
Abstract
In the present investigation, a gene encoding extracellular lipase was cloned from a Bacillus licheniformis. The recombinant protein containing His-tag was expressed as inclusion bodies in Esherichia coli BL21DE3 cells, using pET-23a as expression vector. Expressed protein purified from the inclusion bodies demonstrated ~22 kDa protein band on 12 % SDS-PAGE. It exhibited specific activity of 0.49 U mg−1 and % yield of 8.58. Interestingly, the lipase displayed activity at wide range of pH and temperature, i.e., 9.0–14.0 pH and 30–80 °C, respectively. It further demonstrated ~100 % enzyme activity in presence of various organic solvents. Enzyme activity was strongly inhibited in the presence of β-ME. Additionally, the serine and histidine modifiers also inhibited the enzyme activities strongly at all concentrations that suggest their role in the catalytic center. Enzyme could retain its activity in presence of various detergents (Triton X-100, Tween 20, Tween 40, SDS). Sequence and structural analysis employing in silico tools revealed that the lipase contained two highly conserved sequences consisting of ITITGCGNDL and NLYNP, arranged as parallel β-sheet in the core of the 3D structure. The function of these conserve sequences have not fully understood.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lipases are triacylglycerol hydrolases (E.C. 3.1.1.3) that catalyzes variety of conversion reactions, ranging from interesterification, esterification, alcoholysis, acidolysis and aminolysis (Gupta et al. 2004; Hayes 2004; Kim et al. 2002; Nagao et al. 2001; Saxena et al. 2003). They are frequently used in various industries including detergents, leather, dairy, baking and pharmaceutical (Jaeger and Eggert 2002). Thermostable lipases have received much more attention, as they remain stable at high temperature. Reactions carried out at higher temperature lead to higher diffusion rate, increases solubility of lipids and other hydrophobic substrates in water, can reduce the risk of contamination (Annamalai et al. 2011). They display broader range of substrate specificity, tolerance towards extreme of acidic and alkaline conditions, and solvents (Gupta et al. 2004; Haki and Rakshit 2003). The most remarkable sources of thermostable lipases are bacteria (Jaeger and Eggert 2002). Importantly, the Bacillus sp. is known to produce commercially important lipases (Chakraborty and Raj 2008). Recently, Shariff et al. (2011) characterized a lipase from Bacillus sp. L2 that showed lipase activity at wide range of pH and temperature. In another study, a lipase was characterized from Bacillus licheniformis strain that was isolated from the mangrove sediments. Interestingly, this enzyme showed stability up to 70 °C and pH 12.0 (Annamalai et al. 2011). More recently, Gohel et al. (2013) found better stability and activity of thermophilic lipases in its immobilized form. In general, a number of lipases have been characterized from various bacterial species and cannot be discussed here.
In the present investigation, we report cloning, expression, purification and characterization of lipase from bacteria, and interestingly we observed that the present lipase exhibits facet of industrial relevance.
Materials and methods
Screening of lipase-producing bacterial strain
Soil sample was collected from hot springs of Manikaran (average water temperature: 60–65 °C), Himachal Pradesh, India (32°1′40.34″N and 77°2′52.95″E) in sterile oakridge centrifuge tube. The soil sample was dried in an oven maintained at 50 °C for screening thermostable lipase-producing strain. One gram soil was dissolved in water and diluted appropriately to 10−9 dilutions, followed by spreading on LB (Luria broth) agar plates, containing 1 % emulsified tributyrin. The plates were incubated at 40 °C for 2 days. The colony displaying hydrolytic zone on substrate plate was streaked further for selecting single colony.
DNA extraction and 16S rRNA amplification
The colony displaying zone of hydrolysis in LB agar tributyrin plates was cultured overnight in 5 ml LB at 37 °C in shaker (220 rpm). Genomic DNA was extracted from overnight grown culture employing standard phenol–chloroform method. Purified DNA was used for PCR amplification of the 16S rRNA gene, using universal primers: 27F (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1642R (5′-CGGYTACCTTGTTACGAC-3′), respectively. The following thermal cycling conditions were used to amplify the 16S rRNA gene, 94 °C for 4 min, followed by 30 cycles of 94 °C for 50 s, 55 °C for 50 s, 72 °C for 2 min, with final extension of 7 min at 72 °C. The 25 µl PCR mix contained 1 µl (0.1 µg) DNA, 1 μl each for ward and reverse primers (2.5 µM), 200–400 µM dNTPs mix (Thermo Scientific), 1U Taq DNA polymerase (Thermo Scientific) and 1× reaction buffer (Thermo Scientific). PCR-amplified product was checked on a 1.5 % agarose gel, and purified from gel employing XcelGen DNA Gel/PCR Purification kit (Xcelris genomics). The amplified product was cloned in pGEM-T easy vector (Promega, USA) as per manufacturer’s instruction. The recombinant plasmid DNA was extracted from white colonies employing Hi Yield™ Plasmid DNA Mini Kit (Real Genomics) and sequenced using M13 forward and reverses primer.
