
Abstract
Introduction
This 32-week, open-label, randomized, parallel-group, multinational trial aimed to compare the efficacy and safety of stepwise insulin intensification of biphasic insulin aspart 30 (BIAsp 30) relative to stepwise intensification of a basal–bolus regimen in insulin-naïve adults with type 2 diabetes (T2D) who continued pretrial treatment with metformin and sulfonylurea.
Methods
Adults with T2D were randomized into one of two treatment arms for 32 weeks: (1) BIAsp 30 once daily (OD), with the possibility of stepwise treatment intensification up to BIAsp 30 three times daily (TID); (2) insulin glargine OD, with the possibility of stepwise treatment intensification with insulin aspart up to TID. The primary endpoint was change from baseline in HbA1c after 32 weeks.
Results
After 32 weeks, the estimated mean change in HbA1c from baseline was statistically significantly lower in the BIAsp 30 arm (− 1.18%) versus basal–bolus (− 1.36%) [estimated treatment difference 0.18%; 95% confidence interval (95% CI) 0.01; 0.36; p < 0.05]. The proportion of patients with HbA1c below 7.0% was statistically significantly lower with BIAsp 30 (42.9%) compared with basal–bolus (56.9%) (odds ratio 0.58; 95% CI 0.37; 0.89; p = 0.01). The overall rate of severe or blood glucose (BG)-confirmed hypoglycemic events was numerically lower for BIAsp 30 compared with basal–bolus, and a statistically significantly lower rate in nocturnal severe or BG-confirmed hypoglycemia in the BIAsp 30 arm relative to basal–bolus was observed: estimated rate ratio 0.32 (95% CI 0.13; 0.79), p = 0.0131. The proportion of patients with adverse events was similar in both treatment arms.
Conclusion
Insulin intensification with BIAsp 30 and basal–bolus showed an improvement in glycemic control; the change in HbA1c was statistically significantly lower for BIAsp 30 compared to basal–bolus. Basal–bolus treatment was accompanied by a numerically, and statistically significantly, higher rate of overall and nocturnal severe or BG-confirmed hypoglycemia, respectively, compared with BIAsp 30.
Funding
Novo Nordisk A/S.
Trial Registration
ClinicalTrials.gov identifier, NCT02453685.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The progressive nature of type 2 diabetes (T2D) necessitates initiation and intensification of medication to achieve glycemic control and to reduce the risk of diabetes-related complications [1]. For instance, the 10-year follow-up of the UK Prospective Diabetes Study (UKPDS) showed a sustained legacy effect of early, intensive blood glucose control in significantly decreasing the risk of myocardial infarction, microvascular disease, and death from any cause in patients with T2D [2]. Based on data such as these, recent guidelines recommend lowering HbA1c below 7.0% in most patients and that insulin therapy should not be delayed [3].
After metformin monotherapy, basal insulins, such as the long-acting insulin analogues, are often recommended as initial insulin therapy [3, 4]. However, in many parts of the world, a premix insulin containing both rapid-acting and intermediate-acting components in one formulation is also used to initiate or intensify insulin treatment [5, 6]. Biphasic insulin aspart 30 (BIAsp 30) is one such analogue premix insulin, which contains a mixture of soluble insulin aspart (IAsp) 30% and protaminated IAsp 70%, and is recommended to be injected either once daily (OD), twice daily (BID), or three times daily (TID) [7]. In insulin-naïve patients with T2D who have poor glycemic control, initiation of BIAsp 30 has been shown to significantly reduce HbA1c and has been associated with a slight weight gain, but no increased risk of severe hypoglycemic episodes, compared with basal insulin only regimens [8,9,10,11,12].
