycler (ThermoFischer Scientific) and SYBR Green Master Mix (Bio-Rad Laboratories). Data are shown as fold change relative to control samples using the ΔΔCq method with Rpl4 as an internal control gene. Primers are listed in Table 1.
Statistical Analysis
Student’s unpaired t-test was used to compare single means. Data were analyzed and graphed using the GraphPad Prism software (v. 8.0.2; GraphPad Software). For all comparisons, statistical significance was set at p < 0.05. Asterisks denote level of significance based on p value: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results
Extents of SaCas9 Distribution and Activity
For a visual assessment of the pattern and extent of virally delivered SaCas9 distribution, we performed immunohistochemistry in one of our animal models (Gbe1Y239S) using an anti-SaCas9 antibody. Large numbers of cells expressed the enzyme in brain regions surrounding the cerebral ventricles, namely the cortex, hippocampus, and thalamus/hypothalamus. Expressing cells became sparser the further from the ventricles to almost none in the cerebellum (Fig. 2).

AAV-SaCas9 virus distribution as detected by SaCas9 immunohistochemistry. Representative images show SaCas9 expressing cells in cortex a and hippocampus b. Scale bars, 1 mm
To quantify the number of Gys1 alleles disrupted by the Gys1-targeted SaCas9, we used ddPCR and obtained the percentage of total Gys1 with indel formations in extracts from aliquots of frozen powdered whole hemispheres. In all three mouse models (Gbe1Y239S, Epm2a−/−, and Epm2b−/−), approximately 17% of Gys1 alleles had been edited (Figs. 3a; 4a; and 5a).

Gys1 targeting AAV-SaCas9 disrupts the Gys1 gene, reduces Gys1 mRNA and protein levels, and decreases insoluble glycogen and PB accumulation in brains of the Gbe1Y239S APBD mouse model. Neonatal mice (P2) were injected with PBS (N = 12 for each experiment) or AAV-SaCas9 (N = 12 for each experiment), and mice were sacrificed at 3 months for brain tissue analysis. Indel percentage a and Gys1 mRNA level b were measured by ddPCR. Representative brain GYS1 western blots with stain-free gel as loading control c. Quantification of GYS1 western blots normalized to stain-free gel shown in d. Polyglucosan body (PB) quantification in the hippocampus e and degradation resistant glycogen content f. Representative micrographs of PASD stained hippocampus of PBS g and i vs AAV-SaCas9 h and j treated mice. Scale bars, 300 µm. Data are presented as mean ± SEM. Significance levels are indicated as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001

Gys1 targeting AAV-SaCas9 disrupts the Gys1 gene, reduces Gys1 mRNA and protein levels, and decreases insoluble glycogen and PB accumulation in brains of the Epm2a−/− LD mouse model. Neonatal mice (P2) were injected with PBS (N = 13 for each experiment) or AAV-SaCas9 (N = 8 in WB quantification and N = 10 for other experiments), and mice were sacrificed at 3 months for brain tissue analysis. Indel percentage a and Gys1 mRNA level b were measured by ddPCR. Representative brain GYS1 western blots with stain-free gel as loading control c. Quantification of GYS1 western blots normalized to stain-free gel shown in d. Polyglucosan body (PB) quantification in the hippocampus e and degradation resistant glycogen content f. Representative micrographs of PASD stained hippocampus of PBS g and i vs AAV-SaCas9 h and j treated mice. Scale bars, 300 µm. Data are presented as mean ± SEM. Significance levels are indicated as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001

Gys1 targeting AAV-SaCas9 disrupts the Gys1 gene, reduces Gys1 mRNA and protein levels, and decreases insoluble glycogen and PB accumulation in brains of the Epm2b−/− LD mouse model. Neonatal mice (P2) were injected with PBS (N = 10 for each experiment) or AAV-SaCas9 (N = 14 for each experiment), and mice were sacrificed at 3 months for brain tissue analysis. Indel percentage a and Gys1 mRNA level b were measured by ddPCR. Representative brain GYS1 Western blots with stain-free gel as loading control c. Quantification of GYS1 Western blots normalized to stain-free gel shown in d. Polyglucosan body (PB) quantification in the hippocampus e and degradation resistant glycogen content f. Representative micrographs of PASD stained hippocampus of PBS g and i vs AAV-SaCas9 h and j treated mice. Scale bars, 300 µm. Data presented as mean ± SEM. Significance levels are indicated as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001
Effects on Gys1 mRNA and Protein
To measure the extent of reduction of whole hemisphere Gys1 mRNA, we again used ddPCR on whole hemisphere extracts and found an approximately 15% reduction in the AAV-Cas9-treated mice of all three genotypes (Figs. 3b; 4b; and 5b), a value not dissimilar to the extent of Gys1 DNA rearrangement. Reduction of GYS1 protein measured by quantitative western blotting on whole hemisphere extract was by approximately 50% in all three disease models (Figs. 3c, d; 4c, d; and 5c, d), unexpectedly higher than the mRNA reduction.
