
Abstract
Atractylodes chinensis (fam. Asteraceae) is an important medicinal plant due to its unique pharmacological activity. The species is widely distributed in most areas of northern China. It is difficult to identify different populations of A. chinensis due to their similarity in characteristics. This study was the first investigation to date that assessed the genetic diversity of A. chinensis from different geographical counties of northern China using simple sequence repeat (SSR) markers. Of the 106 SSR primers in the clusters classified in the sesquiterpenoid biosynthesis pathway in the transcriptomic database of A. chinensis, ten with high polymorphism were used to analyze the inter-populations genetic diversity and construct DNA fingerprinting of 19 A. chinensis populations. A total of 78 alleles were detected, with an average number of 6.5 alleles per primer. The PIC value ranged from 0.4748 to 0.8918 with a mean of 0.6265. The neighbor-joining tree was used to classify 19 populations of A. chinensis into three clusters. DNA fingerprinting was performed according to these ten SSR markers. The results revealed that geographic origin is not exactly related to genetic diversity, as populations belonging to different provinces are grouped in the same cluster. The results of this study confirm that SSR markers are effective for genetic diversity analysis. The inter-populations genetic diversity and fingerprinting of A. chinensis in this study could provide a scientific basis for species identification and selective breeding.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Atractylodes chinensis (DC.) Koidz (typically referred to as “Bei Cang Zhu” in Chinese) is a major medicinal plant known as rhizome atractylodes, which are used to treat digestive disorders, rheumatic diseases and night blindness [5]. Modern pharmacological studies have reported that rhizome atractylodes was also used for anti-inflammatory, anti-bacterial [10, 17] and anti-tumor properties [11]. A. chinensis is widely distributed throughout most areas of northern China and is mainly produced in Hebei, Inner Mongolia, Liaoning and other provinces of China [35]. The contents of atractylodin in rhizome atractylodes, an important standard of quality assessment in the Chinese pharmacopeia, vary among provinces and even counties [13], but are similar in characteristics. The utilization of and research on A. chinensis have received less attention worldwide. Additionally, A. chinensis faces an unprecedented threat of even extinction due to its sharp reduction in wild resources as well as increasing medicinal demand. Although cultivation relieved some of this pressure over the past ten years, species of stable and consistent quality have not yet been cultivated due to unclear genetic basis. China is very rich in genetic variability of A. chinensis. Therefore, it is critical to adopt an effective methodology to assess the interpopulations genetic diversity of wild A. chinensis populations.
High-performance liquid chromatography (HPLC) fingerprinting [13], ITS [9, 12] and trnL-F [8, 22] sequences and chloroplast genome variation [30, 33] have been used to analyze interspecific phylogenetic relationships of Atractylodes species. However, these methods are not effective for intraspecific diversity analysis [34]. Simple sequence repeats (SSRs) are the ideal markers due to their high polymorphism, codominance and low cost. SSR markers have been widely used in variety identification, fingerprinting construction and intraspecific genetic diversity analysis [14, 25, 34]. The selection of a set of core SSR primers for germplasm identification and genetic diversity have been conducted for many medicinal plants, such as Glehnia littoralis [27], Glycyrrhiza [16], and Euryale ferox [15]. However, such a marker toolkit is not presently available for A. chinensis genetic diversity analysis.
In this paper, we screened SSR loci in clusters classified into the sesquiterpenoid biosynthesis pathway based on the transcriptomic database of A. chinensis. Ten SSRs with high polymorphism were used to analyze the interpopulations genetic diversity and fingerprinting of 19 A. chinensis populations. Interpopulation genetic diversity and fingerprinting will provide a scientific basis for species identification and selective breeding in A. chinensis.
Materials and methods
DNA extraction and PCR amplification
A. chinensis rhizomes were collected from different counties of northern China (Table 1), including Hebei, Shandong, Inner Mongolia and Jilin Provinces. No permission was required to collect wild resources of A. chinensis. All of the samples used in this study were identified as A. chinensis by Professor Qiaosheng Guo who works at Nanjing Agriculture University (Nanjing, Jiangsu Province, China). Professor Guo identified the experimental species through comparison with specimens inform the institute of botany Jiangsu Province, and the Chinese Academy of Sciences. All the samples were planted in the experimental farm of Hebei Normal University of Science & Technology (Qinhuangdao, Hebei, China). The quality and price of rhizome atractylodes were established according to the counties in the Chinese herbal medicine market. Young leaves of ten randomly selected plants from each population were mixed as one sample, immediately frozen in liquid nitrogen and stored at − 80 °C prior to DNA extraction.
