
Abstract
The unprecedented threats to coral reef ecosystems from global climate change require an urgent response from the aquarium community, which is becoming an increasingly vital coral conservation resource. Unfortunately, many hermatypic corals in aquaria are not identified to species level, which hinders assessment of their conservation significance. Traditional methods of species identification using morphology can be challenging, especially to non-taxonomists. DNA barcoding is an option for species identification of Scleractinian corals, especially when used in concert with morphology-based assessment. This study uses DNA barcodes to try to identify aquarium specimens of the diverse reef-forming genus Acropora from 127 samples. We identified to our best current knowledge, to species name 44% of the analysed samples and provided provisional identification for 80% of them (101/127, in the form of a list of species names with associate confidence values). We highlighted a sampling bias in public nucleotide sequences repertories (e.g. GenBank) towards more charismatic and more studied species, even inside a well-studied genus like Acropora. In addition, we showed a potential “single observer” effect with over a quarter of the reference sequences used for these identifications coming from the same study. We propose the use of barcoding and query matching as an additional tool for taxonomic experts and general aquarists, as an additional tool to increase their chances of making high confidence species-level identifications. We produce a standardised and easily repeatable methodology to increase the capacity of aquariums and other facilities to assess non-ascribed species, emphasising the value of integrating this approach with morphological identification optimising usage of authoritative identification guides and expert opinion.
Similar content being viewed by others

Introduction
It is widely acknowledged that coral reef ecosystems are facing unprecedented threats from global climate change (GCC) e.g. (Pratchett et al. 2013; Hoegh-Guldberg et al. 2017; Cowburn et al. 2018; Hughes et al. 2018; Masson-Delmotte et al. 2018; Brondizio et al. 2019; Sheppard et al. 2020). Warm water reef-building corals create the most biodiverse ecosystems on the planet and have been estimated to support 830,000 species of multi-cellular plants and animals worldwide (Bellwood and Hughes 2001; Mora et al. 2008, 2011; Fisher et al. 2015), providing a variety of habitats for fish, invertebrates and other taxa in shallow tropical seas (Bellwood and Hughes 2001). Acropora corals play a key functional ecosystem role as they are the major reef builders in the majority of warm water reefs ecosystems (Fukami et al. 2000).
Corals in aquaria
Ex situ conservation can play an important role in ensuring the survival of many species (Turley 1999; Hutchins 2003; Raven 2004; Blanco et al. 2009; Fa et al. 2011; Leus et al. 2011; Lacy 2013; Skibins and Powell 2013). Given the severity of the species and ecosystem level climate change threats to corals, and the alarming rates of recent losses (Masson-Delmotte et al. 2018), coral collection can potentially play a valuable role in future conservation efforts. Aquaria institutions therefore have a vitally important conservation remit to support coral restoration strategies allowing for both sexual and asexual coral recruitment, biobanking and a wide range of research, education and thus contribution towards in situ conservation activities—although most of the progress with ex-situ coral recruitment has been done recently (Petersen et al. 2006, 2007; Craggs et al. 2017; O’Neil et al. 2021). Early attempts in the 1980s at keeping corals in aquaria faced difficulties (Borneman 2008; Brunner 2012) it was until 1980 that Acropora was the first genus to be successfully cultured by Stüber (Borneman 2008). From the 1990s onwards coral husbandry boomed, reaching a level where virtually all families of zooxanthellate corals were not only being maintained for many years, but were being propagated and traded between private and public aquaria (Borneman 2008). Specimens conserved in Aquaria thus, became a highly valuable resource for restoration strategies (Rinkevich 1995).
Identification of corals
Coral taxonomy is a traditionally difficult discipline, subject to all the challenges of modern taxonomy (Godfray 2002). The genus Acropora is among the most diverse and geographically widespread reef building corals (Wallace 1999). Research on Acropora suggests that species limits may be sometimes narrower, sometimes broader, than generally perceived (Wallace and Willis 1994). Somewhat blurry species boundaries are confounded by hybridisation (Willis 1997; Hatta et al. 1999; Van Oppen et al. 2002; Vollmer and Palumbi 2002; Wolstenholme 2004; Willis et al. 2006; Wei et al. 2012; Palumbi et al. 2012; Isomura et al. 2013), often worsened by the possibility of synchronised spawning events (Harrison et al. 1984; Babcock et al. 1986; Van Oppen et al. 2002; Márquez et al. 2002). Currently the number of officially recognised Acropora species are between 135 [WORMS (Horton et al. 2020)] and 163 [Corals of the World (Veron et al. 2020)], reaching 186 if we include the “taxon inquirendum” on WORMS (Horton et al. 2020). There have been different attempts at grouping these species within the genus Acropora (Wallace 1999). These attempts lead to the creation of the concept of species group (aka syngameon). The concept of species groups, first defined for convenience of identification and without implying taxonomic relation, has been revised to reflect the evolution and phylogeny of the genus (Wallace 1999; Wallace et al. 2012). The use of genetic tools for species identification has provided another line of evidence for species delimitation, further adding to the blurred boundaries between species groups, species, and sub-species.
Traditionally, identification of coral has relied on morphological skeletal features, with the great majority of taxa originally described following typological species concepts defined around a century ago (Best et al. 1984; Todd 2008). The relationship between morphological variation within-species and the environment has been a source of contention since the late 1800s (Todd 2008). Whether this variability is due to different underlying genotypes or plastic phenotypes is unclear. Evidence supports both scenarios [difference in phenotype e.g. (Willis 1985; Ayre and Willis 1988; Todd et al. 2004)– Plastic phenotypes e.g.: (Foster 1979; Miller 1994; Bruno and Edmunds 1997; Muko et al. 2000; Todd et al. 2004)] and these scenarios are not necessary mutually exclusive but probable to operate simultaneously (Foster 1979; Amaral 1994; Todd 2008). This has particular relevance in the face of continuous GCC that is creating an increasingly unpredictable wild reef environment.
