
Abstract
Poorly controlled diabetes is characterized by premature cardiovascular mortality and morbidity. The mechanisms linking hyperglycemia with accelerated atherosclerotic disease have not been fully elucidated; however, are thought to be mediated through vascular inflammation, oxidative stress and endothelial dysfunction. The advent of incretin-based therapy, whether by increasing endogenous glucagon-like peptide (GLP)-1 and glucose-dependent inhibitory polypeptide by inhibition of their breakdown using di-peptidyl peptidase 4 inhibitors, or augmenting GLP-1 activity using either exendin-4-based drugs or synthetic GLP-1 analogs promised not just improvements in glycemic control, but improvements in endothelial function, lipid profiles and markers of vascular inflammation. As such, it was anticipated they would demonstrate cardiovascular benefit in those with diabetes, indeed early meta-analyses suggested cardiovascular events would be reduced. To date, however, this benefit has failed to materialize, indeed the cardiovascular outcome trials, whilst meeting their primary endpoint of cardiovascular safety, have failed to demonstrate any improvements in stroke or myocardial infarction. This review will explore the data and attempt to answer the question: what went wrong?
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes mellitus currently affects approximately 415 million people worldwide [1]. It is estimated the global health expenditure on diabetes is approximately $673billion; indeed in the USA alone the cost is approximately $320 billion. Of this cost, however, only 12% is spent on directly managing the diabetes itself, with the majority of the expenditure being on the complications of the disease [2]. Cardiovascular disease is the most common cause of death and disability among people with diabetes; the diagnosis conferring a twofold excess risk of cardiovascular disease, independent of the usually accompanying adverse lipid and weight profile [3]. Therefore, reducing the cardiovascular risk of people with diabetes has been at the forefront of diabetes research with the intention of improving the health and wellbeing of the population whilst simultaneously reducing the global financial burden of the disease. At the core of cardiovascular protection, good blood pressure control, weight reduction, improved physical activity and appropriate statin usage have been demonstrated to substantially reduce event rate [4].
Diabetes is diagnosed and characterized by hyperglycemia, and thus for many years, strict glycemic control was thought to be key to improving cardiovascular events. Whereas tight glycemic control unequivocally improves microvascular outcomes, the benefit has not been consistently demonstrated in the macrovasculature [5–7]. Multiple explanations have been presented for this lack of benefit, or in the case of the ACCORD study (ClinicalTrials.gov identifier, NCT00000620) the apparent harm [6]. There is still no unifying accepted theory. Many have suggested the predominantly insulinocentric approach to the management of glucose may have contributed to the lack of benefit [8] as these therapies are associated with significant weight gain [9] and risk of hypoglycemia [10]. Although unlikely to account for the complete lack of benefit seen in these studies it is generally accepted that these recognized complications of treatment mitigate the benefit of good glycemic control. Indeed, the 2.7-fold excess cardiovascular mortality reported in the systematic review and meta-analysis of observational studies was in part attributed to these complications of treatment [11]. As such, it was hoped the advent of incretin-based therapies, designed to support physiological regulation of glycemia without precipitating hypoglycemia or weight gain, would provide an alternative treatment strategy with cardioprotective benefits. Early phase 2 and 3 studies provided promising results, however, as yet, these benefits have failed to be demonstrated.
This review will explore the pre-clinical and early clinical data and compare it with the subsequent larger clinical trials to propose potential explanations for the disconnect between the physiological benefits presented in early work and the lack of apparent benefit demonstrated in large-scale cardiovascular safety studies.