PCR amplification and cloning of the lipase gene
The analysis of the 16 SrRNA gene sequence revealed that the strain producing lipase was Bacillus licheniformis. After careful analysis of the Bacillus licheniformis genome at NCBI, specific forward and reverse primers were designed to amplify the corresponding lipase gene, F:5′ATTCAGCATATGGAAGAGGATTTGAACGAA3′R:5′ATTCAGCTCGAGGCCCTCCAGCCGCCCGTA3′ containing restriction sites Nde1 and Xho1 in forward and reverse primers, respectively. The lipase gene (lip P1) was amplified employing following reaction conditions, initial denaturation at 94 °C for 4 min, followed by subsequent 35 cycles of 94 °C for 50 s, 50 °C for 1 min, 72 °C for 1 min and final extension at 72 °C for 7 min. The amplified gene product was checked on 1.5 % agarose gel and extracted by gel extraction kit. The eluted DNA fragment was ligated into pGEM-T-easy vector, as per manufacturer instructions and transformed into E. coli competent cells. The white colonies (recombinant clones) carrying a recombinant plasmid were cultured overnight in 5 ml LB having ampicillin (100 μg ml−1) followed by plasmid DNA extraction and sequencing of lipase gene using M13 forward and reverse primers.
Protein expression and purification
The lip P1 gene without the terminal signal was further sub-cloned in frame into the pET-23a expression vector, after digesting the amplified DNA fragment and pET 23a plasmid with Nde1 and Xho1. The two DNA fragments were ligated and transformed into the E. coli BL-21 DE3 cells. The recombinant clones were selected on LB agar plates having ampicillin (100 µg/ml) and chloramphenicol (25 µg/ml). Transformed colonies were further selected randomly for the presence of insert by colony PCR methods. The positive colony was cultured overnight initially into 5 ml LB medium, having appropriate antibiotics. Next day 1 % overnight grown culture was inoculated into 500 ml LB containing appropriate antibiotics. The culture was induced by addition of 0.005 M IPTG when OD600nm reached ~0.4. The cells were pelleted down, lysed and centrifuged. The SDS-PAGE was performed (sodium dodecyl sulfate–polyacrylamide gel electrophoresis) with both the pellet and the soluble fractions, to know the localization of the recombinant protein. The expressed protein was recovered from inclusion bodies by dissolving it in 8 M urea, and the denatured protein was refolded by dialyzing it in gradient buffer consisting of 100, 50 and 10 mM sodium phosphate. The clear supernatant obtained after dissolving the inclusion bodies was passed through the equilibrated Ni-NTA column, and the eluted protein was collected in fractions of 1.5 ml in 2-ml centrifuge tubes, and checked on 12 % SDS-PAGE. The fractions showing similar pattern of protein bands in the gel were pooled and dialyzed. The protein concentration was estimated by Bicinchoninic acid method (BCA) kit (Banglore Genei, India) and Bovine serum albumin (BSA) was used as standard.
Note
All steps for the protein purifications were carried out at 4 °C.
Enzyme assay
The enzyme was assayed by modified method of Sigurgisladottir et al. (1993). The final reaction mixture (1 ml) contained 0.8 ml of 0.05 M sodium phosphate buffer (pH 8.0), 0.1 ml enzyme of appropriate dilution, and 0.1 ml of 0.001 M p-nitrophenyl laurate. The reaction was carried out at 60 °C for 1 h, the enzyme reaction was de-activated by addition of 0.1 M Na2CO3 (0.25 ml), followed by centrifugation. The enzymatic activity was recorded at 420 nm in UV/Vis spectrophotometer (JENWAY 6505 UK). One unit of enzyme activity was defined as the amount of enzyme which liberates 1 µmol of p-nitrophenol from pNP-laurate as substrate per min under standard assay conditions. The total enzyme activity was expressed in Unit/ml and specific activity was expressed as Unit/mg of protein.
