
Abstract
Leukemia is an abnormal proliferation of white blood cells that occurs in bone marrow and expands through the blood. It arises from dysregulated differentiation, uncontrolled growth, and inhibition of apoptosis. Glutamine (GLN) is a "conditionally essential" amino acid that promotes growth and proliferation of leukemic cells. Recently, details about the role of GLN and its metabolism in the diagnosis and treatment of acute myeloid, chronic lymphocytic, and acute lymphoblastic leukemia have emerged. The uptake of GLN by leukemia cells and the dynamic changes of glutamine-related indexes in leukemia patients may be able to assist in determining whether the condition of leukemia is in a state of progression, remission or relapse. Utilizing the possible differences in GLN metabolism in different subtypes of leukemia may help to differentiate between different subtypes of leukemia, thus providing a basis for accurate diagnosis. Targeting GLN metabolism in leukemia requires simultaneous blockade of multiple metabolic pathways without interfering with the normal cellular and immune functions of the body to achieve effective leukemia therapy. The present review summarizes recent advances, possible applications, and clinical perspectives of GLN metabolism in leukemia. In particular, it focuses on the prospects of GLN metabolism in the diagnosis and treatment of acute myeloid leukemia. The review provides new directions and hints at potential roles for future clinical treatments and studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Leukemias are a group of aggressive hematologic malignancies (also known as blood cancers) involving clonal proliferation of immature myeloid progenitor cells in the bone marrow and peripheral blood. They are caused by genetic mutations in hematopoietic stem cells. Presently, the treatment of choice includes chemotherapy and allogeneic stem cell transplantation [1]. With the development of time and technology, immunotherapeutic approaches for various leukemias have shown great promise, such as CD33 or CLL-1-specific chimeric antigen receptor (CAR)-T cell therapy[2, 3] and immune checkpoint inhibitor therapies such as TIM3, CD47, and anti-CD70 [4,5,6]. Regardless of therapy, relapse is common and shortens the survival of leukemia patients. Therefore, alternative treatment strategies are needed.
Growing evidence points to the critical role of amino acid metabolism on the diagnosis and treatment of leukemia. The metabolic pathways for GLN, arginine, isoleucine, tryptophan, cysteine, tyrosine, threonine, and L-serine play a crucial role in cancer. Moreover, amino acid metabolism is active in high-risk populations and the corresponding genes are associated with the immune microenvironment in acute myeloid leukemia (AML) patients [7]. Among the various amino acids, GLN metabolism seems to be an effective target against leukemia [8]. Leukemia cells have changes in the uptake and utilization of GLN as well as metabolic pathways. Through the dynamic changes of GLN-related indexes in patients can help to determine whether the leukemia is in progress, remission or relapse, and the differences in glutamine metabolism in different subtypes of leukemia can help to differentiate between subtypes of leukemia. Therefore, this review discusses the role of GLN metabolism in three common types of leukemia: AML, chronic lymphocytic leukemia (CLL), and acute lymphoblastic leukemia (ALL). Furthermore, it discusses the latest advances and developments in the field, as well as the therapeutic opportunities and challenges of GLN targeting.
2 Glutamine metabolism
GLN is a nonessential amino acid with two amino groups, the α-amino group and a readily hydrolysable side-chain amide group, with five carbons, a molecular weight of 146.15 kDa, and a chemical composition of C = 41.09%, H = 6.90%, O = 32.84%, and N = 19.17%. Classified as a neutral at physiological pH, it is the most abundant and versatile amino acid in the body (~ 0.6–0.8 mM) [9]. GLN is converted by glutaminase (GLS) to glutamate, which is then transformed to α-ketoglutarate (α-KG), an intermediate in the tricarboxylic acid (TCA) cycle and a core element in GLN metabolism [9, 10]. Glutamate can be directly converted to α-KG in two ways. The first is via glutamate dehydrogenase (GLDH), which produces the potential autophagy inducer ammonium and NADH or NADPH as cofactors. The second is via a group of transaminases, including glutamate–oxaloacetate transaminase, glutamate-pyruvate transaminase, and phosphoserine aminotransferase. Glutamate serves as a metabolite for the growth and proliferation of cancer cells via the TCA cycle. Moreover, glutamate can be deaminated in a number of reactions, thus providing a source of nitrogen for nonessential amino acids, purines and pyrimidines [11]. At the same time, intracellular glutathione (GSH) derived from GLN effectively scavenges intracellular reactive oxygen species (ROS), mediating ferroptosis and redox homeostasis in cancer cells [8]. Notably, GLN promotes the activation of rapamycin complex 1 (mTORC1), which is associated with apoptosis and autophagy in cancer cells.