Following failure of adequately titrated basal insulin and oral antidiabetic drugs (OADs) to maintain glycemic control, the American Diabetes Association (ADA)’s recommended treatment algorithm is stepwise intensification with a rapid-acting prandial insulin added to a basal insulin OD regimen (“basal plus”), up to three injections of prandial insulin per day (“basal–bolus”) [3, 13, 14]. The ADA also now recommends that patients not achieving optimal glycemic control on a basal only or a basal plus regimen can switch to premix insulin BID, which can be intensified to TID to achieve optimal glycemic control [3, 15]. With regard to basal plus versus premix BID regimens, clinical evidence indicates that both regimens are broadly comparable with regard to efficacy and safety [16,17,18,19]. Equally, when further intensification is needed, both basal–bolus and premix TID regimens are now considered at the same level of efficacy and safety [3, 8, 20]. Overall, this parity emphasizes the important role of patient-centered factors in clinical decision-making; for instance, factors such as regimen simplicity, experience of hypoglycemia, and variability in patient lifestyles can all dictate the success of an insulin intensification regimen [3, 8, 19].
Whilst stepwise intensification of basal–prandial and premix regimens are recommended treatment strategies [3], there is a need to provide further clinical evidence for their use in patients with T2D. Thus, based on the lack of existing data, the aim of the current trial in insulin-naïve patients with T2D was to compare the efficacy and safety of stepwise insulin intensification with BIAsp 30 relative to stepwise intensification with a basal–bolus regimen of insulin glargine (IGlar) and IAsp.
Methods
Design
This was a 32-week, open-label, randomized, parallel-group, multicenter, multinational, phase 4 trial in patients with T2D aged at least 18 years (clinicaltrials.gov identifier, NCT02453685). The trial was conducted in nine countries: Australia, Bulgaria, Hungary, India, Republic of Korea, Serbia, Thailand, Turkey, and United Arab Emirates, between September 2015 and September 2016. The trial period consisted of a screening period (up to 2 weeks before randomization) and a 32-week treatment period divided into four periods of 8 weeks each. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. Informed consent was obtained from all patients for being included in the study.
Eligible participants were insulin-naïve men or women aged at least 18 years and had been clinically diagnosed with T2D for at least 6 months prior to screening, were treated with a stable daily dose (for at least 90 days prior to screening) of metformin (at least 1000 mg or maximum tolerated dose in the patient medical record) and sulfonylurea (SU) and were willing to discontinue any other OADs, including insulin secretagogues (except SU), dipeptidyl peptidase-4 inhibitor (DPP-4i), sodium glucose co-transporter 2 inhibitor (SGLT2i), colesevelam, bromocriptin, and/or combination products at randomization, had an HbA1c 7.0–9.5% (both inclusive) and body mass index (BMI) less than 40.0 kg/m2. Key exclusion criteria included previous participation in the current trial or participation in another clinical trial 1 month before screening, any contraindication to BIAsp 30, IGlar, or IAsp, impaired liver function, high blood pressure, proliferative retinopathy or maculopathy requiring acute treatment, mental incapacity, psychiatric disorder, unwillingness or language barriers precluding adequate understanding or co-operation.
Eligible patients were randomized (stratified by pretrial OAD therapy) into one of two treatment arms: (1) BIAsp 30 OD with the largest meal, with the possibility of treatment intensification up to BIAsp 30 TID; (2) IGlar OD, with the possibility of treatment intensification with IAsp up to TID (basal–bolus).
Treatments, Insulin Dosing and Titration
Patients were instructed in trial product administration and the investigator documented that direction for use was given to the patient orally and in writing at the first dispensing visit. Subsequent directions for use were given at succeeding visits if deemed appropriate by the investigator. It was the investigator’s or delegated staff’s responsibility to assess if the patient was capable of following instructions during training and those in the directions for use.
Pretrial treatment with metformin and SU was continued as background treatment throughout the entire trial, in line with the treatment practice demonstrated in the A1chieve study [21]. The stable dose levels and dosing frequencies prescribed pretrial could be changed for safety reasons only, at the discretion of the investigator. An individualized approach to insulin titration and intensification regimen was used in the current trial as opposed to using fixed insulin doses. All patients underwent a weekly insulin dose titration during the entire treatment period in order to achieve the pre-meal self-monitored plasma glucose (SMPG) target of 4.4–6.1 mmol/L (80–110 mg/dL).