The total DNA of A. chinensis was extracted through the improved CTAB method using plant genomic extraction kits (Cat.No.0419-50 CB, Huayueyang, Beijing, China, http://www.huayueyang.com.cn/product/276782043). The purities of extracted DNA samples were tested in a 2.0% agarose gel with electrophoresis on a horizontal electrophoresis DYCP-31DN apparatus (Liuyi, Beijing, China) and a gel-imaging system (GBOX-HR, Syn-gene, UK). The OD260/280 ratios of DNA were measured by a spectrophotometer (Synergy HT, Gene Company Limited, Hong Kong, China).
For SSR amplification, a 10 μL volume of reaction mixture included 50 ng/μL DNA, 2.5 mM dNTPs, 10 × buffer (Mg2+ included), 5.0 U/μL Taq enzyme, 10 μM of each primer, and ddH2O. Procedures for SSR amplification were carried out in a thermal cycler (BIO-RAD S1000 PCR, California, USA) by the following cycles: an initial 4 min pre denaturation at 94 °C, followed by 35 cycles of a 30-s denaturation at 94 °C, a 30-s annealing phase at 55 °C, and a 1 min extension at 72 °C, and a final extension at 72 °C for 10 min. The PCR products were preserved at 4 °C. PCR products were separated by polyacrylamide gel electrophoresis (6%) at a constant voltage (130 V) for 3 h. A 1000 bp DNA marker (TaKaRa, Japan) was used to determine allele size.
RNA sequencing and core SSR marker screening
RNA extraction and sequencing were performed as described by Zhao et al. (2021) [36]. RNA of A. chinensis was extracted using TRIzol Reagent (Invitrogen). Transcriptome data of A. chinensis were acquired based on the Illumina Hiseq Xten PE150 platform, by Novogene Co. (Beijing, China). All SSR primers used in this study were designed from the transcriptomic database as reported by Zhao et al. 2021 [36], and they are available in the SRA (BioProject ID PRJNA698794, https://www.ncbi.nlm.nih.gov/sra/PRJNA698794). SSR marker detection, identification and primer design were performed as described by Wu et al. (2021) [31].
This study was carried out to analyze the inter-populations genetic diversity based on those markers in clusters classified into sesquiterpenoid biosynthesis pathway. Twenty-five SSR primers in clusters annotated as terpene skeleton biosynthesis and eighty-one primers in clusters annotated as the sesquiterpenoid biosynthesis pathway were screened for polymorphism testing (Supplementary Table S1). SSR primers were constructed by Shanghai Invitrogen Biotechnology Company (Shanghai, China). The core primers, with high allelic frequencies (> 2), were screened by amplification with DNA extracted from 8 A. chinensis populations from different counties. Only ten SSR primers with distinct bands and high polymorphism were used to analyze interpopulations genetic diversity in this study (Table 3).
Data analysis
The amplified bands with good resolution from 10 SSR primers were counted and scored as 1 (present) or 0 (absence). Several genetic diversity assessment parameters such as the observed number of alleles, effective number of alleles, Nei's (1973) gene diversity (h) and Shannon's information index (I) were determined using software POPGENE version 1.32 [19]. The polymorphism information content (PIC) was calculated as described by Botstein et al. (1980) [3]. Similarity coefficients were calculated using the similarity program in PopGene version 1.32.
The clustering of 19 A chinensis populations was performed based on a similarity matrix using an unweighted pair group method with arithmetic average (UPGMA) algorithm following SAHN module of NTSYS version 2.10. The phylogenetic tree was constructed using the neighbor-joining method by MEGA version 7.0.21.
Results and discussion
SSR polymorphism
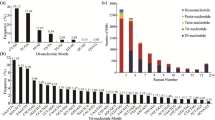
A total of 89,005 SSRs with 3 or more mono, di-, tri-, tetra-, penta-, and hexanucleotide repeat units were identified in the A. chinensis transcriptome. Among the SSRs, 46,188 (51.89%), 30,675 (34.46%), 9,791 (11.00%), 904 (1.02%), 886 (1.00%), and 561 (0.63%) were mono-, di-, tri-, tetra-, penta-, and hexanucleotides SSRs, respectively (Table 2). A/T (18,572, 20.87%), AG/CT (3,274, 3.68%) and ACC/GGT (1,303, 1.46%) were the most abundant of the mononucleotide, dinucleotide and trinucleotide motifs, respectively. Of the tetra-, penta- and hexanucleotide motifs, AAAT/ATTT (151, 0.17%), AAACC/GGTTT (339, 0.38%), and AACCCT/AGGGTT (15, 0.02%) were the most abundant, respectively (Table 2).