GLN is as important in hematologic tumors as one of the nutrients on which cancer cells depend for survival. Given the need of tumor cells for glucose (for anaerobic glycolysis), GLN deficiency has been associated with cell death [12,13,14]. In both healthy and diseased states, immune cells consume as much GLN as possible, and its deprivation holds promise as a new therapeutic tool.
3 Glutaminase
GLS is a key enzyme involved in GLN metabolism. It comprises renal glutaminase-1 (GLS-1) and hepatic glutaminase-2 (GLS-2). GLS-1 has two variable splice isoforms: glutaminase C and renal glutaminase. The TCA cycle yields metabolic intermediates that are involved in the biosynthesis of nucleotides, GSH, and other amino acids [15]. In addition, GLN can be converted to α-KG for oxidative phosphorylation to produce ATP. Elevated expression of GLS-1 is directly or indirectly associated with poor prognosis in stem cell, colorectal, and breast cancers [16].
The GLS-2 gene is located on chromosome 12q13 and contains 18 coding exons. GLS-2 is considered more of a tumor suppressor than GLS-1. GLS-2 has been shown to be a p53 target as it contains two possible p53 binding sites [17]. The tumor suppressor p53 activates GLS-2 expression, regulates intracellular ROS levels and reduced/oxidized GSH ratios, and removes intracellular ROS to protect cells from genomic damage and ROS-sensitive apoptosis [18, 19]. TAp63, TAp73, and long-chain non-coding RNAs can also regulate GLS-2 [20,21,22]. Meanwhile, increased mitochondrial GLS expression enhances GLN catabolism by Myc oncogene inhibition of miR-23, which in turn targets GLS [23]. GLS inhibition decreases the production of GSH in AML cell lines, leading to increased mitochondrial ROS and apoptosis [8, 24]. Thus. GLS-1 and GLS-2 may serve as diagnostic and therapeutic targets for certain cancers. Clinical studies should explore new chemotherapeutic combinations of GLS inhibitors in the treatment of leukemia.
4 Inhibitors of the GLN transporter limit tumor demand for GLN
Strong expression of alanine-serine-cysteine transporter protein 2 (ASCT2), a GLN transporter, helps meet the amino acid needs of tumors [25] (Fig. 1). However, ASCT2 has also anticancer properties [26], because its deletion can lead to apoptosis of leukemia cells [27]. Inhibition of ASCT2-mediated GLN uptake in human cells using a lead compound (V-9302) resulted in attenuation of cancer cell growth and proliferation, frequent cell death, and increased oxidative stress [28]. ASCT2 plays the same role in cell proliferation and apoptosis in several cancers [29,30,31,32,33,34,35,36]. In a study of 25 patients with different clinically aggressive tumors (lung, breast, colon, or lymphoma), who underwent fluorine 18-(2S,4R)-4-fluoroglutamine positron emission tomography, all showed abnormal GLN metabolism [37]. Notably, ASCT2-mediated pharmacological inhibitors significantly reduced GLN uptake by triple-negative basal-like breast cancer cells, while having little effect on luminal breast cancer cells [38]. Taken together, this evidence implies that ASCT2 may serve as a potential target for antitumor drugs. ASCT2 inhibitors and the combination of ASCT2 inhibitors with other antitumor therapies may offer a promising antitumor strategy. However, more research is needed in this area of leukemia. Because GLN may be closely related to leukemogenesis and progression, it may directly or indirectly affect the diagnosis and treatment of leukemia.

Overview of glutamine metabolism in leukemia cells. The increased demand for glutamine by leukemia cells and simultaneous inhibition of the ASCT2 transporter, result in a declining hydrolysis of glutamine to glutamate. α-KG, α-ketoglutarate; ASCT2, alanine-serine-cysteine transporter protein 2; GLS, glutaminase; TCA, tricarboxylic acid. The Illustration was created in Figdraw
5 GLN metabolism in leukemia
Leukemia is a myeloid malignancy characterized by abnormal proliferation and differentiation of hematopoietic precursor cells. Cancer cells are highly dependent on GLN metabolism and availability [39]. GLN metabolism is centered in the mitochondria. Mitochondria play a crucial role in the maintenance of hematopoietic stem cells, whose malignant transformation ultimately leads to leukemic stem cells [40]. The first evidence of impaired mitochondrial metabolism in AML was the presence of mutations in the gene encoding isocitrate dehydrogenase (IDH) in AML patients [41, 42].
