
Abstract
Stroke in China is distinguished by its high rates of morbidity, recurrence, disability, and mortality. The ultra-early administration of rtPA is essential for restoring perfusion in acute ischemic stroke, though it concurrently elevates the risk of hemorrhagic transformation. High-mobility group box 1 (HMGB1) emerges as a pivotal player in neuroinflammation after brain ischemia and ischemia–reperfusion. Released passively by necrotic cells and actively secreted, including direct secretion of HMGB1 into the extracellular space and packaging of HMGB1 into intracellular vesicles by immune cells, glial cells, platelets, and endothelial cells, HMGB1 represents a prototypical damage-associated molecular pattern (DAMP). It is intricately involved in the pathogenesis of atherosclerosis, thromboembolism, and detrimental inflammation during the early phases of ischemic stroke. Moreover, HMGB1 significantly contributes to neurovascular remodeling and functional recovery in later stages. Significantly, HMGB1 mediates hemorrhagic transformation by facilitating neuroinflammation, directly compromising the integrity of the blood–brain barrier, and enhancing MMP9 secretion through its interaction with rtPA. As a systemic inflammatory factor, HMGB1 is also implicated in post-stroke depression and an elevated risk of stroke-associated pneumonia. The role of HMGB1 extends to influencing the pathogenesis of ischemia by polarizing various subtypes of immune and glial cells. This includes mediating excitotoxicity due to excitatory amino acids, autophagy, MMP9 release, NET formation, and autocrine trophic pathways. Given its multifaceted role, HMGB1 is recognized as a crucial therapeutic target and prognostic marker for ischemic stroke and hemorrhagic transformation. In this review, we summarize the structure and redox properties, secretion and pathways, regulation of immune cell activity, the role of pathophysiological mechanisms in stroke, and hemorrhage transformation for HMGB1, which will pave the way for developing new neuroprotective drugs, reduction of post-stroke neuroinflammation, and expansion of thrombolysis time window.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In China, stroke exhibits high incidence, recurrence, disability, and mortality rates, placing the country at the forefront globally in terms of stroke-related deaths and disability-adjusted life years [1]. Analyses for the Global Burden of Disease Study 2019 found that there were 12·2 million (95% UI 11·0–13·6) incident strokes globally and 3·94 million (95% UI 3·43–4·58) incident strokes in China in 2019, with the largest increase in ischemic stroke incidence in China from 1990 to 2019 at 22.5% (21.3 ~ 24.5) [1, 2]. Ultra-early application of rtPA (recombinant tissue plasminogen activator) is essential for the recovery of perfusion to ischemic brain tissue in acute cerebral infarction. Still, it also increases the risk of hemorrhagic transformation, which may result in deterioration of neurological function. Identifying early predictors of hemorrhagic transformation can help reduce its incidence and severity. Stroke-induced inflammation and activation of pro-inflammatory mediators have been the focus of studies on the mechanisms of ischemic stroke-induced brain injury, with neuroinflammation leading to apoptosis, blood–brain barrier disruption, cerebral edema, and hemorrhagic transformation in the acute phase while supporting tissue repair and functional recovery in the late stage [3].
High-mobility group box 1 (HMGB1) is a highly conserved non-histone DNA-binding protein in the nucleus that can be released into the extracellular environment to mediate inflammatory responses [4]. Damage-associated molecular pattern (DAMP) is passively released or actively secreted from cells, activates the innate immune system, and is involved in the direct and rapid activation of the inflammatory response after stroke onset. HMGB1 is a typical DAMP molecule that, in addition to being involved in neuroinflammation, has been found to mediate thrombosis, atherosclerosis, disruption of the blood–brain barrier, and, at a later stage, neurovascular remodeling and stroke recovery [5,6,7]. The complex role of HMGB1 in ischemic stroke and the mechanism of hemorrhagic transformation need to be further investigated and provide potential targets for neuroprotective strategies after stroke.
In this review, we elucidate the structural and redox properties, release mechanisms, receptors, and complex roles of HMGB1 in ischemic stroke, focusing on the part of HMGB1 with related neuroglia and immune cells and in hemorrhagic transformation. Furthermore, we have summarized therapeutic strategies targeting HMGB1, aiming to establish HMGB1 as a novel target for the treatment and prognostic prediction of ischemic stroke and hemorrhagic transformation. This approach could potentially serve as a basis for identifying individuals at risk of hemorrhagic conversion and for the adjunctive use of HMGB1 inhibitors in thrombolytic therapy to reduce the incidence of such transformations.
Structure and Redox Properties of HMGB1
HMGB1 is a non-histone DNA-binding and amphipathic protein containing 215 amino acid residues, including three structural domains in a l-type arrangement of positively charged A and B boxes and a 30 amino acid long negatively charged c-segment tail [8, 9] (Fig. 1). The A box is associated with anti-inflammatory effects. In contrast, the B box is essential for cytokine induction and cell differentiation, and the c-terminal tail is related to regulating the DNA binding affinity of HMGB1 and enhancing the anti-inflammatory effects of the A box [10,11,12,13]. HMGB1 contains two nuclear localization signals, NLS1 (28–44 amino acids) and NLS2 (179–185 amino acids), as well as a nuclear export signal in the DNA-binding region (NES) [14], which contributes to the sublocalization of HMGB1 between the cytoplasm and nucleus.

Structure, function, and redox properties of HMGB1. HMGB1 includes three structural domains in an anti-inflammatory A, a pro-inflammatory B boxes, and a 30 amino acid long c-segment tail. HMGB1 mediates the immune response by interacting with receptors such as toll-like receptor 4 (C89-C108) and late glycosylation end product-specific receptor (C150-C183). The redox state of HMGB1 is essential for the role of HMGB1 in regulating pathophysiological processes like inflammatory responses, autophagy, NET formation, and neurotrophin expression promotion
The redox state of HMGB1 is essential for the role of HMGB1 in regulating inflammatory responses and autophagy. Redox modifications can occur at cysteine residues 23, 45, and 106, which regulate the functional properties of HMGB1 [15]. All three cysteines are present in the fully reduced state together with sulfhydryl residues in resting cells, and nuclear HMGB1 tends to be present in the fully reduced form [9]. Upon extracellular release, the all-sulfhydryl state of HMGB1 synergizes with the chemokine CXCL12 (C-X-C motif chemokine ligand 12) to form chemotactic heterocomplexes [16]. It binds synergistically to the CXCL12 chemokine receptor CXCR4(C-X-C chemokine receptor type 4), which contributes to the chemotactic activity of HMGB1 [16]. It was found that cysteine is not required for chemotactic activity, and serine replaces all of them to be more effective at recruiting leukocytes in vivo than wild-type HMGB1 [17]. An increase in oxygen free radicals leads to the formation of disulfide bonds on cysteine residues 23 and 45, and the cysteine residue 106 in the reduced state is required for HMGB1 to exert its pro-inflammatory action [18]. Disulfided HMGB1 can interact with the TLR4 (Toll-like receptor 4) receptor to activate NF-kB (nuclear factor kappa-B) signaling in microglia/macrophages to stimulate inflammatory responses and increase NMDA (N-methyl-D-aspartate)-induced neuronal cell death [19, 20]. However, it has been shown that the disulfide bond between the C23 and C45 of HMGB1 is required to bind to Beclin 1 to maintain the autophagic process, thereby enhancing cell survival in response to cellular stress [21]. The disulfide bond between C23 and C45 of HMGB1 disulfated HMGB1 also significantly increases platelet aggregation and promotes NETs (neutrophil extracellular traps) formation, which plays a vital role in thrombosis [22, 23]. All cysteine residues in HMGB1 can also be oxidized by sustained ROS (reactive oxygen species) and RNS (reactive nitrogen species) released from mitochondria of apoptotic cells and fully oxidized sulfonylated HMGB1 induces immune tolerance via the Rage pathway [24, 25]. In addition, oxidized HMGB1 was found to promote brain recovery by promoting the expression of neurotrophic factors [26]. The effects of different redox forms of HMGB1 on the complex roles of neuroimmunity and functional recovery in ischemic stroke at different times need further investigation.
Release Mechanism of HMGB1
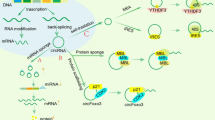
HMGB1 release includes passive release in early post-stroke cell necrosis and active secretion in the late phase of stroke [27]. The secretion and release of HMGB1 are regulated by various factors, including acetylation, methylation, N-glycosylation, phosphorylation, and oxidation [14] (Fig. 2). Post-translational modifications of nuclear localization sequences (NLS) or nuclear export sequences (NES) affect the transfer of proteins from the nucleus to cytoplasmic accumulation and subsequent release, with the most studied mechanism in HMGB1 being lysine hyperacetylation within the NLS site [28, 29]. The acetylation of lysine regulates the cytoplasmic accumulation of HMGB1 through the co-determination of histone acetylase (HATS) and histone deacetylase (HDAC) [29]. Recent studies have explored the lactated modification of HMGB1, where lactated/acetylated HMGB1 is released from macrophages via exosomal secretion, thereby increasing endothelial permeability [30]. The effect of lactate-modified HMGB1 on the blood–brain barrier and hemorrhagic transformation needs further study.

Release mechanism of HMGB1. HMGB1 release is regulated by various factors, including acetylation, methylation, N-glycosylation, phosphorylation, oxidative, and lactation. HMGB1 can be passively released after cell death and actively secreted, including direct secretion of HMGB1 into the extracellular space and packaging of HMGB1 into intracellular vesicles
Under these chemical modifications, the two specific forms of secretion of HMGB1 are as follows. HMGB1 can be passively secreted after cell death (such as necrosis, necroptosis, apoptosis, NETosis, lysosome-mediated cell death, pyroptosis, autophagy-dependent cell death, and ferroptosis) in response to various stimuli stimulation especially oxidative stress [14]. Active forms of secretion include direct secretion of HMGB1 into the extracellular space and packaging of HMGB1 into intracellular vesicles (such as lysosomes or autophagosomes), followed by release of HMGB1 into the extracellular after fusion of the vesicles with the cytoplasmic membrane [14].
After a stroke or other stressful injury, active secretion of HMGB1 was observed in activated monocyte macrophages, vascular endothelial cells, fibroblasts, NK cells, and glial cells [27]. When HMGB1 is secreted from necrotic cells or its secretion by activated macrophages, it induces the recruitment of inflammatory cells and mediates signaling between natural killer cells (NK cells), dendritic cells (DCs), T cells, and macrophages, thereby inducing positive feedback and exacerbating inflammatory injury [25]. In addition to the passive release of HMGB1 from neurons, recent studies have shown that stimulated peripheral sensory neurons actively transfer nuclear HMGB1 to the cytoplasm, where it is eventually released at nerve endings, stimulating the release of proinflammatory factors and exacerbating neuroinflammation in tissues. However, there are still fewer studies on the mechanism of the effect of active neuronal HMGB1 release on neuroinflammation in the central nervous system after stroke and the role of ablation of neuronal HMGB1 on ischemic brain tissue [31, 32]. Platelets are also an essential source of active HMGB1 secretion, and HMGB1 is critical in the acute phase of stroke to produce neutrophil extracellular traps(NETs) that exacerbate worsening post-stroke outcomes [33]. HMGB1 promotes monocyte accumulation and activation via the receptor for advanced glycosylation end products (RAGE) and Toll-like receptor 2 (TLR2). It facilitates RAGE-mediated formation of prethrombotic neutrophil extracellular traps (NETs), where the combined effects of coagulation and inflammation lead to thrombosis [22]. It has been demonstrated that autophagy induction by HMGB1 is a prerequisite for neutrophil cytosolic NET generation, and blocking autophagy reverses HMGB1-induced platelet production. Further elucidation is needed regarding the mechanism of HMGB1-induced autophagosome formation on thrombus formation in ischemic stroke.
In summary, the interplay of HMGB1 release, inflammatory response, and neutrophil extracellular trap generation may be critical for stroke onset and progression, and HMGB1 may serve as a novel target for anti-inflammation and thrombosis, especially in patients with neocoronary stroke. HMGB1 is a crucial mediator of systemic inflammatory response, and observational studies from Brazil have shown that SARS-CoV-2 infection induces the upregulation of HMGB1 in patients with the most severe forms of COVID-19(Coronavirus disease 2019) [34]. The prognosis and mechanisms by which HMGB1 affect the condition of critically ill patients with new coronary strokes or underlying infectious diseases need to be further investigated.
Receptor System of HMGB1
Although there are multiple receptors for HMGB1, post-stroke HMGB1 receptor studies have centered around Toll-like receptors (TLR-2 and TLR-4) and the receptor for advanced glycosylation end products (RAGE) (Fig. 3). MMP9 is a key molecule in blood–brain barrier injury and hemorrhagic transformation, and HMGB1 is involved in MMP9 release and activation through TLR and RAGE receptors.

Receptor system and potential pathway of HMGB1 in stroke. HMGB1 induces BBB disruption and hemorrhagic transformation by interaction with TLR2, TLR4, and RAGE. HMGB1 also promotes MMP9 activation by binding to these receptors. The HMGB1-TLR2-ERK and HMGB1-RAGE-MAPK axis also participate in oligodendrocyte migration and neurovascular remodeling
Toll-like receptors (TLRs) are transmembrane pattern recognition receptors (PRRs) that initiate signaling in response to multiple pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) [35]. In general, Toll-like receptor activation promotes the recruitment of adaptor proteins to activate nuclear factor NF-kB, which induces the expression of pro-inflammatory genes, inflammatory cytokines, adhesion molecules, and the activation of adaptive immunity [36]. A clinical study including patients with ischemic stroke has found that the TLR2 and TLR4 in serum were independently associated with poor prognosis after stroke and correlated with higher serum levels of IL-1β, IL6, tumor necrosis factor a (TNF-α), and the vascular cell adhesion molecule (VCAM1), and that TLR4 in serum was independently associated with lesion volume in patients with ischemic stroke [37]. HMBG1-TLR4 can regulate the activation, proliferation, migration, and survival of microglia, neural stem cells, oligodendrocytes, and neurons. At the early phase of stroke, HMBG1 mediates ischemia–reperfusion injury by activating microglia through TLR-4 signaling, such as increased NF-kB activity, NO production, transcriptional upregulation of COX-2 (cyclooxygenase2), TNF-a and IL-1β [38]. During the delayed phase, HMGB1 plays a beneficial role in stroke through TLR receptors possibly by regulating proliferation and differentiation of neural stem progenitor cells and migration of oligodendrocytes. HMGB1 affects the proliferation of neural stem cells in the subventricular zone and the differentiation of progenitor cells to neuronal progenitors through TLR4 receptors [39]. It has been found that activation of the HMGB1-TLR2 axis is associated with IκB-α degradation, ERK1/2 (extracellular signal-regulated kinase 1/2) phosphorylation, and CREB (CAMP response element binding protein) phosphorylation, promotes oligodendrocyte migration and survival, and plays a vital role in the regeneration of myelin sheaths and functional integrity after cerebral white matter strokes [40, 41]. The signaling pathways activated by TLRs are broadly classified as myeloid differentiation factor 88 (MyD88, the universal bridging protein for recruiting all TLRs except TLR3)-dependent and MyD88-non-dependent pathways [35]. HMGB1 leads to upregulation of Myd88, mediating neuronal injury, and there is a positive correlation between Myd88 expression and NFkB binding activity, pro-inflammatory cytokines TNF-a and IL-1β [42]. After HMGB1 binds to the TLRs, TLR2 or TLR4 uses MAL (MyD88 adaptor-like protein) as a bridging adaptor to recruit MyD88 and to activate the NF-κB pathway and p38 and JNK(Jun N-terminal kinase) and ERK1/2 and MAPK (mitogen-activated protein kinase) pathways [43, 44]. MyD88-independent signaling pathways are not associated with brain injury after acute cerebral ischemia/reperfusion, and several studies have suggested that Myd88-dependent pathways may be more critical than MyD88-independent pathways in stroke [38, 45].