For medicinal plants without a reference genome, SSR molecular marker technology may not be directly used for genetic diversity analysis. High-throughput RNA sequencing effectively provides SSR markers. This is the first time that genetic diversity analysis of A. chinensis populations has been reported using SSR markers. In this paper, among the 106 SSR markers (Supplementary Table S1) screened on 8 randomly selected A. chinensis populations (data not shown), ten SSR markers generated polymorphisms (Table 3, Supplementary Fig S1). These ten SSR markers were used for DNA fingerprinting and genetic diversity analysis of 19 A. chinensis populations.
The size of the amplified fragments was estimated by using the DNA ladder that produced the expected size (100–1000 bp). SSR locus diversity data from ten SSR primers are summarized in Table 4. The overall size of the amplified fragments varied from 200 to 1000 bp. A total of 65 loci in 78 alleles (80.33%) were detected revealing the presence of a large difference. The number of polymorphic alleles per SSR locus ranged from 2 (S4) to 13 (S2) with an average of 6.5 alleles per locus (Table 4), showing that 19 A. chinensis populations exhibited a high level of genetic diversity. The average number of allelic genes in this study was more than that of many other crop species, namely, 3.7 in Euryale ferox [15], 5.1 in Lactuca sativa var capitata [37] and 4.5 in Sesamum indicum [2].
SSRs with PIC values > 0.5 were considered highly informative markers [24]. The PIC values among the 19 A. chinensis populations varied from 0.4908 (S54) to 0.8918 (S2) with an average of 0.6265 (Table 4), which was much higher than 0.5. The calculated average PIC value (0.6265) in A. chinensis was higher than that in some crops, namely, 0.495 in Camellia sinensis [7], 0.32 in Gossypium hirsutum [23], 0.5619 in Sorghum bicolor ssp. bicolor [21], which indicated their high informativeness. The value of PIC related to the relative frequency and number of alleles [24] was proportional to the polymorphic locus. The highest number of polymorphic alleles and PIC value were 13 and 0.8918, respectively, in primer S2. Eight out of ten (80.00%) markers had a PIC value > 0.5, except for S4 (0.4908) and S54 (0.4748), indicating that they were suitable for genetic diversity and fingerprinting studies.
Genetic diversity and relatedness
A dendrogram elucidating the genetic relationships among the 19 A. chinensis populations was constructed using the neighbor-joining method by MEGA version 7.0.21. To better understand their relationships, we divided the tested 19 A. chinensis populations into three clusters (Fig. 1). Populations P11 were grouped into Cluster I. Cluster II consisted of 9 populations distributed into two subgroups. One population, P6, was grouped into the subgroup. The remaining 8 populations were grouped into the second subgroup. Populations belonging to different provinces constituted cluster II. For example, P5 (Shandong Province) and P19 (Jilin Province) were grouped into the Hebei Province cluster. Similarly, cluster III consisted of 9 populations derived from different provinces. Populations P2 and P15 from Inner Mongolia Province were grouped into Hebei Province.

Neighbor-joining tree of 19 A. chenesis populations using MEGA
The 15 populations from Hebei Province were divided into two clusters, and grouped with Shandong and Jilin Provinces or with Inner Mongolia Province. The three clusters formed in the dendrogram revealed that the geographic origin does not exactly corroborate genetic diversity. This phenomenon appeared in many SSR marker-based genetic diversities, such as Sesamum indicum [2, 20], Camellia oleifera [4], Vicia amoena [31] and Trifolium repens [32]. Wu et al. carried out genetic diversity analysis of Trifolium repens using PCoA, UPGMA and STRUCTURE, and indicated that UPGMA analysis was implemented based on genetic distance, which provided more detailed relationships [32]. In this study, we used MEGA software to determine the genetic diversity of A. chinensis based on UPGMA. Weak genetic differentiation was observed in Pennisetum glaucum among the geographical regions, suggesting high seed and pollen-mediated gene flow among the regions [1]. A. chinensis is an often cross-pollinated plant, and 90% of the plants had only female flowers in our investigation (data not shown). Mixed samples for genetic diversity analysis may have resulted in the elimination of genetic variation; moreover, the complex genetic structure in wild resources of A. chinensis decreased the regional differentiation among populations. Additionally, the use of different SSR markers leads to the different dendrograms. Thus, the dendrogram constructed in the present study may not reflect the geographic origins of 19 A. chinensis populations.