Recent in vivo and in vitro studies have shown that GLN is restricted to the cancer cell environment [43]. Glutamine is used as an alternative fuel for the TCA cycle, with plasma concentrations of 0.6–0.8 mM, and is the most common amino acid in blood [9, 10]. As in other cancers, plasma GLN concentrations in AML patients are quite low, 0.3 mM or less, suggesting that GLN is rapidly depleted in AML cells [10, 44, 45]. A study of 55 newly diagnosed AML patients and 45 healthy individuals also showed that GLN levels were much lower in the former than in the latter [46]. AML cells are completely dependent on exogenous GLN, and knockdown of high-affinity ASCT2 leads to apoptosis in AML cell lines and inhibits tumor progression in AML xenografts and primary AML mouse models [47] Indeed, ASCT2 plays pleiotropic roles in cellular metabolism and serves as a promising molecular target for the treatment of leukemia [27].
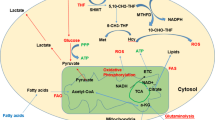
GLS is a rate-limiting factor for TCA activity in AML and is highly expressed in AML patients [48]. The initial step required for glucose-independent oxidative phosphorylation is the conversion of GLN to glutamate. Subsequently, glutamate provides the substrate for the synthesis of α-KG. IDH catalyzes the oxidative decarboxylation of isocitrate to α-KG. Mutations in IDH result in the conversion of α-KG to R2-hydroxyglutarate, which is detected in approximately 2% of adult AML patients [42, 49] (Fig. 2). ATP and metabolic pathways localized to the mitochondria have been shown to play an important role in the progression of AML [50]. Elevated levels of 2-hydroxyglutarate, a metabolite associated with the TCA cycle, may promote tumorigenesis [51]. Meanwhile, the central nervous system is also involved in metabolic processes, posing a major challenge to the treatment of acute leukemia [52]. In addition, in studies on GLN metabolism, N6-methyladenosine (m6A) regulates GLN metabolism through modification of insulin-like growth factor 2 mRNA-binding protein 2 (IGF2BP2), which directly or indirectly promotes AML cell development and self-renewal, and higher levels of IGF2BP2 expression correlate with a poor prognosis for AML [53].

Strategies for targeting glutamine metabolism in AML. l-asparaginase allows the hydrolysis of extracellular glutamine, impeding its synthesis and hydrolysis. Upon translocation to the cell, glutamine is transformed to glutamate by the two isoforms of glutaminase, GLS-1 and GLS-2. Glutamine is synthesized from glutamate and ammonia (NH3) by glutamine synthetase. In this reaction, an ATP is consumed. The reverse reaction yields glutamate and ammonium ions (NH4+). Almost all cells in the body contain glutamine and ammonium ions, and express both glutamine synthetase and GLS. The predominant expression of one or the other of these enzymes will determine whether tissues are more likely to produce or consume glutamine. α-KG, α-ketoglutarate; ASCT2, alanine-serine-cysteine transporter protein 2; GLS, glutaminase; GSH, glutathione; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species; TCA, tricarboxylic acid; XCT, cystine/glutamate transporter
Taken together, changes in glutamine utilization and metabolic pathways in leukemia cells are expected to be potential prognostic markers. For example, GLS is highly expressed in AML patients and IGF2BP2 is associated with prognosis in AML. By detecting dynamic changes in glutamine-related markers, it is possible to understand the progression, remission or relapse of leukemia and provide diagnostic value. Glutaminolysis inhibits the conversion of GLN to circulating TCA metabolites by regulating various enzymes, and GLS is the first step in the process. Therefore, targeting GLS to block GLN degradation is a promising therapeutic strategy. Targeting of the two GLS isoforms, GLS-1 and GLS-2, will provide new insights on the treatment of leukemia. Hereafter, we discuss the relationship between GLN and AML, CLL, and ALL.
6 Glutamine as a therapeutic strategy for leukemia
6.1 Acute myeloid leukemia
Reducing intracellular GLN levels in AML patients is one of the main strategies for AML treatment. The most important step in targeting GLN is the use of GLS to catalyze the deamination reaction of GLN to glutamate. Glutamate is further catabolized and metabolized to α-KG which feeds into the TCA cycle to provide energy. Therefore, glutaminase inhibitors are a popular antitumor strategy [54]. In particular, the renal-type GLS-1 form disrupts GLN-driven oxidative phosphorylation in AML cell lines, thereby preventing tumor growth and inducing apoptosis [55].