RAGE is widely expressed in various cells, including neurons, glial cells, macrophages, neutrophils, and endothelial cells [46,47,48]. Under physiological conditions, RAGE expression is low in cells but increases when its ligand molecules like HMGB1 increase [44]. HMGB1-RAGE axis leads to activation of several signaling pathways like MAPK, phosphatidylinositol 3 kinase/protein kinase B (PI3K/Akt), cell division cycle 42 (Cdc42), Ras-related C3 botulinum toxin substrate (Rac), and just another kinase/signal transducer and activator of transcription 1 (JAK/STAT1)-mediated signal transduction pathways, which finally contribute to the translocation of NF-κB, triggering the expression of inflammatory cytokines and chemokines that help immune cells mature, migrate, and express surface receptors and lead to neuroinflammation [44]. After day 1 of middle cerebral artery occlusion in rats, RAGE levels were higher in the ischemic hemisphere relative to the nonischemic hemisphere [49]. HMGB1, an essential ligand for RAGE, may be involved in stroke progression through the actions of neutrophils and macrophages. After stroke, various leukocytes infiltrate the brain parenchyma, with neutrophils being the first blood-borne immune cells to invade ischemic tissue [50]. HMGB1-dependent neutrophil recruitment and activation pathway requires the functional interplay between Mac-1 and RAGE [48]. Neutrophil migration is critical for stroke prognosis and hemorrhagic transformation. Research has indicated that when tPA was treated after middle cerebral artery occlusion (MCAO), TLR4 and RAGE expression was increased in type 1 diabetic mice, and elevated TLR4 and RAGE expression was positively correlated with hemorrhagic conversion [51]. The studies suggest that the mechanisms by which HMGB1-RAGE may affect neutrophil migration and activation to mediate neuroninflammation and hemorrhagic transformation deserve further elucidation. After 4 ~ 6 h after neutrophil infiltration, monocytes adhere to the vessel wall to enter the ischemic zone and differentiate into macrophages [52]. During cerebral ischemia, macrophages consist partly of activated microglia and mostly of migratory macrophages [46]. HMGB1-RAGE signaling links necrosis to macrophage activation and may mediate post-stroke brain injury [46]. Research has indicated that HMGB1 initiates endocytosis through RAGE- and dynamin-dependent signaling, which in turn induces cellular pyroptosis [53]. It is well known that cellular pyroptosis, a mode of programmed cell death accompanied by inflammation, is closely associated with neuroinflammation and neuronal death in the early stages of stroke. Significant gaps remain in our understanding of the effect of HMGB1-RAGE axis-activated macrophages on neuroinflammation and neural focal death after ischemic stroke. Notably, plasma levels of both soluble RAGE (sRAGE) and HMGB1 were significantly increased 48 h after ischemic stroke, and sRAGE levels were an independent predictor of functional outcome 3 months after stroke [54]. Howerver, administration of recombinant sRAGE significantly improved the outcome of injury in mice, protected cultured neurons against oxygen and glucose deprivation, and ameliorated the detrimental effect of recombinant HMGB1,which indicates sRAGE may compete with cell surface RAGE for HMGB1 [15, 54].
MMPs are a group of proteases with more than 20 members that can lead to blood–brain barrier opening, brain edema, hemorrhage, and cell death, with MMP9 being the predominant form in the brain [55]. Delayed tPA (tissue plasminogen activator) treatment activates MMP9 in the ischemic brain, and activation of MMP9 subsequently disrupts extracellular matrix and tight junction proteins and mediates blood–brain barrier leakage and hemorrhagic transformation [55]. HMGB1 was significantly increased in plasma after tPA treatment in stroke patients, promoting the release of MMP9, and leading to blood–brain barrier disruption and hemorrhagic transformation [4, 56]. Studies further elucidated that HMGB1 is released from necrotic neurons and promotes MMP9 activation by binding to its receptors, including RAGE, TLR2, and TLR4 [57,58,59].
Regulation of Immune Cell Phenotype and Immunoreactivity by HMGB1
HMGB1 plays a vital role in the regulation of immunophenotypes and immunoreactivity in microglia, astrocytes, neutrophils, monocyte macrophages, and T cells through TLR, RAGE, MAC1 (Alphambeta2), and CXCR receptors, which modulate processes such as inflammation, excitotoxicity, and autophagy (Fig. 4). The study of the immune mechanisms of HMGB1 in ischemic stroke provides new insights into neuroprotective therapy.

Regulation of immune cell phenotype and immunoreactivity by HMGB1. HMGB1 plays a vital role in regulating immunophenotypes and immunoreactivity in microglia, astrocytes, neutrophils, monocyte macrophages, and T cells. HMGB1 participates in neuroinflamation, excitotoxicity, autophagy, neurovascular reconstruction, NET formation, and atherosclerosis through binding with TLR, RAGE, MAC1, and CXCR receptors on these immune cell
Microglia
Microglia are critical cells in post-stroke immunoregulation, shifting between pro-inflammatory M1 and anti-inflammatory M2 phenotypes. M1 activation is considered a pro-inflammatory and neurotoxic state induced by Toll-like receptor (TLR) and interferon-gamma (IFN-γ) signaling pathways and is associated with an increase in pro-inflammatory mediators (IFNγ, IL-1β, TNFα, IL-6, CXCL10, etc.) in protein synthesis, ROS, and NO production, and an increase in protein hydrolases (MMP9, MMP3) that act on the extracellular matrix to cause blood–brain barrier breakdown [60, 61]. M2 activation is an immunosuppressive, pro-angiogenic, and tissue-healing state that produces anti-inflammatory cytokines (IL-10 and TGF-β), growth factors (i.e., insulin-like growth factor 1, colony-stimulating factor 1, vascular endothelial growth factor), and neurotrophic factors (glial cell-derived neurotrophic factor (GDNF) and brain-derived neurotrophic factor (BDNF)) [60]. After the acute stroke, microglia are activated within minutes, predominantly M2 cells in the early stages. Still, around the infarct zone, M2 cells are continuously recruited and converted to M1 cells, mediating neuroinflammation and disrupting the blood–brain barrier, increasing the risk of hemorrhagic transformation [4]. It has been shown that activated microglia directly engulf endothelial cells via phagocytosis and lead to vascular disintegration, ultimately disrupting the blood–brain barrier (BBB) [62].
High-mobility group protein 1 (HMGB1) is one of the essential proinflammatory mediators promoting M1 polarization of microglia, interacting and activating microglia through the receptor for advanced glycosylation end products (RAGE), Toll-like receptors (TLRs), scavenger receptor MAC1, and chemokine receptor CXCR7 [63]. HMGB1 leads to translocation of NF-κB, a significant influence factor on M1 polarization because of its role in regulating pro-inflammatory mediators and blood–brain barrier leakage, including IL-1, IL-2, IL-6, IL-12, TNF-a, iNOS (inducible nitric oxidesynthase), and COX-2 and MMP9 [44, 64]. Through selective internalization, HMGB1 is cleared by activated microglia and infiltrating monocyte macrophages, which limits cascading inflammatory injury [65]. CD36 and scavenger receptor A mediate microglia internalization [66, 67], but the unexplored pathways by which activated microglia mediate HMGB1 clearance and suppression of immune responses in stroke require attention. In addition to the internalized clearance of HMGB1, autophagy as a critical process for immune regulation in ischemic stroke needs to be further studied. Autophagic flux was induced in early OGD/R and inhibited in late OGD/R; the inhibition of autophagic flux in microglia is vital in ischemic stroke primarily by regulating microglia to M1 type and promoting inflammatory responses [68]. A study of neuroinflammation in the etiology of stress hypertension found that the HMGB1/RAGE axis mediates stress-induced impairment of mitochondrial autophagic flux and immunophenotypic switching of microglia, leading to neuroinflammatory responses such as NFκB activation and PIC release [69]. Similarly, progesterone regulates microglia activation, inflammatory response, and neuronal loss in the ischemic brain after stress by reducing HMGB1 release and NLRP3 inflammatory vesicle activation while enhancing microglia autophagy [70]. The involvement of HMGB1 in the autophagy process of microglia in stroke and as an immune target for therapeutic purposes needs to be further investigated.
Several studies have shown that glycyrrhizic acid and its hydrolyzed product glycyrrhetinic acid can reduce the size of cerebral infarction, restore motor function, inhibit M1 microglia activation, enhance M2 activation, and induce neural regeneration by inhibiting HMGB1 [63, 71]. The inhibition of HMGB1 can be an effective strategy for treating cerebral infarction injury.
Monocyte Macrophages
Monocyte macrophages are a double-edged sword in ischemic stroke, playing an essential role in removing debris and inflammation, causing tissue damage, and promoting tissue remodeling [72]. Unlike microglia, which peak 2–3 days after ischemic stroke, monocyte-derived macrophages are not recruited in large numbers to the injured brain until 3–7 days after ischemic stroke [73, 74]. There are functional differences in monocyte subtypes in ischemic stroke, with early proinflammatory monocytes limiting ischemic stroke injury by eliminating necrotic cellular debris and maintaining cerebral microvascular stability, whereas anti-inflammatory monocytes are implicated in tissue remodeling and healing during the subacute and chronic phases of ischemic stroke [75]. Less is known about CNS border-associated macrophages, a small population of specialized macrophages localized in the choroid plexus, meninges, and perivascular spaces, which have prominent functional roles in promoting vascular leakage, leukocyte attraction and infiltration into the ischemic brain, and promoting inflammation [76].
Monocyte macrophages release HMGB1 in response to cellular injury. In turn, activated HMGB1 signaling activates macrophages to release TNF-α, IL-1, IL-6, and other pro-inflammatory molecules via TLR4 [77, 78]. H2O2 may stimulate the release of HMGB1 from macrophages and monocytes through MAPK- and crm1-dependent pathways [79]. ROS oxidized low-density lipoprotein (OxLDL), a risk factor for stroke, stimulates oxidative stress-induced secretion of HMGB1 in macrophages, and HMGB1 promotes macrophage-derived foam cell formation via the endoplasmic reticulum stress (ERS)/C/EBP-homologous protein (CHOP) pathway [80]. HMGB1 promotes monocyte-macrophage activation, amplifies inflammatory processes, and is a critical molecule in atherosclerosis. It has been shown that stroke-induced up-regulation of monocyte hexokinase 2 (Hk2) depends on HMGB1, thus mediating vascular inflammation and atherosclerosis progression after stroke [81]. However, infiltrating anti-inflammatory monocyte macrophages directly enhances the expression of the scavenger receptor Msr1 (macrophage scavenger receptor 1) via the transcription factor MAFB (MAF BZIP transcription factor B), which induces the internalized clearance of HMGB1, and impaired clearance leads to more severe inflammation and exacerbates neuronal damage [65]. In addition, HMGB1 co-localized with the autophagy protein Beclin 1 in macrophages near the necrotic core of carotid plaques, suggesting that HMGB1 may be involved in regulating macrophage autophagy [82]. Autophagy plays an essential role in the stabilization of atherosclerotic plaques by inhibiting oxidative stress, inflammation, and foam cell formation, and promoting the transformation of anti-inflammatory macrophages within the plaque [83], suggesting that HMGB1 may play a beneficial role in the progression of atherosclerosis and the prognosis of ischemic stroke through macrophage autophagy. Thus, HMGB1 as a possible target for macrophages needs to be further investigated.
Astrocytes
Reactive astrocytes after ischemic stroke have pro-inflammatory and neuroprotective functions [84]. Reactive astrocyte proliferation occurs in the peri-infarct region, forming a glial scar to maintain CNS (central nervous system) homeostasis and isolate the lesion [85]. After undergoing reactive astrocyte proliferation, astrocytes produce and release proinflammatory mediators such as IL6, TNF-α, IL-1α, IL-1β, and IFNγ, as well as free radicals such as NO, superoxide, and peroxynitrite that can lead to neuronal death and infarct progression [86]. Astrocytes are critical for the integrity of the blood–brain barrier and may be implicated in hemorrhagic transformation. Ischemic neurons activate astrocytes to increase vascular endothelial growth factor (VEGF) production, leading to disruption of the endothelial barrier [87]. Activation of the VEGF signaling pathway is a key factor in hemorrhagic transformation after tPA, and combination treatment with tPA and anti-VEGF neutralizing antibody significantly reduced VEGF expression in blood–brain barrier, MMP9 activation, and degradation of blood–brain barrier components in a rat thromboembolic model [88].
HMGB1 plays a role in the dual action of astrocytes in ischemic stroke. HMGB1 is involved in inflammatory activation and excitatory amino acid toxicity in astrocytes. Under various stress conditions, extracellular RNA released by hypoxia/ischemia in combination with the TLR2 ligands Pam2CSK4 and HMGB1 stimulates strong activation of astrocytes and significantly promotes the expression and release of pro-inflammatory cytokines [89]. PNX-14 and the benzene derivative CD21 inhibit HMGB1-mediated activation of NLRP3 inflammatory vesicles and production of inflammatory factors such as IL-1β, IL-18, IL-6, or TNF-α, thereby exerting a neuroprotective effect [90, 91]. It has been found that cerebral ischemia/reperfusion injury activates the HMGB1/TLR4 axis by inhibiting the expression of glutamate transporter (GLAST) in primary astrocytes, thereby decreasing glutamate clearance activity and increasing excitatory amino acid-mediated ischemic injury [92]. However, some studies have demonstrated the role of HMGB1 in astrocytes in limiting brain injury and functional remodeling. Treatment with the astrocytic metabolic inhibitor fluorocitrate significantly decreased HMGB1-expressing astrocytes, suppressed neurovascular markers, and correspondingly worsened neurological outcomes [93].
Neutrophils
Neutrophils are involved in early peripheral inflammatory infiltration and play an important role in developing ischemic stroke and hemorrhagic transformation. Increased circulating neutrophils are observed as early as 4–6 h after ischemic stroke, peaking at 1–3 days and then declining over time, with NETs detected 2–3 days after stroke [94]. Neutrophil infiltration exacerbates ischemic stroke and hemorrhagic transformation, and lower neutrophil–lymphocyte ratios are associated with independent predictors of successful reperfusion and good clinical outcome after endovascular thrombectomy, as well as a reduced risk of symptomatic cerebral hemorrhage and death [95]. The ischemic environment and the interaction of neutrophils with endothelial adhesion molecules shift the neutrophil phenotype from a protective N2 to a damaging N1 phenotype [94]. Neutrophils synthesize and secrete pro-inflammatory factors such as TNF-α, IL-1β, and IL-6 and promote the expression of MMP9, which aggravates cerebrovascular endothelial cell injury after stroke and increases the risk of hemorrhagic complications [4, 96].
NETs are composed of DNA, histones, and granule proteins, and NETs promote thrombosis and vascular occlusion by activating platelets, as well as scaffolding platelets and erythrocytes, and procoagulant molecules to promote coagulation. They are associated with reperfusion resistance in acute stroke [97,98,99]. It has been shown that HMGB1 is detected in all thrombus emboli and co-localizes with neutrophils and NETs, participating in NETs-mediated thrombosis and ischemic stroke development [100]. HMGB1-induced oxidative stress is associated with neutrophil activation and NET formation, and HMGB-1 influences NETs formation through its receptors TLR2, TLR4, and RAGE binding [101, 102]. In addition to this, it has been shown that activated platelets deliver HMGB1 to neutrophils and engage them in autophagy and NET generation, exacerbating thrombotic inflammatory lesions [103]. HMGB1-mediated formation and release of NETs exacerbated inflammation and neuronal damage in the ischemic brain [23]. Targeting HMGB1 is efficacious in improving clinical outcomes in stroke. Platelet depletion or platelet-specific knockdown of HMGB1 significantly reduced plasma HMGB1 and NET levels after stroke and greatly improved stroke prognosis [33]. The HMGB1/TLR4 signaling pathway inhibition promotes endogenous anti-inflammatory defenses and inhibits pro-inflammatory responses by targeting circulating neutrophils, which may be actively involved in cerebral ischemic neuroprotection [104]. In addition to this, HMGB1 increased the risk of mediating hemorrhagic transformation and reperfusion resistance after recanalization, and the effect of HMGB1 preconditioning on mechanical thrombolysis and thrombolysis needs to be further investigated.