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
The Incretin System in Health and Disease
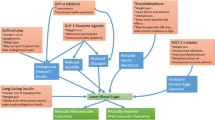
The pathophysiology of diabetes includes attenuated pancreatic α and β cell function resulting in inappropriate glucagon release and diminished insulin production, cellular insulin resistance in the muscles, liver, brain and adipose tissues, increased renal glucose reabsorption and impairments of the incretin system [12]. Incretins are peptides secreted from the gut in response to ingestion of food. The two peptides, glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like polypeptide 1 (GLP-1) account for as much as 90% of the “incretin effect” first described by Jean LaBarre in 1930 [13]. They are responsible for as much as half of the glucose-dependent insulin release after food ingestion in addition to suppressing glucagon release. Further, in health they have been attributed with several preparatory responses to nutrition, including suppressing hepatic gluconeogenesis, promoting satiety and inhibiting gastric emptying. People with diabetes have reduced incretin production and enhanced degradation by the enzyme di-peptidyl peptidase-4 (DPP-4) [14]. Research into these pathophysiological deficits has resulted in the successful development and use of therapies either increasing endogenous GLP-1 by inhibiting its breakdown by DPP-4 or administering synthetic GLP-1 receptor agonists. The latter class of drugs is further subdivided into agents based on exendin-4, originally isolated from the Gila monster of the Southwestern United States, which has approximately 50–53% homology with human native GLP-1 making it resistant to DPP-4 degradation, but interacts with the GLP-1 receptor, or the synthetic GLP-1 analogs which share up to 97% homology with endogenous GLP-1, but have been engineered to resist DPP-4 breakdown. These agents stimulate appropriate insulin secretion, suppress inappropriate glucagon release and thus regulate glucose with a low risk of hypoglycemia. There is substantial interest, however, in the non-glycemic effects these agents had.
Cardiovascular Effects of Incretin-Based Therapy
In vitro studies exploring the effect of GLP-1 on endothelial function demonstrate that there are vascular benefits which are not mediated through improvements in hyperglycemia, weight loss or the accompanying blood pressure reduction. Exendin-4 stimulates proliferation of endothelial cells, a critical step in endothelial repair, arterial healing, and angiogenesis, by a GLP-1 receptor-dependent mechanism [15]. The endothelial dysfunction that characterizes premature atherosclerosis is attenuated in human umbilical vein endothelial cells (HUVECs) exposed to the GLP-1 analog liraglutide similarly through GLP-1 receptor-dependent mechanisms [16]. Liraglutide also shows marked anti-oxidative, anti-inflammatory, and anti-apoptotic effects on HUVECs exposed to tumor necrosis factor alpha (TNFα) [17], and attenuates the accompanying endothelial cell dysfunction [18]. Additionally, liraglutide reduced hyperglycemia-induced endoplasmic reticulum stress in HUVECs via mitochondrial fusion processes, thereby reducing apoptosis [19]. Interestingly this effect may be a direct mitochondrial effect rather than a GLP-1 receptor-mediated effect. Exenatide, however, failed to protect rat femoral arterial ring endothelial cells from triglyceride-induced cellular dysfunction [20].
In the animal models GLP-1 also appears to have favorable effects on vascular function, independent of its glucose lowering effects. GLP-1 administration mediates endothelial-dependent relaxation in the rat pulmonary artery [21, 22], which was attenuated in the presence of a nitric oxide synthase antagonist suggesting the involvement of nitric oxide (NO) in mediating its vascular effects. This is supported by observations that GLP-1 promotes NO-dependent relaxation of mouse mesenteric arteries [23]. The GLP-1 vascular effect appears to vary by vascular bed, causing endothelial-independent relaxation, via the GLP-1 receptor, in femoral arteries [24] but having no impact on rat aorta isolates [21]. GLP-1 is also protective against ischemia–reperfusion injury in isolated rat hearts [23, 25–28] and has renoprotective (reducing proteinuria and microalbuminuria) effects, in addition to the cardioprotective effects in Dahl salt-sensitive hypertensive rats [29]. Whether these effects are mediated directly via the GLP-1 receptor is unclear, as the vasodilatory effects have been observed to be both dependent [24] and independent [23] of the GLP-1 receptor. In the latter of these studies, GLP-1(9-36), which is the product from the degradation of GLP-1 by DPP-4, mediated relaxation of mouse mesenteric arteries [23]. Thus, it is clearly evident that GLP-1 acts as a vasodilator in animal models potentially having cardioprotective properties, although whether this is dependent on GLP-1 receptor or another mechanism remains to be elucidated.
Early work in humans replicated these findings. Acute administration of GLP-1 increased flow-mediated dilatation (endothelial dependent) in type 2 diabetic male subjects with coronary artery disease but had no significant effect on young healthy, lean male subjects in whom no existing failure of endogenous GLP-1 activity would be anticipated [30]. In a broader general population sample aged 18–50 years, GLP-1 did improve forearm blood flow by approximately 30% and augmented endothelial-dependent forearm blood flow response to acetylcholine by up to 40% [31]. Conversely, endothelial-independent function was not influenced by the acute administration of GLP-1 in either diabetic or healthy individuals suggesting GLP-1 improves function rather than structure [30, 31]. GLP-1 infusions were also shown to improve regional and global left ventricular function when administered within 6 h of an acute myocardial infarction and improve systolic function after successful primary angioplasty in those with severe left ventricular dysfunction [32].