Biochemical characterization
Effect of temperature and pH on lipase activity and stability
To determine the optimum temperature, purified lipase was assayed at various temperatures (30–80 °C). The enzyme stability was studied by incubating the purified enzyme at 60 °C. The enzyme without incubation was taken as control (100 %). The lipase activity was measured at intervals of 20, 40, 60 and 120 min. Reaction mix without enzyme served as a blank. Next, the pH optimum was determined by assaying the purified lipase in presence of a range of pH (6.0–14.0) at 60 °C.
Effect of additives and chemical modifiers on lipase activity
The effect of various metal ions (1 and 10 mM each), detergents (1, 5, and 10 % W/V each), enzyme inhibitors (1, 5, 10, and 20 mM each) and organic solvents (1, 5, and 10 % V/V each) were studied by adding these modifiers and additives separately into the reaction mix. The enzyme assay was performed according to standard assay conditions, and carried out in both the presence and absence of chemical modifiers. The reaction mix with respective additives/chemical modifiers but without enzyme served as blank. The sample without any additive/modifier was taken as control (100 %).
Nucleotide sequencing and gene submission
Both 16S rRNA and lipase gene were sequenced using universal M13 forward and reverse primers. The nucleotide sequencing was carried out by Bangalore Genei, India using an automated AB1 3100 genetic analyzer that uses fluorescent label dye terminator, based on dideoxy chain termination method. The 16S rRNA gene was submitted to NCBI under the accession number KM438034, and the lipase nucleotide sequence submitted was assigned gene accession number KF815062.
In silico characterization
To gain structural insights, homology studies were carried out. Using ConSeq server (http://consurf.tau.ac.il/), the homologues were collected from the UNIREF90, and best hits were considered for further studies. Multiple Sequence Alignment (MSA) was performed using MAFFT, using default values of ConSeq server. Conservation scores were calculated by the Bayesian method. Further the secondary and tertiary structure analysis was carried out in PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/) and Swiss Model (http://swissmodel.expasy.org) server respectively. The target lipase sequence displayed poor sequence similarity with most of the earlier reported lipases for which X-ray structures were available, and could be modeled using template PDB ID: 3BZW of a putative lipase reported from Bacteroides thetaiotaomicron that showed ~20 % homology with the lipase under investigation.
Results
Screening and identification of the lipase-producing strain
Screening of hot spring soil for the lipase-producing strain resulted in isolation of bacterial strain that showed a clear zone of hydrolysis around the colonies (Fig. 1). Genomic DNA extracted showed an intact band (>23 kb) on 0.8 % agarose gel (data not shown). Further, the 16S rRNA gene amplified from the genomic DNA showed amplified band corresponding to ~1.5 kb. BLAST N analysis of the 16S rNRA nucleotide revealed >95 % homology to the 16S rRNA sequences of the Bacillus licheniformis.

Isolated strain demonstrating the extracellular lipase activity in 1 % emulsified tributyrin plate
Cloning, sequencing, analysis and expression studies
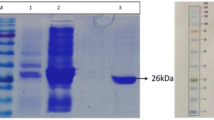
Lipase gene Lip P1 corresponding to a size of ~0.6 kb was amplified cloned and sequenced. The sequencing of the lipase gene confirmed an ORF (open reading frame) consisting of 0.636 kb that encodes an extracellular lipase of 211 amino acids long. BlastN at NCBI demonstrated that it had shared ~99 % identity with B. licheniformis ATCC 14580 (NCBI accession number: CP000002.3), B. licheniformis DSM 13 (AE017333.1), B. licheniformis Pb-WC11006 (JX164087.1), B. licheniformis Pb-WC09005 (HM006905.1), B. licheniformis N22 (GU086425.1). Further analysis of the gene using Signal P 4.1 server (Petersen et al. 2011) revealed a terminal signal sequence of 30 amino acids, and hence the mature lipase consists of 181 amino acids. The lipase gene Lip P1 without terminal signal expressed in E. coli BL21 (DE3) showed an induced protein band at ~22 kDa, as judged by Coomassie brilliant blue staining of SDS-PAGE (Fig. 2). The purification profile of the protein obtained through the Ni–NTA affinity chromatography is presented in Table 1. From purification table it becomes evident that protein had a specific activity of 0.49 U mg−1, and had yield of 8.5 %.