T Cells
T cells infiltrate significantly as early as 24 h after the middle cerebral artery occlusion model, reach peak infiltration 3–5 days after stroke, and persist for a more extended period [105, 106]. Notably, the accumulation of T cells in the post-ischemic brain is primarily driven by increased local T cell proliferation rather than T cell invasion [107]. T cells are involved in post-stroke inflammation in an antigen-dependent and late antigen-independent manner in the early phase, and the early T cell response in an antigen-independent manner is strongly correlated with the development of infarct volume [106]. After ischemic stroke, invading CD4 and CD8 T cells interact with reactive astrocytes and exhibit increased expression of T cell activation markers, proinflammatory cytokines, and corresponding transcription factors [108]. CD8 + T cells exert cytotoxicity through the release of granzyme and perforin, which form holes in target cells and induce apoptosis; on the other hand, they degrade post-stroke blood–brain barrier tight junctions through the activation of VEGF signaling, which promotes blood–brain barrier permeability [109,110,111]. Treg cells participate in the inflammatory response and neuroplasticity process in ischemic stroke through various mechanisms, such as secretion of anti-inflammatory factors, inhibition of pro-inflammatory factors, induction of cell lysis, involvement in microvascular dysfunction and thrombosis, production of factors promoting neuroregeneration, and modulation of microglia and macrophage polarization, and thus exert both beneficial and adverse effects in ischemic stroke [112, 113].
HMGB1 has been shown to induce the proliferation of CD3 + T cells, which can lead to Th1 polarization as well as IL-2, IL-12, and IFN-γ production, and another study showed polarization of CD4 + T cells to a Th2 phenotype through dendritic cell activation [8, 114,115,116].
HMGB1 plays a crucial role in T cell infiltration and proliferation, and the HMGB1 inhibitor glycyrrhizin prevents ischemia and reduces infarct size in part by inhibiting the infiltration and proliferation of T cells and their subtypes into the ischemic brain [117]. Similarly, the HMGB1 competitive inhibitory protein, HMGB1 A box, significantly inhibited the infiltration of almost all T cells, including Th17, and microglia-induced differentiation of naïve T cells to Th17 cells [118]. In addition, HMGB1 may be a critical factor in Treg cell activity through TLR4 receptor and RAGE receptor. HMGB1 promotes atherosclerosis by negatively regulating the Treg/Th17 ratio [119]. HMGB1 significantly reduced the expression of CTLA4 and Foxp3 and the secretion of IL-10 in Treg cells, and TLR4-neutralizing antibody abrogated HMGB1-induced alterations in Treg phenotype and function [120]. However, some studies have also found that HMGB1 may stimulate Treg immunosuppressive activity by binding to RAGE receptor [121, 122]. HMGB1 signaling maintains high autophagic activity, survival, and immune tolerance of Treg cells through the RAGE-ERK and mTOR pathways [123]. Taken together, the underlying mechanisms in the immune function of HMGB1 on Treg after ischemic stroke remains to be elucidated. Treg cell-derived bone-bridging proteins have been found to promote tissue-repairing microglial cell responses, thereby promoting oligodendrocyte regeneration and myelin re-formation after stroke [124]. HMGB1 has been implicated as a downstream of bone-bridging proteins in other diseases, and further discoveries are needed to determine whether HMGB1 is involved in brain repair by Treg cells.
Role of HMGB1 in the Progression of Ischemic Stroke
HMGB1 plays complex and biphasic roles in the onset and progression of ischemic stroke. HMGB1 is released in large quantities during acute injury induced by excitotoxicity and is the main upstream inflammatory mediator within the neurovascular unit in ischemic stroke during the hyper-acute(within 24 h post-stroke) and acute phase (especially within 4–5 days post-stroke), exacerbating neuronal death and disruption of the blood–brain barrier. However, HMGB1 may exert a significant role in promoting vascular remodeling and neurological recovery during the acute (4–5 days to 1 week after stroke) and subacute(1–3 weeks after stroke), and chronic (> 3 weeks after stroke) phases. HMGB1 can be significantly translocated from the nucleus to the cytoplasm of neuronal cells at 2–4 h after ischemia–reperfusion, but the extracellular release is reduced at 12 h after reperfusion [125]. Furthermore, HMGB1 is actively secreted by activated glial and other cells 2 days after stroke, peaks on day 4, and can persist for weeks or even up to 1 month [59, 126, 127].
HMGB1 may promote ischemic stroke progression during the acute phase through excitatory amino acid toxicity, oxidative/nitrification stress, and modulation of neuroinflammation. In the rat transient MCAO model, glutamate concentration increased rapidly during cerebral ischemia, decreased during reperfusion, and increased again 1 h after reperfusion and persisted until 6 h after reperfusion; whereas anti-HMGB1 antibody prevented the sustained increase in glutamate concentration after reperfusion [125]. Post-stroke can cause a large release of reactive oxygen species in addition to an increase in excitatory amino acids. NADPH oxidase activity was significantly increased after cerebral ischemia–reperfusion, which decreased HDAC4 and HDAC5 expression and promoted apoptosis, at least to some extent, through the HMGB1 signaling pathway [128]. As a response to ischemic brain injury, cells rapidly express stress-induced heat shock proteins (Hsp), among which Hsp72 is abundantly induced in neurons in the penumbra or ischemic core and plays a protective role against oxidative stress [129, 130]. In addition to inhibiting LPS- or TNFα-induced HMGB1 release, overexpression of Hsp72 strongly inhibited HMGB1-induced expression and release of cytokines (TNFα and IL-1), which was closely related to inhibition of MAP kinases (p38, JNK, and ERK) and NF-kB pathway inhibition [131]. A recent study has found that microglial myeloperoxidase(MPO) -containing exosomes increase HOCl production in neighboring neurons and mediate disulfide HMGB1 translocation, thereby exacerbating neurological deficits in ischemic brain injury and cerebral ischemia/reperfusion injury [132]. As for HMGB1’s role in neuroinflammation, short hairpin-mediated HMGB1 knockdown or HMGB1 monoclonal antibody-mediated inhibition of HMGB1 during the hyperacute phase after stroke inhibited increased blood–brain barrier permeability, microglia activation, inflammatory factor release, and iNOS expression [126, 133].
However, key mediators of the penumbral in the acute phase mediate ischemic injury and cell death but contribute to neurovascular remodeling in the recovery phase (weeks later) [134]. In an in vivo model of cerebral ischemia, injection of HMGB1 siRNA into the ventricle at 5 d after stroke significantly suppressed endothelial progenitor cell (EPC) accumulation and peri-infarct microvessel density and exacerbated the deterioration of neurological prognosis 14 days after stroke [135]. Similarly, intraperitoneal injection of the HMGB1 inhibitor glycyrrhizin 4–14 days after tMCAO blocked the beneficial effects of improved vascular remodeling and neurobehavior after human peripheral blood-derived (hPB) -EPC transplantation [136].
HMGB1 Promotes Atherosclerosis and Thrombosis, Leading to Stroke
HMGB1 is strongly associated with atherosclerosis and thrombosis (Fig. 5), which may be involved in the development and progression of ischemic stroke. HMGB1 is involved in LDL transport through the SREBP2 (sterol regulatory element binding protein 2)-SR-BI (scavenger receptor class B type 1) axis and contributes to atherosclerosis [137]. It was found that anti-HMGB1 neutralizing antibodies led to reduced accumulation of macrophages, dendritic cells, and CD4 + T cells in atherosclerotic lesions, as well as reduced expression of the vascular cell adhesion molecule, VCAM-1, and the monocyte chemotactic protein (MCP) [137, 138]. Smooth muscle cells, endothelial cells, foam cells, macrophages, and activated platelets in atherosclerotic plaques may secrete HMGB1, exacerbating the multiple inflammatory effects of HMGB1 on endothelial cells, smooth muscle cells, and macrophages and the progression of atherosclerosis [6]. In atherosclerosis, HMGB1 promotes smooth muscle cell proliferation, migration to the intimal layer, and release of increased amounts of HMGB1 and C-reactive protein, as well as the expression of MMP2, MMP3, and MMP9 [8]. Recombinant HMGB1 activates vascular endothelial cells and increases proinflammatory expression of intercellular adhesion molecule 1 (ICAM-1), VCAM-1, RAGE, TNFα, monocyte chemotactic protein 1 (MCP-1), IL-8, tPA, and plasminogen activator inhibitor 1 [PAI-1], which is mediated by early TNFα secretion and involves the activation of stress mitogen–activated protein(MAP) kinase pathways and the transcription factors NF-kB and Sp1 [139]. HMGB1 promoted the formation and apoptosis of macrophage-derived foam cells via activation of ERS/CHOP pathway, which may be involved in atherosclerotic plaque formation and rupture [80]. After macrophages are transformed into foam cells, these cells release growth factors, cytokines, matrix metalloproteinases (MMP), reactive oxygen species, and HMGB1 and amplify inflammatory process [8]. Taken together, HMGB1 is involved in low-density lipoprotein transport, recruitment of inflammatory cells, and exacerbation of the multiple inflammatory effects of HMGB1 on endothelial cells, smooth muscle cells, and macrophages, thereby inducing the onset and progression of atherosclerosis. The progression of carotid and intracranial atherosclerosis may lead to plaque rupture and thrombosis, thus rendering the development of stroke. Oil acetic acid significantly reduces plaque HMGB1, can attenuate carotid plaque instability, and may potentially reduce the risk of ischemic stroke [140]. HMGB1 is also a critical component of the thromboembolism that leads to large-vessel occlusive stroke and is strongly associated with neutrophil and platelet counts [100]. Extracellular HMGB1 derived from activated platelets or NETosed neutrophils induces thrombosis, further aggravating NETosis and neuronal death, thereby exacerbating inflammation and subsequent damage in the ischemic brain [23, 98].

Role of HMGB1 in the progression of ischemic stroke. a HMGB1 promotes atherosclerosis and thrombosis, leading to stroke. HMGB1 exacerbates the progression of atherosclerosis by participating in the transport of LDL, aggregating inflammatory cells, and exacerbating the multiple inflammatory effects of HMGB1 on endothelial cells, smooth muscle cells, and macrophages. HMGB1 derived from activated platelets or NETosed neutrophils induces thrombosis and further aggravating NETosis. b HMGB1 promotes harmful inflammation in the early phase of ischemic stroke while plays a role in promoting vascular remodeling and functional recovery late after stroke
HMGB1 as a Prognostic Marker for Cerebral Ischemia
HMGB1 has good predictive value for cerebral ischemia–reperfusion injury in patients with ischemic stroke, which correlates with leukocyte infiltration, extensive cerebral infarction, and worsened prognosis [141, 142]. Serum HMGB1 levels above 7.5 ng/mL were an independent risk factor for poor prognosis, and MMP9 activation was significantly associated with increased HMGB1 [143, 144]. HMGB1 was released in large quantities from neurons during acute injury induced by excitotoxicity and cerebral ischemia (1 h after MCAO). It was slowly but extensively induced in microglia, astrocytes, and microvascular endothelial cells of the brain after ischemia (2 days after MCAO/reperfusion), peaking on the fourth day [59]. Some studies have also suggested that the secondary release of HMGB1 is actively secreted by activated immune cells in the brain and peripheral immune systems such as microglia and invasive monocytes and macrophages in response to tissue injury, as these cells are known to actively secrete large amounts of HMGB1 upon activation [15, 27, 145]. A recent study has found that platelet depletion or platelet-specific knockdown of HMGB1 significantly reduced plasma levels after stroke and greatly improved stroke prognosis, demonstrating that platelets are also a key source of HMGB1 in the plasma during ischemic stroke brain injury [33]. Extracellular HMGB1 may act as a proinflammatory cytokine, activating microglia, and other inflammation-associated cells, stimulating the release of other cytokines and exacerbating brain injury [126]. HMGB1 participates in and amplifies the inflammatory response after ischemia, leading to activation of the inflammatory cascade through MAP kinase and NF-kB,TLR2, TLR4, and RAGE signaling, as well as the expression of TNFα, IL-1β, ICAM-1, VCAM-1, E-selectin, and iNOS in different cell types [146]. HMGB1 induces activation of glial cells and secretion of cytokines in the acute phase, then induces second infiltration of immune cells, and transmits signals between NK cells, DCs, T cells, and macrophages during the later progression, thereby amplifying the initial injury [27]. Kim et al. reported that microinjection of short hairpin RNA in the striatum within 24 h after MCAO reduced neuronal death and restricted the expression of IL-1β, COX-2, TNFα, and iNOS in rats by inhibiting the expression of HMGB1 [126]. Injection of anti-HMGB1 monoclonal antibody immediately and 6 h later after cerebral ischemia–reperfusion inhibited the increase of blood–brain barrier permeability, microglia activation, TNFα and iNOS expression, and inhibited MMP9 activity during the acute phase of stroke; conversely, intracerebroventricular injection of HMGB1 aggravated the severity of the infarction [133]. Similarly, the anti-HMGB1 antibody and HMGB1 box A (a functional antagonist of HMGB1 interaction with RAGE) significantly reduced infarct size in the MCAO mouse model [46]. Qiu et al. demonstrated that TNFa and the ICAM-1 were increased by the addition of recombinant HMGB1 in glial and endothelial cells cultured in vitro [57]. Recently, it has been found that higher HMGB1 levels in the acute phase of ischemic stroke are also associated with an increased risk of post-stroke depression and stroke-related pneumonia [147, 148]. In addition to this, HMGB1 mediates enhanced pro-inflammatory effects of lipopolysaccharides and exacerbates TLR4-dependent systemic and brain inflammation, and there is a positive feedback loop between the enhancement of LPS function by HMGB1 and the subsequent release of HMGB1 [149].
Role of HMGB1 in Vascular and Functional Remodeling During Stroke Recovery
Interestingly, HMGB1 plays a role in promoting vascular remodeling and functional recovery late after stroke. Using a model of focal cerebral ischemia in mice, Hayakawa et al. found that reactive astrocytes expressing HMGB1 increased in the peri-infarct cortex from day 7 after cerebral ischemia in parallel with the increase of neurovascular remodeling markers CD31, synaptophysin, and PSD95 [93]. They further demonstrated that fluorocitrate had significantly decreased HMGB1-positive reactive astrocytes and neurovascular remodeling, with a corresponding deterioration in behavioral recovery in ischemic mice [93]. Vascular endothelial growth factor VEGF-A promoted the proliferation and differentiation of neural precursor cells in vitro by upregulating HMGB1 secretion from astrocytes [150]. IL-8, IL-6, and IL-1β are involved in HMGB1-mediated neuroprotection and functional recovery of astrocytes [136, 151, 152]. HMGB1-mediated activation of TLR4 is necessary to maintain the increased proliferation of neural stem cells and to promote differentiation and migration of neuroblastoma cells [39]. In addition to this, HMGB1 promotes the proliferation of EPC and the regeneration of blood vessels in ischemic areas during the chronic phase of stroke and reduces the volume of brain atrophy through the mitogen-activated protein/extracellular regulated protein kinases (MEK/ERK) pathway initiated by endothelial progenitor cell RAGE receptors [135]. Using a standard lysophosphatidylcholine injection model to induce focal demyelination of the mouse corpus callosum, Hayakawa et al. found that HMGB1 expression was upregulated in astrocytes, along with EPCs expressed pro-recovery mediators such as brain-derived neurotrophic factor and basic fibroblast growth factor within the focal white matter lesions [153]. Human peripheral blood-derived-EPC (hPB-EPC) transplantation improved neurobehavioral outcomes, reduced brain atrophy volume, and enhanced neovascularization in MCAO mice, while intraperitoneal injection of the HMGB1 inhibitor glycyrrhizin 4–14 days after tMCAO blocked the beneficial effects of hPB-EPC transplantation [136]. HMGB1/TLR2 activates autocrine trophic signaling pathways in oligodendrocytes and maintains white matter’s structural and functional integrity under ischemic conditions [41]. In an oligodendrocyte oxygen–glucose deprivation model, Choi et al. demonstrated that siRNA knockdown of HMGB1 or application of glycyrrhizin exacerbated OGD-induced oligodendrocyte death, and recombinant HMGB1 application reduced oligodendrocyte death in a TLR2-dependent manner [41]. They also confirmed inhibition of HMGB1 by glycyrrhizin amplifies demyelinating lesions in a TLR2-dependent manner with exacerbation of sensory-motor behavioral deficits in an endothelin-1-induced focal cerebral white matter stroke model [41]. Notably, some studies have found that HMGB1 may play a deleterious role in chronic cerebral ischemic white matter lesions [154, 155]. In a rat model of chronic cerebral hypoperfusion, anti-HMGB1 antibody attenuated white matter damage in the optic tract, which was associated with the downregulation of inflammatory responses characterized by glial cell activation and TLR4/NF-κB signaling downstream of HMGB1 [155].