Observations from early clinical trials suggested these benefits would extend into improvements in cardiovascular outcomes. A meta-analysis of the GLP-1 receptor agonist use demonstrated a reduction in blood pressure of 3.6/1.4 mmHg, weight of 2.9 kg and total cholesterol of 0.1 mmol/L [33]. A retrospective analysis of obese people with type 2 diabetes suggested an improvement in both blood pressure and C-reactive protein with the exendin-4-based GLP-1 Receptor agonist, exenatide [34]. A meta-analysis of the prospective liraglutide effect and action in diabetes (LEAD) studies, the registration program for the GLP-1 analog, liraglutide, demonstrated improvements in lipid profile, B-type natriuretic peptide, high-sensitivity C-reactive protein and plasminogen activator inhibitor-1 all of which have been associated with cardiovascular outcomes [35]. These benefits have also been demonstrated with exenatide and the DPP-4 inhibitor sitagliptin, suggesting this is a class effect [36, 37]. Reductions in postprandial triglycerides and apoB48, again both tightly associated with cardiovascular risk, have been demonstrated with both the DPP-4 inhibitor, vildagliptin [38], and the GLP-1 analog, liraglutide [39].
Cardiovascular Outcome Data
During pre-clinical investigation of incretin therapies, most trials record and adjudicate cardiovascular events. Although differing in protocols, the standardized recording and reporting of major adverse cardiac events (MACE) allows for meaningful meta-analyses. In a meta-analysis of 25 trials lasting >6 months and reporting at least one cardiovascular event, GLP-1 receptor agonists demonstrated a significant 49% reduction in MACE (p < 0.03) compared to placebo and a trend towards a 22% improvement versus active comparator. This failed to reach statistical significance due to the small numbers of events, short-term nature of the studies and the low-risk nature of the participants [40]. A similar benefit was seen in DPP-4 inhibitor trials, although with 70 trials to analyze, the 29% reduction in MACE was highly significant [41]. Again this analysis was limited by the relatively short duration of the studies and the relatively pure and healthy populations that were studied in these regulatory trials.
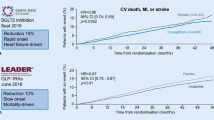
Since the concern over rosiglitazone potentially increasing cardiovascular events (subsequently refuted), licensing with the Food and Drug Administration (FDA), however, requires a priori cardiovascular outcome trials (CVOTs) to demonstrate cardiovascular safety in anti-glycemic agents. As such there are currently in excess of 100,000 people with diabetes worldwide participating in CVOTs using incretin-based therapies. To date, four of the trials have reported the trial to evaluate cardiovascular outcomes after treatment with sitagliptin (TECOS, ClinicalTrials.gov identifier, NCT00790205) [42], saxagliptin and cardiovascular outcomes in patients with type 2 diabetes (SAVOR-TIMI 53, ClinicalTrials.gov identifier, NCT01107886) [43] and the examination of cardiovascular outcomes with alogliptin versus standard of care (EXAMINE ClinicalTrials.gov identifier, NCT00968708) [44], using DPP-4 inhibitors, and the evaluation of lixisenatide in acute coronary syndrome (ELIXA, ClinicalTrials.gov identifier, NCT01147250) using lixisenatide. Whilst meeting all of the safety requirements of the FDA, many have felt disappointment regarding the lack of benefit achieved in these trials. The TECOS study randomized 14,671 people with diabetes and established cardiovascular disease to receive sitagliptin or placebo. After a median of 3.0 years, there was no difference in the composite of cardiovascular outcome namely cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, or hospitalization for unstable angina, with a hazard ratio (HR) of 0.98 (95% confidence interval [CI] 0.88–1.09). The EXAMINE study explored the effect of alogliptin in people with type 2 diabetes post-myocardial infarction or acute coronary syndrome for a median of 18 months and demonstrated a similar HR of 0.96 (95% upper limit of confidence interval 1.16) compared to placebo. SAVOR-TIMI demonstrated an HR of exactly 1.0 (95% CI 0.89–1.12) when comparing saxagliptin with placebo in 16,492 people at risk of or with a history of cardiovascular events for a median 2.1 years. The ELIXA study differed from the DPP-4-based studies, in that it was originally designed to demonstrate the superiority of the exendin-4-based GLP-1 receptor agonist, lixisenatide, over placebo after acute coronary syndrome. The 6068 participants recruited after acute coronary syndromes took either lixisenatide or placebo for a median of 25 months. Again, there was no difference in cardiovascular events (HR 1.02, 95% CI 0.89–1.17) although this still met the FDA’s requirements for demonstrating cardiovascular safety.