Blocking GLN metabolism with the GLS inhibitor CB-839 results in GSH depletion [56, 57]. In AML, where GSH acts as an antioxidant, a decrease in GSH leads to the accumulation of mitochondrial ROS and subsequent apoptosis [58, 59]. At the same time, the inhibitory effect of CB-839 makes AML cells more sensitive to adjuvants of the mitochondrial redox state, such as arsenic trioxide and hypertriglyceride [8]. Therefore, CB-839 applied together with the above adjuvants induces apoptosis in AML cells. In addition to inducing apoptosis in AML cells, CB-839 inhibits also the mTOR signaling pathway [54]. In AML cells, GLN condenses with cysteine and glycine to produce GSH, which maintains redox homeostasis, prevents ROS-induced damage, and provides a nitrogen source for DNA replication [55, 60]. In addition, many AML gene mutations have been shown to be associated with a number of mutations. In addition, many AML gene mutants are specific for GLN metabolism. For example, the glutaminase inhibitor BPTES was able to target and inhibit the unique metabolic profile of primary AML cells with IDH mutations (i.e., glutamine addiction), which in turn slowed the growth of primary AML cells with mutant IDH [49]. The fact that there is a unique selective inhibitory effect of interfering with glutamine metabolism on AML cells with IDH mutations is demonstrated. Other than this, aberrant expression of the FMS-like tyrosine kinase 3 (FLT3) gene in AML also leads to disorders of glutamate metabolism [61]. Approximately 25–30% of AML cases show hyperactivation due to mutations in tandem duplications within genes (FLT3–ITD) or in the structural domain of tyrosine kinase (FLT3–TKD) [62]. The FLT3 inhibitor AC220 (also known as Quizartinib) decreases GLN uptake and GSH production in AML cells, while increasing sensitivity to oxidative stress [63]. In addition, GLN is also used as a parenteral nutrient to assist in the treatment of AML. In a randomized, double-blind, controlled study including 45 adult AML patients and 127 cycles of chemotherapy, GLN improved the clinical course of patients after bone marrow transplantation and parenteral nutrition [64]. If in AML, the degree of GLN dependence of AML cells with specific gene mutations (e.g. IDH mutations and FLT3 mutations) is investigated. It is possible to assist in the diagnosis of such subtypes of AML with specific gene mutations by detecting GLN-related metabolic markers.
At present, the specific quantitative indicators and the exact extent of GLN dependence in AML subtypes with specific genetic mutations need to be investigated in further studies. In general, however, this dependence may be manifested by an increased rate of cellular uptake and utilization of GLN, increased activity of enzymes involved in intracellular GLN metabolism, and a more critical role of the GLN pathway in maintaining cell survival, proliferation, and energy supply. In conclusion, GLN is essential for the treatment of leukemia and is an effective therapeutic strategy. It is also important in medical research as a nutrient to support cell growth and repair, and as a potential antitumor agent for the treatment of leukemia.
6.2 Chronic lymphocytic leukemia
The 13q deletion is the most common cytogenetic mutation in CLL. Bruton’s tyrosine kinase and B-cell lymphoma-2 inhibitors are widely used in the clinic for the treatment of CLL; however, CLL cells have developed resistance to these drugs. CB-839, a small-molecule GLS-1 inhibitor, decreases GLS-1 activity and inhibits CLL cell proliferation; however, the efficacy of CB-839 is limited in combination with conventional CLL drugs [65]. In addition, CLL lymphocytes in del11q-positive CLL cells exhibit altered glutamine metabolism [66]. Mitochondria in CLL have been reported to increase ROS production [67]. The role of GLN in preventing the overproduction of ROS underscores its importance in tumor growth and energy production [68].
6.3 Acute lymphoblastic leukemia
Acute lymphoblastic leukemia is a heterogeneous malignancy of immature B or T lymphoblastoid cells that is most prevalent in children [69, 70]. l-asparaginase (ASNase) is the first-line therapy for childhood ALL [71, 72], as well as adult ALL [73]. ASNase hydrolyzes GLN to produce glutamate and may be considered for patients with Notch1 ALL positivity [74]. The notch1 receptor is effective in the treatment of ALL. Notably, when GLN is secreted by adipocytes, its cytotoxicity towards ALL cells is blocked [75]. Therefore, targeting GLN and ASNase may also serve in the development of novel therapeutic agents [76, 77]. In addition, GLN nutritional therapy during chemotherapy can effectively improve and enhance the systemic nutritional status and immunity of pediatric patients with ALL [78]. Notably, there are genomic differences in relapsed ALL during treatment [79]. Among these differences, reduced dependence on GLN is an important cause of drug resistance in leukemia cells [80].