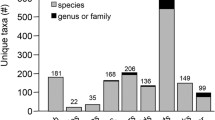
Notwithstanding the taxonomic difficulties, the identification of coral species can be challenging even in optimal ‘wild’ conditions. However, identification requirements are far more than just for well documented field collections, but also cover aquaria collections, that in some cases are held in conditions dramatically different from the wild. Furthermore, customs officials rely in proper taxonomic identification even at the genera level for threatened taxa by the international wildlife trade (AC25 Doc. 23 CITES 2011), although currently Acropora are covered by the blanket listing of all Scleractinia under CITES appendix II. A status of these identification difficulties in aquaria can be demonstrated by an analysis of Species360 Zoological Information Management Software (ZIMS), adopted by more than 95 aquariums in 24 countries. Assuming correct identification ZIMS indicates c. 42.9% of corals of the orders Scleractinia, Alcyonacea, Helioporacea, Antipatharia, Corallimorpharia, Pennatulacea, and Zoanthidea in aquaria are identified to genus or higher taxonomic level (ZIMS list of species holdings—4th June 2020). Most of the records correspond to the order Scleractinia, which is also the order with more species represented in aquariums. Corallimorpharia is, however, the order with the highest number of reported individuals in aquariums members of the Species360 network (Fig. 1). In addition to this the non-species associated genera (e.g.: Acropora sp.) is usually recorded once in the ZIMS database and most other aquarium inventory systems, which possibly significantly underestimates the actual number of non-identified specimens. Moreover, there are currently no confidence values ascribed to any taxonomic level a collection allocates to a specimen which means that there is likely to mean that a significant number of species ascribed specimens are actually at the lower end of the confidence spectrum and would benefit from being reassessed.

Number of records from member institutions of the Species360 network at genus or higher level in light blue (i.e. order, family and genus) and at species or lower level in purple (i.e. species and subspecies). This information is based on the rank field from the holdings information reported by each institution through the ZIMS software. Information downloaded from ZIMS (Species360, 2020) on 4th June 2020
In many aquariums’, corals come from three sources: fragments from other aquaria, via ornamental trade, or from confiscated and seized shipments (which aquariums are usually asked to accommodate). All three sources present identification challenges as often exact knowledge from their wild origin is absent. This specially a challenge for confiscated specimens since they have none or relatively poor source records, sample integrity, and any other additional background information that might be of assistance for the identification process. This, together with the difficulties caused by environmental phenotypic plasticity makes coral identification in aquariums a particularly challenging task. Data from the Convention on the International Trade of Endangered Species of Flora and Fauna (CITES) shows a total of 4557 instances of coral confiscation from 1982 to 2018 worldwide, for a total of 27,027 individual items and 5461 kg per year (CITES data—https://trade.cites.org 02/07/2020). Of these, less than 20% are reported at species level, meaning that for most cases we know only the genus, family or even order of the specimens confiscated. Given these challenges CITES currently requires identification only to the order level.
DNA barcoding
An alternative or complementary diagnostic option to morphological identification is genetic analysis. DNA Barcodes have been proposed by Hebert et al. (2003) as a way to circumvent the “limitations inherent in morphology-based identification systems”. The core concept of DNA barcoding is the use of a standard sequence corresponding to a single homologous region that can be amplified with universal primers that is sufficiently variable to distinguish between species—usually represented by the mitochondrially encoded cytochrome c oxidase I (MT-CO1 or CO1 or COX1) (Hebert et al. 2003).
Beside the inherent limitations (Ferguson 2002; Shearer et al. 2002; Moritz and Cicero 2004; DeSalle et al. 2005; Meyer and Paulay 2005; Prendini 2005; Hickerson et al. 2006; Meier et al. 2006, 2008; Cognato 2006; Little and Stevenson 2007), DNA barcoding is especially attractive as an option for species identification of scleractinian corals, as this method bypasses the problems caused by phenotypic plasticity or life stage (larva, juvenile or adult). Unfortunately, the proposed ‘universal’ Barcoding region CO1 is at least 10–20 times slower in Anthozoa than the standard vertebrate mtDNA mutation rate, and 2–5 times slower than the average Anthozoan nuclear sequence(Van Oppen et al. 1999a, b; Shearer et al. 2002; Hellberg 2006; Huang et al. 2008; Chen et al. 2009; Ortman et al. 2010) with zero interspecific divergence for many species (Shearer and Coffroth 2008). This has led to the use of a number of different barcoding regions for corals (Shearer et al. 2002).
The diverse, reef forming Acropora group are widely grown in aquaria and have been a target for barcoding work. Potential barcodes were reviewed by Shearer et al. (2002) examining both nuclear and mitochondrial regions. Substitution rates of mitochondrial genes in Acropora tend to be lower relative to the nuclear counterparts mentioned by Shearer (i.e. Pax-C intron; internal transcribed spacer 1 and 2 (ITS) (Shearer et al. 2002). Although, Van Oppen et al. (1999a, b) and Vollmer et al. (2002) have highlighted the mitochondrial putative control region (mtCR) is more variable than the other mitochondrial regions. Subsequently Shearer et al. (2002) compared the genetic divergence of mitochondrial and nuclear regions in Scleractinia, highlighting how the mtCR region has a similar range of divergence to nuclear coding regions like the Pax-C intron, but is much slower than the hyper-variable ITS1-2. Notwithstanding all the above mentioned work, Acropora’s molecular taxonomy remains to be resolved, and to do so it would require extensive taxonomic work and genome-wide markers (Cowman et al. 2020).
This study seeks to build on these studies by investigating the utility of DNA barcoding as a method for provisional species identification of Acropora collections in aquaria, as an aid to morphological taxonomy rather than an alternative, and as part of an integrated diagnostic approach.
Material and methods
Study area and collection
As part of the “idhelper” project we collected 224 samples across seven institutions (Aquarium de Paris (France), Chester’s Zoo (UK), Haus des Meeres-Vienna (Austria), Horniman Museum and Gardens (UK), Royal Burgers’ Zoo (Netherlands), Tierpark Hagenbeck Aquarium (Germany), ZSL London Zoo (UK)). Branch tip fragments of 2 cm were collected and stored in single ethanol (+ 95%) vials labelled with a unique identifier. During the sample collection the colonies were tagged and photographed with collection dates and tank locations were recorded. The photographs and tags were also recorded in the “idhelper” database [database—idhelper.net (2020)]. After collection samples were stored at − 20 °C until extraction, with an initial change of ethanol after 1–2 days aimed at reducing degradation. We analysed 127 Acropora samples from these collections before COVID-19 restrictions stopped further progress on this project.
DNA extraction, amplification and sequencing
The DNA extractions were undertaken using the DNeasy PowerSoil Pro Kit from Qiagen© with a modified version of the manufacturer’s protocol. An incubation step with proteinase K was added and the physical breaking of the cell structure with a chemical dissolution was removed from the protocol as it resulted in excessive shearing of the DNA (Online Resource 1). Extracted DNA was then preserved at − 20 °C, and the DNA concentration and quality tested with both Nanodrop assay and gel electrophoresis. The DNA was diluted to standardise the concentration to between 5 and 20 μg/ml.