Of these studies, the result that generated most attention was the post hoc analysis of the non-adjudicated outcome for admissions due to heart failure in the SAVOR-TIMI study. In the first 6 months of the study there was an increase of 0.7% in hospitalization due to heart failure, which amounted to a 27% relative risk increase [45]. Despite the fact these were non-adjudicated outcomes in patients with pre-existing heart failure, the highly significant increase in events raised considerable concerns. As a result, existing studies were required to perform an analysis of heart failure admissions. The EXAMINE study showed a similar trend of 19% increase in admissions, although due to small numbers this did not reach significance [46]. TECOS, reassuringly, showed no increase in heart failure signal with an HR of 1.0 (95% CI 0.83–1.20). There are several potential reasons that have been hypothesized for this. The first simply suggests that the treatment of hyperglycemia reduces the osmotic diuresis effect of glucose, thereby increasing the symptoms of pre-existing heart failure. Others have hypothesized that this is simple statistical chance, although the similar trend in EXAMINE would appear to refute this. Other potential mechanisms include the potential for drug interactions, notably with angiotensin-converting enzyme (ACE)-inhibitors or diuretics, which are more likely with saxagliptin which does interact with the CYP3A4/A5 inhibitors of the cytochrome p450 pathway. This, however, does not account for the trend seen with alogliptin. An alternative explanation is found in the other metabolites of DPP-4; B-type natriuretic peptide (BNP), neuropeptide Y (NPY), peptide YY (PYY), substance P and stromal cell-derived factor 1 alpha (SDF-1α) [47]. These are all cardioactive substrates that influence vascular function. In isolation and in animal models increasing these by inhibition of DPP-4 should result in improved function; however, it is unknown whether interaction between and the relative balance of such factors paradoxically diminish function. Further, long-term DPP-4 inhibition may upregulate alternative compensatory degradation mechanisms, such as neutral endopeptidases. This is analogous to the increased chymase activity in long-term ACE-inhibitor use that sees angiotensin 2 levels rise to pre-treatment levels within 12 months. Neutral endopeptidases preferentially degrade BNP, thereby reducing the cardioprotective natriuretic effect in those with heart failure. The neutral effect of sitagliptin in TECOS, however, despite similar populations makes the latter explanation less likely.
What Causes the Disconnect Between the Early Clinical Work and the Randomized Controlled Trials?
There has been much deliberation as to why the potential benefit promised in the pre-clinical and early clinical work has not been realized in the definitive randomized controlled trials. One hypothesis explores the possibility that this may be simply due to the duration of the studies. Indeed, if the heart protection study (HPS) or the Scandinavian simvastatin survival study (4S) had been censored at 18 months or 2 years they would have not demonstrated any benefit from simvastatin [48, 49], yet statin therapy is widely accepted as a core element of cardiovascular risk reduction. If this is the case, studies such as the liraglutide effect and action in diabetes: evaluation of cardiovascular outcome results (LEADER, ClinicalTrials.gov identifier, NCT01179048) [50] and the cardiovascular outcome trial of linagliptin versus glimepiride in type 2 diabetes (CAROLINA, ClinicalTrials.gov identifier, NCT01243424) [51] trials that have a study minimum duration of 3.5 years (the point at which the HPS curves were clearly diverged) should show benefit. The reduction in cardiovascular events suggested by the early meta-analyses, however, was present in studies with a mean duration of 44.1 weeks [41]. Thus, duration of exposure to agents may not be the only factor.
Another significant difference between the regulatory studies that contributed to the meta-analyses and the subsequent cardiovascular outcome trials is the duration of diabetes in those studied. Regulatory studies tend to be performed in otherwise well participants, early in diabetes. Complex multi-morbid patients, such as those enrolled in TECOS, EXAMINE, SAVOR-TIMI and ELIXA are rarely used in these studies to minimize inter-participant variability and maximize the ability to detect differences. Event driven cardiovascular outcome trials, however, prefer high-risk patients to maximize the event rate and increase study power. This necessitates a longer duration of diabetes and multi-morbidity driven polypharmacy.