Mitochondria are one of the major sources of ROS production. Redox dysfunction plays a crucial role in leukemogenesis in ALL, and inhibition of ROS production via NADPH oxidases is a novel therapeutic tool for the treatment of ALL [81]. Many enzymes neutralize ROS, including superoxide dismutase, catalase, glutathione peroxidase (GPX), thioredoxin, peroxiredoxin, and glutathione transferase [82]. In addition, activating mutations in NOTCH1 are common in T-cell ALL, and inhibition of NOTCH1 signaling suppresses and promotes autophagy during GLN catabolism [83]. GLN metabolism regulates the expression of mitochondrial uncoupling protein 2 (UCP2) in T-cell ALL cell lines, and UCP2 is required for T-cell ALL proliferation [84]. A link between UCP2 and ROS production has been demonstrated [85]. Therefore, promoting GSH production by blocking GLN metabolism and indirectly preventing ROS production is a novel therapeutic strategy for ALL.
7 GLN causes cellular ferroptosis in an indirect way
In clinical settings, radiotherapy remains the mainstay treatment for leukemia, although GLN-targeting agents (e.g., CB-839) have been developed to indirectly induce ROS production [8]. It is well known that ROS production and lipid peroxidation is a key feature and an important step in iron death, and the generation of ROS promotes lipid peroxidation, which in turn triggers iron death. However, the mechanism of iron death involves a variety of factors, including antioxidant system factors, such as GSH and GPX4, which are important mechanisms of iron death [86, 87]; iron metabolism factors, such as Fe2+ which promotes the production of ROS through the Fenton reaction and so on, which in turn promotes lipid peroxidation [88]; lipid metabolism-related factors, such as lipoxygenase enzymes (LOXs), which can directly oxidize unsaturated fatty acids on biological membranes (PUFAs) and PUFA-containing lipids on biological membranes, which may induce iron death [87]; signaling pathway factors, such as cystathionine-glutamate transporter receptor (system Xc -), p53, and other pathways can regulate iron death. The mechanism underlying the role of GLN in leukemia remains unclear, and its use in clinical practice is relatively rare. Notably, p53-dependent activation of GLS-2 expression correlates with ROS, while elevated ROS levels lead to p53 stabilization and activation [89]. Sawako et al. demonstrated that GLS-2 reduces cellular sensitivity to ROS-related apoptosis [19]. Increased mitochondrial production of ROS and lipid peroxidation, along with decreased expression of GSH and GPX4, lead to ferroptosis [90, 91]. The accumulation of intracellular iron during ferroptosis is important in leukemic cells. GLN metabolism enhances ROS production in the TCA cycle [92,93,94]. GLN catabolism inhibits intracellular GSH depletion and subsequent ROS generation [49, 95], as well as affecting the TCA cycle [96]. The accumulation of lipid ROS can lead to ferroptosis [96]. Notably, GLS-2 is present on the cell surface of human neutrophils [97], which promotes lipid ROS production and enhances ferroptosis by catalyzing the generation of α-KG from glutamate [98, 99]. GLN increases α-KG levels and can activate amino acid sensor kinases, leading to formation of mTORC1 [100, 101], which then regulates ferroptosis sensitivity [102, 103]. In conclusion, increasing ROS levels through GLN metabolism promotes ferroptosis by blocking GSH synthesis (Fig. 3), which may provide new therapeutic guidelines for ferroptosis-based clinical treatment.

Glutamine induces ferroptosis. α-KG, α-ketoglutarate; ASCT2, alanine-serine-cysteine transporter protein 2; GLS, glutaminase; GPX4, glutathione peroxidase 4; GSH, glutathione; OXPHOS, oxidative phosphorylation; PLs, phospholipids; PUFA, polyunsaturated fatty acids; ROS, reactive oxygen species; TCA, tricarboxylic acid; XCT, cystine/glutamate transporter
8 Discussion
GLN and its metabolites have significant antileukemic effects. Because of the close association between GLN and leukemia prognosis, we have summarized the latest developments on GLN and leukemia-related drugs or other studies by publication date (in no particular order), category, and content (Table 1).
There are multiple pathways involving GLN in leukemia, along with multiple factors that regulate and interfere with each pathway. GLN metabolic pathway: Supplies carbon for TCA cycle intermediates and nitrogen for nucleotide and amino acid biosynthesis, and plays an important role in hematopoietic tumors and hematologic neoplasms [116]; Bypass pathway: when leukemia cells are deprived of Gln, the serine pathway upregulates the key serine enzymes phosphoglycerate dehydrogenase (PHGDH) and phosphoribosyltransferase (PSAT), leading to an increased demand for serine and exacerbation of serine dependence in leukemia cells [117]; Lipid-related metabolism: AML cells are dependent on OXPHOS [118,119,120], AML cells obtain free fatty acids from bone marrow adipocytes and utilize fatty acid oxidation (FAO) and OXPHOS to maintain AML cell survival and growth [121,122,123]. However, OXPHOS-deficient cells accelerate the utilization of GLN, and GLN depletion promotes the accumulation of ROS [124]. We also note that targeting GLN and GLN metabolism (or parts of it) may have a limited impact on leukemia therapy. For example, ASCT2 is not the sole transporter for GLN [125]. This suggests that GLN influences the development and progression of leukemia. Therefore, future studies related to GLN should focus on the inhibition of multiple metabolic pathways for the effective treatment of leukemia.