Two genes, PaxC and mtCR were selected as barcodes based on an assessment of their variability and availability as reference sequences (Van Oppen et al. 2001; Shearer et al. 2002). The ITS1-2 region was excluded due to the lower number of taxa available as reference sequences. PCR amplification was conducted with Q5® High-Fidelity DNA Polymerase mixed following manufacturer’s indication. 3 μl of DNA template and 0.5 μl of Bovine Serum Albumin (BSA) 1 μg were added, reaching a final volume of 25 μl with RNA free water. For the PaxC gene the primer used were PaxC FP1 5′-TCC AGA GCA TTA GAG ATG CTG G-3′ and PaxC RP1 5′-GGC GAT TTG AGA ACC AAA CCT GTA-3′ (Van Oppen et al. 2001) with a protocol of 98 °C for 30 s, followed by 31 cycles at 98 °C for 10 s, 62 °C for 30 s and 72 °C for 60 s, ending with a final phase of 72 °C for 2 min. For the mtCR the primer used were RNS2: 5′-CAG AGT AAG TCG TAA CAT AG-3′ and GR: 5′-AAT TCC GGT GTG TGT TCT CT-3′ (Suzuki et al. 2008) with a protocol of 98 °C for 30 s, followed by 40 cycles at 98 °C for 10 s, 62 °C for 15 s and 72 °C for 60 s, ending with a final phase of 72 °C for 5 min.
Successful PCR amplifications (assessed by gel electrophoresis) were sent to Eurofins Genomics for PCR cleaning and custom Sanger sequencing on both the forward and the reverse primers using cycle sequencing technology (dideoxy chain termination/cycle sequencing) on ABI 3730XL sequencing machines. The returned sequences were manually trimmed to remove poor quality sections and assembled using Geneious Prime® (2019.1.2.).
Identification
Taxonomy
Both the barcode gap and specimen identifications were initially performed at the species level, but were also performed at the level of species group, based on the revision of Acropora by Wallace et al. (Wallace et al. 2012), to provide an additional layer of information in the final integrated approach.
Barcoding gap
Reference sequences for the two targeted regions were collated. FASTA files were created with the records matching the following query on GenBank: mtCR = ““Acroporidae” [Organism] AND mitochondrial “complete genome” OR “control region” OR “putative control region” OR mitochondrial control region”; PaxC = ““Acroporidae” [Organism] AND PaxC [gene] OR pax-c [gene]”. Two multiple alignments were performed, one for each gene, using the Geneious® algorithm (progressive pairwise aligner) and trimmed. For both the produced references alignments distance matrices were calculated using the function dist.dna [R package apex (Jombart et al. 2017)] and with different models (Raw; JC69; K80; K81; F84; BH87; paralin; indel Y; indelblock). Successively the frequency distribution of intra/inter-specific distances was plotted. The quantile function (R package “stats”) was used to define the 99; 50; 1% quantile confidence interval used in the blast match analysis. Note: the two alignments were constructed and used only for the specific purposes of examining the barcoding gap for the targeted region and determining potential confidence interval for the blast match analysis.
Phylogenetic tree
Separate unrooted Maximum-likelihood (ML) phylogenetic trees were created, one for each targeted region. The references obtained from the previously mentioned query were cleaned for duplicated sequences and subsequently aligned. The two phylogenetic analyses were performed using the program IQ-TREE, with default settings and automatic model selection (Minh et al. 2013; Nguyen et al. 2015) with analysis conducted on the usegalaxy.eu public server (Afgan et al. 2018).
Blast match
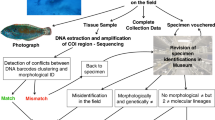
Query based specimen identification with blast match (Benson 2000) was determined using a custom Blastn database. All records from the query ““Acroporidae” [Organism]” on GenBank were downloaded and included in the local database built with the packages rBLAST (Hahsler and Nagar 2019) and rentrez (Winter 2017). A local Blast match (Camacho et al. 2009) was ultimately performed. To filter for species match with identical scores a decision tree (Fig. 2) was designed relying on Bit score, Percentage identity, and number of mismatches. Matches shorter than 2/3 of the original sample sequence query were removed. The decision tree prioritised 100% pairwise identity matches where possible, then to the highest bit score, then lowest number of base mismatches. 100% pairwise identity matches to a species group rather than a single species name were prioritised over single species name matches with lower pairwise identity with higher bit score, both possible IDs were retained. We confronted the match from the two targeted regions to obtain a single most probable ID. To account for conflicting matches between the two region we relied on the 99; 50; 1% quantile distribution of the intraspecific distances obtained from the barcoding gap analysis. Using pairwise identity threshold as confidence intervals to discern which region offers the better match for the ID (R-scripts as Online Resource 3 and 4).

Decision tree for conflict solving based on Pairwise percentage identity, Bit score and number of mismatches
Results
From the 127 treated samples all were successfully extracted and, 107 mtCR and 100 PaxC had a successful DNA amplification, respectively 84% and 79% with 76 samples overlap between the two regions (Table 1). 348 reads were successfully sequenced, 84 mtCR sample and 90 PaxC. The median sequence lengths were 734 post assembly for the mtCR region and 649 for the PaxC region (Table 1).
Barcoding
Our GenBank query produced a total of 1348 mtCR sequences and 427 PaxC sequences. The mtCR references query, once all sequence duplicates with the same species name were removed, produced a 579-sequence alignment of length 914 bp. The PaxC references query, after similar cleaning produced a 352-sequence alignment of length 770 bp.
The mtCR alignment of 579 sequences resulted in a matrix of 335,241 pairwise genetic distances, grouped by 57 species names. 6.4% of these (21,471) are intraspecific distances, while 93.6% (313,770) are interspecific distances (Fig. 3). The PaxC alignment of 352 sequences resulted in a matrix of 123,904 genetic distances, grouped by 55 species names. 3% of these (3726) are intraspecific distances, while 97% (120,178) are interspecific distances (Fig. 3). Between the two groups there is an overlap of 49 species names. Tables 3 and 4 with the sources of the sequences used in these alignments.

Barcoding gap (raw distance matrix frequency distribution)—mtCR max distance = 0.0877 minimum distance = 0; PaxC max distance = 0.5895 minimum distance = 0. (Bin size = 0.002) (A) Minimum interspecific distance and maximum intraspecific distance (B) all distances
Figure 3 highlights in blue the intraspecific variation (sequences with the same name on GenBank) and in the light green the interspecific distances (sequences with different names on GenBank). In both Fig. 3A) and B), and both barcoding regions, we see no discernable barcoding gap.
The quantile within species distribution produce the following distance threshold for our confidence interval: mtCR 0.027 (99%); 0.0068 (50%); 0 (1%) and PaxC 0.17 (99%); 0.014 (50%); 0 (1%).
In Fig. 4, for the mtCR and the PaxC respectively, we can see the above-mentioned uneven distribution of the GenBank records towards specific species and species groups. With example like species group humilis showing 4 times the frequency of less represented species group like loripes.