At the end of each 8-week period (weeks 8, 16, and 24), insulin treatment was intensified in a stepwise manner by adding an additional injection of BIAsp 30 in the BIAsp 30 arm, and IAsp in the basal–bolus arm, on the basis of whether patients had achieved target HbA1c below 7.0%. The insulin treatment could only be stepwise intensified at the appropriate 8-week period. Patients randomized to BIAsp 30 had the possibility of receiving up to three daily injections of BIAsp 30 and patients randomized to basal–bolus had one daily injection of IGlar plus the possibility of up to three daily injections of IAsp. In the BIAsp 30 arm, patients received BIAsp 30 OD at a starting dose of 12 U administered with the largest meal. In the event of intensification to twice- or thrice-daily injections of BIAsp 30, the second or third injection was given at the second or third largest meal, respectively. Patients randomized to the basal–bolus arm received IGlar OD at a starting dose of 10 U administrated at the same point of time every day. For patients receiving IGlar OD who were intensified to once-, twice-, or thrice-daily insulin injections with IAsp, the first, second, or third injection was given at the first, second, or third largest meal, respectively, with a starting dose of 4 U.
Efficacy and Safety Endpoints
The primary endpoint was change from baseline in HbA1c after 32 weeks of treatment. Secondary endpoints included the proportion of patients achieving HbA1c below 7% after 32 weeks of treatment (total and without severe hypoglycemia during the last 12 weeks of treatment), change from baseline in fasting plasma glucose (FPG) after 32 weeks of treatment, mean 7-point SMPG profiles and prandial plasma glucose (PG) increments after 32 weeks of treatment, total daily insulin dose during the 32 weeks of treatment, and change in patient-reported diabetes treatment satisfaction from baseline to 32 weeks of treatment assessed by the diabetes treatment satisfaction questionnaire (DTSQ) [22]. Safety endpoints included the number of treatment-emergent hypoglycemic episodes classified according to the Novo Nordisk definition of confirmed symptomatic hypoglycemia during 32 weeks of treatment, change in body weight from baseline to 32 weeks of treatment, incidence of adverse events (AEs) during 32 weeks of treatment, and change in clinical safety parameters (physical examination, vital signs, laboratory parameters) from baseline to 32 weeks of treatment.
Statistics
Sample size was based on the half-width of a 95% confidence interval (CI) for the difference between the two means of change from baseline in HbA1c for the full analysis set (FAS) (i.e., all randomized patients). The sample size was determined by requiring that half the length of the 95% confidence interval was less than 0.27 with 90% probability. To ensure this, 336 patients needed to be randomized. Thus, assuming a screening failure rate of 30%, 480 patients were needed to be screened for inclusion in the trial.
Change from baseline in HbA1c after 32 weeks of randomized treatment was analyzed using a mixed model repeated measurement (MMRM) where all calculated changes in HbA1c from baseline at weeks 4, 7, 12, 15, 20, 23, 28, and 32 were included in the analysis. This model included treatment, region, and strata (two levels: pretrial metformin and SU, with/without other OAD) as fixed effects, HbA1c at baseline as covariate, and interactions between all fixed effects and visit, and between the covariate and visit. A dichotomous (responder/non-responder) endpoint was defined on the basis of whether a patient had met HbA1c below 7.0% after 32 weeks of randomized treatment (overall and without severe hypoglycemia during the last 12 weeks of treatment). As a conservative approach, patients withdrawn before 32 weeks were defined as non-responders. It was analyzed using a logistic regression model with treatment, strata, and region as factors, and baseline HbA1c as covariate. Changes from baseline in FPG after 32 weeks of randomized treatment were analyzed on the basis of all planned post-baseline measurements until or at 32 weeks using an MMRM similar to the model used for analysis of the primary endpoint, except with the respective baseline value as covariate. Prandial PG increment for each meal (breakfast, lunch, main evening meal) was derived from the 7-point SMPG profile as the difference between prandial 90-min PG values and the PG value before meal in each separate profile. Mean 90-min postprandial glucose increments over all meals were derived as the mean of all corresponding meal increments. Prandial PG increment endpoints (mean and each separate meal) were analyzed on the basis of all planned post-baseline measurements until or at 32 weeks using an MMRM similar to the model used for analysis of the primary endpoint and with the corresponding baseline value as covariate.