Research on human leukemia therapy and GLN continues. The body's immune cells are also the focus of our interference with GLN, both in terms of the dependence of cancer cells on GLN and as a component of the TCA cycle. For example, immune cells use GLN to grow rapidly and gain immunity [126]. Reprogrammed GLN metabolism plays an important role in the antitumor immune response of immune cells, such as T cells, B cells, macrophages, and natural killer cells [127]. Parenteral GLN supplements may be relevant in the anti-tumor immune response, as they enhance neutrophil phagocytosis and maintain the nutritional status [128]. Inhibition of GLN metabolism can lead to immune escape for cancer cells, as observed with the GLN inhibitor V-9302 in human breast cancer cells [129]. Therefore, further experiments should be conducted to determine whether GLN inhibitors and related drugs disrupt the anticancer effects of immune cells in the bone marrow microenvironment during diagnosis and treatment. Targeting GLN metabolism in cancer cells without interfering with the immune response is a major challenge for future research.
9 Conclusion
Existing evidence points to the attractiveness of GLN as a new target for the treatment of leukemia. This strategy is already exemplified by CB-839 and V-9302, but includes also mTOR signaling and apoptosis in AML cells. Expression of an abnormal gene (FLT3) can cause GLN metabolic disorders. In addition, GLN plays an important role in CLL and ALL. The key enzyme in GLN metabolism is the GLS-2 isoform, which acts as a tumor suppressor. It activates and promotes hepatic glutamine catabolism and triggers the activation of mTORC1, a signal that also controls ferroptosis.
Inhibitors of GLN metabolism have also some limitations in the treatment of leukemia. First, a clear framework for clinical care has not been established, and there is a lack of additional empirical support for the use of GLN inhibitors to improve the prognosis of leukemia. More experimental studies are needed to provide better empirical data to improve the prognostic gap in GLN treatment of leukemia. Second, numerous studies have been conducted on AML, while relatively few have considered other types of leukemia (including, but not limited to, leukemia with rare genotypes and phenotypes). Such studies could determine whether the mechanism of action is the same in AML as in other types of leukemia.
In conclusion, available studies suggest that GLN is an attractive new strategy for the treatment of leukemia.
10 Future directions
The study of GLN in leukemia is in its preliminary stages, and its mechanism of action and clinical applications need further investigation. Future studies can focus on the following aspects: (1) the regulatory effect of GLN on various functional molecules in leukemia cells, (2) the regulatory mechanism of GLN on the relevant functional molecules in leukemia cells, (3) the application of GLN in leukemia treatment, (4) the effects of GLN on leukemia cell functions, such as cellular energy supply mechanisms, essential molecules, intracellular redox homeostasis mechanisms, and related signaling pathways for survival and proliferation, and (5) the effect of GLN on the prognosis of leukemia patients. The accrued knowledge will provide a new perspective for the effective clinical treatment of different types of leukemia.
Data availability
No datasets were generated or analysed during the current study.
References
Pollyea DA, Bixby D, Perl A, Bhatt VR, Altman JK, Appelbaum FR, de Lima M, Fathi AT, Foran JM, Gojo I, Hall AC, Jacoby M, Lancet J, Mannis G, Marcucci G, Martin MG, Mims A, Neff J, Nejati R, Olin R, Percival M-E, Prebet T, Przespolewski A, Rao D, Ravandi-Kashani F, Shami PJ, Stone RM, Strickland SA, Sweet K, Vachhani P, Wieduwilt M, Gregory KM, Ogba N, Tallman MS. NCCN guidelines insights: acute myeloid leukemia, Version 2.2021. J Natl Compr Cancer Netw JNCCN. 2021;19:16–27. https://doi.org/10.6004/jnccn.2021.0002.
Jin X, Zhang M, Sun R, Lyu H, Xiao X, Zhang X, Li F, Xie D, Xiong X, Wang J, Lu W, Zhang H, Zhao M. First-in-human phase I study of CLL-1 CAR-T cells in adults with relapsed/refractory acute myeloid leukemia. J Hematol OncolJ Hematol Oncol. 2022;15:88. https://doi.org/10.1186/s13045-022-01308-1.
Wang Q, Wang Y, Lv H, Han Q, Fan H, Guo B, Wang L, Han W. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015;23:184–91. https://doi.org/10.1038/mt.2014.164.