Number of mtCR and PaxC references sequences with the same species name by species group
Phylogeny
Both the mtCR (Online Resource 5) and the PaxC (Online Resource 6) phylogenies show clear sign of polyphyly. Looking at our best matches (i.e.: 100% match to a reference), most of our sample sequences match to single species—63% of mtCR 100% matches and all of the PaxC 100% matches. Focusing on just these sequences with 100% matches to a reference, we see that 0/19 mtCR sequences and 1/4 PaxC match to single-species clades (a monophyletic group composed of more than one sample of a single taxon name). However, some of these match to species that a polyphyletic within the tree (All of 100% mtCR matches tie to polyphyletic taxa and 75% for PaxC).
Blast match
The local database for blast matching contained 510,826 sequences. In Table 2 we see that 23 of our 76 samples (with both regions sequenced) were uniquely identified to a single species, while 13 samples matched to conflicting names. 20 of our samples had a unique species name match for the PaxC region but mtCR region matched to 2 or more names with all but 6 of these including the matching PaxC name. Very few samples matched to multiple species for both regions (4).
We have 47% of samples matching to a single species name with both regions, 5% with the same identical match. While we have 42% of the matches to a single species name that are in conflict, 25% of these conflicts are resolvable (i.e.: one of the two region with a match better fitting than the other one, based on the defined confidence thresholds of each region, rather than directly on the pairwise identity). Similar results for the other sample matching for two or more in at least one of the two regions. Out of the 76 samples, in 15 cases we observe the second region adding information to the first ones (Table 5 as Online Resource 2). Mainly from a mtCR multi name ID to a single PaxC name (i.e.: sample P269 has 7 mtCR identical matches to 7 different species names, and the PaxC matches at a lower rate to only one of them). In addition, the second region functions as backup option whenever one of the two falls too short (5/76 sample have too low-quality match to be trusted independently).
The complete list of our current best species name match, with species group information and detailed genetic info is provided as Online Resource (Table 5—Online Resource 2).
Discussion
Barcoding gap
Using DNA barcodes to identify unknown samples is only achievable if a well- studied, well-sampled reference sequence database is available (Meyer and Paulay 2005), and even though databases such as GenBank (which comprises the DNA DataBank of Japan (DDBJ), the European Nucleotide Archive (ENA), and GenBank at NCBI—http://www.ncbi.nlm.nih.gov/genbank) and BOLD (Barcode Of Life Data System—http://www.boldsystems.org) contain an ever increasing number of species-level identified sequences (i.e.: 216,531,829 total sequences on GenBank as April 2020), these databases are far from complete. The open nature of GenBank is destined to produce a bias towards species with a higher research interest [i.e.: Human sequences constitute 56% of the total sequences on GenBank (Benson 2000)]. The genus focus of our study, Acropora, is one of the more commonly studied and widespread hard corals in the ocean (Wallace 1999; Fukami et al. 2000) making it one of the most covered coral genera in GenBank. The reality is that even for a widely studied group such as Acropora we can observe a disparity in the number of sequences available for each species name (Fig. 4). The best example of this is the delta between two species group: group humilis being the most sampled (251 mtCR and 61 PaxC references sequences from our query) and group robusta one of the least sampled (11 mtCR and 13 PaxC references sequences from our query). Note that both species group are formed by of the same number of species (8 species each (Wallace et al. 2012). However, while counting the number of sequences available for a group may be an indication of the level of research that group receives, it does not mean that groups with the most sequences have the best reference collection for a DNA barcoding project. The most important quality for a reference sequence collection is it’s representation of the underlying species diversity, ideally covering a wide geographical and morphological range (Vellend and Geber 2005; Rozenfeld et al. 2007; Ma et al. 2017), all-the-while using a marker that shows sufficient variability to permit robust identifications. Sampling bias is a problem for all specimen collections and this must always be kept in mind when interpreting results (Leray et al. 2019).
From our analysis we could not identify a clear barcoding gap in both our targeted regions (Fig. 3), and furthermore we can observe a total overlap of the intra-specific distances by the inter-specific distances. For both our targeted regions, even if more evident in the mtCR (Fig. 3A), we see that the inter-specific distances follow a distribution reminiscent of a more common and more expected barcoding distribution (Fig. 3B, mtCR). We performed the same barcoding gap analysis at the higher taxonomic level using the species group information without obtaining a noticeable difference. With the lack of a clear Barcoding gap, and accounting for the extensive criticism present in the literature: For the use of distances in taxonomy and systematics (Ferguson 2002; Lee 2004; Moritz and Cicero 2004; DeSalle et al. 2005; Knapp et al. 2005; Meyer and Paulay 2005; Prendini 2005; Hickerson et al. 2006; Cognato 2006; Little and Stevenson 2007; Meier et al. 2008); for the drawbacks of using GenBank data (Meier et al. 2006); for the evidence of different rates of evolution in Scleractinia (Van Oppen et al. 1999a, b; Shearer et al. 2002; Kitahara et al. 2010). We cannot establish a simple distance base threshold to use for our identification purposes, leading us to depend more on the blast match.
Another potential issue with the use of these methods of identification is the source of the reference sequences. With databases like GenBank, with little or no review on the uploaded information, the question is the confidence that the initial identification of the now available reference sequences is indeed correct (James Harris 2003). In our specific case after the processing we performed for the mtCR reference alignment, out of the total 579 included 42% (242 sequences) come from a single doctoral thesis project, while 5.4% (33 sequences) are from unpublished papers (Table 3). For the PaxC 16% (58 sequences) are from unpublished papers, and another 39% (138 sequences) from a single paper (Richards et al. 2008) (Table 4). Highlighting clearly the need for more study on the subject to increase the number of potential sources and alleviate the possible bias of a “single observer”.
Phylogeny
While the construction of phylogenetic trees is a more refined and, by some, recommended form of species identification, it is more demanding in terms of time and expertise, and has stages such as sequence alignment and phylogenetic interpretation that often require significant manual interpretation and intervention (Nixon and Wheeler 1990; Minh et al. 2013; Nguyen et al. 2015). We observed high levels of polyphyly across all of our highest confidence matches, with only 1 of evident case in which the 100% match was to a monophyletic species (Acropora humilis PaxC region). Our preference is to employ a standardised, quickly repeatable, objective process, while keeping in mind that the results are imperfect and designed to inform but not dictate identification efforts.
Blast match
The use of query match with blast is far from being a complete solution, with 26 out of 76 samples without a single species or species group resolved ID (Table 5), we have proposed a theoretical decision tree that standardise the choice when there is a need of sorting through the output of a multi-species name blast match (Fig. 2). In addition we used two different regions, a mitochondrial one and a nuclear one to increase the confidence in our results (Suzuki et al. 2008). Our proposed species ID are provisional and subject to revision when more species and sequences are available, hence the value of a repeatable process for matching and re-matching.