SDS-PAGE gel stained with Coomassie brilliant blue showing B. licheniformis lipase. Lane 1 Purified lipase, lanes 2–7 expression of lipase gene induced with IPTG, lane M Protein molecular weight marker
Biochemical characterization of the purified lipase
Effect of temperature and pH on lipase activity and stability
The purified lipase from Bacillus licheniformis demonstrated optimum temperature activity at 60 °C (Fig. 3), and retained ~99 and 98 % of its original activity when assayed at intervals of 20,40,60,80 and 120 min at 60 °C (Fig. 4). Next, lipase retained activity at wide pH range (6.0–14.0) and was observed to be stable for 1 h under alkaline pH conditions. However, the enzyme activity was considerably at pH 6.0, ~50 % activity was retained after 1 h (Fig. 5).

Relative activity of purified lipase from B. licheniformis at different temperature ranges

Thermostability profile of purified lipase from B. licheniformis at 60 °C

Relative activity of purified lipase from B. licheniformis at different pH ranges
Effect of organic solvents and metal ion on enzyme activity
One of the unique properties of the enzymes is their activity and stability in presence of the organic solvents.Lipase under investigation has shown ~100 % activity in presence of isopropanol and methanol. In presence of acetone and toluene ~60–90 % enzyme activity was retained. Next, effect of metal ions on lipase activity demonstrated that in 1 mM concentration each of Ca2+, Mg2+, Ni2+, and Cu2+ lipase displayed 81, 71, 56, and 41 % activity, respectively; whereas, at 10 mM concentration, Mg2+ had shown 90 %, Ca2+ displayed 85 %, while both Ni2+ and Cu2+ showed 70 and 63 % enzyme activity.
Effect of inhibitors and denaturants on lipase activity
Enzyme activity in presence of inhibitors, β-mercaptoethanol (β-ME), phenylmethylsulfonyl fluoride (PMSF) and diethylpyrocarbonate (DEPC) was tested. At 1 mM concentration both DEPC and PMSF resulted in 98.9 and 86.4 % inhibition, respectively. In contrary, β-ME, an enzyme denaturant has shown 91.8 % inhibition. At 5 mM and 10 mM concentration, the inhibition shown by DEPC, PMSF, β-ME was 94.9, 91, 88 and 93 %, 93 %, 90 %, respectively. PMSF almost completely inhibited enzyme activity at 20 mM concentration, while β-ME and DEPC showed 55.1 and 88.1 % inhibition. Enzyme activity tested in presence of detergent and denaturants revealed that it was able to maintain enzyme activity at 1 and 10 % of Triton X-100 and Tween-20, respectively. Lipase activity was increased from 65.3 to 71.5 % and 63.8 to 67.7 % when treated with Tween-40 and SDS, respectively.
In silico analysis
Sequence and structural analysis of the lipase gene revealed two highly conserved sequences of ITITGCGNDL and NLYNP, the structurally and functionally important residues were also depicted (Fig. 6a). The secondary structure predicted showed coils helixes and sheets in the structure (Fig. 6b). In addition to this, the 3D model predicted demonstrated a partial α–β hydrolase fold, which has been observed in most of the lipases, interestingly highly conserved sequences observed during MSA were observed to be arranged in a parallel β-sheet at the core of protein. Over all superimposition of the target lipase onto the template showed a scaffold having a core structure and disorder structure in the loop region (Fig. 7a, b).

a Functionally and structurally important residues identified using ConSeq server, b predicted secondary structure using the PSIPRED server

3D comparative model structure: a the overall 3D structure and highly conversed region, b superimposed structure of model and template structure
Discussion
Recombinant DNA technology has brought great revolution in the field of enzyme industries (Schmidt-Dannert 1999). With the advent of this technology, a number of industrially important enzymes have been cloned and characterized from thermophilic organisms. Interestingly, the thermostable enzymes are preferred more in industrial processes, as most of the industrial processes are carried out at temperatures above 50 °C (Sharma et al. 2002). Higher temperatures offer superior conversion rate, annihilate microbial contamination, increase substrate solubility, help in reducing the viscosity of the reaction medium, favoring mass transfer (Guncheva and Zhiryakova 2011). The present work report cloning, expression, purification and characterization of thermostable lipase from B. licheniformis isolated from hot spring soil of Manikaran area of Himachal Pradesh. The mature lipase has 211 amino acids out of which thirty amino acids were coding a terminal signal sequence, as determined by Signal P 4.1 server. This terminal signal sequence helps in secretion of mature lipase out of the cells (Carlsson et al. 2006). Based on the sequence analysis, the lipase gene showed high similarities (~99 %) with most of the Bacillus licheniformis lipase. Interestingly, the BlastX analysis revealed that the mature lipase has variation in its amino acid sequence, having replacement of a positively charged amino acid lysine (K) to glutamic acid (a negatively charge amino acid). High level expression of the recombinant protein was achieved by inducing T7 promoter by addition of 0.005M IPTG (pET system). The recombinant lipase purified to homogeneity employing Ni–NTA affinity chromatography demonstrated single band of ~22 kDa on SDS-PAGE. The molecular weight of B. licheniformis lipase reported from other studies was also observed in the range of 19–25 kDa (Mishra and Madan 2009; Nthangeni et al. 2001; Sharma and Kanwar 2012). Low molecular weight lipases exhibit smaller changes in their tertiary structure during the harsh processes carried out in industries, and hence preferred more (Gray 1993). The low yield of lipases in the present investigation may be attributed to its aggregation behavior, and was also well documented previously (Kambourova et al. 2003; Lesuisse et al. 1993).