Results
Of the 481 patients screened, 146 were screening failures. A total of n = 335 patients were randomized and all randomized patients were exposed, except for three patients [all three unexposed patients were included in the FAS, but none of them were included in the safety analysis set (i.e., all patients receiving at least one dose of the investigational product or its comparator)]. Overall, n = 166 patients were exposed to the BIAsp 30 arm and n = 166 patients were exposed to the basal–bolus arm. Baseline characteristics were comparable across treatment arms (Table 1).
After 32 weeks of treatment, the observed mean HbA1c was reduced to 7.1% in the BIAsp 30 arm and 6.9% in the basal–bolus arm (Fig. 1). The estimated mean change in HbA1c from baseline to week 32 was statistically significantly lower in the BIAsp 30 arm (− 1.18%) compared with the basal–bolus arm (− 1.36%) [estimated treatment difference (ETD) 0.18%; 95% confidence interval (95% CI) 0.01; 0.36; p < 0.05]. In Fig. 2, the observed mean HbA1c over time can be seen for patients on the basis of the regimen and treatment that they were on after 24 weeks of treatment. The resulting HbA1c levels are a consequence of the trial design, as treatments were intensified on the basis of the patients’ glycemic target throughout the trial. Hence, patients that were in good control on average at intensification visits did not intensify their treatment, whereas the opposite was the case for patients with HbA1c above 7%. As a result of this trial design, as the initial randomization was not preserved, no conclusions on treatment comparisons can be made.

Mean HbA1c over time. BIAsp 30, biphasic insulin aspart 30

Observed mean HbA1c over time by treatment regimen. Full analysis set, observed data. BIAsp 30 biphasic insulin aspart 30, BID twice daily, IAsp insulin aspart, IGlar insulin glargine, OD once daily, TID three times a day
The proportion of patients who achieved HbA1c below 7.0% after 32 weeks of treatment was statistically significantly lower in the BIAsp 30 arm (42.9%) compared with the basal–bolus arm (56.9%) [odds ratio (OR) 0.58; (95% CI) 0.37; 0.89; p = 0.01]. The proportion of patients who achieved HbA1c below 7.0% after 32 weeks of treatment without severe hypoglycemia during the last 12 weeks of treatment was statistically significantly lower in the BIAsp 30 arm (42.3%) compared with the basal–bolus arm (56.3%) [OR 0.58; (95% CI 0.37; 0.89); p = 0.01].
The estimated mean reduction in FPG was statistically significantly lower at week 32 in the BIAsp 30 arm (− 1.93 mmol/L) compared with the basal–bolus arm (− 2.72 mmol/L) [ETD 0.79 (95% CI 0.23; 1.35); p < 0.01].
The estimated mean of the mean prandial PG increment was statistically significantly lower at week 32 in the BIAsp 30 arm (1.29 mmol/L) compared with the basal–bolus arm (1.85 mmol/L) [ETD − 0.55 mmol/L (95% CI − 0.97; − 0.14); p < 0.01]. The estimated mean prandial PG increments were statistically significantly lower at breakfast [− 0.61 mmol/L (95% CI − 1.20; − 0.02); p < 0.05] and dinner [– 0.77 mmol/L (95% CI − 1.43; − 0.12); p < 0.05] for BIAsp 30 compared with basal–bolus, whereas the difference at lunch [− 0.13 mmol/L (95% CI − 0.70; 0.44); p = 0.66] was not statistically significant.