All methods of identification that rely on DNA barcodes (e.g.: Blast matching, Barcoding Gap analysis, etc.) use genetic divergence thresholds to assign individuals to correct species. This based on two fundamental assumptions (1) monophyly of species with respect to the molecular marker used, and (2) intraspecific genetic divergence is much smaller than interspecific genetic differences (Toffoli et al. 2008). The first assumption is more dependent on the experimental choices made (i.e. sequence region selected). The second assumption is where most of the criticism of barcodes methods lies, on not comparing sister species or geographical distribution thus, underestimating intraspecific genetic variability (Moritz and Cicero 2004; Toffoli et al. 2008). Fundamentally, the issue of using thresholds lies in the fact that species naturally embodies an evolutionary process, being subject to demographic and selective processes that will act on the genetic diversity [e.g. (Avise et al. 2000; Hey 2001; Coyne and Orr 2004; Toffoli et al. 2008)]. Since species are real evolutionary groups and not categories which are created as a direct function of perceived distinction (Hey 2001), the use of thresholds in “discovering” new species is overly simplistic, and in some cases even misleading (Toffoli et al. 2008). We argue against simply substituting morphology based taxonomy with DNA barcoding based taxonomy, as each system is more adapt to answer different questions (Toffoli et al. 2008). Correct assignment is thus only possible by complementing DNA barcoding with other data types, such as morphological, and ecological characters as part of an integrated diagnostic approach (Toffoli et al. 2008).
While the simple blast match is inheritably less complex than other traditional DNA barcodes identification methods [i.e.: the above mentioned barcoding gap analysis and, the potentially more direct Automatic Barcoding Gap Discovery (ABGD) (Puillandre et al. 2012)]. The argument we make is that given the shared limitation and the common need for additional data in an integrated approach, a faster and simpler blast match provides a relatively similar additional amount of information for the species identification while being more easily repeatable, especially considering the value of standardized data among aquariums (da Silva et al. 2019) and in light of the ever increasing quantity of reference materials being added to GenBank and BOLD.
Available sequences libraries are not exhaustive, and probably never will be. That should not stop us from attempting identifications based on the best available knowledge. All identifications must fit within the context of our evolving understanding of taxonomy, where new descriptions invalidate existing ones and often we do not have unambiguous taxonomic descriptions for every species (Simpson 1951; Wiley 1978; Mayr 1982, 2000; Nixon and Wheeler 1990; Medlin et al. 1995). The reality is that barcoding as an independent tool of identification has its clear limitation (Van Oppen et al. 1999a, b; Ferguson 2002; Shearer et al. 2002; Lee 2004; Moritz and Cicero 2004; DeSalle et al. 2005; Knapp et al. 2005; Meyer and Paulay 2005; Prendini 2005; Hickerson et al. 2006; Meier et al. 2006, 2008; Cognato 2006; Little and Stevenson 2007; Kitahara et al. 2010), and coral barcoding is no exception. With some of the concern on the use of genetic repertories in common of all the methods explored in this study, we strongly recommend an integrated diagnostic approach, combining morphological and genetic means of identification, in agreement with C. Moritz in “DNA Barcoding: Promise and Pitfalls” (Moritz and Cicero 2004). Proposing the use of barcoding and query matching as an additional tool for identification, increasing confidence of experts and both confidence and capacity of non-experts’ taxonomist. Producing this way reliably identified sequences that can potentially become new reference, adding to the repertories already freely available. Considering the specific needs of the aquarium community and their critical role to support corals conservation [i.e., by hosting at least 24% of the corals assessed as highly vulnerable to climate change (da Silva et al. 2019)], we emphasise the value of integrating standardised barcoding analysis, and specimen morphology photographs to optimise usage of authoritative identification guides and expert opinion.
Data availability
All genetics data will be uploaded to appropriate repertories, all the coral specimen sampled are housed in their respective institutions.
Code availability
Custom code available as supplementary online material.
References
Afgan E, Baker D, Batut B et al (2018) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46:W537–W544. https://doi.org/10.1093/nar/gky379
Amaral FD (1994) Morphological variation in the reef coral Montastrea cavernosa in Brazil. Coral Reefs 13:113–117. https://doi.org/10.1007/BF00300771
Avise JC, Baker RJ, Porter CA et al (2000) Phylogeography: the history and formation of species. Harvard University Press, Cambridge
Ayre DJ, Willis BL (1988) Population structure in the coral Pavona cactus: clonal genotypes show little phenotypic plasticity. Mar Biol 99:495–505. https://doi.org/10.1007/BF00392557
Babcock RC, Bull GD, Harrison PL et al (1986) Synchronous spawnings of 105 scleractinian coral species on the Great Barrier Reef. Mar Biol 90:379–394. https://doi.org/10.1007/BF00428562
Bellwood DR, Hughes TP (2001) Regional-scale assembly rules and biodiversity of coral reefs. Science 292:1532–1534. https://doi.org/10.1126/science.1058635
Benson DA (2000) GenBank. Nucleic Acids Res 28:15–18. https://doi.org/10.1093/nar/28.1.15
Best BM, Boekschoten GJ, Oosterbaan A (1984) Species concept and ecomorph variation in living and fossil Scleractinia. Palaeontogr Am 54:70–79
Blanco JM, Wildt DE, Höfle U et al (2009) Implementing artificial insemination as an effective tool for ex situ conservation of endangered avian species. Theriogenology 71:200–213. https://doi.org/10.1016/j.theriogenology.2008.09.019
Borneman E (2008) Introduction to the husbandry of corals in aquariums: a review. In: Leewis RJ, Janse M (eds) Advances in coral husbandry in public aquariums, vol 2. Burgers’ Zoo, Arnhem, pp 3–14