To date, the only trial to demonstrate cardiovascular benefit from glycemic control is the UKPDS (ClinicalTrials.gov identifier, NCT01099865) [5], which took newly diagnosed people with diabetes. The subsequent glycemic control trials, ACCORD, ADVANCE (ClinicalTrials.gov number, NCT00145925) and VADT (ClinicalTrials.gov identifier, NCT00032487) failed to demonstrate benefit [6, 7, 52, 53]. The failure to achieve benefit in these studies has been attributed to existing bad legacy effect from the preceding 8–12 years of poor glycemic control. Until recently it was felt that this legacy effect was purely an incremental accumulation of vascular damage, rather than an ongoing process. Recent work has demonstrated early adverse glycemic exposure triggers acute changes in protein expression, notably p66SHC in the mitochondria. This adaptor protein functions as a redox enzyme within the mitochondria responsible for reactive oxygen species generation and subsequent cellular apoptosis, vascular inflammation and endothelial dysfunction [54]. Gene silencing of p66SHC blunts persistent endothelial dysfunction and oxidative stress in the vasculature of diabetic mice [55]. Elsewhere, hyperglycemia has been demonstrated to trigger epigenetic changes. Deacetylation of the SIRT-1 gene and tumor suppressor p53 occurs [56]. These have both been demonstrated to control p66SHC transcription [57, 58]. Thus, the epigenetic changes in the SIRT-1 and p53 genes may perpetuate the original hyperglycemic effects. Interestingly metformin has been demonstrated to reverse the effects of SIRT-1 [59], in keeping with the observed benefit in UKPDS whereby metformin was substantially superior to sulphonylurea and insulin-based therapy [60]. If the failure of benefit in the existing trials is attributable to this, the vildagliptin efficacy in combination with metformin for early treatment of type 2 diabetes (VERIFY, ClinicalTrials.gov identifier, NCT01528254), which is taking newly diagnosed people with diabetes and randomizing them to receive either immediate DPP-4 combination with metformin or metformin alone, may demonstrate a benefit. Unfortunately, the primary endpoint of this study; time to initial treatment failure rate and rate of loss in glycemic control over time, may result in the study being terminated before being able to demonstrate a cardiovascular benefit.
Finally, these complex multi-morbid patients are characterized by polypharmacy, whereas regulatory trials are usually performed with as few concomitant prescriptions as possible. It is known that glibenclamide, and to a lesser extent glimepiride, attenuates the vascular benefit of GLP-1 [31]. This is most likely mediated through effects on mitochondrial ATP-sensitive potassium (KATPase) channels. These KATPase channels are also responsible for myocardial ischaemic preconditioning, and their inhibition by sulphonylurea use has been attributed, at least in part, to the historic poorer prognosis after myocardial infarction of those with diabetes [61]. This interaction between sulphonylurea and GLP-1 is well established, however, is not accounted for in any of the existing studies with the exception of CAROLINA, comparing DPP-4 inhibitor to sulphonylurea [51]. The possibility of other interactions in clinical practice attenuating the endothelial benefits of GLP-1 is possible, if not probable given the multiple mechanisms of action on the endothelium of many cardioprotective drugs used post-myocardial infarction.
Conclusion
Incretin-based therapies have demonstrated endothelial benefit in people with diabetes. To date, these have failed to translate into cardiovascular benefit in outcome trials. Potentially this may be due to the short duration of trials, the complex multi-morbid patients participating in these trails, a failure to tackle diabetes prior to an epigenetic bad glycemic legacy, or potential interactions with concomitant medications. This not withstanding, the profile of incretin-based drugs providing good glycemic control, with low risk of hypoglycemia and weight neutrality or benefit render them a suitable option after metformin, compared to alternatives associated with higher rates of hypoglycemia and weight gain, both of which have been demonstrated to adversely impact cardiovascular outcomes. Future work, exploring cardiovascular outcomes over longer time frames, such as the LEADER trial, earlier in diabetes such as VERIFY, or in the absence of potential interacting therapies such as CAROLINA may demonstrate benefits and help elucidate the ideal candidate for incretin-based intervention.