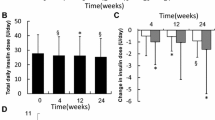
The mean total daily insulin dose increased throughout the treatment period with no pronounced differences between BIAsp 30 and basal–bolus; from 0.157 U/kg at baseline to 0.700 U/kg at week 32 for BIAsp 30 and from 0.127 U/kg at baseline to 0.708 U/kg at week 32 for basal–bolus.
At week 8, the proportion of patients who intensified treatment was 73.5% in the BIAsp 30 arm and 71.7% in the basal–bolus arm. At week 16, 46.4% and 48.2% of patients had their treatment intensified in the BIAsp 30 arm and basal–bolus arms, respectively. At week 24, the proportion was 13.9% and 35.5% in the BIAsp 30 and basal–bolus arms, respectively. The low proportion of patients in the BIAsp 30 arm who had their insulin treatment intensified at week 24 can potentially be explained by there being no further options for insulin intensification for patients who already had been intensified to the maximum number of three injections at week 16.
At week 24, 87 out of 149 patients in the BIAsp 30 arm were prescribed the maximum number of injections (three injections) and 42 out of 156 patients in the basal–bolus arm were prescribed the maximum number of injections (four injections). There were 18 out of 149 patients receiving BIAsp 30 OD and 31 out of 156 patients receiving IGlar OD, with 42 out of 156 patients receiving IGlar OD plus IAsp OD. Forty-four out of 149 patients were receiving BIAsp 30 BID and 41 out of 156 patients were receiving IGlar OD plus IAsp BID.
The overall rate of severe or BG-confirmed hypoglycemia (Novo Nordisk classification) was 4.24 episodes/patient-year of exposure (PYE) in the BIAsp 30 group and 5.2 episodes/PYE in the basal–bolus group: estimated rate ratio 0.83 (95% CI 0.58; 1.19); p = 0.3061 (Table 2). There were four severe hypoglycemia episodes in the BIAsp 30 arm and 20 episodes reported in the basal–bolus arm (Table 2). A mean cumulative plot of severe or BG-confirmed hypoglycemia is reported in Fig. 3. The observed rate of severe or BG-confirmed nocturnal hypoglycemia [defined as an episode that occurred between 00:01 and 05.59 (both inclusive)] was 0.53 episodes/PYE for BIAsp 30 and 0.98 episodes per PYE for basal–bolus. A post hoc analysis showed that the rate of severe or BG-confirmed nocturnal hypoglycemia was statistically significantly different: mean rate ratio 0.32 (95% CI 0.13; 0.79), p = 0.0131.

Severe or BG-confirmed treatment-emergent hypoglycemia cumulative plot. BIAsp 30, biphasic insulin aspart 30
After 32 weeks of treatment, an increase in observed mean body weight was seen in both treatment arms: + 2.49 kg in the BIAsp 30 arm and + 2.36 kg in the basal–bolus arm.
Overall, the proportion of patients with AEs was similar in both the BIAsp 30 (48.8%) and basal–bolus (46.4%) arms (Table 3). The number of severe AEs was lower in the BIAsp 30 arm than the basal–bolus arm (Table 3). Of the nine severe AEs observed in the BIAsp 30 arm, none were related to hypoglycemia whereas eight of the 15 severe AEs observed in the basal–bolus arm were related to hypoglycemia. There were three episodes of hypoglycemic unconsciousness in the basal–bolus arm and none in the BIAsp 30 arm. One patient in the basal–bolus arm died during the trial from cerebellar infarction, which was considered unlikely related to the trial product.