Brondizio ES, Settele J, Dìaz S et al (2019) Global assessment report on biodiversity and ecosystem services of the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services. IPBES, Bonn
Brunner B (2012) The Ocean at Home: an illustrated history of the aquarium. Reaktion Books, London
Bruno JF, Edmunds PJ (1997) Clonal variation for phenotypic plasticity in the coral Madracis mirabilis. Ecology 78:2177–2190. https://doi.org/10.2307/2265954
Camacho C, Coulouris G, Avagyan V et al (2009) BLAST+: architecture and applications. BMC Bioinform 10:1–9. https://doi.org/10.1186/1471-2105-10-421
Chen I-P, Tang C-Y, Chiou C-Y et al (2009) Comparative analyses of coding and noncoding DNA regions indicate that Acropora (Anthozoa: Scleractina) possesses a similar evolutionary tempo of nuclear vs. mitochondrial genomes as in plants. Mar Biotechnol 11:141–152. https://doi.org/10.1007/s10126-008-9129-2
Cognato AI (2006) Standard percent DNA sequence difference for insects does not predict species boundaries. J Econ Entomol 99:1037–1045. https://doi.org/10.1093/jee/99.4.1037
Cowburn B, Samoilys MA, Obura D (2018) The current status of coral reefs and their vulnerability to climate change and multiple human stresses in the Comoros Archipelago, Western Indian Ocean. Mar Pollut Bull 133:956–969. https://doi.org/10.1016/j.marpolbul.2018.04.065
Cowman PF, Quattrini AM, Bridge TCL et al (2020) An enhanced target-enrichment bait set for Hexacorallia provides phylogenomic resolution of the staghorn corals (Acroporidae) and close relatives. Mol Phylogenet Evol 153:106944. https://doi.org/10.1016/j.ympev.2020.106944
Coyne JA, Orr HA (2004) Speciation. Sinauer Associates, Sunderland
Craggs J, Guest JR, Davis M et al (2017) Inducing broadcast coral spawning ex situ: closed system mesocosm design and husbandry protocol. Ecol Evol 7:11066–11078. https://doi.org/10.1002/ece3.3538
da Silva R, Pearce-Kelly P, Zimmerman B et al (2019) Assessing the conservation potential of fish and corals in aquariums globally. J Nat Conserv 48:1–11. https://doi.org/10.1016/j.jnc.2018.12.001
DeSalle R, Egan MG, Siddall M (2005) The unholy trinity: taxonomy, species delimitation and DNA barcoding. Philos Trans R Soc B:Biol Sci 360:1905–1916. https://doi.org/10.1098/rstb.2005.1722
Fa JE, Funk SM, O’Connell D (2011) Zoo conservation biology. Cambridge University Press
Ferguson JWH (2002) On the use of genetic divergence for identifying species. Biol J Lin Soc 75:509–516. https://doi.org/10.1046/j.1095-8312.2002.00042.x
Fisher R, O’Leary RA, Low-Choy S et al (2015) Species richness on coral reefs and the pursuit of convergent global estimates. Curr Biol 25:500–505. https://doi.org/10.1016/j.cub.2014.12.022
Foster AB (1979) Phenotypic plasticity in the reef corals Montastraea annularis (Ellis & Solander) and Siderastrea siderea (Ellis & Solander). J Exp Mar Biol Ecol 39:25–54. https://doi.org/10.1016/0022-0981(79)90003-0
Fukami H, Omori M, Hatta M (2000) Phylogenetic relationships in the coral family Acroporidae, reassessed by inference from Mitochondrial genes. Zoolog Sci 17:689–696. https://doi.org/10.2108/zsj.17.689
Godfray HCJ (2002) Challenges for taxonomy. Nature 417:17–19. https://doi.org/10.1038/417017a
Hahsler M, Nagar A (2019) rBLAST: interface to the basic local alignment search tool (BLAST)
Harrison PL, Babcock RC, Bull GD et al (1984) Mass spawning in tropical reef corals. Science 223:1186–1189. https://doi.org/10.1126/science.223.4641.1186
Hatta M, Fukami H, Wang W et al (1999) Reproductive and genetic evidence for a reticulate evolutionary history of mass-spawning corals. Mol Biol Evol 16:1607–1613. https://doi.org/10.1093/oxfordjournals.molbev.a026073
Hebert PDN, Cywinska A, Ball SL, DeWaard JR (2003) Biological identifications through DNA barcodes. Proc R Soc B Biol Sci 270:313–321. https://doi.org/10.1098/rspb.2002.2218
Hellberg ME (2006) No variation and low synonymous substitution rates in coral mtDNA despite high nuclear variation. BMC Evol Biol 6:1–8. https://doi.org/10.1186/1471-2148-6-24
Hey J (2001) Genes, categories, and species: the evolutionary and cognitive cause of the species problem. Oxford University Press, Oxford
Hickerson MJ, Meyer CP, Moritz C (2006) DNA barcoding will often fail to discover new animal species over broad parameter space. Syst Biol 55:729–739. https://doi.org/10.1080/10635150600969898
Hoegh-Guldberg O, Poloczanska ES, Skirving W, Dove S (2017) Coral reef ecosystems under climate change and ocean acidification. Front Mar Sci 4:158. https://doi.org/10.3389/fmars.2017.00158
Horton T, Kroh A, Ahyong S et al (2020) World register of marine species (WoRMS)
Huang D, Meier R, Todd PA, Chou LM (2008) Slow mitochondrial COI sequence evolution at the base of the Metazoan tree and its implications for DNA barcoding. J Mol Evol 66:167–174. https://doi.org/10.1007/s00239-008-9069-5
Hughes TP, Connolly SR, Eakin CM et al (2018) Global warming transforms coral reef assemblages. Nature 556:492–496. https://doi.org/10.1038/s41586-018-0041-2
Hutchins M (2003) Zoo and aquarium animal management and conservation: current trends and future challenges. Int Zoo Yearb 38:14–28. https://doi.org/10.1111/j.1748-1090.2003.tb02060.x
idhelper.net (2020) In: Version 0.01 Beta. https://idhelper.net
Isomura N, Iwao K, Fukami H (2013) Possible natural hybridization of two morphologically distinct species of Acropora (Cnidaria, Scleractinia) in the Pacific: fertilization and larval survival rates. PLoS ONE 8:1–8. https://doi.org/10.1371/journal.pone.0056701
James Harris D (2003) Can you bank on GenBank? Trends Ecol Evol 18:317–319. https://doi.org/10.1016/S0169-5347(03)00150-2
Jombart T, Kamvar ZN, Schliep K et al (2017) apex: phylogenetic methods for multiple gene data
Kitahara MV, Cairns SD, Stolarski J et al (2010) A comprehensive phylogenetic analysis of the Scleractinia (Cnidaria, Anthozoa) based on mitochondrial CO1 sequence data. PLoS ONE. https://doi.org/10.1371/journal.pone.0011490
Knapp S, Lughadha EN, Paton A et al (2005) Taxonomic inflation, species concepts and global species lists (multiple letters). Trends Ecol Evol 20:7–8. https://doi.org/10.1016/j.tree.2004.11.001
Lacy RC (2013) Achieving true sustainability of zoo populations. Zoo Biol 32:19–26. https://doi.org/10.1002/zoo.21029
Lee MSY (2004) The molecularisation of taxonomy. Invertebr Syst 18:1–6