Interestingly, the lipase was observed to be quite thermostable and displayed optimum activity and high stability at 60 °C, higher than previously reported lipases from thermostable B. licheniformis, where the optimum enzyme activity was observed in the range of 40–55 °C (Annamalai et al. 2011; Chakraborty and Raj 2008; Mishra and Madan 2009, Sangeetha et al. 2010, Sharma and Kanwar 2012). Interestingly, the temperature optimum of Lip1 displayed comparable activity to the lipase reported by Nthangeni et al. (2001). In addition, the pH optima displayed by the lipase was comparable to the earlier reported studies on B. licheniformis (Annamalai et al. 2011; Chakraborty and Raj 2008; Mishra and Madan 2009; Sangeetha et al. 2010; Sharma and Kanwar 2012). The most interesting finding about the pH study is retention of enzyme activity till pH 14, indicating its potential application in detergent-making industry, such observation may be attributed to the ionization of certain amino acids in polypeptide that may favor activity and stability of enzyme at these pH values (Fullbrook 1996), whereas the stability of enzyme at higher temperature is correlated to number of hydrophobic interactions (Goldsack 1970). One unique property of the lipases for which it is explored most is its activity in presence of organic solvent, as enzymes in non-aqueous systems are reported to possess several advantages over the aqueous environment (Klibanov 2001; Sharma and Kanwar 2014). The lipase Lip P1 demonstrated ~100 % activity in presence of the isopropanol and methanol, whereas insignificant inhibition was observed in presence of acetone and toluene. On the contrary, lipase from one of the previous study reported less activity in isopropanol, methanol and acetone, respectively (Chakraborty and Raj 2008); however, the solvent stable features shown by current enzyme was in line with one of the study carried out previously (Rahman et al. 2005). The solvent stability of the lipase may be accredited to retention of the native conformation of the enzyme by forming a protective sheath of water molecules along the hydrophilic surface of enzyme (Nawani and Kaur 2000) that may shield it against the denaturation processes, while increasing the structural flexibility and conformational mobility for optimal catalysis (Klibanov 2001).
Additionally, the metal ion presence in the reaction mix revealed mix effect, Cu2+ and Ni2+ were relatively showed more inhibition compared to Ca2+ and Mg2+. The decrease in the enzyme activity suggests that metal ion might be competing at the active site of the enzyme, as also documented in previous studies (Ghori et al. 2011). Previous studies have also shown miscellaneous effect of metal ions on enzyme activity (Annamalai et al. 2011; Chakraborty and Raj 2008). Transition metals (Cu2+ and Ni2+) reported to hinder the enzyme activity via interacting with side chain groups of surface amino acids that may affect the conformational stability of enzyme (Rahman et al. 2006). Inhibition by Cu2+ evinces the presence of sulphydryl group in the protein (Enujiugha et al. 2004; Sanders and Pattee 1975). Numerous studies have also talked about the inhibition of lipase activity in presence of Ni2+ and Cu2+ (Kumar et al. 2005; Ma et al. 2006). In contrast to this, the higher activity of lipase in presence of Ca2+ may be ascribed to the formation of long chain insoluble fatty acid calcium salts during hydrolysis, plus formation of bridges at the active site that help in improving the enzyme stability (Kim et al. 2000; Wills 1960). Further analysis of the effect of the enzyme inhibitors on enzyme activity demonstrated that both PMSF and DEPC, resulted in inhibition of the enzyme activity that principally indicates presence of serine and histidine in the active center. The catalytic center is suggested to be consisting of a catalytic triad with Ser–His–Asp/Glu (Gupta et al. 2004; Shariff et al. 2011).