HMGB1 and Hemorrhagic Transformation
HMGB1 and the Blood–Brain Barrier Injury
The blood–brain barrier consists of the terminal and basement membranes of vascular endothelial cells, pericytes, and astrocytes, and HMGB1 is involved in disrupting the blood–brain barrier and increasing permeability. HMGB1-mediated neuroinflammation is critical in promoting blood–brain barrier disruption and inducing hemorrhagic transformation. HMGB1 mediates the secretion of MMP9, which is involved in early blood–brain barrier degradation and plasma leakage during ischemia via pericytes [156]. Intravenous injection of anti-HMGB1 monoclonal antibody significantly protected rats from ischemia-induced blood–brain barrier disruption, which was associated with inhibiting the expression of proinflammatory factors like iNOS, TNF-α and MMP9 and microglial cell activation [133, 157]. Notoginseng leaf triterpenes also play a role in reducing blood–brain barrier and ischemia–reperfusion injury by inhibiting HMGB1 signaling, suppressing the activation of MAPKs and NF-κB, and down-regulating the concentration of inflammatory cytokines in the ischemic brain, including VCAM-1, MMP9, MMP2, and ICAM-1. Hyperglycemia activates the HMGB1-RAGE signaling pathway and induces the release of inflammatory factors and neutrophil infiltration, given that Xiao-Xu-Ming decoction inhibits RAGE-mediated neuroinflammation, blood–brain barrier disruption, and hemorrhagic transformation [158]. These studies suggest that HMGB1 can mediate blood–brain barrier damage by promoting the release of inflammatory factors such as MMP9 and the activation and infiltration of glial cells and neutrophils. HMGB1 also directly affects the cells that constitute the blood–brain barrier. Astrocyte end-feet swellings are evident around capillaries 3 h after the onset of reperfusion, and the end feet are often detached from capillary basement membranes, and tight junctions between vascular endothelial cells are dissociated (Fig. 6) [125]. These blood–brain barrier disruptions are consistent with up-regulation of pericapillary astrocyte end-foot aquaporin-4 expression, which is inhibited by anti-HMGB1 monoclonal antibodies [125]. In addition to the HMGB1 monoclonal antibody, it has now been found that histidine-rich glycoprotein (HRG) inhibits HMGB1 release and HMGB1-mediated neutrophil adhesion and endothelial barrier function to improve survival in septic mice, and further studies are needed to investigate the effect of HRG on the blood–brain barrier and as a new therapeutic option for stroke [159]. Furthermore, HMGB1 acts on target receptors on endothelial progenitor cells (EPCs) to promote peri-infarct angiogenesis, associated with ischemia/reperfusion-induced hemorrhagic transformation(HT) [135, 160].

HMGB1 and hemorrhagic transformation. HMGB1 disrupts the blood–brain barrier and induces hemorrhagic transformation through direct damage to the structure of the blood–brain barrier, release of inflammatory factors, and neovascularization. In addition to this, HMGB1 binds to tissue-type plasminogen activator (tPA), which amplifies the action of plasmin, promotes the release of TNF-α and MMP9, and participates in the injury through oxidative and nitrosative stress and direct disruption of the blood–brain barrier after tPA thrombolysis
The underlying pathophysiological mechanism of hemorrhagic transformation after mechanical thrombectomy is the disruption of the blood–brain barrier secondary to ischemic and mechanical endothelial injury, resulting in increased tissue permeability [161]. Reperfusion injury can lead to disruption of the blood–brain barrier, including endothelial cell activation, oxygen-free radical overproduction, inflammatory response, leukocyte recruitment, increased cytokine production, and edema formation [161]. In a rat cerebral venous sinus thrombosis model, mechanical thrombectomy combined with glycyrrhizin treatment inhibited the extracellular transport of HMGB1, suppressed the HMGB1-RAGE inflammatory pathway and its downstream inflammatory factors (TNF-α, IL-1β, and IL–6) and oxidative stress [162]. However, mechanisms of HMGB1 in neuroinflammation and oxidative stress leading to blood–brain barrier disruption and hemorrhagic transformation after mechanical thrombectomy in stroke models need further investigation. In addition, poor collateral circulation and recanalization failure also predispose to hemorrhagic conversion after mechanical thrombectomy. Analysis of cerebral artery thrombus emboli after mechanical thrombectomy revealed that areas containing NETs showed strong reactivity to von Willebrand factor (VWF), platelets, and HMGB1 and may make the thrombus less susceptible to dissolution [163]. Excessive NETs release exacerbates the inflammatory response, exacerbates thrombosis, disrupts the blood–brain barrier, and indicates poorer collateral circulation [164, 165]. Further research is needed to address the gaps in understanding the associated pathways between HMGB1 and NETs and their roles in hemorrhagic transformation after mechanical thrombectomy.
HMGB1 and tPA-Related Hemorrhagic Transformation
Serum HMGB1 was increased in both patients and rats after tPA treatment, and blocking HMGB1 signaling significantly reduced neurovascular complications and tPA-induced hemorrhage transformation [160]. HMGB1 may bind to fibrinogen and tPA to amplify fibrinolytic enzymes and promote the release of MMP9, leading to the disruption of the blood–brain barrier and elevated hemorrhage transformation [4, 27]. Taken together, positive feedback activation of tPA with HMGB1 is critical for proinflammatory factor release and hemorrhagic transformation. Li et al. reported that early tPA infusion for 2 h did not induce significant hemorrhagic transformation, whereas after 4.5 h of delayed tPA treatment, significant HT was seen in the ischemic brain [160]. Meanwhile, they found that HMGB1-binding heptamer peptide (HBHP) treatment significantly down-regulated protein expression of delayed tPA-treated rat cerebral ischemia hemisphere-inducible nitric oxide synthase, COX-2, and IL-1β, which was beneficial in attenuating neurovascular complications [160]. Glycyrrhizin down-regulated the expression of NADPH oxidase and iNOS in ischemic brain tissues; inhibited the production of superoxide and peroxynitrite; decreased the expression of HMGB1, TLR2, and MMP9; and preserved type IV collagen and the tight junction protein claudin-5 thereby decreasing hemorrhagic transformation and mortality in a rat model of ischemic stroke after delayed thrombolysis (tPA infusion time as 5 h after MCAO) [56]. These studies demonstrate the important role of oxidative/nitrosative stress and immune response activation in hemorrhagic transformation after tPA thrombolysis. Reactive oxygen species and reactive nitrogen increase after reperfusion, facilitate the HMGB1 activation and release, and interact with HMGB1 downstream immune receptors such as TLR2/4 and RAGE, which is a crucial pathological mechanism to amplify cerebral ischemia–reperfusion injury [55, 166]. Significantly elevated oxygen levels during reperfusion overdrive mitochondria, increasing the ratio of oxidants to antioxidants and exacerbating oxidative stress and positive feedback injury [167]. However, the specific pathways of oxidative stress and mitochondrial dysfunction with HMGB1-mediated hemorrhagic transformation in tPA-treated hemorrhage still lack direct evidence for further in-depth studies. In addition, HMGB1 is also involved in direct endothelial barrier damage and infiltration of inflammatory cells after tPA thrombolysis. Inhibition of the hypoxia-inducible factor HIF1 protects the integrity of the blood–brain barrier by inhibiting HMGB1/TLR4/NF-κB-mediated neutrophil infiltration, thereby reducing the risk of delayed tPA (administered at 6 h after MCAO)-induced hemorrhagic transformation [168].
Hyperglycemia and hypertension are risk factors for stroke, and the role played by HMGB1 in hemorrhagic transformation after rtPA thrombolysis needs to be further elucidated. In hyperglycemic animals, tPA-induced blood–brain barrier toxicity was associated with a significant increase in TXNIP/NLRP3/IL-1β activation and an increase in HMGB-1/NF-κB/TNF-α levels [169]. HMGB-1 was also strongly associated with NLRP3 inflammatory vesicle activation, which together amplified the risk of hyperglycemia-mediated hemorrhagic transformation [169]. The expression of TLR4 and RAGE is increased in type 1 diabetic mice when treated with tPA 2 h after middle cerebral artery occlusion (MCAO) and is positively correlated with hemorrhagic transformation [51]. Thus, blocking HMGB1 signaling can help prevent complications associated with thrombolysis in ischemic stroke, especially in those with combined hyperglycemia. Hypertension is a crucial factor in hemorrhagic conversion after thrombolysis. Some studies found that hypertensive episodes affect the upregulation of RAGE in mice’s cerebral cortex and hippocampal vasculature and that telmisartan reduces blood HMGB1 levels in hemorrhagic-converted patients [170,171,172]. The mechanism by which the HMGB1-RAGE pathway may influence hemorrhagic conversion after thrombolysis in hypertensive patients requires further investigation.
Therapeutic Strategies Modulating HMGB1 Signaling Pathway
Physical Therapy
Physical therapies after ischemic stroke include remote Ischemic preconditioning, hyperbaric oxygen, and hypothermia treatment, which may be involved in inhibiting HMGB1-mediated ischemic brain injury. Inhibition of HMGB1 remote ischemic preconditioning and post-ischemic treatment reduced plasma HMGB1 levels and enhanced its neuroprotective effects against cerebral ischemia–reperfusion injury by inhibiting autophagic processes [173]. Hyperbaric oxygen ameliorated ischemic brain injury by modulating a DNA-dependent histone deacetylase SIRT1-induced HMGB1 deacetylation and inhibiting MMP9 [174]. Hypothermia inhibits infarct volume expansion and reduces IL-1β and TNF-α expression in the peri-infarct region by preventing HMGB1 release from post-ischemic neurons [175].
Lifestyle Modification
Diet and exercise may be involved in the regulation of HMGB1 in ischemic stroke, and a low-fat diet and moderate exercise are beneficial for stroke prognosis. A chronic high-fat diet was found to exacerbate pyroptosis and necroptosis and worsen ischemic brain pathology by enhancing the HMGB1/TLR4/NF-κB signaling pathway after cerebral ischemia–reperfusion injury and leading to poor outcomes after stroke [176]. Treadmill exercise inhibits autophagy in the ischemic semi-dark band, inhibits HMGB1 translocation and binds to Beclin1, and reduces apoptosis and infarct volume [177].
Targeted Therapy
HMGB1-targeted therapy improves the prognosis of ischemic stroke. IVIg protects neurons from HMGB1-induced neuronal cell death by regulating the expression of TLR and RAGE, the expression of the anti-apoptotic protein Bcl-2, and the phosphorylation of cell death-associated kinases, such as JNK, MAPK, and NF-κB [178]. In a 2-h MCAO rat model, administration of anti-HMGB1 monoclonal antibody immediately and 6 h later after cerebral ischemia–reperfusion had a reducing effect on infarct volume in both the cerebral cortex and striatum of rats, with reductions of 90% and 75% at 24 and 48 h, respectively [133]. In a 4-h MCAO mouse model, the anti-HMGB1 antibody reduced infarct size and swelling and improved neurological impairment and motor coordination without hemorrhagic complications compared with the IgG and delayed tPA treatment groups [179]. In addition, injection of HMGB1-binding heptamer peptide (HBHP) within 30 min of delayed tPA treatment significantly attenuated mortality, neurological scores, brain swelling, blood–brain barrier permeability, and hemorrhagic transformation in rats in a 4.5-h MCAO rat model [160]. Administration of HMGB1 A box at 1, 3, or 6 h after MCAO reduced mean infarct volume by 81.3%, 42.6%, and 30.7%, respectively, compared with untreated MCAO brains and significantly improved neurologic deficits [180]. When siRNA was administered intranasally 24, 12, or 3 h before MCAO or 1, 3, or 6 h after MCAO, HMGB1 siRNA administered 6 h after MCAO significantly reduced infarct volume by 73.8 ± 7.2% of that in the PBS-treated model [181]. These studies demonstrate that the application of HMGB1-targeted therapy during the hyperacute period after stroke may be a potential neuroprotective strategy for patients beyond the tPA treatment time window. Nanotechnology provides a new direction to improve the efficiency of therapeutic agent delivery and targeted stroke treatment. Nanomedicines can be used as carriers to enhance the delivery and transfection efficiency of anti-HMGB1 siRNAs [182]. The reactive oxygen species-sensitive nanomedicines can effectively alleviate stroke pathology by controlling the release of therapeutic agents, inhibiting the translocation of nuclear HMGB1, and modulating microglial polarization to reduce infarct volume and enhance neurogenesis [63, 182]. The cerebroprotective effects of a brain-targeted drug-loaded nanoformulation co-mediated by low-density lipoprotein receptor and neutrophil receptor are on the one hand used to improve the blood–brain barrier transport capacity and on the other hand are associated with significant down-regulation of inflammatory cytokines, neutrophil infiltration, and intracellular calcium overload, and blockade of the inflammatory signaling pathway HMGB1/TLRs/MyD88/TRIF/NF-kB [183]. Furthermore, it is noteworthy to explore the differences between blocking HMGB1 directly versus inhibiting its individual receptors. Direct blockade of HMGB1 can more completely inhibit the inflammatory response and cellular damage mediated by HMGB1 during the acute phase of stroke, but may affect other physiological processes related to HMGB1, such as DNA repair and gene transcription leading to adverse reactions or side effects. Selective inhibition of HMGB1 binding to specific receptors can more precisely modulate specific signaling pathways, thereby reducing interference with other physiological processes and providing a better safety profile; however, inhibition of a single receptor may not completely block HMGB1-mediated inflammatory responses, and therapeutic effects may be relatively weak.
Clinical Medications and Compounds with Therapeutic Potential
Atorvastatin significantly attenuated ischemia-induced overexpression of HMGB1, RAGE, TLR4, and NF-κB, significantly improving neurological deficits and reducing cerebral edema and infarct size 24 h after stroke [184]. Melatonin and the neuropeptide PNX-14 inhibited microglia activation-mediated inflammation after stroke by suppressing the high expression and release of HMGB1 and modulating the subsequent activation of the TLR4/MyD88/NF-κB signaling pathway [185, 186]. The antitumor activity and acute myeloid leukocyte drug methylisoindigo attenuated ischemic stroke-induced brain injury by blocking the activation of NLRP3 inflammatory vesicles and modulating microglia/macrophage polarization through inhibition of the TLR4/NF-κB signaling pathway [187]. Uric acid reduced hypoxia-induced proinflammatory cytokine release and attenuated microglia activation in vivo to reduce the volume of cerebral infarction by inhibiting HMGB1-TLR4-NF-kB signaling [188]. HMGB1 has also been found to be a target for antithrombotic drugs, with salicylic acid inhibiting the chemoattractant activity of fully reduced HMGB1 as well as the increased expression of proinflammatory COX-2 induced by disulfide HMGB1. Aspirin has been found to block the release of HMGB1 from thrombin significantly- and collagen-induced activated platelets [189, 190]. Antithrombotic drugs using HMGB1 as a target in the prevention and treatment of ischemic stroke need to be further investigated. Recombinant human thrombomodulin (rhsTM) improves cerebral ischemic injury in mice without hemorrhagic complications through HMGB1 inhibition and has a longer time window to improve the prognosis of ischemic stroke potentially, and its mechanism of action needs to be further investigated [191].