Leray M, Knowlton N, Ho SL et al (2019) GenBank is a reliable resource for 21st century biodiversity research. Proc Natl Acad Sci USA 116:22651–22656. https://doi.org/10.1073/pnas.1911714116
Leus K, Traylor-Holzer K, Lacy RC (2011) Genetic and demographic population management in zoos and aquariums: recent developments, future challenges and opportunities for scientific research. Int Zoo Yearb 45:213–225. https://doi.org/10.1111/j.1748-1090.2011.00138.x
Little DP, Stevenson DW (2007) A comparison of algorithms for the identification of specimens using DNA barcodes: examples from gymnosperms. Cladistics 23:1–21. https://doi.org/10.1111/j.1096-0031.2006.00126.x
Ma B, Liao L, Peng Q et al (2017) Reduced representation genome sequencing reveals patterns of genetic diversity and selection in apple. J Integr Plant Biol 59:190–204. https://doi.org/10.1111/jipb.12522
Márquez LM, Van Oppen MJHH, Willis BL et al (2002) The highly cross-fertile coral species, Acropora hyacinthus and Acropora cytherea, constitute statistically distinguishable lineages. Mol Ecol 11:1339–1349. https://doi.org/10.1046/j.1365-294X.2002.01526.x
Masson-Delmotte V, Zhai P, Pörtner H-O et al (2018) IPCC Special Report on the impacts of global warming of 1.5°C above pre-industrial levels and related global greenhouse gas emission pathways. The context of strengthening the global response to the threat of climate change, sustainable development. World Meteorological Organization, Geneva
Mayr E (1982) The growth of biological thought: diversity, evolution, and inheritance. Harvard University Press, Cambridge
Mayr E (2000) The biological species concept. Species concepts and phylogenetic theory: a debate. Columbia University Press, New York, pp 17–29
Medlin LK, Lange M, Barker GLA, Hayes PK (1995) Can molecular techniques change our ideas about the species concept? Molecular ecology of aquatic microbes. Springer, Berlin, Heidelberg, pp 133–152
Meier R, Shiyang K, Vaidya G, Ng PKL (2006) DNA barcoding and taxonomy in diptera: a tale of high intraspecific variability and low identification success. Syst Biol 55:715–728. https://doi.org/10.1080/10635150600969864
Meier R, Zhang G, Ali F (2008) The use of mean instead of smallest interspecific distances exaggerates the size of the “barcoding gap” and leads to misidentification. Syst Biol 57:809–813. https://doi.org/10.1080/10635150802406343
Meyer CP, Paulay G (2005) DNA barcoding: error rates based on comprehensive sampling. PLoS Biol 3:e422. https://doi.org/10.1371/journal.pbio.0030422
Miller KJ (1994) Morphological variation in the coral genus Platygyra—environmental influences and taxonomic implications. Mar Ecol Prog Ser 110:19–28. https://doi.org/10.3354/meps110019
Minh BQ, Nguyen MAT, von Haeseler A (2013) Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol 30:1188–1195. https://doi.org/10.1093/molbev/mst024
Mora C, Tittensor DP, Myers RA (2008) The completeness of taxonomic inventories for describing the global diversity and distribution of marine fishes. Proc R Soc B Biol Sci 275:149–155. https://doi.org/10.1098/rspb.2007.1315
Mora C, Tittensor DP, Adl S et al (2011) How many species are there on earth and in the ocean? PLoS Biol 9:1–8. https://doi.org/10.1371/journal.pbio.1001127
Moritz C, Cicero C (2004) DNA barcoding: promise and pitfalls. PLoS Biol 2:e354. https://doi.org/10.1371/journal.pbio.0020354
Muko S, Kawasaki K, Sakai K et al (2000) Morphological plasticity in the coral Porites sillimaniani and its adaptive significance. Bull Mar Sci 66:225–239
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. https://doi.org/10.1093/molbev/msu300
Nixon KC, Wheeler QD (1990) An amplification of the phylogenetic species concept. Cladistics 6:211–223. https://doi.org/10.1111/j.1096-0031.1990.tb00541.x
O’Neil KL, Serafin RM, Patterson JT, Craggs JRK (2021) Repeated ex situ spawning in two highly disease susceptible corals in the family Meandrinidae. Front Mar Sci. https://doi.org/10.3389/fmars.2021.669976
Ortman BD, Bucklin A, Pagès F, Youngbluth M (2010) DNA barcoding the Medusozoa using mtCOI. Deep Sea Res Part II 57:2148–2156. https://doi.org/10.1016/j.dsr2.2010.09.017
Palumbi SR, Vollmer S, Romano S et al (2012) The role of genes in understanding the evolutionary ecology of reef building corals. Evol Ecol 26:317–335. https://doi.org/10.1007/s10682-011-9517-3
Petersen D, Laterveer M, van Bergen D et al (2006) The application of sexual coral recruits for the sustainable management of ex situ populations in public aquariums to promote coral reef conservation—SECORE Project. Aquat Conserv Mar Freshwat Ecosyst 16:167–179. https://doi.org/10.1002/aqc.716
Petersen D, Falcato J, Gilles P, Jones R (2007) Sexual reproduction of scleractinian corals in public aquariums: current status and future perspectives. Int Zoo Yearb 41:122–137. https://doi.org/10.1111/j.1748-1090.2007.00006.x
Pratchett MS, Pisapia C, Sheppard CRC (2013) Background mortality rates for recovering populations of Acropora cytherea in the Chagos Archipelago, central Indian Ocean. Mar Environ Res 86:29–34. https://doi.org/10.1016/j.marenvres.2013.02.007
Prendini L (2005) Comment on “Identifying spiders through DNA barcodes.” Can J Zool 83:498–504. https://doi.org/10.1139/z05-025
Puillandre N, Lambert A, Brouillet S, Achaz G (2012) ABGD, automatic barcode gap discovery for primary species delimitation. Mol Ecol 21:1864–1877. https://doi.org/10.1111/j.1365-294X.2011.05239.x
Raven PH (2004) Ex situ plant conservation: supporting species survival in the wild. Island Press
Richards ZT, van Oppen MJH, Wallace CC et al (2008) Some rare Indo-Pacific coral species are probable hybrids. PLoS ONE 3:1–8. https://doi.org/10.1371/journal.pone.0003240
Rinkevich B (1995) Restoration strategies for coral reefs damaged by recreational activities: the use of sexual and asexual recruits. Restor Ecol 3:241–251. https://doi.org/10.1111/j.1526-100X.1995.tb00091.x
Rozenfeld AF, Arnaud-Haond S, Hernández-García E et al (2007) Spectrum of genetic diversity and networks of clonal organisms. J R Soc Interface 4:1093–1102. https://doi.org/10.1098/rsif.2007.0230
Shearer TL, Coffroth MA (2008) DNA BARCODING: barcoding corals: limited by interspecific divergence, not intraspecific variation. Mol Ecol Resour 8:247–255. https://doi.org/10.1111/j.1471-8286.2007.01996.x
Shearer TL, van Oppen MJH, Romano SL, Wörheide G (2002) Slow mitochondrial DNA sequence evolution in the Anthozoa (Cnidaria). Mol Ecol 11:2475–2487. https://doi.org/10.1046/j.1365-294X.2002.01652.x