References
International Diabetes Federation. IDF diabetes atlas. 7th ed. Brussels: International Diabetes Federation; 2015.
American Diabetes A. Economic costs of diabetes in the U.S. in 2012. Diabetes Care. 2013;36:1033–46.
Emerging Risk Factors C, Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215–22.
Buse JB, Ginsberg HN, Bakris GL, Clark NG, Costa F, Eckel R, et al. Primary prevention of cardiovascular diseases in people with diabetes mellitus: a scientific statement from the american heart association and the american diabetes association. Circulation. 2007;115:114–26.
Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (ukpds 35): prospective observational study. BMJ. 2000;321:405–12.
Gerstein HC, Miller ME, Byington RP, Goff DC Jr, Bigger JT, Buse JB, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–59.
Zoungas S, Chalmers J, Neal B, Billot L, Li Q, Hirakawa Y, et al. Follow-up of blood-pressure lowering and glucose control in type 2 diabetes. N Engl J Med. 2014;371:1392–406.
Dluhy RG, McMahon GT. Intensive glycemic control in the accord and advance trials. N Engl J Med. 2008;358:2630–3.
UKPDS 33. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (ukpds 33). Uk prospective diabetes study (ukpds) group. Lancet. 1998;352:837.
Bonds DE, Miller ME, Bergenstal RM, Buse JB, Byington RP, Cutler JA, et al. The association between symptomatic, severe hypoglycaemia and mortality in type 2 diabetes: retrospective epidemiological analysis of the accord study. BMJ. 2010;340:b4909.
Forst T, Hanefeld M, Jacob S, Moeser G, Schwenk G, Pfutzner A, et al. Association of sulphonylurea treatment with all-cause and cardiovascular mortality: a systematic review and meta-analysis of observational studies. Diabetes Vasc Dis Res. 2013;10:302–14.
Defronzo RA. Banting lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–95.
la Barre J, Still E. Studies on the physiology of secretin. Am J Physiol. 1930;91:649–53.
Holst JJ, Vilsboll T, Deacon CF. The incretin system and its role in type 2 diabetes mellitus. Mol Cell Endocrinol. 2009;297:127–36.
Erdogdu O, Nathanson D, Sjoholm A, Nystrom T, Zhang Q. Exendin-4 stimulates proliferation of human coronary artery endothelial cells through enos-, pka- and pi3k/akt-dependent pathways and requires glp-1 receptor. Mol Cell Endocrinol. 2010;325:26–35.
Ding L, Zhang J. Glucagon-like peptide-1 activates endothelial nitric oxide synthase in human umbilical vein endothelial cells. Acta Pharmacol Sin. 2012;33:75–81.
Shiraki A, Oyama J, Komoda H, Asaka M, Komatsu A, Sakuma M, et al. The glucagon-like peptide 1 analog liraglutide reduces tnf-alpha-induced oxidative stress and inflammation in endothelial cells. Atherosclerosis. 2012;221:375–82.
Liu H, Dear AE, Knudsen LB, Simpson RW. A long-acting glucagon-like peptide-1 analogue attenuates induction of plasminogen activator inhibitor type-1 and vascular adhesion molecules. J Endocrinol. 2009;201:59–66.
Schisano B, Harte AL, Lois K, Saravanan P, Al-Daghri N, Al-Attas O, et al. Glp-1 analogue, liraglutide protects human umbilical vein endothelial cells against high glucose induced endoplasmic reticulum stress. Regul Pept. 2012;174:46–52.
Nathanson D, Erdogdu O, Pernow J, Zhang Q, Nystrom T. Endothelial dysfunction induced by triglycerides is not restored by exenatide in rat conduit arteries ex vivo. Regul Pept. 2009;157:8–13.
Golpon HA, Puechner A, Welte T, Wichert PV, Feddersen CO. Vasorelaxant effect of glucagon-like peptide (7-36) amide and amylin on the pulmonary circulation in the rat. Regul Pept. 2001;102:81–6.
Richter G, Feddersen O, Wagner U, Barth P, Goke R, Goke B. Glp-1 stimulates secretion of macromolecules from airways and relaxes pulmonary artery. Am J Physiol Lung Cell Mol Physiol. 1993;265:L374–81.
Ban K, Noyan-Ashraf H, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and independent pathways. Circulation. 2008;117:2340–50.