Medicinal Herbs
Medicinal herbs may be a new possibility for the treatment of ischemic stroke, and HMGB1 targeting may be an essential mechanism involved. Glycyrrhizin, a direct inhibitor of HMGB1, reduces pro-inflammatory M1-type polarization of microglia, IFN-γ mediated T-cell activity, inhibits cytochrome C release and cysteine asparaginase three activity-mediated anti-apoptotic effects, attenuates the expression levels of inflammatory and oxidative stress-related molecules such as TNF-α, iNOS, IL-1β, and IL-6 that are overexpressed in ischemic reperfusion cerebral infarction and attenuated the hemorrhagic transformation of tPA treatment by inhibiting the ONOO−/HMGB1/TLR2 signaling cascade [56, 71, 117, 192]. The modulation of the interaction between HMGB1, TLR4, and NF-κB by berberine should be considered a sound ischemic preconditioning strategy to reduce infarct size, neurological deficits, pathological changes, cerebral edema, and inflammatory mediators in serum and ischemic cortical tissues, and the combination of berberine and glycyrrhizin had a more substantial inhibitory effect on the HMGB1/TLR4/NF-κB as compared with either berberine alone or the glycyrrhizin pathway was more potent in inhibiting HMGB1/TLR4/NF-κB [193]. The inhibitory effect of dioscin on HMGB-1/TLR4 signaling and subsequent inhibition of inflammation showed powerful neuroprotective effects [194]. Notoginseng leaf triterpenes reduced HMGB1 expression, inhibited activation of MAPKs and NF-κB to suppress neuroinflammation, and suppressed microglia activation in the hippocampus and cortex, thereby facilitating cerebral ischemia–reperfusion-induced neuropathological changes [195]. Ginseng Rg1 inhibits HMGB1 and oxidative stress-induced activation of glial cells and MMP-induced disruption of the blood–brain barrier by regulating MAPK, thereby exerting significant neuroprotective effects against cerebral ischemic injury [196]. Salvianolic acid D inhibited nucleoplasmic translocation of HMGB1 and its downstream TLR4/MyD88/NF-κB signaling, thereby attenuating ischemia–reperfusion brain injury [197].
In summary, physical therapy, lifestyle modification, targeted therapy, clinical medications, specific compounds, medicinal herbs, or their extracts can promote neurological recovery and attenuate ischemia–reperfusion injury and infarct size after ischemic stroke through the HMGB1-mediated signaling pathway (Fig. 7). However, given the complex role of HMGB1 at different times of ischemic stroke, the time window for targeting HMGB1 therapeutic modalities needs to be further investigated. HMGB1 participates in blood–brain barrier disruption and inflammatory response, and its potential as a therapeutic target for mitigating post-ischemic stroke complications such as hemorrhagic transformation after thrombolysis or infection needs to be explored.

Therapeutic strategies modulating HMGB1 signaling pathway. Physical therapy, lifestyle modification, targeted therapy, clinical medications, specific compounds, medicinal herbs, or their extracts can promote neurological recovery and attenuate ischemia–reperfusion injury and infarct size after ischemic stroke through the HMGB1-mediated signaling pathway
Conclusion
HMGB1-mediated inflammation is closely related to the development of ischemic stroke and hemorrhagic transformation, including glial cell activation, peripheral inflammatory cell infiltration, and release of inflammatory factors. In addition, HMGB1 directly affects astrocytes and vascular endothelial cells that form the blood–brain barrier and bind to tPA to amplify fibrinolytic enzymes and promote the release of MMP9, which promotes the development of hemorrhagic transformation after thrombolysis. HMGB1 promotes necroptosis and harmful inflammation in the early phase of ischemic stroke, while it plays an important role in the remodeling of the neurovascular system and the recovery of function in the late phase of stroke. Different redox states of HMGB1 regulate inflammatory responses, thrombosis, and secretion of neurotrophic factors. Immune cells and glial cells are involved in the pathogenesis of ischemia. HMGB1 plays a vital role in polarizing their different isoforms, mediating excitatory amino acid toxicity, autophagy, release of MMP9 formation of NETs, and autocrine trophic pathways. HMGB1 is an important therapeutic target and prognostic predictor of ischemic stroke and hemorrhagic transformation; however, based on the complex biphasic role of HMGB1 and the variability in treatment times between clinical and experimental models, the efficiency and safety of targeting HMGB1 in the acute treatment of cerebral infarction and long-term rehabilitation remain a major challenge in this clinical area at present.
Data Availability
Not applicable.
Abbreviations
- BBB:
-
Blood-brain barrier
- BDNF:
-
Brain-derived neurotrophic factor
- Cdc42:
-
Cell division cycle 42
- CHOP:
-
C/EBP-homologous protein
- CNS:
-
Central nervous system
- COVID-19:
-
Coronavirus disease 2019
- COX2:
-
Cyclooxygenase2
- CREB:
-
CAMP response element binding protein
- CXCL12:
-
C-X-C motif chemokine ligand 12
- CXCR4:
-
C-X-C chemokine receptor type 4
- DAMP:
-
Damage-associated molecular pattern
- DCs:
-
Dendritic cells
- EPCs:
-
Endothelial progenitor cells
- ERK1/2:
-
Extracellular signal-regulated kinase 1/2
- ERS:
-
Endoplasmic reticulum stress
- GDNF:
-
Glial cell-derived neurotrophic factor
- GLAST:
-
Glutamate transporter
- HATS:
-
Histone acetylase
- HBHP:
-
HMGB1-binding heptamer peptide
- HDAC:
-
Histone deacetylase
- Hk2:
-
Hexokinase 2
- HMGB1:
-
High-mobility group box
- HRG:
-
Histidine-rich glycoprotein
- HT:
-
Hemorrhagic transformation
- hPB-EPC:
-
Human peripheral blood-derived-EPC
- ICAM-1:
-
Intercellular adhesion molecule 1
- IFN-γ:
-
Interferon-gamma
- IL:
-
Interleukin
- iNOS:
-
Inducible nitric oxide synthase
- JAK/STAT1:
-
Just another kinase/signal transducer and activator of transcription 1
- JNK:
-
Jun N-terminal kinase
- MAC1:
-
Alphambeta2
- MAFB:
-
MAF BZIP Transcription Factor B
- MAL:
-
MyD88 adaptor-like protein
- MAP:
-
Mitogen–activated protein
- MAPK:
-
Mitogen-activated protein kinase
- MCAO:
-
Middle cerebral artery occlusion
- MCP-1:
-
Monocyte chemotactic protein 1
- Msr1:
-
Macrophage scavenger receptor 1
- MMP9:
-
Matrix metalloproteinase 9
- MMPs:
-
Matrix metalloproteinases
- MyD88:
-
Myeloid differentiation factor 88
- NES:
-
Nuclear Export Signal
- NETs:
-
Neutrophil extracellular traps
- NF-kB:
-
Nuclear factor kappa-B
- NK:
-
Natural killer cells
- NLS1:
-
Nuclear Localization Signal 1
- NLS2:
-
Nuclear Localization Signal 2
- NMDA:
-
N-Methyl-D-aspartate
- OGD:
-
Oxygen–glucose deprivation
- OxLDL:
-
Oxidized low-density lipoprotein
- PAI-1:
-
Plasminogen activator inhibitor 1
- PAMPs:
-
Pathogen-associated molecular patterns
- PI3K/Akt:
-
Phosphatidylinositol 3 kinase/protein kinase B
- PRRs:
-
Pattern recognition receptors
- RAGE:
-
Receptor for advanced glycosylation end products
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- Rac:
-
Ras-related C3 botulinum toxin substrate
- RhsTM:
-
Recombinant human thrombomodulin
- rtPA:
-
Recombinant tissue plasminogen activator
- sRAGE:
-
Soluble RAGE
- SR-BI:
-
Scavenger receptor class B type 1
- SREBP2:
-
Sterol regulatory element binding protein 2
- TGF-β:
-
Transforming growth factor beta
- TLR4:
-
Toll-like receptor 4
- TLR2:
-
Toll-like receptor 2
- TLRs:
-
Toll-like receptors
- TNF-α:
-
Tumor necrosis factor a
- tPA:
-
Tissue plasminogen activator
- VCAM1:
-
Vascular cell adhesion molecule
- VEGF:
-
Vascular endothelial growth factor
- VWF:
-
Von Willebrand factor
References
Ma Q, Li R, Wang L, Yin P, Wang Y, Yan C, Ren Y, Qian Z, Vaughn MG, McMillin SE, et al. Temporal trend and attributable risk factors of stroke burden in China, 1990–2019: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health. 2021;6:e897–906. https://doi.org/10.1016/s2468-2667(21)00228-0.
Global, regional, and national burden of stroke and its risk factors, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurol 2021, 20:795–820. https://doi.org/10.1016/s1474-4422(21)00252-0.
Jurcau A, Simion A: Neuroinflammation in cerebral ischemia and ischemia/reperfusion injuries: from pathophysiology to therapeutic strategies. Int J Mol Sci 2021, 23. https://doi.org/10.3390/ijms23010014.
Ma G, Pan Z, Kong L, Du G. Neuroinflammation in hemorrhagic transformation after tissue plasminogen activator thrombolysis: potential mechanisms, targets, therapeutic drugs and biomarkers. Int Immunopharmacol. 2021;90:107216. https://doi.org/10.1016/j.intimp.2020.107216.
Wu H, Li R, Pei LG, Wei ZH, Kang LN, Wang L, Xie J, Xu B. Emerging role of high mobility group box-1 in thrombosis-related diseases. Cell Physiol Biochem. 2018;47:1319–37. https://doi.org/10.1159/000490818.
Richard SA, Sackey M, Su Z, Xu H: Pivotal neuroinflammatory and therapeutic role of high mobility group box 1 in ischemic stroke. Biosci Rep 2017, 37. 10.1042/bsr20171104.
Nishibori M, Wang D, Ousaka D, Wake H: High mobility group box-1 and blood-brain barrier disruption. Cells 2020, 9. https://doi.org/10.3390/cells9122650.
de Souza AW, Westra J, Limburg PC, Bijl M, Kallenberg CG. HMGB1 in vascular diseases: its role in vascular inflammation and atherosclerosis. Autoimmun Rev. 2012;11:909–17. https://doi.org/10.1016/j.autrev.2012.03.007.
Andersson U, Yang H, Harris H. High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin Immunol. 2018;38:40–8. https://doi.org/10.1016/j.smim.2018.02.011.
Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–42. https://doi.org/10.1038/nri1594.
Sparatore B, Patrone M, Passalacqua M, Pedrazzi M, Gaggero D, Pontremoli S, Melloni E. Extracellular processing of amphoterin generates a peptide active on erythroleukaemia cell differentiation. Biochem J. 2001;357:569–74. https://doi.org/10.1042/0264-6021:3570569.
Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, Harris HE, Czura CJ, Wang H, Ulloa L, et al. Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med. 2003;9:37–45.
Gong W, Zheng Y, Chao F, Li Y, Xu Z, Huang G, Gao X, Li S, He F. The anti-inflammatory activity of HMGB1 A box is enhanced when fused with C-terminal acidic tail. J Biomed Biotechnol. 2010;2010:915234. https://doi.org/10.1155/2010/915234.
Chen R, Kang R, Tang D. The mechanism of HMGB1 secretion and release. Exp Mol Med. 2022;54:91–102. https://doi.org/10.1038/s12276-022-00736-w.
Singh V, Roth S, Veltkamp R, Liesz A. HMGB1 as a key mediator of immune mechanisms in ischemic stroke. Antioxid Redox Signal. 2016;24:635–51. https://doi.org/10.1089/ars.2015.6397.
Schiraldi M, Raucci A, Muñoz LM, Livoti E, Celona B, Venereau E, Apuzzo T, De Marchis F, Pedotti M, Bachi A, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. 2012;209:551–63. https://doi.org/10.1084/jem.20111739.
Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med. 2012;209:1519–28. https://doi.org/10.1084/jem.20120189.
Yang H, Antoine DJ, Andersson U, Tracey KJ. The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol. 2013;93:865–73. https://doi.org/10.1189/jlb.1212662.
Yang H, Wang H, Ju Z, Ragab AA, Lundbäck P, Long W, Valdes-Ferrer SI, He M, Pribis JP, Li J, et al. MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med. 2015;212:5–14. https://doi.org/10.1084/jem.20141318.
Balosso S, Liu J, Bianchi ME, Vezzani A. Disulfide-containing high mobility group box-1 promotes N-methyl-D-aspartate receptor function and excitotoxicity by activating Toll-like receptor 4-dependent signaling in hippocampal neurons. Antioxid Redox Signal. 2014;21:1726–40. https://doi.org/10.1089/ars.2013.5349.
Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–80. https://doi.org/10.1038/cdd.2010.191.
Stark K, Philippi V, Stockhausen S, Busse J, Antonelli A, Miller M, Schubert I, Hoseinpour P, Chandraratne S, von Brühl ML, et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood. 2016;128:2435–49. https://doi.org/10.1182/blood-2016-04-710632.
Kim SW, Lee H, Lee HK, Kim ID, Lee JK. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathol Commun. 2019;7:94. https://doi.org/10.1186/s40478-019-0747-x.
Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 2008;29:21–32. https://doi.org/10.1016/j.immuni.2008.05.013.
Hubert P, Roncarati P, Demoulin S, Pilard C, Ancion M, Reynders C, Lerho T, Bruyere D, Lebeau A, Radermecker C, et al: Extracellular HMGB1 blockade inhibits tumor growth through profoundly remodeling immune microenvironment and enhances checkpoint inhibitor-based immunotherapy. J Immunother Cancer 2021, 9. https://doi.org/10.1136/jitc-2020-001966.
Tian X, Sun L, Feng D, Sun Q, Dou Y, Liu C, Zhou F, Li H, Shen H, Wang Z, Chen G. HMGB1 promotes neurovascular remodeling via Rage in the late phase of subarachnoid hemorrhage. Brain Res. 2017;1670:135–45. https://doi.org/10.1016/j.brainres.2017.06.001.
Hayakawa K, Qiu J, Lo EH. Biphasic actions of HMGB1 signaling in inflammation and recovery after stroke. Ann N Y Acad Sci. 2010;1207:50–7. https://doi.org/10.1111/j.1749-6632.2010.05728.x.
Hao N, Budnik BA, Gunawardena J, O’Shea EK. Tunable signal processing through modular control of transcription factor translocation. Science. 2013;339:460–4. https://doi.org/10.1126/science.1227299.
Tang Y, Zhao X, Antoine D, Xiao X, Wang H, Andersson U, Billiar TR, Tracey KJ, Lu B. Regulation of posttranslational modifications of HMGB1 during immune responses. Antioxid Redox Signal. 2016;24:620–34. https://doi.org/10.1089/ars.2015.6409.
Yang K, Fan M, Wang X, Xu J, Wang Y, Tu F, Gill PS, Ha T, Liu L, Williams DL, Li C. Lactate promotes macrophage HMGB1 lactylation, acetylation, and exosomal release in polymicrobial sepsis. Cell Death Differ. 2022;29:133–46. https://doi.org/10.1038/s41418-021-00841-9.
Yang H, Andersson U, Brines M: Neurons are a primary driver of inflammation via release of HMGB1. Cells 2021, 10. https://doi.org/10.3390/cells10102791.
Yang H, Zeng Q, Silverman HA, Gunasekaran M, George SJ, Devarajan A, Addorisio ME, Li J, Tsaava T, Shah V, et al: HMGB1 released from nociceptors mediates inflammation. Proc Natl Acad Sci U S A 2021, 118. https://doi.org/10.1073/pnas.2102034118.
Denorme F, Portier I, Rustad JL, Cody MJ, de Araujo CV, Hoki C, Alexander MD, Grandhi R, Dyer MR, Neal MD, et al: Neutrophil extracellular traps regulate ischemic stroke brain injury. J Clin Invest 2022, 132. https://doi.org/10.1172/jci154225.
Passos FRS, Heimfarth L, Monteiro BS, Corrêa CB, Moura TR, Araújo AAS, Martins-Filho PR, Quintans-Júnior LJ, Quintans JSS. Oxidative stress and inflammatory markers in patients with COVID-19: potential role of RAGE, HMGB1, GFAP and COX-2 in disease severity. Int Immunopharmacol. 2022;104:108502. https://doi.org/10.1016/j.intimp.2021.108502.
Okun E, Griffioen KJ, Mattson MP. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011;34:269–81. https://doi.org/10.1016/j.tins.2011.02.005.
Akira S. TLR signaling. Curr Top Microbiol Immunol. 2006;311:1–16. https://doi.org/10.1007/3-540-32636-7_1.