Sheppard C, Sheppard A, Fenner D (2020) Coral mass mortalities in the Chagos Archipelago over 40 years: regional species and assemblage extinctions and indications of positive feedbacks. Mar Pollut Bull 154:111075. https://doi.org/10.1016/j.marpolbul.2020.111075
Simpson GG (1951) The species concept. Evolution 5:285. https://doi.org/10.2307/2405675
Skibins JC, Powell RB (2013) Conservation caring: measuring the influence of zoo visitors’ connection to wildlife on pro-conservation behaviors. Zoo Biol 32:528–540. https://doi.org/10.1002/zoo.21086
Suzuki G, Hayashibara T, Shirayama Y, Fukami H (2008) Evidence of species-specific habitat selectivity of Acropora corals based on identification of new recruits by two molecular markers. Mar Ecol Prog Ser 355:149–159. https://doi.org/10.3354/meps07253
Todd PA (2008) Morphological plasticity in scleractinian corals. Biol Rev 83:315–337. https://doi.org/10.1111/j.1469-185X.2008.00045.x
Todd PA, Sidle RC, Lewin-Koh NJI (2004) An aquarium experiment for identifying the physical factors inducing morphological change in two massive scleractinian corals. J Exp Mar Biol Ecol 299:97–113. https://doi.org/10.1016/j.jembe.2003.09.005
Toffoli D, Hrbek T, de Araújo MLG et al (2008) A test of the utility of DNA barcoding in the radiation of the freshwater stingray genus Potamotrygon (Potamotrygonidae, Myliobatiformes). Genet Mol Biol 31:324–336. https://doi.org/10.1590/s1415-47572008000200028
Turley SK (1999) Conservation and tourism in the traditional UK zoo. J Tour Stud 10:2
van Oppen MJH, Hislop NR, Hagerman PJ, Miller DJ (1999a) Gene content and organization in a segment of the mitochondrial genome of the Anthozoan subclass Zoantharia. Mol Biol Evol 16:1812–1815
Van Oppen MJHH, Willis BL, Miller DJ (1999b) Atypically low rate of cytochrome b evolution in the scleractinian coral genus Acropora. Proc R Soc Lond B 266:179–183. https://doi.org/10.1098/rspb.1999.0619
Van Oppen MJHH, McDonald BJ, Willis B, Miller DJ (2001) The evolutionary history of the coral genus acropora (Scleractinia, Cnidaria) based on a mitochondrial and a nuclear marker: reticulation, incomplete lineage sorting, or morphological convergence? Mol Biol Evol 18:1315–1329. https://doi.org/10.1093/oxfordjournals.molbev.a003916
Van Oppen MJH, Willis BL, Van Rheede T, Miller DJ (2002) Spawning times, reproductive compatibilities and genetic structuring in the Acropora aspera group: evidence for natural hybridization and semi-permeable species boundaries in corals. Mol Ecol 11:1363–1376. https://doi.org/10.1046/j.1365-294X.2002.01527.x
Vellend M, Geber MA (2005) Connections between species diversity and genetic diversity. Ecol Lett 8:767–781. https://doi.org/10.1111/j.1461-0248.2005.00775.x
Veron JEN, Stafford-Smith MG, Turak E, DeVantier LM (2020) Corals of the World. Available at http://www.coralsoftheworld.com. Accessed on 26 Apr 2020
Vollmer SV (2002) Hybridization and the evolution of reef coral diversity. Science 296:2023–2025. https://doi.org/10.1126/science.1069524
Vollmer SV, Palumbi SR (2002) Hybridization and the evolution of reef coral diversity. Science 296:2023–2025. https://doi.org/10.1126/science.1069524
Wallace CC (1999) Staghorn corals of the world: a revision of the coral genus Acropora (Scleractinia; Astrocoeniina; Acroporidae) worldwide, with emphasis on morphology, phylogeny and biogeography. CSIRO publishing, Clayton
Wallace CC, Willis BL (1994) Systematics of the coral genus Acropora: implications of new biological findings for species concepts. Annu Rev Ecol Syst 25:237–262
Wallace CC, Done BJ, Muir PR (2012) Revision and catalogue of worldwide staghorn corals acropora and isopora (scleractinia: Acroporidae) in the museum of tropical Queensland. Mem Qld Mus 57:1–257
Wei NV, Hsieh HJ, Dai C, Wallace CC, Baird AH, Chen CA (2012) Reproductive isolation among Acropora species (Scleractinia: Acroporidae) in a marginal coral assemblage. Mar Biol Res 51(1):85–92
Wiley EO (1978) The evolutionary species concept reconsidered. Syst Zool 27:17. https://doi.org/10.2307/2412809
Willis BL, van Oppen MJH, Miller DJ et al (2006) The role of hybridization in the evolution of reef corals. Annu Rev Ecol Evol Syst 37:489–517. https://doi.org/10.1146/annurev.ecolsys.37.091305.110136
Willis BL (1985) Phenotypic plasticity versus phenotypic stability in the reef corals Turbinaria mesenterina and Pavona cactus. In: Proceedings of the Fifth International Coral Reef Symposium Vol. 4
Willis BL (1997) Hybridization and breeding incompatibilities within the mating systems of mass spawning reef corals. In: Proceedings of the 8th International Coral Reef Symposium. pp 81–90
Winter DJ (2017) Rentrez: an R package for the NCBI eUtils API. R Journal 9:520–526. https://doi.org/10.32614/rj-2017-058
Wolstenholme JK (2004) Temporal reproductive isolation and gametic compatibility are evolutionary mechanisms in the Acropora humilis species group (Cnidaria; Scleractinia). Mar Biol 144:567–582. https://doi.org/10.1007/s00227-003-1209-2
Acknowledgements
Genetic analysis costs were partly covered by a FAITAG genetics fund grant an initiative from the EAZA (European Association of Zoos and Aquaria) and EUAC (European Union of Aquarium Curators) Fish and Aquatic Invertebrate Taxon Advisory Group (FAITAG).
Funding
Genetic analysis costs were partly covered by a FAITAG genetics fund grant an initiative from the EAZA (European Association of Zoos and Aquaria) and EUAC (European Union of Aquarium Curators) Fish and Aquatic Invertebrate Taxon Advisory Group (FAITAG).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical approval
This project has passed the ethical review process by the appropriate board at the Zoological society of London (ZSL).
Consent to participate
Not applicable.
Consent for publication
All the authors, institutions supplying the sample, and third parties involved have given consent for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Colin, L., Abed-Navandi, D., Conde, D.A. et al. What’s left in the tank? Identification of non-ascribed aquarium’s coral collections with DNA barcodes as part of an integrated diagnostic approach. Conservation Genet Resour 14, 167–182 (2022). https://doi.org/10.1007/s12686-021-01250-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12686-021-01250-3