Effects on Hippocampal PBs and Whole Hemisphere Degradation-Resistant Glycogen
In all three disease models, PBs are more or less evenly distributed across the brain [21,22,23]. In most studies, PB measures are obtained in the hippocampus, because this region’s characteristic structure permits demarcation of a uniform tissue surface area for reliable measurements across experimental animals [13, 17, 26]. Hippocampal PB loads measured as % PASD signal per hippocampal area were reduced by 40 to 60% in the AAV-Cas9-treated mice in the three models (Figs. 3e, g, h; 4e, g, h; and 5e, g, h), a reduction that is in the range of GYS1 protein diminution.
In oxygen and glucose-deprived states, normal brain glycogen is very swiftly digested to generate glucose in an attempt to protect the brain [39, 40]. This is so rapid that within minutes of sacrifice normal murine brain glycogen all but disappears [37]. In the APBD and LD mouse models, brain glycogen is a mix of normally branched soluble and consumable glycogen and the ever-accumulating abnormally branched precipitated and digestion-resistant polyglucosans (PBs). Within 60 min from sacrifice, the soluble glycogen disappears while the precipitated glycogen remains, and can be biochemically quantified as a measure of PBs [3, 37, 41]. This degradation-resistant glycogen was reduced by 40 to 60% in whole hemisphere extracts in AAV-Cas9-treated mice in the three genotypes (Figs. 3f; 4f; and 5f), a value similar to the reduction in PBs as quantified histochemically.
Effects on PB-Associated Immune Activation
In advanced murine LD (over 11 months of age), immunohistochemical studies showed activation and proliferation of astroglia and microglia. Recent transcriptomic analyses showed that 94% of genes upregulated in the disease encode proteins of inflammatory and immune system pathways [14]. Finally, downregulation of GYS1 activity by transgenic or other means resulting in PB reductions partially corrected these abnormalities [15,16,17,18,19,20]. Of the several hundred immune system genes shown to be dysregulated in the transcriptomic studies, a subset (less than 10) was tested and confirmed by qRT-PCR, of which several were shown to already be overexpressed at earlier stages of the disease [14, 20]. To our knowledge, similar transcriptomic and generally RNA-based studies of immune markers have not been performed in APBD mice, although the latter are known to exhibit astrogliosis and microgliosis in immunohistochemical studies [13].
We selected the four most studied immune marker genes and quantified their expression in the current cohorts. At the age of sacrifice (3 months), three (Cxcl10, Lcn2, and C3) of the four were upregulated in the APBD mice and all three were improved with the Gys1-targeting AAV-Cas9 (Fig. 6a). Two genes (Cxcl10 and Ccl5) were upregulated in the Epm2a−/− mice and ameliorated with the treatment (Fig. 6b). One gene (Cxcl10) was upregulated in the Epm2b−/− mice and improved with the treatment.