Brea D, Blanco M, Ramos-Cabrer P, Moldes O, Arias S, Pérez-Mato M, Leira R, Sobrino T, Castillo J. Toll-like receptors 2 and 4 in ischemic stroke: outcome and therapeutic values. J Cereb Blood Flow Metab. 2011;31:1424–31. https://doi.org/10.1038/jcbfm.2010.231.
Yang QW, Lu FL, Zhou Y, Wang L, Zhong Q, Lin S, Xiang J, Li JC, Fang CQ, Wang JZ. HMBG1 mediates ischemia-reperfusion injury by TRIF-adaptor independent Toll-like receptor 4 signaling. J Cereb Blood Flow Metab. 2011;31:593–605. https://doi.org/10.1038/jcbfm.2010.129.
Palma-Tortosa S, Hurtado O, Pradillo JM, Ferreras-Martín R, García-Yébenes I, García-Culebras A, Moraga A, Moro M, Lizasoain I. Toll-like receptor 4 regulates subventricular zone proliferation and neuroblast migration after experimental stroke. Brain Behav Immun. 2019;80:573–82. https://doi.org/10.1016/j.bbi.2019.05.002.
Choi JY, Jin X, Kim H, Koh S, Cho HJ, Kim BG. High mobility group box 1 as an autocrine chemoattractant for oligodendrocyte lineage cells in white matter stroke. Stroke. 2023;54:575–86. https://doi.org/10.1161/strokeaha.122.041414.
Choi JY, Cui Y, Chowdhury ST, Kim BG. High-mobility group box-1 as an autocrine trophic factor in white matter stroke. Proc Natl Acad Sci U S A. 2017;114:E4987-e4995. https://doi.org/10.1073/pnas.1702035114.
Li W, Ling HP, You WC, Ji XJ, Tang Y, Zhao JB, Su XF, Hang CH. Recombinant high-mobility group box 1 protein (HMGB-1) promotes myeloid differentiation primary response protein 88 (Myd88) upregulation in mouse primary cortical neurons. Neurol Sci. 2013;34:847–53. https://doi.org/10.1007/s10072-012-1131-9.
O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–64. https://doi.org/10.1038/nri2079.
Ye Y, Zeng Z, Jin T, Zhang H, Xiong X, Gu L. The role of high mobility group box 1 in ischemic stroke. Front Cell Neurosci. 2019;13:127. https://doi.org/10.3389/fncel.2019.00127.
Hua F, Wang J, Sayeed I, Ishrat T, Atif F, Stein DG. The TRIF-dependent signaling pathway is not required for acute cerebral ischemia/reperfusion injury in mice. Biochem Biophys Res Commun. 2009;390:678–83. https://doi.org/10.1016/j.bbrc.2009.10.027.
Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, Bendszus M, Rossetti G, Nawroth PP, Bierhaus A, Schwaninger M. The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci. 2008;28:12023–31. https://doi.org/10.1523/jneurosci.2435-08.2008.
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–13. https://doi.org/10.1038/nm890.
Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP, Chavakis T. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. Embo j. 2007;26:1129–39. https://doi.org/10.1038/sj.emboj.7601552.
Zhai DX, Kong QF, Xu WS, Bai SS, Peng HS, Zhao K, Li GZ, Wang DD, Sun B, Wang JH, et al. RAGE expression is up-regulated in human cerebral ischemia and pMCAO rats. Neurosci Lett. 2008;445:117–21. https://doi.org/10.1016/j.neulet.2008.08.077.
Jayaraj RL, Azimullah S, Beiram R, Jalal FY, Rosenberg GA. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation. 2019;16:142. https://doi.org/10.1186/s12974-019-1516-2.
Ning R, Chopp M, Yan T, Zacharek A, Zhang C, Roberts C, Cui X, Lu M, Chen J. Tissue plasminogen activator treatment of stroke in type-1 diabetes rats. Neuroscience. 2012;222:326–32. https://doi.org/10.1016/j.neuroscience.2012.07.018.
Alsbrook DL, Di Napoli M, Bhatia K, Biller J, Andalib S, Hinduja A, Rodrigues R, Rodriguez M, Sabbagh SY, Selim M, et al. Neuroinflammation in acute ischemic and hemorrhagic stroke. Curr Neurol Neurosci Rep. 2023;23:407–31. https://doi.org/10.1007/s11910-023-01282-2.
Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, Li Y, Scott MJ, Xiao G, Li S, et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. 2014;21:1229–39. https://doi.org/10.1038/cdd.2014.40.
Tang SC, Wang YC, Li YI, Lin HC, Manzanero S, Hsieh YH, Phipps S, Hu CJ, Chiou HY, Huang YS, et al. Functional role of soluble receptor for advanced glycation end products in stroke. Arterioscler Thromb Vasc Biol. 2013;33:585–94. https://doi.org/10.1161/atvbaha.112.300523.
Chen H, Chen X, Luo Y, Shen J. Potential molecular targets of peroxynitrite in mediating blood-brain barrier damage and haemorrhagic transformation in acute ischaemic stroke with delayed tissue plasminogen activator treatment. Free Radic Res. 2018;52:1220–39. https://doi.org/10.1080/10715762.2018.1521519.
Chen H, Guan B, Wang B, Pu H, Bai X, Chen X, Liu J, Li C, Qiu J, Yang D, et al. Glycyrrhizin prevents hemorrhagic transformation and improves neurological outcome in ischemic stroke with delayed thrombolysis through targeting peroxynitrite-mediated HMGB1 Signaling. Transl Stroke Res. 2020;11:967–82. https://doi.org/10.1007/s12975-019-00772-1.
Qiu J, Nishimura M, Wang Y, Sims JR, Qiu S, Savitz SI, Salomone S, Moskowitz MA. Early release of HMGB-1 from neurons after the onset of brain ischemia. J Cereb Blood Flow Metab. 2008;28:927–38. https://doi.org/10.1038/sj.jcbfm.9600582.
Qiu J, Xu J, Zheng Y, Wei Y, Zhu X, Lo EH, Moskowitz MA, Sims JR. High-mobility group box 1 promotes metalloproteinase-9 upregulation through Toll-like receptor 4 after cerebral ischemia. Stroke. 2010;41:2077–82. https://doi.org/10.1161/strokeaha.110.590463.
Kim JB, Lim CM, Yu YM, Lee JK. Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J Neurosci Res. 2008;86:1125–31. https://doi.org/10.1002/jnr.21555.
Wang Y, Leak RK, Cao G. Microglia-mediated neuroinflammation and neuroplasticity after stroke. Front Cell Neurosci. 2022;16:980722. https://doi.org/10.3389/fncel.2022.980722.
Peng L, Hu G, Yao Q, Wu J, He Z, Law BY, Hu G, Zhou X, Du J, Wu A, Yu L. Microglia autophagy in ischemic stroke: a double-edged sword. Front Immunol. 2022;13:1013311. https://doi.org/10.3389/fimmu.2022.1013311.
Jolivel V, Bicker F, Binamé F, Ploen R, Keller S, Gollan R, Jurek B, Birkenstock J, Poisa-Beiro L, Bruttger J, et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015;129:279–95. https://doi.org/10.1007/s00401-014-1372-1.
Jin L, Zhu Z, Hong L, Qian Z, Wang F, Mao Z. ROS-responsive 18β-glycyrrhetic acid-conjugated polymeric nanoparticles mediate neuroprotection in ischemic stroke through HMGB1 inhibition and microglia polarization regulation. Bioact Mater. 2023;19:38–49. https://doi.org/10.1016/j.bioactmat.2022.03.040.
Zeng J, Bao T, Yang K, Zhu X, Wang S, Xiang W, Ge A, Zeng L, Ge J. The mechanism of microglia-mediated immune inflammation in ischemic stroke and the role of natural botanical components in regulating microglia: a review. Front Immunol. 2022;13:1047550. https://doi.org/10.3389/fimmu.2022.1047550.
Shichita T, Ito M, Morita R, Komai K, Noguchi Y, Ooboshi H, Koshida R, Takahashi S, Kodama T, Yoshimura A. MAFB prevents excess inflammation after ischemic stroke by accelerating clearance of damage signals through MSR1. Nat Med. 2017;23:723–32. https://doi.org/10.1038/nm.4312.
Grajchen E, Wouters E, van de Haterd B, Haidar M, Hardonnière K, Dierckx T, Van Broeckhoven J, Erens C, Hendrix S, Kerdine-Römer S, et al. CD36-mediated uptake of myelin debris by macrophages and microglia reduces neuroinflammation. J Neuroinflammation. 2020;17:224. https://doi.org/10.1186/s12974-020-01899-x.
Zhang H, Su YJ, Zhou WW, Wang SW, Xu PX, Yu XL, Liu RT. Activated scavenger receptor A promotes glial internalization of aβ. PLoS ONE. 2014;9:e94197. https://doi.org/10.1371/journal.pone.0094197.
Xia CY, Zhang S, Chu SF, Wang ZZ, Song XY, Zuo W, Gao Y, Yang PF, Chen NH. Autophagic flux regulates microglial phenotype according to the time of oxygen-glucose deprivation/reperfusion. Int Immunopharmacol. 2016;39:140–8. https://doi.org/10.1016/j.intimp.2016.06.030.
Zhang S, Hu L, Jiang J, Li H, Wu Q, Ooi K, Wang J, Feng Y, Zhu D, Xia C. HMGB1/RAGE axis mediates stress-induced RVLM neuroinflammation in mice via impairing mitophagy flux in microglia. J Neuroinflammation. 2020;17:15. https://doi.org/10.1186/s12974-019-1673-3.
Espinosa-Garcia C, Atif F, Yousuf S, Sayeed I, Neigh GN, Stein DG: Progesterone attenuates stress-induced NLRP3 inflammasome activation and enhances autophagy following ischemic brain injury. Int J Mol Sci 2020, 21. https://doi.org/10.3390/ijms21113740.
Sun Y, Hei M, Fang Z, Tang Z, Wang B, Hu N. High-mobility group box 1 contributes to cerebral cortex injury in a neonatal hypoxic-ischemic rat model by regulating the phenotypic polarization of microglia. Front Cell Neurosci. 2019;13:506. https://doi.org/10.3389/fncel.2019.00506.
Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T, Shimohata T: Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int J Mol Sci 2017, 18. https://doi.org/10.3390/ijms18102135.
Denes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA, Allan SM. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab. 2007;27:1941–53. https://doi.org/10.1038/sj.jcbfm.9600495.
Breckwoldt MO, Chen JW, Stangenberg L, Aikawa E, Rodriguez E, Qiu S, Moskowitz MA, Weissleder R. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc Natl Acad Sci U S A. 2008;105:18584–9. https://doi.org/10.1073/pnas.0803945105.
ElAli A, Jean LeBlanc N. The role of monocytes in ischemic stroke pathobiology: new avenues to explore. Front Aging Neurosci. 2016;8:29. https://doi.org/10.3389/fnagi.2016.00029.
Gerganova G, Riddell A, Miller AA. CNS border-associated macrophages in the homeostatic and ischaemic brain. Pharmacol Ther. 2022;240:108220. https://doi.org/10.1016/j.pharmthera.2022.108220.
Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107:11942–7. https://doi.org/10.1073/pnas.1003893107.
Yang H, Lundbäck P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, Al-Abed Y, Andersson U, Tracey KJ. Redox modifications of cysteine residues regulate the cytokine activity of HMGB1. Mol Med. 2021;27:58. https://doi.org/10.1186/s10020-021-00307-1.
Tang D, Shi Y, Kang R, Li T, Xiao W, Wang H, Xiao X. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J Leukoc Biol. 2007;81:741–7. https://doi.org/10.1189/jlb.0806540.
Wu H, Chen Z, Chen JZ, Pei LG, Xie J, Wei ZH, Kang LN, Wang L, Xu B. High Mobility Group B-1 (HMGB-1) Promotes apoptosis of macrophage-derived foam cells by inducing endoplasmic reticulum stress. Cell Physiol Biochem. 2018;48:1019–29. https://doi.org/10.1159/000491970.
Sun Y, Zhang L, Cao Y, Li X, Liu F, Cheng X, Du J, Ran H, Wang Z, Li Y, et al. Stroke-induced hexokinase 2 in circulating monocytes exacerbates vascular inflammation and atheroprogression. J Thromb Haemost. 2023. https://doi.org/10.1016/j.jtha.2023.02.021.
Umahara T, Uchihara T, Hirao K, Shimizu S, Hashimoto T, Kohno M, Hanyu H. Essential autophagic protein Beclin 1 localizes to atherosclerotic lesions of human carotid and major intracranial arteries. J Neurol Sci. 2020;414:116836. https://doi.org/10.1016/j.jns.2020.116836.
Lin L, Zhang MX, Zhang L, Zhang D, Li C, Li YL. Autophagy, pyroptosis, and ferroptosis: new regulatory mechanisms for atherosclerosis. Front Cell Dev Biol. 2021;9:809955. https://doi.org/10.3389/fcell.2021.809955.
Xu S, Lu J, Shao A, Zhang JH, Zhang J. Glial cells: role of the immune response in ischemic stroke. Front Immunol. 2020;11:294. https://doi.org/10.3389/fimmu.2020.00294.
Choudhury GR, Ding S. Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiol Dis. 2016;85:234–44. https://doi.org/10.1016/j.nbd.2015.05.003.
Liu Z, Chopp M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog Neurobiol. 2016;144:103–20. https://doi.org/10.1016/j.pneurobio.2015.09.008.
Li YN, Pan R, Qin XJ, Yang WL, Qi Z, Liu W, Liu KJ. Ischemic neurons activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. J Neurochem. 2014;129:120–9. https://doi.org/10.1111/jnc.12611.
Kanazawa M, Igarashi H, Kawamura K, Takahashi T, Kakita A, Takahashi H, Nakada T, Nishizawa M, Shimohata T. Inhibition of VEGF signaling pathway attenuates hemorrhage after tPA treatment. J Cereb Blood Flow Metab. 2011;31:1461–74. https://doi.org/10.1038/jcbfm.2011.9.
Fischer S, Nasyrov E, Brosien M, Preissner KT, Marti HH, Kunze R. Self-extracellular RNA promotes pro-inflammatory response of astrocytes to exogenous and endogenous danger signals. J Neuroinflammation. 2021;18:252. https://doi.org/10.1186/s12974-021-02286-w.
Wang J, Zheng B, Yang S, Tang X, Wang J, Wei D. The protective effects of phoenixin-14 against lipopolysaccharide-induced inflammation and inflammasome activation in astrocytes. Inflamm Res. 2020;69:779–87. https://doi.org/10.1007/s00011-020-01355-9.
Li X, Shi MQ, Chen C, Du JR. Phthalide derivative CD21 ameliorates ischemic brain injury in a mouse model of global cerebral ischemia: involvement of inhibition of NLRP3. Int Immunopharmacol. 2020;86:106714. https://doi.org/10.1016/j.intimp.2020.106714.
Lin CH, Chen HY, Wei KC: Role of HMGB1/TLR4 axis in ischemia/reperfusion-impaired extracellular glutamate clearance in primary astrocytes. Cells 2020, 9. https://doi.org/10.3390/cells9122585.
Hayakawa K, Nakano T, Irie K, Higuchi S, Fujioka M, Orito K, Iwasaki K, Jin G, Lo EH, Mishima K, Fujiwara M. Inhibition of reactive astrocytes with fluorocitrate retards neurovascular remodeling and recovery after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2010;30:871–82. https://doi.org/10.1038/jcbfm.2009.257.
Bui TA, Jickling GC, Winship IR. Neutrophil dynamics and inflammaging in acute ischemic stroke: a transcriptomic review. Front Aging Neurosci. 2022;14:1041333. https://doi.org/10.3389/fnagi.2022.1041333.
Aly M, Abdalla RN, Batra A, Shaibani A, Hurley MC, Jahromi BS, Potts MB, Ansari SA. Follow-up neutrophil-lymphocyte ratio after stroke thrombectomy is an independent biomarker of clinical outcome. J Neurointerv Surg. 2021;13:609–13. https://doi.org/10.1136/neurintsurg-2020-016342.