Nei's unbiased measures of genetic similarity are shown in Table 5. The similarity coefficient ranged from 0.46 to 0.90 among 19 A. chinensis populations based on ten SSR primer amplification results. P8 and P10 showed the highest similarity (0.90), and the lowest similarity (0.46) was estimated between P2 and P11.
SSR marker analysis is an effective method for genetic diversity analysis and molecular marker-assisted selection breeding [6, 28]. In the present study, we used ten well-chosen SSR markers in clusters annotated as sesquiterpenoid biosynthesis to analyze 19 A. chinensis populations in northern China. The results showed that these markers were highly polymorphic. The SSR marker analyses revealed the presence of genetic diversity among 19 A. chinensis populations which could be helpful for selective breeding in the future.
Establishment of DNA fingerprinting
According to the amplification results, the set of SSR markers used here provided a discernible assessment of the ability of SSR primers to produce unique DNA profiles of A. chinensis populations. The ten SSR markers were able to differentiate 19 A. chinensis populations. DNA fingerprints of the 19 A. chinensis populations were constructed according to the original data matrix of amplification results (Supplementary Table S2).
DNA fingerprinting is a popular technique for identifying species. The genus Atractylodes comprises species of perennial herbs used as important crude drugs prescribed in Chinese, Japanese, Korean and Thai traditional medicine, including Atractylodes lancea, A. chinensis, Atractylodes japonica and Atractylodes macrocephala [35]. A. lancea and A. chinensis are known as Cangzhu in Chinese and Sojutsu in Japanese. A. japonica is recorded in the Japanese and Korean Pharmacopoeias but not in the Chinese pharmacopeia. The plants of the genus Atractylodes showed similar morphological features of stems, leaves and rhizomes, leading to disagreement regarding whether they are unique species and to their frequent misuses in medical products [29]. DNA fingerprinting is immensely helpful in detecting populations with high similarity. The results of the present study revealed that SSR marker-based fingerprinting databases are useful to detect genetic polymorphisms representing a method for analyzing unique populations. Marker-based fingerprinting provides a desirable reference for species and germplasm identification in the genus Atractylodes.
Unique alleles
SSR markers, in contrast to morphological markers, have strong species specificity [26]. Seventeen populations produced unique bands with certain SSR markers (Table 6). For P11, five SSR markers generated unique bands, and three markers generated unique bands for P14. Four SSR markers received unique bands for P6, and three markers received unique bands for P7.
Among the ten SSR primers used in the present study, seven were detected to generate unique fragments in certain populations (Table 7). Numerous specific SSR loci enabled us to select markers that yield highly specific amplifications independently (Supplementary Fig. S1, Table 7). S54 generated unique fragments of 320 and 350 bp specific to P6. Similarly, S99 produced bands sizes of 200, 750 and 1000 bp in P1. The specific bands were generated from S53 (320 and 350 bp) in P6, S52 (750 bp) and S63 (380 and 500 bp) in P11. S74 generated unique fragments 420 and 850 bp in P2, 480 bp in P3 and 650 bp in P11. The unique fragment generated through natural selection [18] was utilized for the evaluation of germplasm resources and molecular marker-assisted selection breeding.
Conclusion
The selection of set of core SSR primers is a crucial step for genetic diversity, DNA fingerprinting and germplasm identification. The ten SSR markers used in this study enable conclusions regarding the overall polymorphism and number of alleles observed in the 19 studied A. chinensis populations but do not relate explicitly to functional diversity and specific traits. The genetic diversity combination of agronomic traits (such as yield and quality traits) and SSR markers can be a key source of information to exploit superior A. chinensis germplasm resources for selective breeding.