Quality of life was assessed by the DTSQ that was completed by patients at randomization and after 32 weeks of treatment. The DTSQ contained eight questions, of which six related to the overall treatment satisfaction and two to glycemic control. For the overall treatment satisfaction, a higher score (0–36) was related to a better perception of treatment satisfaction. For hypoglycemia and hyperglycemia, a lower score (0–6) was related to a better blood glucose control. The median (min; max) DTSQ score for overall treatment satisfaction at week 32 was 33 (6; 36) in the BIAsp 30 arm and 32 (6; 36) in the basal–bolus arm and the median change from baseline was 3 (− 24; 26) and 4 (− 27; 30) in the BIAsp 30 and basal–bolus arms, respectively. The median (min; max) DTSQ score for hyperglycemia at week 32 was 2.0 (0; 6) in the BIAsp 30 arm and 1.0 (0; 6) in the basal–bolus arm and the median change from baseline was − 1 (− 6; 5) and − 2 (− 6; 4) in the BIAsp 30 and basal–bolus arms, respectively. The median (min; max) DTSQ score for hypoglycemia at week 32 was 2.0 (0; 6) in the BIAsp 30 arm and 2.0 (0; 6) in the basal–bolus arm and the median change from baseline was 1 (− 6; 6) and 1 (− 6; 6) in the BIAsp 30 and basal–bolus arms, respectively.
Discussion
In this population of insulin-naïve patients with T2D who continued pretrial treatment with metformin and SU, insulin intensification with both BIAsp 30 and basal–bolus showed an improvement in glycemic control as measured by reductions in HbA1c. Compared with BIAsp 30, basal–bolus treatment resulted in a greater reduction in HbA1c from baseline, a higher proportion of patients achieving HbA1c below 7% (overall and without severe hypoglycemia during the last 12 weeks of treatment), along with a greater reduction in FPG. However, the estimated mean prandial PG increments were statistically significantly lower at breakfast and dinner for the BIAsp 30 arm compared with the basal–bolus arm, whereas the difference at lunch was not statistically significant. The overall rate of severe or BG-confirmed hypoglycemic events was numerically lower for BIAsp 30 compared with basal–bolus, with a post hoc analysis indicating that the overall rate of severe or BG-confirmed nocturnal hypoglycemia was statistically significantly lower with BIAsp 30 compared with basal–bolus. Overall, BIAsp 30 and basal–bolus treatments were safe and well-tolerated and no unexpected safety issues were identified, with the proportion of patients with AEs and the number of AEs being similar for both treatments.
On the basis of clinical evidence, the ADA now state that premix BID has parity with basal plus as an intensification strategy following failure of basal insulin (usually with metformin ± other non-insulin agent) to achieve glycemic control [3, 16,17,18,19]. Furthermore, premix TID and basal–bolus regimens are now viewed as having comparable efficacy and safety, where patients may switch between the two to achieve optimal glycemic control [3, 8, 20]. When appraising the results of the current trial within the patient-centered model of treatment of T2D, the importance of achieving optimal control of HbA1c may be overridden by the importance of preventing hypoglycemia for certain patients, with hypoglycemia having a number of deleterious physical and psychological outcomes that can significantly impair patient functioning [23, 24].
When intensifying treatment with a patient who has failed to achieve glycemic control with basal insulin OD and OADs, a full basal–bolus regimen provides a more flexible possibility to fine-tune dosing in relation to meals etc. However, many clinicians find that premix insulin has advantages over a basal–bolus insulin regimen. For instance, current guidance suggests that premix insulin regimens may have greater simplicity, require fewer injections and self-measured blood glucose monitoring, and be more practical for patients with physical disabilities [25]. A premix regimen may be more suitable for patients with regular meals and a large, consistent carbohydrate intake [19]. Premix insulin regimens require only one injection device and this has the potential to reduce the risk of mix-ups that can often be observed when patients have to manage multiple devices during basal–bolus therapy.
There are limitations of the current trial that should be discussed. For example, on the basis of our clinical experience, it is likely that achieving HbA1c below 7% in 8 weeks with a premix regimen alongside metformin and SU, in the absence of hypoglycemia, would be a challenge for many patients with T2D. Further, although the A1chieve study in real-world clinical practice showed a significant reduction in HbA1c with a relatively low rate of hypoglycemia when patients began or changed to insulin analogue therapy and continued metformin and SU, the design of the current trial differed. Specifically, in A1chieve, the participant and physician determined the choice and starting dose of insulin, administration frequency, and any later changes to either dose or frequency [21]. An additional limitation could also be argued to be the open-label nature of the trial, which was required as blinding of the insulin interventions was considered inappropriate and would have resulted in a requirement for placebo injections. Lastly, because of the way that hypoglycemia was assessed in the current trial, episodes of nocturnal hypoglycemia may not have been recorded if the patient was asleep (i.e., nocturnal hypoglycemia unawareness).