Rosell A, Cuadrado E, Ortega-Aznar A, Hernández-Guillamon M, Lo EH, Montaner J. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke. 2008;39:1121–6. https://doi.org/10.1161/strokeaha.107.500868.
Sørensen OE, Borregaard N. Neutrophil extracellular traps - the dark side of neutrophils. J Clin Invest. 2016;126:1612–20. https://doi.org/10.1172/jci84538.
Kim SW, Lee JK: Role of HMGB1 in the interplay between NETosis and thrombosis in ischemic stroke: a review. Cells 2020, 9. https://doi.org/10.3390/cells9081794.
Ducroux C, Di Meglio L, Loyau S, Delbosc S, Boisseau W, Deschildre C, Ben Maacha M, Blanc R, Redjem H, Ciccio G, et al. Thrombus neutrophil extracellular traps content impair tPA-induced thrombolysis in acute ischemic stroke. Stroke. 2018;49:754–7. https://doi.org/10.1161/strokeaha.117.019896.
Essig F, Babilon L, Vollmuth C, Kollikowski AM, Pham M, Solymosi L, Haeusler KG, Kraft P, Stoll G, Schuhmann MK: High mobility group box 1 protein in cerebral thromboemboli. Int J Mol Sci 2021, 22. https://doi.org/10.3390/ijms222011276.
Maugeri N, Rovere-Querini P, Baldini M, Baldissera E, Sabbadini MG, Bianchi ME, Manfredi AA. Oxidative stress elicits platelet/leukocyte inflammatory interactions via HMGB1: a candidate for microvessel injury in sytemic sclerosis. Antioxid Redox Signal. 2014;20:1060–74. https://doi.org/10.1089/ars.2013.5298.
Ma YH, Ma TT, Wang C, Wang H, Chang DY, Chen M, Zhao MH. High-mobility group box 1 potentiates antineutrophil cytoplasmic antibody-inducing neutrophil extracellular traps formation. Arthritis Res Ther. 2016;18:2. https://doi.org/10.1186/s13075-015-0903-z.
Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, Maiuri L, Maseri A, D’Angelo A, Bianchi ME, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12:2074–88. https://doi.org/10.1111/jth.12710.
Chang CY, Kao TK, Chen WY, Ou YC, Li JR, Liao SL, Raung SL, Chen CJ. Tetramethylpyrazine inhibits neutrophil activation following permanent cerebral ischemia in rats. Biochem Biophys Res Commun. 2015;463:421–7. https://doi.org/10.1016/j.bbrc.2015.05.088.
Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, Orthey E, Arumugam TV, Leypoldt F, Simova O, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. 2012;120:3793–802. https://doi.org/10.1182/blood-2012-02-412726.
Zhang D, Ren J, Luo Y, He Q, Zhao R, Chang J, Yang Y, Guo ZN. T cell response in ischemic stroke: from mechanisms to translational insights. Front Immunol. 2021;12:707972. https://doi.org/10.3389/fimmu.2021.707972.
Heindl S, Ricci A, Carofiglio O, Zhou Q, Arzberger T, Lenart N, Franzmeier N, Hortobagyi T, Nelson PT, Stowe AM, et al: Chronic T cell proliferation in brains after stroke could interfere with the efficacy of immunotherapies. J Exp Med 2021, 218. https://doi.org/10.1084/jem.20202411.
Xie L, Li W, Hersh J, Liu R, Yang SH. Experimental ischemic stroke induces long-term T cell activation in the brain. J Cereb Blood Flow Metab. 2019;39:2268–76. https://doi.org/10.1177/0271678x18792372.
Suidan GL, Dickerson JW, Chen Y, McDole JR, Tripathi P, Pirko I, Seroogy KB, Johnson AJ. CD8 T cell-initiated vascular endothelial growth factor expression promotes central nervous system vascular permeability under neuroinflammatory conditions. J Immunol. 2010;184:1031–40. https://doi.org/10.4049/jimmunol.0902773.
Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. 2015;15:388–400. https://doi.org/10.1038/nri3839.
Voskoboinik I, Dunstone MA, Baran K, Whisstock JC, Trapani JA. Perforin: structure, function, and role in human immunopathology. Immunol Rev. 2010;235:35–54. https://doi.org/10.1111/j.0105-2896.2010.00896.x.
Kleinschnitz C, Kraft P, Dreykluft A, Hagedorn I, Göbel K, Schuhmann MK, Langhauser F, Helluy X, Schwarz T, Bittner S, et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood. 2013;121:679–91. https://doi.org/10.1182/blood-2012-04-426734.
Wang H, Wang Z, Wu Q, Yuan Y, Cao W, Zhang X. Regulatory T cells in ischemic stroke. CNS Neurosci Ther. 2021;27:643–51. https://doi.org/10.1111/cns.13611.
Liu QY, Yao YM, Yan YH, Dong N, Sheng ZY. High mobility group box 1 protein suppresses T cell-mediated immunity via CD11c(low)CD45RB(high) dendritic cell differentiation. Cytokine. 2011;54:205–11. https://doi.org/10.1016/j.cyto.2011.01.008.
Huang LF, Yao YM, Meng HD, Zhao XD, Dong N, Yu Y, Sheng ZY. The effect of high mobility group box-1 protein on immune function of human T lymphocytes in vitro. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2008;20:7–13.
Sundberg E, Fasth AE, Palmblad K, Harris HE, Andersson U. High mobility group box chromosomal protein 1 acts as a proliferation signal for activated T lymphocytes. Immunobiology. 2009;214:303–9. https://doi.org/10.1016/j.imbio.2008.09.006.
Xiong X, Gu L, Wang Y, Luo Y, Zhang H, Lee J, Krams S, Zhu S, Zhao H. Glycyrrhizin protects against focal cerebral ischemia via inhibition of T cell activity and HMGB1-mediated mechanisms. J Neuroinflammation. 2016;13:241. https://doi.org/10.1186/s12974-016-0705-5.
Tian Y, Cao Y, Chen R, Jing Y, Xia L, Zhang S, Xu H, Su Z. HMGB1 A box protects neurons by potently inhibiting both microglia and T cell-mediated inflammation in a mouse Parkinson’s disease model. Clin Sci (Lond). 2020;134:2075–90. https://doi.org/10.1042/cs20200553.
Ding JW, Zheng XX, Zhou T, Tong XH, Luo CY, Wang XA. HMGB1 modulates the Treg/Th17 ratio in atherosclerotic patients. J Atheroscler Thromb. 2016;23:737–45. https://doi.org/10.5551/jat.31088.
Zhu XM, Yao YM, Liang HP, Xu CT, Dong N, Yu Y, Sheng ZY. High mobility group box-1 protein regulate immunosuppression of regulatory T cells through toll-like receptor 4. Cytokine. 2011;54:296–304. https://doi.org/10.1016/j.cyto.2011.02.017.
Huang LF, Yao YM, Zhang LT, Dong N, Yu Y, Sheng ZY. The effect of high-mobility group box 1 protein on activity of regulatory T cells after thermal injury in rats. Shock. 2009;31:322–9. https://doi.org/10.1097/SHK.0b013e3181834070.
Wild CA, Bergmann C, Fritz G, Schuler P, Hoffmann TK, Lotfi R, Westendorf A, Brandau S, Lang S. HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int Immunol. 2012;24:485–94. https://doi.org/10.1093/intimm/dxs051.
Cheng LS, Li J, Liu Y, Wang FP, Wang SQ, She WM, Wu SD, Qi XL, Zhou YP, Jiang W. HMGB1-induced autophagy: a new pathway to maintain Treg function during chronic hepatitis B virus infection. Clin Sci (Lond). 2017;131:381–94. https://doi.org/10.1042/cs20160704.
Shi L, Sun Z, Su W, Xu F, Xie D, Zhang Q, Dai X, Iyer K, Hitchens TK, Foley LM, et al. Treg cell-derived osteopontin promotes microglia-mediated white matter repair after ischemic stroke. Immunity. 2021;54:1527-1542.e1528. https://doi.org/10.1016/j.immuni.2021.04.022.
Zhang J, Takahashi HK, Liu K, Wake H, Liu R, Maruo T, Date I, Yoshino T, Ohtsuka A, Mori S, Nishibori M. Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke. 2011;42:1420–8. https://doi.org/10.1161/strokeaha.110.598334.
Kim JB, Sig Choi J, Yu YM, Nam K, Piao CS, Kim SW, Lee MH, Han PL, Park JS, Lee JK. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J Neurosci. 2006;26:6413–21. https://doi.org/10.1523/jneurosci.3815-05.2006.
Schulze J, Zierath D, Tanzi P, Cain K, Shibata D, Dressel A, Becker K. Severe stroke induces long-lasting alterations of high-mobility group box 1. Stroke. 2013;44:246–8. https://doi.org/10.1161/strokeaha.112.676072.
He M, Zhang B, Wei X, Wang Z, Fan B, Du P, Zhang Y, Jian W, Chen L, Wang L, et al. HDAC4/5-HMGB1 signalling mediated by NADPH oxidase activity contributes to cerebral ischaemia/reperfusion injury. J Cell Mol Med. 2013;17:531–42. https://doi.org/10.1111/jcmm.12040.
Tang D, Kang R, Zeh HJ 3rd, Lotze MT. High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal. 2011;14:1315–35. https://doi.org/10.1089/ars.2010.3356.
Qiao H, Xu Q, Xu Y, Zhao Y, He N, Tang J, Zhao J, Liu Y. Molecular chaperones in stroke-induced immunosuppression. Neural Regen Res. 2023;18:2638–44. https://doi.org/10.4103/1673-5374.373678.
Tang D, Kang R, Xiao W, Wang H, Calderwood SK, Xiao X. The anti-inflammatory effects of heat shock protein 72 involve inhibition of high-mobility-group box 1 release and proinflammatory function in macrophages. J Immunol. 2007;179:1236–44. https://doi.org/10.4049/jimmunol.179.2.1236.
Chen S, Pan J, Gong Z, Wu M, Zhang X, Chen H, Yang D, Qi S, Peng Y, Shen J. Hypochlorous acid derived from microglial myeloperoxidase could mediate high-mobility group box 1 release from neurons to amplify brain damage in cerebral ischemia-reperfusion injury. J Neuroinflammation. 2024;21:70. https://doi.org/10.1186/s12974-023-02991-8.
Liu K, Mori S, Takahashi HK, Tomono Y, Wake H, Kanke T, Sato Y, Hiraga N, Adachi N, Yoshino T, Nishibori M. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. Faseb j. 2007;21:3904–16. https://doi.org/10.1096/fj.07-8770com.
Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500. https://doi.org/10.1038/nm1735.
Hayakawa K, Pham LD, Katusic ZS, Arai K, Lo EH. Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proc Natl Acad Sci U S A. 2012;109:7505–10. https://doi.org/10.1073/pnas.1121146109.
Chen C, Lin X, Wang J, Tang G, Mu Z, Chen X, Xu J, Wang Y, Zhang Z, Yang GY. Effect of HMGB1 on the paracrine action of EPC promotes post-ischemic neovascularization in mice. Stem Cells. 2014;32:2679–89. https://doi.org/10.1002/stem.1754.
Ghaffari S, Jang E, Naderinabi F, Sanwal R, Khosraviani N, Wang C, Steinberg BE, Goldenberg NM, Ikeda J, Lee WL. Endothelial HMGB1 is a critical regulator of LDL transcytosis via an SREBP2-SR-BI axis. Arterioscler Thromb Vasc Biol. 2021;41:200–16. https://doi.org/10.1161/atvbaha.120.314557.
Kanellakis P, Agrotis A, Kyaw TS, Koulis C, Ahrens I, Mori S, Takahashi HK, Liu K, Peter K, Nishibori M, Bobik A. High-mobility group box protein 1 neutralization reduces development of diet-induced atherosclerosis in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31:313–9. https://doi.org/10.1161/atvbaha.110.218669.
Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, Suffredini AF. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–60. https://doi.org/10.1182/blood-2002-05-1300.
Filipek A, Czerwińska ME, Kiss AK, Polański JA, Naruszewicz M: Oleacein may inhibit destabilization of carotid plaques from hypertensive patients. Impact on high mobility group protein-1. Phytomedicine 2017, 32:68–73. https://doi.org/10.1016/j.phymed.2017.06.004.
Wang J, Jiang Y, Zeng D, Zhou W, Hong X. Prognostic value of plasma HMGB1 in ischemic stroke patients with cerebral ischemia-reperfusion injury after intravenous thrombolysis. J Stroke Cerebrovasc Dis. 2020;29:105055. https://doi.org/10.1016/j.jstrokecerebrovasdis.2020.105055.
Schuhmann MK, Kollikowski AM, März AG, Bieber M, Pham M, Stoll G. Danger-associated molecular patterns are locally released during occlusion in hyper-acute stroke. Brain Behav Immun Health. 2021;15:100270. https://doi.org/10.1016/j.bbih.2021.100270.
Tsukagawa T, Katsumata R, Fujita M, Yasui K, Akhoon C, Ono K, Dohi K, Aruga T. Elevated serum high-mobility group box-1 protein level is associated with poor functional outcome in ischemic stroke. J Stroke Cerebrovasc Dis. 2017;26:2404–11. https://doi.org/10.1016/j.jstrokecerebrovasdis.2017.05.033.
Sapojnikova N, Kartvelishvili T, Asatiani N, Zinkevich V, Kalandadze I, Gugutsidze D, Shakarishvili R, Tsiskaridze A. Correlation between MMP-9 and extracellular cytokine HMGB1 in prediction of human ischemic stroke outcome. Biochim Biophys Acta. 2014;1842:1379–84. https://doi.org/10.1016/j.bbadis.2014.04.031.
Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. Embo j. 2003;22:5551–60. https://doi.org/10.1093/emboj/cdg516.
Gelderblom M, Sobey CG, Kleinschnitz C, Magnus T. Danger signals in stroke. Ageing Res Rev. 2015;24:77–82. https://doi.org/10.1016/j.arr.2015.07.004.
Shan W, Xu L, Qiu Z, Wang J, Shao J, Feng J, Zhao J. Increased high-mobility group box 1 levels are associated with depression after acute ischemic stroke. Neurol Sci. 2022;43:3131–7. https://doi.org/10.1007/s10072-021-05571-x.
E Y, Deng Q, Shi G, Li Z, Liu C, Wang S, Lian H, Cao H, Zhang X, Zhang Y, et al: The association between high mobility group box 1 and stroke-associated pneumonia in acute ischemic stroke patients. Brain Sci 2022, 12. https://doi.org/10.3390/brainsci12111580.
Kim ID, Lee H, Kim SW, Lee HK, Choi J, Han PL, Lee JK. Alarmin HMGB1 induces systemic and brain inflammatory exacerbation in post-stroke infection rat model. Cell Death Dis. 2018;9:426. https://doi.org/10.1038/s41419-018-0438-8.
Wu X, Liu S, Hu Z, Zhu G, Zheng G, Wang G. Enriched housing promotes post-stroke neurogenesis through calpain 1-STAT3/HIF-1α/VEGF signaling. Brain Res Bull. 2018;139:133–43. https://doi.org/10.1016/j.brainresbull.2018.02.018.
Chen JY, Yu Y, Yuan Y, Zhang YJ, Fan XP, Yuan SY, Zhang JC, Yao SL. Enriched housing promotes post-stroke functional recovery through astrocytic HMGB1-IL-6-mediated angiogenesis. Cell Death Discov. 2017;3:17054. https://doi.org/10.1038/cddiscovery.2017.54.
Hayakawa K, Arai K, Lo EH. Role of ERK map kinase and CRM1 in IL-1beta-stimulated release of HMGB1 from cortical astrocytes. Glia. 2010;58:1007–15. https://doi.org/10.1002/glia.20982.
Hayakawa K, Miyamoto N, Seo JH, Pham LD, Kim KW, Lo EH, Arai K. High-mobility group box 1 from reactive astrocytes enhances the accumulation of endothelial progenitor cells in damaged white matter. J Neurochem. 2013;125:273–80. https://doi.org/10.1111/jnc.12120.