Effect of AAV-SaCas9 treatment on polyglucosan body associated immune system activation. Neonatal mice (P2) were injected with PBS or AAV-SaCas9 and mice were sacrificed at 3 months for brain tissue analysis. WT indicates the wild type control group. For each panel, from left to right, Cxcl10, Ccl5, Lcn2, and C3 were used as neuroinflammation markers, and relative mRNA expression levels were analyzed by qRT-PCR for Gbe1Y239S a, Epm2a−/− b, and Epm2b−/− c mice. In panel a, WT (N = 13), PBS (N = 12), and AAV-SaCas9 (N = 12). In panel b, WT (N = 8), PBS (N = 13), and AAV-SaCas9 (N = 10). In panel c, WT (N = 11), PBS (N = 10), and AAV-SaCas9 (N = 14). Data are presented as mean ± SEM. Significance levels are indicated as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001
Discussion
Given its complexity, the brain utilizes the lion’s share of the genome for its development and function [42]. As such, thousands of single gene defects present with neurological problems in childhood, the corollary of which is that child neurology in large part consists of numerous rare monogenic diseases. A much smaller number of single gene defect disorders present in adulthood, but many adult neurological and neuropsychiatric diseases are still genetic, albeit complex. These include common forms of Alzheimer’s disease, schizophrenia, Parkinson’s disease, and ALS. As such, gene-based therapies are a definite part of the future of clinical neurosciences.
At present, CRISPR/Cas-based technologies are the most promising in editing the genome back to health, albeit with unknown timeline to clinical applicability due to several hurdles yet to be overcome. These hurdles include transport of the CRISPR/Cas9 molecular machinery into vast swathes of the brain, ensuring accurate target editing, Cas9 immunogenicity, and off-target effects. The first two difficulties are interrelated because replacing mutations with correct DNA sequences require inclusion of components that makes the cargo to be delivered that much more complex and larger. There is, however, a simple form of editing, namely gene disruption, achievable with only two components; a guide RNA; and a Cas enzyme. The diseases studied here represented an opportunity to test this basic possibility. The AAV9 virus accommodated the comparatively small SaCas9 enzyme and a Gys1-targeting guide in a single plasmid that was delivered and was functional on 17% of Gys1 alleles across the brain.
The above ~ 17% disruption was achieved by ICV injection in neonatal mice, of all known situations the most permissive given the yet immature nature of intercompartmental barriers at this age [43,44,45]. Treatments at older ages through any route (ICV, intrathecal, intra-cisterna magna, or intravenous) are expected to be less widespread and effective and will require enhanced viral or other delivery systems.
The most commonly used streptococcal Cas9 (~ 4 kb) does package in AAV9 but cannot accommodate the guide sequence, thus requiring separate viruses delivering the two necessary components to the same cells, which greatly reduces editing efficiency. The SaCas9 ortholog used here (~ 3.2 kb) does accommodate the guide within one virus [46]. The extent of viral distribution is greatly enhanced with self-complementary vector designs, where two copies of the cargo sequences are incorporated [47, 48]. This of course could not be applied here, given the size even of SaCas9. Future developments in obtaining much smaller Cas enzymes or larger packaging capacity would improve self-complementation-based spread of effective delivery.
The JeTI promoter used in our construct to drive SaCas9 expression is a short artificial promoter designed to make room to fit larger genes within AAV9′s packaging capacity [49, 50]. Its expression potency is low compared to standard gene therapy promoters, which in this case may be an advantage. Cas9 is needed to act twice in each cell, once on each autosome target. Any activity beyond that would be off-target and potentially detrimental, and as such weak promoters are likely preferable.