Gallazzi M, Ucciero MAM, Faraci DG, Mahmoud AM, Al Essa W, Gaidano G, Mouhssine S, Crisà E. New frontiers in monoclonal antibodies for the targeted therapy of acute myeloid leukemia and myelodysplastic syndromes. Int J Mol Sci. 2022;23:7542. https://doi.org/10.3390/ijms23147542.
Abaza Y, Zeidan AM. Immune checkpoint inhibition in acute myeloid leukemia and myelodysplastic syndromes. Cells. 2022;11:2249. https://doi.org/10.3390/cells11142249.
Riether C, Pabst T, Höpner S, Bacher U, Hinterbrandner M, Banz Y, Müller R, Manz MG, Gharib WH, Francisco D, Bruggmann R, van Rompaey L, Moshir M, Delahaye T, Gandini D, Erzeel E, Hultberg A, Fung S, de Haard H, Leupin N, Ochsenbein AF. Targeting CD70 with cusatuzumab eliminates acute myeloid leukemia stem cells in patients treated with hypomethylating agents. Nat Med. 2020;26:1459–67. https://doi.org/10.1038/s41591-020-0910-8.
Zhou H, Wang F, Niu T. Prediction of prognosis and immunotherapy response of amino acid metabolism genes in acute myeloid leukemia. Front Nutr. 2022;9:1056648. https://doi.org/10.3389/fnut.2022.1056648.
Gregory MA, Nemkov T, Park HJ, Zaberezhnyy V, Gehrke S, Adane B, Jordan CT, Hansen KC, D’Alessandro A, DeGregori J. Targeting glutamine metabolism and redox state for leukemia therapy. Clin Cancer Res Off J Am Assoc Cancer Res. 2019;25:4079–90. https://doi.org/10.1158/1078-0432.CCR-18-3223.
Yang L, Venneti S, Nagrath D. Glutaminolysis: a hallmark of cancer metabolism. Annu Rev Biomed Eng. 2017;19:163–94. https://doi.org/10.1146/annurev-bioeng-071516-044546.
Darmaun D, Matthews DE, Bier DM. Glutamine and glutamate kinetics in humans. Am J Physiol. 1986;251:E117-126. https://doi.org/10.1152/ajpendo.1986.251.1.E117.
Rex MR, Williams R, Birsoy K, Ta Llman MS, Stahl M. Targeting mitochondrial metabolism in acute myeloid leukemia. Leuk Lymphoma. 2022;63:530–7. https://doi.org/10.1080/10428194.2021.1992759.
Wang M, Zhao A, Li M, Niu T. Amino acids in hematologic malignancies: current status and future perspective. Front Nutr. 2023;10:1113228. https://doi.org/10.3389/fnut.2023.1113228.
Emadi A. Exploiting AML vulnerability: glutamine dependency. Blood. 2015;126:1269–70. https://doi.org/10.1182/blood-2015-07-659508.
Petronini PG, Urbani S, Alfieri R, Borghetti AF, Guidotti GG. Cell susceptibility to apoptosis by glutamine deprivation and rescue: survival and apoptotic death in cultured lymphoma-leukemia cell lines. J Cell Physiol. 1996;169:175–85. https://doi.org/10.1002/(SICI)1097-4652(199610)169:1%3c175::AID-JCP18%3e3.0.CO;2-C.
Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491:364–73. https://doi.org/10.1038/nature11706.
Saha SK, Islam SMR, Abdullah-AL-Wadud M, Islam S, Ali F, Park KS. Multiomics analysis reveals that GLS and GLS2 differentially modulate the clinical outcomes of cancer. J Clin Med. 2019;8:355. https://doi.org/10.3390/jcm8030355.
Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA. 2010;107:7455–60. https://doi.org/10.1073/pnas.1001006107.
Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–6. https://doi.org/10.1038/358015a0.
Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, Sugano S, Sato E, Nagao T, Yokote K, Tatsuno I, Prives C. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA. 2010;107:7461–6. https://doi.org/10.1073/pnas.1002459107.
Giacobbe A, Bongiorno-Borbone L, Bernassola F, Terrinoni A, Markert EK, Levine AJ, Feng Z, Agostini M, Zolla L, Agrò AF, Notterman DA, Melino G, Peschiaroli A. p63 regulates glutaminase 2 expression. Cell Cycle. 2013;12:1395–405. https://doi.org/10.4161/cc.24478.
Velletri T, Romeo F, Tucci P, Peschiaroli A, Annicchiarico-Petruzzelli M, Niklison-Chirou MV, Amelio I, Knight RA, Mak TW, Melino G, Agostini M. GLS2 is transcriptionally regulated by p73 and contributes to neuronal differentiation. Cell Cycle. 2013;12:3564–73. https://doi.org/10.4161/cc.26771.