Nystrom T, Gonon AT, Sjoholm A, Pernow J. Glucagon-like peptide relaxes rat conduit arteries via an endothelium-independent mechanism. Regul Pept. 2005;125:173–7.
Bose AK, Mocanu MM, Carr RD, Brand CL, Yellon DM. Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes. 2005;54:146–51.
Bose AK, Mocanu MM, Carr RD, Yellon DM. Myocardial ischaemia-reperfusion injury is attenuated by intact glucagon like peptide-1 (glp-1) in the vitro rat heart and may involve the p70s6k pathway. Cardiovasc Drugs Ther. 2007;21:253–6.
Huisamen B, Gende S, Lochner A. Signalling pathways activated by glucagon-like-1 (7-36) amide in the rat heart and their role in protection against ischaemia. Cardiovasc J Afr. 2008;19:77–83.
Sonne DP, Engstrom T, Treiman M. Protective effects of glp-1 analogues exendin-4 and glp-1(9-36) amide against ischaemia-reperfusion injury in rat heart. Regul Pept. 2008;146:243–9.
Yu M, Moreno C, Hoaglund KM, Dahly A, Ditter K, Mistry M, et al. Antihypertensive effects of glucagon-like peptide-1 in dahl salt-sensitive rats. J Hypertens. 2003;21:1125–35.
Nystrom T, Guniak MK, Zhang Q, Zhang F, Holst JJ, Ahren B, et al. Effects of glucagon-like peptide-1 on endothelial function in type 2 diabetes patients with stable coronary artery disease. Am J Physiol Endocrinol Metab. 2004;287:E1209–15.
Basu A, Charkoudian N, Schrage WG, Rizza RA, Basu R, Joyner MJ. Beneficial effects of glp-1 on endothelial function in humans: dampening by glyburide but not glimepiride. Am J Physiol Endocrinol Metab. 2007;293:E1289–95.
Nikolaidis LA, Mankad S, Sokos GG, Mikse G, Shah A, Elahi D, et al. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation. 2004;109:962–5.
Vilsboll T, Christensen M, Junker AE, Knop FK, Gluud LL. Effects of glucagon-like peptide-1 receptor agonists on weight loss: systematic review and meta-analyses of randomised controlled trials. BMJ. 2012;344:d7771.
Viswanathan P, Chaudhuri A, Bhatia R, Al-Atrash F, Mohanty P, Dandona P. Exenatide therapy in obese patients with type 2 diabetes mellitus treated with insulin. Endocr Pract. 2007;13:444–50.
Fonseca VA, DeVries J, Bain SC. Liraglutide improves the profile of lipid and cardiovascular risk biomarkers from baseline. In: 21st World Diabetes Congress of the International Diabetes Federation; 2011.
Chaudhuri A, Ghanim H, Vora M, Sia CL, Korzeniewski K, Dhindsa S, et al. Exenatide exerts a potent antiinflammatory effect. J Clin Endocrinol Metab. 2012;97:198–207.
Makdissi A, Ghanim H, Vora M, Green K, Abuaysheh S, Chaudhuri A, et al. Sitagliptin exerts an antiinflammatory action. J Clin Endocrinol Metab. 2012;97:3333–41.
Matikainen N, Manttari S, Schweizer A, Ulvestad A, Mills D, Dunning BE, et al. Vildagliptin therapy reduces postprandial intestinal triglyceride-rich lipoprotein particles in patients with type 2 diabetes. Diabetologia. 2006;49:2049–57.
Hermansen K, Baekdal TA, During M, Pietraszek A, Mortensen LS, Jorgensen H, et al. Liraglutide suppresses postprandial triglyceride and apolipoprotein b48 elevations after a fat-rich meal in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, cross-over trial. Diabetes Obes Metab. 2013;15:1040–8.
Monami M, Dicembrini I, Nardini C, Fiordelli I, Mannucci E. Effects of glucagon-like peptide-1 receptor agonists on cardiovascular risk: a meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2014;16:38–47.
Monami M, Ahren B, Dicembrini I, Mannucci E. Dipeptidyl peptidase-4 inhibitors and cardiovascular risk: a meta-analysis of randomized clinical trials. Diabetes Obes Metab. 2013;15:112–20.
Green JB, Bethel MA, Armstrong PW, Buse JB, Engel SS, Garg J, et al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2015;373:232–42.
Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, et al. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317–26.