Inhibition by PMSF and DEPC illustrates easy accessibility of inhibitors to the active site serine and histidine that may attribute to lid structure absence in the present lipase (lid structure covers the catalytic site) (Lesuisse et al. 1993). The β-ME also inhibited the enzyme activity to the marked level, which further ruled out the possibility of disulfide linkage in the protein structure (Abramic et al. 1999). Similar inhibitory effects of PMSF, DEPC and β-ME on lipase activity were also reported from earlier studies (Castro-Ochoa et al. 2005; Chakraborty and Raj 2008; Lee et al. 1999; Mishra and Madan 2009; Nthangeni et al. 2001; Shariff et al. 2011). In sharp contrast to this, 100 % lipase activity of B. licheniformis VSG1 in presence of PMSF was reported by Sangeetha et al. (2010); in many studies, β-ME was also not reported to inhibit the lipase activity (Castro-Ochoa et al. 2005; Kanjanavas et al. 2010; Lee et al. 1999; Sharma et al. 2012).
More interestingly, effect of denaturants/detergents on lipase activity were quite dissimilar from other studies, as Triton X-100 and Tween 20 were observed to be inhibitory, and Tween 40 and SDS showed enhanced enzyme activity, as concentration of the chemical modifiers was raised from 1 to 10 %. Previous studies have reported 100 % lipase activity in presence of Triton X-100 (Annamalai et al. 2011; Balan et al. 2013; Castro-Ochoa et al. 2005; Sangeetha et al. 2010; Sharma et al. 2002), Mishra and Madan (2009) reported 90 % lipase activity, whereas Kanjanavas et al. (2010) reported 41.6 % lipase activity. More interestingly, the activity in presence of SDS was higher in contrast to previous studies carried out with Bacillus sp. (Annamalai et al. 2011; Castro-Ochoa et al. 2005; Lee et al. 1999; Mishra and Madan 2009). These observations make this enzyme fit for the detergent-making industries. The effect of Tween 20 on lipase activity was observed to be analogous as reported in other studies (Kanjanavas et al. 2010; Mishra and Madan 2009; Sharma et al. 2002). This may be attributed to the ability of the detergents to hamper enzyme aggregation by weakening hydrophobic interaction and improving the substrate accessibility to the enzyme (Polizelli et al. 2008).
The predicted secondary structure of the lipase contains helix, coil and sheet. Interestingly, important residues predicted through ConSeq server showed number of amino acids that were exposed and buried, and further divulge that the residues present towards the N terminus and C terminus of the protein were highly exposed, and might be interacting with the aqueous environment to form network of hydrogen bonds that help in stabilizing the 3D structure; in silico analysis of the lipase revealed no significant similarity with the previously reported X-ray crystal structure. However, crystal structure of a putative lipase from Bacteroides thetaiotaomicron (PDB ID: 3BZW) showed ~20 % similarity with the lipase under investigation and directed us to predict the 3D structure. Two highly conserved sequences were observed in the protein that contains amino acid sequences consisting of ITITGCGNDL and NLYNP, respectively, which were arranged as parallel β-sheet in the core structure, further analysis of the conserved sequences revealed a defined secondary structure, with loop region observed to be disordered. Such observation may be attributed to the presence of amino acids which confer structural flexibility. Another reason could be the incomplete prediction of 3D structure on a template that had shared only 20 % structural similarity, nonetheless, the predicted structure can be explored in future to under- stand the structure and function of this lipase.
Conclusion
In this investigation, a lipase produced by B. licheniformis was cloned, overexpressed and purified. Of particular note, the lipase was observed to be active at wide range of temperature and pH and displayed activity in presence of organic solvent, metal ions and detergents. By virtue of all these properties, this thermo-alkali-tolerant lipase can be explored for its industrial applications. Next, we aimed at improving the structure and functional stability of the lipase by site-directed mutagenesis.