Hei Y, Chen R, Yi X, Long Q, Gao D, Liu W. HMGB1 neutralization attenuates hippocampal neuronal death and cognitive impairment in rats with chronic cerebral hypoperfusion via suppressing inflammatory responses and oxidative stress. Neuroscience. 2018;383:150–9. https://doi.org/10.1016/j.neuroscience.2018.05.010.
Hei Y, Zhang X, Chen R, Zhou Y, Gao D, Liu W. High-mobility group box 1 neutralization prevents chronic cerebral hypoperfusion-induced optic tract injuries in the white matter associated with down-regulation of inflammatory responses. Cell Mol Neurobiol. 2019;39:1051–60. https://doi.org/10.1007/s10571-019-00702-7.
Underly RG, Levy M, Hartmann DA, Grant RI, Watson AN, Shih AY. Pericytes as Inducers of rapid, matrix metalloproteinase-9-dependent capillary damage during ischemia. J Neurosci. 2017;37:129–40. https://doi.org/10.1523/jneurosci.2891-16.2016.
Nishibori M, Mori S, Takahashi HK. Anti-HMGB1 monoclonal antibody therapy for a wide range of CNS and PNS diseases. J Pharmacol Sci. 2019;140:94–101. https://doi.org/10.1016/j.jphs.2019.04.006.
Liu N, Liu C, Yang Y, Ma G, Wei G, Liu S, Kong L, Du G. Xiao-Xu-Ming decoction prevented hemorrhagic transformation induced by acute hyperglycemia through inhibiting AGE-RAGE-mediated neuroinflammation. Pharmacol Res. 2021;169:105650. https://doi.org/10.1016/j.phrs.2021.105650.
Gao S, Wake H, Sakaguchi M, Wang D, Takahashi Y, Teshigawara K, Zhong H, Mori S, Liu K, Takahashi H, Nishibori M: Histidine-rich glycoprotein inhibits high-mobility group box-1-mediated pathways in vascular endothelial cells through CLEC-1A. iScience 2020, 23:101180. https://doi.org/10.1016/j.isci.2020.101180.
Li M, Chen S, Shi X, Lyu C, Zhang Y, Tan M, Wang C, Zang N, Liu X, Hu Y, et al. Cell permeable HMGB1-binding heptamer peptide ameliorates neurovascular complications associated with thrombolytic therapy in rats with transient ischemic stroke. J Neuroinflammation. 2018;15:237. https://doi.org/10.1186/s12974-018-1267-5.
Krishnan R, Mays W, Elijovich L. Complications of mechanical thrombectomy in acute ischemic stroke. Neurology. 2021;97:S115-s125. https://doi.org/10.1212/wnl.0000000000012803.
Mu SW, Dang Y, Fan YC, Zhang H, Zhang JH, Wang W, Wang SS, Gu JJ. Effect of HMGB1 and RAGE on brain injury and the protective mechanism of glycyrrhizin in intracranial-sinus occlusion followed by mechanical thrombectomy recanalization. Int J Mol Med. 2019;44:813–22. https://doi.org/10.3892/ijmm.2019.4248.
Pir GJ, Parray A, Ayadathil R, Pananchikkal SV, Mir FA, Muhammad I, Abubakar A, Amir N, Hussain S, Haroon KH, et al: Platelet-neutrophil association in NETs-rich areas in the retrieved AIS patient thrombi. Int J Mol Sci 2022, 23. https://doi.org/10.3390/ijms232214477.
Luo H, Guo H, Zhou Y, Fang R, Zhang W, Mei Z. Neutrophil extracellular traps in cerebral ischemia/reperfusion injury: friend and foe. Curr Neuropharmacol. 2023;21:2079–96. https://doi.org/10.2174/1570159x21666230308090351.
Chen X, Wang L, Jiang M, Lin L, Ba Z, Tian H, Li G, Chen L, Liu Q, Hou X, et al. Leukocytes in cerebral thrombus respond to large-vessel occlusion in a time-dependent manner and the association of NETs with collateral flow. Front Immunol. 2022;13:834562. https://doi.org/10.3389/fimmu.2022.834562.
Chen H, He Y, Chen S, Qi S, Shen J. Therapeutic targets of oxidative/nitrosative stress and neuroinflammation in ischemic stroke: applications for natural product efficacy with omics and systemic biology. Pharmacol Res. 2020;158:104877. https://doi.org/10.1016/j.phrs.2020.104877.
Yang H, Qi C, Su F, Shan W, Guo A, Wu J, Wang Y, You H, Wang Q. Cerebral ischemia/reperfusion injury and pharmacologic preconditioning as a means to reduce stroke-induced inflammation and damage. Neurochem Res. 2022;47:3598–614. https://doi.org/10.1007/s11064-022-03789-5.
Kong L, Ma Y, Wang Z, Liu N, Ma G, Liu C, Shi R, Du G. Inhibition of hypoxia inducible factor 1 by YC-1 attenuates tissue plasminogen activator induced hemorrhagic transformation by suppressing HMGB1/TLR4/NF-κB mediated neutrophil infiltration in thromboembolic stroke rats. Int Immunopharmacol. 2021;94:107507. https://doi.org/10.1016/j.intimp.2021.107507.
Ismael S, Nasoohi S, Yoo A, Ahmed HA, Ishrat T. Tissue plasminogen activator promotes TXNIP-NLRP3 inflammasome activation after hyperglycemic stroke in mice. Mol Neurobiol. 2020;57:2495–508. https://doi.org/10.1007/s12035-020-01893-7.
Carnevale D, Mascio G, D’Andrea I, Fardella V, Bell RD, Branchi I, Pallante F, Zlokovic B, Yan SS, Lembo G. Hypertension induces brain β-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension. 2012;60:188–97. https://doi.org/10.1161/hypertensionaha.112.195511.
Nakamura T, Sato E, Fujiwara N, Kawagoe Y, Yamada S, Ueda Y, Koide H. Changes in urinary albumin excretion, inflammatory and oxidative stress markers in ADPKD patients with hypertension. Am J Med Sci. 2012;343:46–51. https://doi.org/10.1097/MAJ.0b013e31821f0552.
Kikuchi K, Tancharoen S, Ito T, Morimoto-Yamashita Y, Miura N, Kawahara K, Maruyama I, Murai Y, Tanaka E. Potential of the angiotensin receptor blockers (ARBs) telmisartan, irbesartan, and candesartan for inhibiting the HMGB1/RAGE axis in prevention and acute treatment of stroke. Int J Mol Sci. 2013;14:18899–924. https://doi.org/10.3390/ijms140918899.
Wang J, Han D, Sun M, Feng J. A combination of remote ischemic perconditioning and cerebral ischemic postconditioning inhibits autophagy to attenuate plasma HMGB1 and induce neuroprotection against stroke in Rat. J Mol Neurosci. 2016;58:424–31. https://doi.org/10.1007/s12031-016-0724-9.
Zhao PC, Xu SN, Huang ZS, Jiang GW, Deng PC, Zhang YM. Hyperbaric oxygen via mediating SIRT1-induced deacetylation of HMGB1 improved cReperfusion inj/reperfusion injury. Eur J Neurosci. 2021;54:7318–31. https://doi.org/10.1111/ejn.15458.
Lee JH, Yoon EJ, Seo J, Kavoussi A, Chung YE, Chung SP, Park I, Kim CH, You JS. Hypothermia inhibits the propagation of acute ischemic injury by inhibiting HMGB1. Mol Brain. 2016;9:81. https://doi.org/10.1186/s13041-016-0260-0.
Yawoot N, Chumboatong W, Sengking J, Tocharus C, Tocharus J. Chronic high-fat diet consumption exacerbates pyroptosis- and necroptosis-mediated HMGB1 signaling in the brain after ischemia and reperfusion injury. J Physiol Biochem. 2022;78:833–44. https://doi.org/10.1007/s13105-022-00906-4.
Pan G, Jin L, Shen W, Zhang J, Pan J, Cheng J, Xie Q, Hu Q, Wu S, Zhang H, Chen X. Treadmill exercise improves neurological function by inhibiting autophagy and the binding of HMGB1 to Beclin1 in MCAO juvenile rats. Life Sci. 2020;243:117279. https://doi.org/10.1016/j.lfs.2020.117279.
Lok KZ, Basta M, Manzanero S, Arumugam TV. Intravenous immunoglobulin (IVIg) dampens neuronal toll-like receptor-mediated responses in ischemia. J Neuroinflammation. 2015;12:73. https://doi.org/10.1186/s12974-015-0294-8.
Nakano T, Tagashira Y, Egashira S, Morimoto M, Irie K, Hosokawa M, Hayashi T, Egawa T, Hayakawa K, Mishima K. Therapeutic effect of anti-HMGB1 antibody in a mouse model of 4-h middle cerebral artery occlusion: comparison with tissue plasminogen activator. NeuroReport. 2022;33:297–303. https://doi.org/10.1097/wnr.0000000000001780.
Jin YC, Kim SW, Cheng F, Shin JH, Park JK, Lee S, Lee JE, Han PL, Lee M, Kim KK, et al. The effect of biodegradable gelatin microspheres on the neuroprotective effects of high mobility group box 1 A box in the postischemic brain. Biomaterials. 2011;32:899–908. https://doi.org/10.1016/j.biomaterials.2010.09.054.
Kim ID, Shin JH, Kim SW, Choi S, Ahn J, Han PL, Park JS, Lee JK. Intranasal delivery of HMGB1 siRNA confers target gene knockdown and robust neuroprotection in the postischemic brain. Mol Ther. 2012;20:829–39. https://doi.org/10.1038/mt.2011.291.
Kim ID, Lim CM, Kim JB, Nam HY, Nam K, Kim SW, Park JS, Lee JK. Neuroprotection by biodegradable PAMAM ester (e-PAM-R)-mediated HMGB1 siRNA delivery in primary cortical cultures and in the postischemic brain. J Control Release. 2010;142:422–30. https://doi.org/10.1016/j.jconrel.2009.11.011.
Dang Y, An C, Li Y, Han D, Liu X, Zhang F, Xu Y, Zhong H, Karim Khan MK, Zou F, Sun X. Neutrophil-mediated and low density lipoprotein receptor-mediated dual-targeting nanoformulation enhances brain accumulation of scutellarin and exerts neuroprotective effects against ischemic stroke. RSC Adv. 2019;9:1299–318. https://doi.org/10.1039/c8ra06688d.
Wang L, Zhang X, Liu L, Yang R, Cui L, Li M. Atorvastatin protects rat brains against permanent focal ischemia and downregulates HMGB1, HMGB1 receptors (RAGE and TLR4). NF-kappaB expression Neurosci Lett. 2010;471:152–6. https://doi.org/10.1016/j.neulet.2010.01.030.
Chibaatar E, Le K, Abdoulaye IA, Wu S, Guo Y. Melatonin ameliorates lipopolysaccharide-induced microglial inflammation via triggering SIRT1/HMGB1 signaling axis. J Mol Neurosci. 2021;71:691–701. https://doi.org/10.1007/s12031-020-01699-1.
Ma H, Su D, Wang Q, Chong Z, Zhu Q, He W, Wang W. Phoenixin 14 inhibits ischemia/reperfusion-induced cytotoxicity in microglia. Arch Biochem Biophys. 2020;689:108411. https://doi.org/10.1016/j.abb.2020.108411.
Ye Y, Jin T, Zhang X, Zeng Z, Ye B, Wang J, Zhong Y, Xiong X, Gu L. Meisoindigo protects against focal cerebral ischemia-reperfusion injury by inhibiting NLRP3 inflammasome activation and regulating microglia/macrophage polarization via TLR4/NF-κB signaling pathway. Front Cell Neurosci. 2019;13:553. https://doi.org/10.3389/fncel.2019.00553.
Wang Q, Zhao H, Gao Y, Lu J, Xie D, Yu W, He F, Liu W, Hisatome I, Yamamoto T, et al. Uric acid inhibits HMGB1-TLR4-NF-κB signaling to alleviate oxygen-glucose deprivation/reoxygenation injury of microglia. Biochem Biophys Res Commun. 2021;540:22–8. https://doi.org/10.1016/j.bbrc.2020.12.097.
Choi HW, Tian M, Song F, Venereau E, Preti A, Park SW, Hamilton K, Swapna GV, Manohar M, Moreau M, et al. Aspirin’s active metabolite salicylic acid targets high mobility group box 1 to modulate inflammatory responses. Mol Med. 2015;21:526–35. https://doi.org/10.2119/molmed.2015.00148.
Goetzl EJ, Goetzl L, Karliner JS, Tang N, Pulliam L. Human plasma platelet-derived exosomes: effects of aspirin. Faseb j. 2016;30:2058–63. https://doi.org/10.1096/fj.201500150R.
Nakamura Y, Nakano T, Irie K, Sano K, Tanaka J, Yamashita Y, Satho T, Matsuo K, Fujioka M, Ishikura H, Mishima K. Recombinant human soluble thrombomodulin ameliorates cerebral ischemic injury through a high-mobility group box 1 inhibitory mechanism without hemorrhagic complications in mice. J Neurol Sci. 2016;362:278–82. https://doi.org/10.1016/j.jns.2016.01.047.
Gong G, Xiang L, Yuan L, Hu L, Wu W, Cai L, Yin L, Dong H. Protective effect of glycyrrhizin, a direct HMGB1 inhibitor, on focal cerebral ischemia/reperfusion-induced inflammation, oxidative stress, and apoptosis in rats. PLoS ONE. 2014;9: e89450. https://doi.org/10.1371/journal.pone.0089450.
Zhu JR, Lu HD, Guo C, Fang WR, Zhao HD, Zhou JS, Wang F, Zhao YL, Li YM, Zhang YD, et al. Berberine attenuates ischemia-reperfusion injury through inhibiting HMGB1 release and NF-κB nuclear translocation. Acta Pharmacol Sin. 2018;39:1706–15. https://doi.org/10.1038/s41401-018-0160-1.
Tao X, Sun X, Yin L, Han X, Xu L, Qi Y, Xu Y, Li H, Lin Y, Liu K, Peng J. Dioscin ameliorates cerebral ischemia/reperfusion injury through the downregulation of TLR4 signaling via HMGB-1 inhibition. Free Radic Biol Med. 2015;84:103–15. https://doi.org/10.1016/j.freeradbiomed.2015.03.003.
Xie W, Zhu T, Dong X, Nan F, Meng X, Zhou P, Sun G, Sun X: HMGB1-triggered inflammation inhibition of notoginseng leaf triterpenes against cerebral ischemia and reperfusion injury via MAPK and NF-κB signaling pathways. Biomolecules 2019, 9. https://doi.org/10.3390/biom9100512.
Xie W, Zhou P, Sun Y, Meng X, Dai Z, Sun G, Sun X: Protective effects and target network analysis of ginsenoside Rg1 in cerebral ischemia and reperfusion injury: a comprehensive overview of experimental studies. Cells 2018, 7. https://doi.org/10.3390/cells7120270.
Zhang W, Song J, Li W, Kong D, Liang Y, Zhao X, Du G. Salvianolic acid D alleviates cerebral ischemia-reperfusion injury by suppressing the cytoplasmic translocation and release of HMGB1-triggered NF-κB activation to inhibit inflammatory response. Mediators Inflamm. 2020;2020:9049614. https://doi.org/10.1155/2020/9049614.
Li H, Wu W, Sun Q, Liu M, Li W, Zhang XS, Zhou ML, Hang CH. Expression and cell distribution of receptor for advanced glycation end-products in the rat cortex following experimental subarachnoid hemorrhage. Brain Res. 2014;1543:315–23. https://doi.org/10.1016/j.brainres.2013.11.023.
Acknowledgements
Figures were created with Biorender.com.
Funding
This work was supported by the National Natural Science Foundation of China (82271312 and 82071314).
Author information
Authors and Affiliations
Contributions
JML and ZXW conceptualized the review. JML drafted the manuscript and drew the figures. ZXW and JML worked on manuscript editing. QFM and HPZ revised the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable as no patients/participants were involved in this review.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, J., Wang, Z., Li, J. et al. HMGB1: A New Target for Ischemic Stroke and Hemorrhagic Transformation. Transl. Stroke Res. (2024). https://doi.org/10.1007/s12975-024-01258-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12975-024-01258-5