Abbreviations
- SSR:
-
Simple sequence repeats
- HPLC:
-
High-performance liquid chromatography
- UPGMA:
-
Unweighted pair group method with arithmetic average
- h :
-
Gene diversity
- I :
-
Shannon's information index
- PIC :
-
Polymorphism information content
References
Bashir EMA, Ali AM, Ali AM, Mohamed ETI, Melchinger AE, Parzies HK, Haussmann BIG. Genetic diversity of Sudanese pearl millet (Pennisetum glaucum (L.) R. Br.) landraces as revealed by SSR markers, and relationship between genetic and agro-morphological diversity. Genet Resour Crop Evol. 2015;62(4):579–91. https://doi.org/10.1007/s10722-014-0183-5.
Bhattacharjee M, Prakash SH, Roy S, Soumen S, Begum T, Dasgupta T. SSR-based DNA fingerprinting of 18 elite Indian varieties of sesame (Sesamum indicum L.). Nucleus. 2019;63(4):67–73. https://doi.org/10.1007/s13237-019-00290-3.
Botstein D, White LR, Sholnick M, Davis RW. Construction of a genetic linkage map in man using restriction fragment length polymorphism. Am J Hum Genet. 1980;32(3):314. https://doi.org/10.1016/0165-1161(81)90274-0.
Chen YN, Dai XG, Hou J, Guan HW, Wang YX, Li Y, Yin TM. DNA fingerprinting of oil camellia cultivars with SSR markers. Tree Genet Genomes. 2016;12:7. https://doi.org/10.1007/s11295-015-0966-7.
Committee SP. Pharmacopoeia of the People’s Republic of China. Beijing: People’s Medical Publishing House; 2020. p. 168–9.
Ercisli S, Ipek A, Barut E. SSR marker-based DNA fingerprinting and cultivar identification of olives (Olea europaea). Biochem Genet. 2011;49(9–10):555. https://doi.org/10.1007/s10528-011-9430-z.
Fang W, Li X, Cheng H, Duan Y, Jiang X. Genetic diversity and relationship of clonal tea (Camellia sinensis) cultivars in China as revealed by SSR markers. Plant Syst Evol. 2012;298:469–83. https://doi.org/10.1007/s00606-011-0559-3.
Ge YF, Hang YY, Xia B, Wei YL. Sequencing of trnL-F and analysis of interspecifc genetic relationship of five medicinal species in Atractylodes DC. J Plant Resour Environ. 2007;16(2):12–6 (in Chinses).
Guo YH, Kondo K, Terabayashi S, Yutaka Y, Shimada H, Fujita M, Kawasaki T, Maruyama T, Goda Y, Mizukami H. DNA authentication of So-jutsu (Atractylodes lancea rhizome) and Byaku-jutsu (Atractylodes rhizome) obtained in the market based on the nucleotide sequence of the 18S–5.8S rDNA internal transcribed spacer region. J Nat Med. 2006;60(2):149–56. https://doi.org/10.1007/s11418-006-0032-8.
Hossen MJ, Chou JY, Li SM, Fu XQ, Yin C, Guo H, Amin A, Chou GX, Yu ZL. An ethanol extract of the rhizome of Atractylodes chinensis exerts anti-gastritis activities and inhibits Akt/NF-kappa B signaling. J Ethnopharmacol. 2019;228:18–25. https://doi.org/10.1016/j.jep.2018.09.015.
Ishii T, Okuyama T, Noguchi N, Nishizawa M. Antiinfammatory constituents of Atractylodes chinensis rhizome improve glomerular lesions in immunoglobulin a nephropathy model mice. J Nat Med. 2020;74:51–64. https://doi.org/10.1007/s11418-019-01342-3.
Kim JH, Doh EJ, Lee G. Evaluation of medicinal categorization of Atractylodes japonica Koidz. by using internal transcribed spacer sequencing analysis and HPLC fingerprinting combined with statistical tools. Evid-Based Compl Altern Med. 2016. https://doi.org/10.1155/2016/2926819.
Kim JH, Doh EJ, Lee G. Chemical differentiation of genetically identified Atractylodes japonica, A. macrocephala, and A. chinensis rhizomes using high-performance liquid chromatography with chemometric analysis. Evid-Based Compl Altern Med. 2018;2018:4860371. https://doi.org/10.1155/2018/4860371.
Kim S, Jo N, Gil J, Koo SC, Um Y, Hong CP, Park SG, Kim OT, Kim SC, Kim HB, Lee DH, Jeong BH, Lee Y. Development of genome-wide simple sequence repeat markers in Codonopsis lanceolata using next-generation sequencing. Hortic Environ Biotechnol. 2021;62:985–93. https://doi.org/10.1007/s13580-021-00389-0.