Conclusions
Insulin intensification with BIAsp 30 and basal–bolus showed an improvement in glycemic control; the change in HbA1c was statistically significantly lower for BIAsp 30 compared to basal–bolus. A numerically greater rate of overall severe or BG-confirmed hypoglycemia and a statistically significantly higher rate of severe or BG-confirmed nocturnal hypoglycemia was observed with basal–bolus compared with BIAsp 30. Viewed within the context of the patient-centered model of treatment, these results may translate to clinically relevant considerations for patients with T2D.
References
Meneghini LF. Intensifying insulin therapy: what options are available to patients with type 2 diabetes? Am J Med. 2013;126:S28–37.
Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–89.
American Diabetes Association. Standards of medical care in diabetes—2017. Diabetes Care. 2017;40:S1–135.
Garber AJ, Abrahamson MJ, Barzilay JI, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm–2016 executive summary. Endocr Pract. 2016;22:84–113.
IDF Clinical Guidelines Task Force. Global guideline for type 2 diabetes. 2012. www.idf.org/sites/default/files/IDF-Guideline-for-Type-2-Diabetes.pdf. Accessed July 2017.
Royal Australian College of General Practitioners and Diabetes (RAGCP). General practice management of type 2 diabetes, 2016–18. https://static.diabetesaustralia.com.au/s/fileassets/diabetes-australia/5d3298b2-abf3-487e-9d5e-0558566fc242.pdf. Accessed July 2017.
NovoNordisk A/S. EU SmPC for Insulin NovoMix® 30.2010. http://www.ema.europa.eu/docs/en_GB/document_908library/EPAR_-_Product_Information/human/000390908/WC500029441.pdf. Accessed June 2017.
Giugliano D, Chiodini P, Maiorino MI, Bellastella G, Esposito K. Intensification of insulin therapy with basal-bolus or premixed insulin regimens in type 2 diabetes: a systematic review and meta-analysis of randomized controlled trials. Endocrine. 2016;51:417–28.
Raskin P, Allen E, Hollander P, et al. INITIATE Study Group. Initiating insulin therapy in type 2 diabetes: a comparison of biphasic and basal insulin analogs. Diabetes Care. 2005;28:260–5.
Rys P, Wojciechowski P, Siejka S, Małecki P, Hak L, Malecki MT. A comparison of biphasic insulin aspart and insulin glargine administered with oral antidiabetic drugs in type 2 diabetes mellitus–a systematic review and meta-analysis. Int J Clin Pract. 2014;68:304–13.
Strojek K, Bebakar WM, Khutsoane DT, et al. Once-daily initiation with biphasic insulin aspart 30 versus insulin glargine in patients with type 2 diabetes inadequately controlled with oral drugs: an open-label, multinational RCT. Curr Med Res Opin. 2009;25:2887–94.
Qayyum R, Bolen S, Maruthur N, et al. Systematic review: comparative effectiveness and safety of premixed insulin analogues in type 2 diabetes. Ann Intern Med. 2008;149:549–59.
Owens DR. Stepwise intensification of insulin therapy in type 2 diabetes management—exploring the concept of the basal-plus approach in clinical practice. Diabet Med. 2013;30:276–88.
Rodbard HW, Visco VE, Andersen H, Hiort LC, Shu DH. Treatment intensification with stepwise addition of prandial insulin aspart boluses compared with full basal-bolus therapy (FullSTEP Study): a randomised, treat-to-target clinical trial. Lancet Diabetes Endocrinol. 2014;2:30–7.