White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Bakris GL, et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327–35.
Scirica BM, Braunwald E, Raz I, Cavender MA, Morrow DA, Jarolim P, et al. Heart failure, saxagliptin, and diabetes mellitus: observations from the savor-timi 53 randomized trial. Circulation. 2014;130:1579–88.
Zannad F, Cannon CP, Cushman WC, Bakris GL, Nissen SE, Heller S, et al. Alogliptin in patients with type 2 diabetes after acute coronary syndromes: Heart failure outcomes and cardiovascular safety in heart failure patients. J Am Coll Cardiol. 2014;63:12_S.
Ussher JR, Drucker DJ. Cardiovascular biology of the incretin system. Endocr Rev. 2012;33:187–215.
Collins R, Armitage J, Parish S, Sleight P, Peto R, Heart Protection Study Collaborative Group. MRC/BHF heart protection study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003;361(9374):2005–16.
Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the scandinavian simvastatin survival study (4s). Lancet. 1994;344(8934):1383–89.
Marso SP, Poulter NR, Nissen SE, Nauck MA, Zinman B, Daniels GH, et al. Design of the liraglutide effect and action in diabetes: evaluation of cardiovascular outcome results (leader) trial. Am Heart J. 2013;166(823–830):e825.
Marx N, Rosenstock J, Kahn SE, Zinman B, Kastelein JJ, Lachin JM, et al. Design and baseline characteristics of the cardiovascular outcome trial of linagliptin versus glimepiride in type 2 diabetes (carolina(r)). Diabetes Vasc Dis Res. 2015;12:164–74.
Macisaac RJ, Jerums G. Intensive glucose control and cardiovascular outcomes in type 2 diabetes. Heart Lung Circ. 2011;20:647–54.
Ahren B, Burke B. Using albumin to improve the therapeutic properties of diabetes treatments. Diabetes Obes Metab. 2012;14(2):121–9.
Cosentino F, Francia P, Camici GG, Pelicci PG, Luscher TF, Volpe M. Final common molecular pathways of aging and cardiovascular disease: role of the p66shc protein. Arterioscler Thromb Vasc Biol. 2008;28:622–8.
Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M, et al. Gene silencing of the mitochondrial adaptor p66(shc) suppresses vascular hyperglycemic memory in diabetes. Circ Res. 2012;111:278–89.
El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205:2409–17.
Zhou S, Chen HZ, Wan YZ, Zhang QJ, Wei YS, Huang S, et al. Repression of p66shc expression by sirt1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ Res. 2011;109:639–48.
Kim CS, Jung SB, Naqvi A, Hoffman TA, DeRicco J, Yamamori T, et al. P53 impairs endothelium-dependent vasomotor function through transcriptional upregulation of p66shc. Circ Res. 2008;103:1441–50.
Zheng Z, Chen H, Li J, Li T, Zheng B, Zheng Y, et al. Sirtuin 1-mediated cellular metabolic memory of high glucose via the lkb1/ampk/ros pathway and therapeutic effects of metformin. Diabetes. 2012;61:217–28.
UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 1998;352(9131):854–65.
Lee TM, Chou TF. Impairment of myocardial protection in type 2 diabetic patients. J Clin Endocrinol Metab. 2003;88:531–7.
Acknowledgments
No funding or sponsorship was received for this study or publication of this article.
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
David Strain reports personal fees from Bayer, Boehringer-Ingelheim, Jannsen, MSD and Pfizer, grants and personal fees from Novo Nordisk and Novartis. WD Strain would like to acknowledge the support of the National Institute for Health Research (NIHR) Exeter Clinical Research Facility and the NIHR Biomedical Research Centre scheme. WD Strain is in receipt of a Higher Education Funding Council for England “New-Blood” Clinical Senior Lectureship award. The views expressed in this publication are those of the authors and not necessarily those of the National Health Service, the NIHR, or the Department of Health.
Christine Smith is an employee of Novo Nordisk UK Ltd. The opinions expressed in the publication are of the author and may not reflect that of Novo Nordisk.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content To view enhanced content for this article go to www.medengine.com/Redeem/2984F0604272C927.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Strain, W.D., Smith, C. Cardiovascular Outcome Studies in Diabetes: How Do We Make Sense of These New Data?. Diabetes Ther 7, 175–185 (2016). https://doi.org/10.1007/s13300-016-0165-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-016-0165-z