Kumar N, Shikha D, Kumari S, Choudhary BK, Kumar L, Singh IS. SSR-Based DNA fingerprinting and diversity assessment among Indian germplasm of Euryale ferox: an aquatic underutilized and neglected food crop. Appl Biochem Biotechnol. 2018;185:34–41. https://doi.org/10.1007/s12010-017-2643-9.
Liu Y, Geng Y, Song M, Zhang PF, Hou JL, Wang WQ. Genetic structure and diversity of Glycyrrhiza populations based on transcriptome SSR markers. Plant Mol Biol Rep. 2019;37:401–12. https://doi.org/10.1007/s11105-019-01165-2.
Lyu Z, Ji XF, Chen G, An BY. Atractylodin ameliorates lipopolysaccharide and D-galactosamine-induced acute liver failure via the suppression of infammation and oxidative stress. Inter Immunopharmacol. 2019;72:348–57. https://doi.org/10.1016/j.intimp.2019.04.005.
Mousadik A, Petit RJ. High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa(L.) Skeels] endemic to Morocco. Theor Appl Genet. 1996;92:832–9. https://doi.org/10.1007/BF00221895.
Nei M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics. 1978;89(3):583–90. https://doi.org/10.1093/genetics/89.3.583.
Pandey SK, Das A, Rai P, Dasgupta T. Morphological and genetic diversity assessment of sesame (Sesamum indicum L.) accessions differing in origin. Physiol Mol Biol Plants. 2015;21:519–29. https://doi.org/10.1007/s12298-015-0322-2.
Pei Z, Gao J, Chen Q, Wei J, Li Z, Luo F, Shi L, Ding B, Sun S. Genetic diversity of elite sweet sorghum genotypes assessed by SSR markers. Biol Plant. 2010;54(4):653–8. https://doi.org/10.1007/s10535-010-0116-x.
Peng HS, Yuan QJ, Li QQ, Huang LQ. Molecular systematics of genus Atractylodes (Compositae, Cardueae): evidence from internal transcribed spacer (ITS) and trnL-F sequences. Int J Mol Sci. 2012;13(11):14623–33. https://doi.org/10.3390/ijms131114623.
Rakshit A, Rakshit S, Santhy V, Gotmare VP, Mohan P, Singh VV, Singh S, Singh J, Balyan HS, Gupta PK, Bhat SR. Evaluation of SSR Markers for the assessment of genetic diversity and fingerprinting of Gossypium hirsutum accessions. J Plant Biochem Biotechnol. 2010;19:153–60. https://doi.org/10.1007/BF03263335.
Romero-Severson L, Smith JSC, Ziegle J, Hauser J, Joe L, Hookstra G. Pedigree analysis and haplotype sharing within diverse groups of Zea mays L. inbreds. Theor Appl Genet. 2001;103:567–74. https://doi.org/10.1007/PL00002911.
Sun CZ, Li Y, Zhang SN, Zheng JS. Production of intergeneric allotetraploid between autotetraploid nonheading Chinese cabbage (Brassica campestris ssp. chinensis Makino) and autotetraploid radish (Raphanus sativus L.). Acta Soc Bot. 2013;83(1):75–9. https://doi.org/10.5586/asbp.2013.037.
Sun JW, Liu T, Guo BT, Jin DM, Wang B. Development of SSR primers from EST sequences and their application in germplasm identification of Porphyra lines (Rhodophyta). Eur J Phycol. 2006;41(3):329–36. https://doi.org/10.1080/09670260600740906.
Tamura Y, Kubo N, Ohsako T. Genetic diversity among Japanese local populations of an edible and medicinal coastal plant Glehnia littoralis F. Schmidt ex Miq. Genet Resour Crop Evol. 2022;69:85–97. https://doi.org/10.1007/s10722-021-01205-5.
Varshney RK, Marcel TC, Ramsay L, Russell J, Röder MS, Stein N, Waugh R, Langridge P, Niks RE, Graner A. A high density barley microsatellite consensus map with 775 SSR loci. Theor Appl Genet. 2007;114:1091–103. https://doi.org/10.1007/s00122-007-0503-7.
Wang LQ, Zhang H, Wu X, Wang ZY, Fang WW, Jiang M, Chen HM, Huang LF, Liu C. Phylogenetic relationships of Atractylodes lancea, A. chinensis and A. macrocephala, revealed by complete plastome and nuclear gene sequences. PLoS ONE. 2020;15(1):e0227610. https://doi.org/10.1371/journal.pone.0227610.