Garber AJ, Wahlen J, Wahl T, et al. Attainment of glycaemic goals in type 2 diabetes with once-, twice-, or thrice-daily dosing with biphasic insulin aspart 70/30 (The 1-2-3 study). Diabetes Obes Metab. 2006;8:58–66.
Jin SM, Kim JH, Min KW, et al. Basal-prandial versus premixed insulin in patients with type 2 diabetes requiring insulin intensification after basal insulin optimization: a 24-week randomized non-inferiority trial. J Diabetes. 2016;8:405–13.
Tinahones FJ, Gross JL, Onaca A, Cleall S, Rodríguez A. Insulin lispro low mixture twice daily versus basal insulin glargine once daily and prandial insulin lispro once daily in patients with type 2 diabetes requiring insulin intensification: a randomized phase IV trial. Diabetes Obes Metab. 2014;16:963–70.
Vora J, Cohen N, Evans M, Hockey A, Speight J, Whately-Smith C. Intensifying insulin regimen after basal insulin optimization in adults with type 2 diabetes: a 24-week, randomized, open-label trial comparing insulin glargine plus insulin glulisine with biphasic insulin aspart (LanScape). Diabetes Obes Metab. 2015;17:1133–41.
Downie M, Kilov G, Wong J. Initiation and intensification strategies in type 2 diabetes management: a comparison of basal plus and premix regimens. Diabetes Ther. 2016;7:641–57.
Bowering K, Reed VA, Felicio JS, Landry J, Ji L, Oliveira J. A study comparing insulin lispro mix 25 with glargine plus lispro therapy in patients with type 2 diabetes who have inadequate glycaemic control on oral anti-hyperglycaemic medication: results of the PARADIGM study. Diabet Med. 2012;29:e263–72.
Home P, Naggar NE, Khamseh M, et al. An observational non-interventional study of people with diabetes beginning or changed to insulin analogue therapy in non-Western countries: the A1chieve study. Diabetes Res Clin Pract. 2011;94:352–63.
Bradley C. Diabetes treatment satisfaction questionnaire. In: Bradley C, editor. Handbook of psychology and diabetes: a guide to psychological measurement in diabetes research and practice. Chur: Harwood Academic; 1994. p. 111–32.
Brod M, Pohlman B, Wolden M, Christensen T. Non-severe nocturnal hypoglycemic events: experience and impacts on patient functioning and well-being. Qual Life Res. 2013;22:997–1004.
Cryer PE, Davis SN, Shamoon H. Hypoglycemia in diabetes. Diabetes Care. 2003;26:1902–12.
Wu T, Betty B, Downie M, et al. Practical guidance on the use of premix insulin analogs in initiating, intensifying, or switching insulin regimens in type 2 diabetes. Diabetes Ther. 2015;6:273–87.
Acknowledgements
Sponsorship for this study and article processing charges were funded by Novo Nordisk A/S, Bagsværd, Denmark.
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Medical writing and submission support were provided by Liam Gillies PhD and Beverly La Ferla of Watermeadow Medical, an Ashfield company, part of UDG Healthcare PLC, funded by Novo Nordisk A/S.
Disclosures
Sultan Linjawi has attended advisory panels and a speaker’s bureau for Novo Nordisk A/S. Salahedeen Abusnana has been an investigator on Novo Nordisk A/S clinical trials and attended Novo Nordisk A/S advisory boards. Susanna Lövdahl is an employee of Novo Nordisk A/S. Shanti Werther is an employee of Novo Nordisk A/S. Ömür Tabak and Byung-Wan Lee have nothing to disclose.
Compliance with Ethics Guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. Informed consent was obtained from all patients for being included in the study.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/D0DCF06010705822.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Linjawi, S., Lee, BW., Tabak, Ö. et al. A 32-Week Randomized Comparison of Stepwise Insulin Intensification of Biphasic Insulin Aspart (BIAsp 30) Versus Basal–Bolus Therapy in Insulin-Naïve Patients with Type 2 Diabetes. Diabetes Ther 9, 1–11 (2018). https://doi.org/10.1007/s13300-017-0334-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-017-0334-8