In our study, the extent of Gys1 mRNA reduction corresponded to that of Gys1 gene editing (15–17%). Surprisingly, the degree of GYS1 protein reduction was substantially higher (~ 50%). We observed a similar greater GYS1 protein than mRNA reduction in our previous work where we knocked out the Gys1 gene through a conditional transgenic approach [20]. Generally, disconnect between mRNA versus protein relative quantities is not surprising and well-documented, with a multiplicity of post-transcriptional mechanisms invoked and under study [51]. In the present case, the disconnect is favourable: treatments for APBD and LD with finite impacts on GYS1 mRNA would have a greater impact on the ultimate enzyme target.
The recent recognition of the inflammatory component in the pathogenesis of LD opened novel anti-inflammatory therapeutic possibilities. These are presently being studied, while more permanent root cause-based therapies are developed [14,15,16, 52]. A side result of our study is the identification of similar transcription-level immune abnormalities, and their improvement, in APBD. These data supplement our recent immunohistochemical findings of astrogliosis and microgliosis in APBD and their correction following transgenic targeting of the Gys1 gene [13]. Collectively, these results suggest that the immune pathology, and its response to GYS1 and PB lowering therapies, may be shared across polyglucosan storage diseases. In fact, the immune dysregulation appears to be greater in the APBD mouse model than in the LD models, with more immune markers affected at the early age of three months. This greater immune disturbance correlates with the larger number of PBs and the greater accumulation of digestion-resistant glycogen in the APBD versus LD models (compare panels e and f across Figs. 3, 4, and 5), further supporting a relationship between PB accumulation and immunopathology. Longer-term studies in animal models, and eventually in possible clinical trials, are needed to confirm whether CRISPR/Cas9-mediated GYS1 knockdown will alter behavioural and clinical disease progression.
In conclusion, it is possible to achieve amelioration of pathological bases of neurological diseases of the brain through AAV9-delivered Cas9 genome editing. Immunopathology may be shared across brain polyglucosan storage diseases. A single therapy, targeting GYS1, may be possible for multiple glycogen storage diseases of the brain.
References
Roach PJ. Glycogen and its metabolism. Curr Mol Med. 2002;2(2):101-20.
Cenci U, Nitschke F, Steup M, et al. Transition from glycogen to starch metabolism in Archaeplastida. Trends Plant Sci. 2014;19(1):18-28.
Sullivan MA, Nitschke S, Skwara EP, et al. Skeletal Muscle Glycogen Chain Length Correlates with Insolubility in Mouse Models of Polyglucosan-Associated Neurodegenerative Diseases. Cell Rep. 2019;27(5):1334–44 e6.
Andersen DH. Familial cirrhosis of the liver with storage of abnormal glycogen. Lab Invest. 1956;5(1):11-20.
Lossos A, Meiner Z, Barash V, et al. Adult polyglucosan body disease in Ashkenazi Jewish patients carrying the Tyr329Ser mutation in the glycogen-branching enzyme gene. Ann Neurol. 1998;44(6):867-72.
Dainese L, Monin ML, Demeret S, et al. Abnormal glycogen in astrocytes is sufficient to cause adult polyglucosan body disease. Gene. 2013;515(2):376-9.
Tagliabracci VS, Girard JM, Segvich D, et al. Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J Biol Chem. 2008;283(49):33816-25.
Gentry MS, Worby CA, Dixon JE. Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci U S A. 2005;102(24):8501-6.
Nitschke F, Ahonen SJ, Nitschke S, et al. Lafora disease - from pathogenesis to treatment strategies. Nat Rev Neurol. 2018;14(10):606-17.
Minassian BA, Lee JR, Herbrick JA, et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20(2):171-4.
Chan EM, Young EJ, Ianzano L, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35(2):125-7.
Turnbull J, DePaoli-Roach AA, Zhao X, et al. PTG depletion removes Lafora bodies and rescues the fatal epilepsy of Lafora disease. PLoS Genet. 2011;7(4):e1002037.
Chown EE, Wang P, Zhao X, et al. GYS1 or PPP1R3C deficiency rescues murine adult polyglucosan body disease. Ann Clin Transl Neurol. 2020;7(11):2186-98.