Wang YH, Wang S, Liu YL, Yuan QJ, Sun JH, Guo LP. Chloroplast genome variation and phylogenetic relationships of Atractylodes species. BMC Genomics. 2021;22:103. https://doi.org/10.1186/s12864-021-07394-8.
Wu FF, Zhang SX, Gao Q, Liu F, Wang JL, Wang XG. Genetic diversity and population structure analysis in a large collection of Vicia amoena in China with newly developed SSR markers. BMC plant biol. 2021;21:544. https://doi.org/10.1186/s12870-021-03330-w.
Wu FF, Ma SN, Zhou J, Han CY, Hu RC, Yang XY, Nie G, Zhang XQ. Genetic diversity and population structure analysis in a large collection of white clover (Trifolium repens L.) germplasm worldwide. Peer J. 2021;9:e11325. https://doi.org/10.7717/peerj.11325.
Xie HB, Shi MM, Shi LC, Liu JX, Zhao CY. The complete chloroplast genome of Atractylodes koreana (Nakai) Kitam and its phylogenetic analysis. Mitochondrial DNA part B. 2021;6(7):2041–3. https://doi.org/10.1080/23802359.2021.1928561.
Zhang L, Cai R, Yuan M, Tao A, Xu J, Lin L, Qi J. Genetic diversity and DNA fingerprinting in jute (Corchorus spp.) based on SSR markers. Crop J. 2015;3:416–22. https://doi.org/10.1016/j.cj.2015.05.005.
Zhang WJ, Zhao ZY, Chang LK, Gao Y, Wang S, Kang CZ, Wang HY, Zhou L, Huang LQ, Guo LP. Atractylodis rhizoma: a review of its traditional uses, phytochemistry, pharmacology, toxicology and quality control. J Ethnopharmacol. 2021;266(3): 113415. https://doi.org/10.1016/j.jep.2020.113415.
Zhao JH, Sun CZ, Shi FY, Ma SS, Zheng JS, Du X, Zhang LP. Comparative transcriptome analysis reveals sesquiterpenoid biosynthesis among 1-, 2- and 3-year old Atractylodes chinensis. BMC plant boil. 2021;21:354. https://doi.org/10.1186/s12870-021-03131-1.
Zhou HY, Zhang PH, Luo J, Liu XY, Fan SX, Liu CJ, Han YY. The establishment of a DNA fingerprinting database for 73 varieties of Lactuca sativa capitate L. using SSR molecular markers. Hortic Environ Biotechnol. 2019;60:95–103. https://doi.org/10.1007/s13580-018-0102-3.
Acknowledgements
This work was supported financially by Bureau of Science and Technology of Hebei Province, China (Grant Number H2019407120), Scientific research project of colleges and universities in Hebei Province, China (Grant Number ZD2022014), Key Research and Development Project of Hebei Province, China (Grant Number 19226354D).
Author information
Authors and Affiliations
Contributions
JZ: designed the experiment and revised the manuscript; JZ and WS: wrote the manuscript; JZ and SM: conducted experiments and analysed the data. SZ; WZ and SS: organized and collated the references.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Corresponding Editor: Maumita Bandyopadhyay; Reviewers: Sreetama Bhadra, Palaniyandi Karuppaiya, Abhishek Sadhu.
Supplementary Information
Below is the link to the electronic supplementary material.
13237_2022_398_MOESM1_ESM.xlsx
Supplementary Table S1 Summary of 106 SSR primers in clusters classified into sesquiterpenoid biosynthesis and terpene skeleton biosynthesis pathways in the A. chinensis transcriptomic database (XLSX 19 KB)
13237_2022_398_MOESM3_ESM.rar
Supplementary Fig. S1 Gel images of ten SSR markers in 19 A. chinensis populations. Large letters indicate the analyzed bands in gel images. S1A: S2, S1B: S4, S1C: S52, S1D:S53, S1E:S54, S1F:S63, S1G: S74, S1H: S77, S1I: S87, S1J: S99 (RAR 42814 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, S., Zhao, J., Su, W. et al. Transcriptome-derived SSR markers for DNA fingerprinting and inter-populations genetic diversity assessment of Atractylodes chinensis. Nucleus 65, 321–329 (2022). https://doi.org/10.1007/s13237-022-00398-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13237-022-00398-z