References
Abramic M, Lescic I, Korica T, Vitale L, Saenger W, Pigac J (1999) Purification and properties of extracellular lipase from Streptomyces rimosus. Enz Microb Technol 25:522–529
Annamalai N, Elayaraja S, Vijayalakshmi S, Balasubramanian T (2011) Thermostable alkaline tolerant lipase from Bacillus licheniformis using peanut oil cake as a substrate. Afr J Biochem Res 5:176–181
Balan A, Ibrahim D, Rahim RA (2013) Organic-solvent and surfactant tolerant thermostable lipase, isolated from a thermophilic bacterium, Geobacillus thermodenitrificans IBRL-nra. Adv Stud Biol 5:389–401
Carlsson F, Stalhammar-Carlemalm M, Flardh K, Sandin C, Carlemalm E, Lindahl G (2006) Signal sequence directs localized secretion of bacterial surface proteins. Nature 442:943–946
Castro-Ochoa LD, Rodriguez-Gomez C, Valerio-Alfaro G, Ros RO (2005) Screening, purification and characterization of the thermoalkalophilic lipase produced by Bacillus thermoleovorans CCR11. Enzyme Microb Tech 37:648–654
Chakraborty K, Raj P (2008) An extra-cellular alkaline metallolipase from Bacillus licheniformis MTCC 6824: purification and biochemical characterization. Food Chem 109:727–736
Enujiugha VN, Thani FA, Sanni TM, Abigor RD (2004) Lipase activity in dormant seeds of the african oil bean (Pentaclethra macrophylla Benth). Food Chem 88:405–410
Fullbrook PD (1996) Practical applied kinetics. In: Godfrey T, West S (eds) Industrial enzymology, 2nd edn. Stockholm Press, New York, pp 483–540
Ghori MI, Iqbal MJ, Hameed A (2011) Characterization of a novel lipase from Bacillus sp. isolated from tannery wastes. Braz J Microbiol 42:22–29
Gohel HR, Ghosh SK, Bragazna VJ (2013) Production, purification and immobilization of extracellular lipases from thermophilic Bacillus subtilis XRF11 and Bacillus licheniformis XRF12 for production of alkyl esters. Int J Lifesci Biotechnol Pharma Res 2:2239–2250
Goldsack DE (1970) Relation of hydrophobicity index to the thermal stability of homologous proteins. Biopolymers 9:247–252
Gray CJ (1993) Stabilization of enzymes with soluble additives. Thermostability Enzymes 8:124–143
Guncheva M, Zhiryakova D (2011) Catalytic properties and potential applications of Bacillus lipases. J Mol Catal B Enzym 68:1–25
Gupta R, Gupta N, Rathi P (2004) Bacterial lipases: an overview of production, purification and biochemical properties. Appl Microbiol Biotechnol 64:763–781
Haki GD, Rakshit SK (2003) Developments in industrially important thermostable enzymes: a review. Bioresour Technol 89:17–34
Hayes DG (2004) Enzyme-catalyzed modification of oilseed materials to produce eco-friendly products. J Am Oil Chem Soc 81:1077–1103
Jaeger KE, Eggert T (2002) Lipases for biotechnology. Curr Opin Biotechnol 13:390–397
Kambourova M, Kirilova N, Mandeva R, Derekova A (2003) Purification and properties of thermostable lipase from a thermophilic Bacillus stearothermophilus MC 7. J Mol Catal B Enzym 22:307–313
Kanjanavas P, Khuchareontaworn S, Khawsak P, Pakpitcharoen A, Pothivejkul K, Santiwatanakul S, Matsui K, Kajiwara T, Chansiri K (2010) Purification and characterization of organic solvent and detergent tolerant lipase from Thermotolerant bacillus sp. RN2. Int J Mol Sci 11:3783–3792
Kim MH, Kim HK, Lee JK, Park SY, Oh TK (2000) Thermostable lipase of Bacillus stearothermophilus: high level production, purification, and calcium-dependent thermostability. Biosci Biotechnol Biochem 64:280–286
Kim IH, Kim H, Lee KT, Chung SH, Ko SN (2002) Lipase-catalyzed acidolysis of perilla oil with caprylic acid to produce structured lipids. J Am Oil Chem Soc 79:363–367
Klibanov AM (2001) Improving enzyme by using them in organic solvents. Nature 409:241–246
Kumar S, Kikon K, Upadhyay A, Kanwar SS, Gupta R (2005) Production, purification, and characterization of lipase from thermophilic and alkaliphilic Bacillus coagulans BTS-3. Protein Expr Purif 41:38–44
Lee DW, Koh YS, Kim KJ, Kim BC, Choi HJ, Kim DS, Suhartono MT, Pyun YR (1999) Isolation and characterization of a thermophilic lipase from Bacillus thermoleovorans ID-1. FEMS Microbiol Lett 179:393–400