Lahuerta M, Gonzalez D, Aguado C, et al. Reactive Glia-Derived Neuroinflammation: a Novel Hallmark in Lafora Progressive Myoclonus Epilepsy That Progresses with Age. Mol Neurobiol. 2020;57(3):1607-21.
Sanz P, Serratosa JM. Neuroinflammation and progressive myoclonus epilepsies: from basic science to therapeutic opportunities. Expert Rev Mol Med. 2020;22:e4.
Lopez-Gonzalez I, Viana R, Sanz P, et al. Inflammation in Lafora Disease: Evolution with Disease Progression in Laforin and Malin Knock-out Mouse Models. Mol Neurobiol. 2017;54(5):3119-30.
Varea O, Duran J, Aguilera M, et al. Suppression of glycogen synthesis as a treatment for Lafora disease: Establishing the window of opportunity. Neurobiol Dis. 2021;147:105173.
Sinha P, Verma B, Ganesh S. Trehalose Ameliorates Seizure Susceptibility in Lafora Disease Mouse Models by Suppressing Neuroinflammation and Endoplasmic Reticulum Stress. Mol Neurobiol. 2021;58(3):1088-101.
Israelian L, Nitschke S, Wang P, et al. Ppp1r3d deficiency preferentially inhibits neuronal and cardiac Lafora body formation in a mouse model of the fatal epilepsy Lafora disease. J Neurochem. 2020. https://doi.org/10.1111/jnc.15176
Nitschke S, Chown EE, Zhao X, et al. An inducible glycogen synthase-1 knockout halts but does not reverse Lafora disease progression in mice. J Biol Chem. 2020. https://doi.org/10.1074/jbc.RA120.015773
Ganesh S, Delgado-Escueta AV, Sakamoto T, et al. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet. 2002;11(11):1251-62.
Turnbull J, Wang P, Girard JM, et al. Glycogen hyperphosphorylation underlies lafora body formation. Ann Neurol. 2010;68(6):925-33.
Orhan Akman H, Emmanuele V, Kurt YG, et al. A novel mouse model that recapitulates adult-onset glycogenosis type 4. Hum Mol Genet. 2015;24(23):6801-10.
Pederson BA, Turnbull J, Epp JR, et al. Inhibiting glycogen synthesis prevents Lafora disease in a mouse model. Ann Neurol. 2013;74(2):297-300.
Turnbull J, Epp JR, Goldsmith D, et al. PTG protein depletion rescues malin-deficient Lafora disease in mouse. Ann Neurol. 2014;75(3):442-6.
Duran J, Gruart A, Garcia-Rocha M, et al. Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum Mol Genet. 2014;23(12):3147-56.
Raikwar SP, Kikkeri NS, Sakuru R, et al. Next Generation Precision Medicine: CRISPR-mediated Genome Editing for the Treatment of Neurodegenerative Disorders. J Neuroimmune Pharmacol. 2019;14(4):608-41.
Young CS, Pyle AD, Spencer MJ. CRISPR for Neuromuscular Disorders: Gene Editing and Beyond. Physiology (Bethesda). 2019;34(5):341-53.
Karimian A, Gorjizadeh N, Alemi F, et al. CRISPR/Cas9 novel therapeutic road for the treatment of neurodegenerative diseases. Life Sci. 2020;259:118165.
Hsu PD, Scott DA, Weinstein JA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827-32.
Ran FA, Cong L, Yan WX, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520(7546):186-91.
Karumuthil-Melethil S, Nagabhushan Kalburgi S, Thompson P, et al. Novel Vector Design and Hexosaminidase Variant Enabling Self-Complementary Adeno-Associated Virus for the Treatment of Tay-Sachs Disease. Hum Gene Ther. 2016;27(7):509-21.
Clement N, Grieger JC. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol Ther Methods Clin Dev. 2016;3:16002.
Kawasaki H, Kosugi I, Sakao-Suzuki M, et al. Intracerebroventricular and Intravascular Injection of Viral Particles and Fluorescent Microbeads into the Neonatal Brain. J Vis Exp. 2016(113). https://doi.org/10.3791/54164
Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671-5.
Berg S, Kutra D, Kroeger T, et al. ilastik: interactive machine learning for (bio)image analysis. Nat Methods. 2019;16(12):1226-32.
Nitschke S, Petkovic S, Ahonen S, et al. Sensitive quantification of alpha-glucans in mouse tissues, cell cultures, and human cerebrospinal fluid. J Biol Chem. 2020;295(43):14698-709.
Lowry O, Passonneau JV. A Flexible System of Enzymatic Analysis. 2012.
Suh SW, Bergher JP, Anderson CM, et al. Astrocyte glycogen sustains neuronal activity during hypoglycemia: studies with the glycogen phosphorylase inhibitor CP-316,819 ([R-R*,S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethyl)pro pyl]-1H-indole-2-carboxamide). J Pharmacol Exp Ther. 2007;321(1):45-50.
Lopez-Ramos JC, Duran J, Gruart A, et al. Role of brain glycogen in the response to hypoxia and in susceptibility to epilepsy. Front Cell Neurosci. 2015;9:431.
Nitschke F, Sullivan MA, Wang P, et al. Abnormal glycogen chain length pattern, not hyperphosphorylation, is critical in Lafora disease. EMBO Mol Med. 2017;9(7):906-17.
Bae BI, Jayaraman D, Walsh CA. Genetic changes shaping the human brain. Dev Cell. 2015;32(4):423-34.
Liu D, Zhu M, Zhang Y, et al. Crossing the blood-brain barrier with AAV vectors. Metab Brain Dis. 2021;36(1):45-52.
Miyake N, Miyake K, Yamamoto M, et al. Global gene transfer into the CNS across the BBB after neonatal systemic delivery of single-stranded AAV vectors. Brain Res. 2011;1389:19-26.
Johnson TB, White KA, Brudvig JJ, et al. AAV9 Gene Therapy Increases Lifespan and Treats Pathological and Behavioral Abnormalities in a Mouse Model of CLN8-Batten Disease. Mol Ther. 2021;29(1):162-75.
Friedland AE, Baral R, Singhal P, et al. Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol. 2015;16:257.
McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8(16):1248-54.
McCarty DM, Fu H, Monahan PE, et al. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther. 2003;10(26):2112-8.
Bailey RM, Armao D, Nagabhushan Kalburgi S, et al. Development of Intrathecal AAV9 Gene Therapy for Giant Axonal Neuropathy. Mol Ther Methods Clin Dev. 2018;9:160-71.
Tornoe J, Kusk P, Johansen TE, et al. Generation of a synthetic mammalian promoter library by modification of sequences spacing transcription factor binding sites. Gene. 2002;297(1-2):21-32.
Maier T, Guell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583(24):3966-73.
Molla B, Heredia M, Sanz P. Modulators of Neuroinflammation Have a Beneficial Effect in a Lafora Disease Mouse Model. Mol Neurobiol. 2021. https://doi.org/10.1007/s12035-021-02285-1
Acknowledgements
B.A.M. holds the University of Texas Southwestern Jimmy Elizabeth Westcott Chair in Pediatric Neurology. We thank Drs. Feng Zhang (Massachusetts Institute of Technology and Harvard University) and Steven Gray (University of Texas Southwestern) and team members for their help in vector design, construction, and packaging.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article
Funding
This work was funded by the National Institutes of Health under award P01NS097197.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gumusgoz, E., Guisso, D.R., Kasiri, S. et al. Targeting Gys1 with AAV‐SaCas9 Decreases Pathogenic Polyglucosan Bodies and Neuroinflammation in Adult Polyglucosan Body and Lafora Disease Mouse Models. Neurotherapeutics 18, 1414–1425 (2021). https://doi.org/10.1007/s13311-021-01040-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-021-01040-7