Lesuisse E, Schanck K, Colson C (1993) Purification and preliminary characterization of the extracellular lipase of Bacillus subtilis 168, an extremely basic pH-tolerant enzyme. Eur J Biochem 216:155–160
Ma J, Zhang Z, Wang B, Kong X, Wang Y, Cao S, Feng Y (2006) Overexpression and characterization of a lipase from Bacillus subtilis. Protein Expr Purif 45:22–29
Mishra B, Madan P (2009) Overexpression, purification and characterization of organic solvent stable lipase from Bacillus licheniformis RSP-09. J Mol Microbiol Biotechnol 17:118–123
Nagao T, Shimada Y, Sugihara A, Murata A, Komemushi S, Tominaga Y (2001) Use of thermostable Fusarium heterosporum lipase for production of structured lipid containing oleic and palmitic acids in organic solvent- free system. J Am Oil Chem Soc 78:167–172
Nawani N, Kaur J (2000) Purification, characterization and thermostability of a lipase from a thermophilic Bacillus sp. J33. Mol Cell Biochem 206:91–96
Nthangeni MB, Patterton HG, van Tonder A, Vergeer WP, Litthauer D (2001) Over-expression and properties of a purified recombinant Bacillus licheniformis lipase: a comparative report on Bacillus lipases. Enzyme Microb Tech 28:705–712
Petersen TN, Brunak S, von Heijne G, Nielson H (2011) Signal P 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8:785–786
Polizelli PP, Tiera MJ, Bonilla-Rodriguez GO (2008) Effect of surfactants and polyethylene glycol on the activity and stability of a lipase from oilseeds of Pachira aquatica. J Am Oil Chem Soc 85:749–753
Rahman RNZRA, Baharum SN, Basri M, Salleh AB (2005) High yield purification of an organic solvent-tolerant lipase from Pseudomonas sp. Strain S5. Anal Biochem 341:267–274
Rahman RNZRA, Baharum SN, Salleh AB, Basri M (2006) S5 lipase: an organic solvent tolerant enzyme. J Microbiol 44:583–590
Sanders TH, Pattee HE (1975) Peanut alkaline lipase. Lipids 10:50–54
Sangeetha R, Geetha A, Arulpandi I (2010) Concomitant production of protease and lipase by Bacillus licheniformis VSG1: production, purification and characterization. Braz J Microbiol 41:179–185
Saxena RK, Sheoran A, Giri B, Davidson WS (2003) Purification strategies for microbial lipases. J Microbiol Methods 52:1–18
Schmidt-Dannert C (1999) Recombinant microbial lipases for biotechnological applications. Biorg Med Chem 7:2123–2130
Shariff FM, Rahman RNZRA, Basri M, Salleh AB (2011) A newly isolated thermostable lipase from Bacillus sp. Int J Mol Sci 12:2917–2934
Sharma CK, Kanwar SS (2012) Purification of a novel thermophilic lipase from B. licheniformis MTCC-10498. Int Res J Biol Sci 1:43–48
Sharma S, Kanwar SS (2014) Organic Solvent Tolerant Lipases and Applications. Sci World J 2014:625258. doi:10.1155/2014/625258
Sharma R, Soni SK, Vohra RM, Gupta LK, Gupta JK (2002) Purification and characterisation of a thermostable alkaline lipase from a new thermophilic Bacillus sp. RSJ-1. Process Biochem 37:1075–1084
Sharma PK, Singh K, Singh R, Capalash N, Ali A, Mohammad O, Kaur J (2012) Characterization of a thermostable lipase showing loss of secondary structure at ambient temperature. Mol Biol Rep 39:2795–2804
Sigurgisladottir S, Konraosdottir M, Jonsson A, Kristjansson JK, Matthiasson E (1993) Lipase activity of thermophilic bacteria from Icelandic hotsprings. Biotechnol Lett 15:361–366
Wills ED (1960) The relation of metals and –SH groups to the activity of pancreatic lipase. Biochim Biophys Acta 40:481–490
Acknowledgments
Dr. P.K.S. would like to thank Science and Engineering Research Board New Delhi for financial assistance conferred to him under Young scientist scheme (FTYS) No. SB/YS/LS-63/2013.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare no conflict of interests publishing this article in 3 Biotech.
Additional information
Gagandeep Kaur and Amninder Singh have contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kaur, G., Singh, A., Sharma, R. et al. Cloning, expression, purification and characterization of lipase from Bacillus licheniformis, isolated from hot spring of Himachal Pradesh, India. 3 Biotech 6, 49 (2016). https://doi.org/10.1007/s13205-016-0369-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-016-0369-y