Hung C-L, Wang L-Y, Yu Y-L, Chen H-W, Srivastava S, Petrovics G, Kung H-J. A long noncoding RNA connects c-Myc to tumor metabolism. Proc Natl Acad Sci USA. 2014;111:18697–702. https://doi.org/10.1073/pnas.1415669112.
Gao P, Tchernyshyov I, Chang T-C, Lee Y-S, Kita K, Ochi T, Zeller K, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV. c-Myc suppression of miR-23 enhances mitochondrial glutaminase and glutamine metabolism. Nature. 2009;458:762–5. https://doi.org/10.1038/nature07823.
Dernie F. Characterisation of a mitochondrial glutamine transporter provides a new opportunity for targeting glutamine metabolism in acute myeloid leukaemia. Blood Cells Mol Dis. 2021;88: 102422. https://doi.org/10.1016/j.bcmd.2020.102422.
Cormerais Y, Massard PA, Vucetic M, Giuliano S, Tambutté E, Durivault J, Vial V, Endou H, Wempe MF, Parks SK, Pouyssegur J. The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J Biol Chem. 2018;293:2877–87. https://doi.org/10.1074/jbc.RA117.001342.
Teixeira E, Silva C, Martel F. The role of the glutamine transporter ASCT2 in antineoplastic therapy. Cancer Chemother Pharmacol. 2021;87:447–64. https://doi.org/10.1007/s00280-020-04218-6.
Ni F, Yu W-M, Li Z, Graham DK, Jin L, Kang S, Rossi MR, Li S, Broxmeyer HE, Qu C-K. Critical role of ASCT2-mediated amino acid metabolism in promoting leukaemia development and progression. Nat Metab. 2019;1:390–403. https://doi.org/10.1038/s42255-019-0039-6.
Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, Kondo J, Coffey RJ, Johnson MO, Rathmell JC, Sharick JT, Skala MC, Smith JA, Berlin J, Washington MK, Nickels ML, Manning HC. Pharmacological blockade of ASCT2-dependent glutamine transport leads to anti-tumor efficacy in preclinical models. Nat Med. 2018;24:194–202. https://doi.org/10.1038/nm.4464.
Huang F, Zhao Y, Zhao J, Wu S, Jiang Y, Ma H, Zhang T. Upregulated SLC1A5 promotes cell growth and survival in colorectal cancer. Int J Clin Exp Pathol. 2014;7:6006–14.
Hassanein M, Hoeksema MD, Shiota M, Qian J, Harris BK, Chen H, Clark JE, Alborn WE, Eisenberg R, Massion PP. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res Off J Am Assoc Cancer Res. 2013;19:560–70. https://doi.org/10.1158/1078-0432.CCR-12-2334.
Wang Q, Hardie R-A, Hoy AJ, van Geldermalsen M, Gao D, Fazli L, Sadowski MC, Balaban S, Schreuder M, Nagarajah R, Wong JJ-L, Metierre C, Pinello N, Otte NJ, Lehman ML, Gleave M, Nelson CC, Bailey CG, Ritchie W, Rasko JEJ, Holst J. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J Pathol. 2015;236:278–89. https://doi.org/10.1002/path.4518.
Nikkuni O, Kaira K, Toyoda M, Shino M, Sakakura K, Takahashi K, Tominaga H, Oriuchi N, Suzuki M, Iijima M, Asao T, Nishiyama M, Nagamori S, Kanai Y, Oyama T, Chikamatsu K. Expression of amino acid transporters (LAT1 and ASCT2) in patients with stage III/IV laryngeal squamous cell carcinoma. Pathol Oncol Res POR. 2015;21:1175–81. https://doi.org/10.1007/s12253-015-9954-3.
Honjo H, Kaira K, Miyazaki T, Yokobori T, Kanai Y, Nagamori S, Oyama T, Asao T, Kuwano H. Clinicopathological significance of LAT1 and ASCT2 in patients with surgically resected esophageal squamous cell carcinoma. J Surg Oncol. 2016;113:381–9. https://doi.org/10.1002/jso.24160.
Ren P, Yue M, Xiao D, Xiu R, Gan L, Liu H, Qing G. ATF4 and N-Myc coordinate glutamine metabolism in MYCN-amplified neuroblastoma cells through ASCT2 activation. J Pathol. 2015;235:90–100. https://doi.org/10.1002/path.4429.
Ye J, Huang Q, Xu J, Huang J, Wang J, Zhong W, Chen W, Lin X, Lin X. Targeting of glutamine transporter ASCT2 and glutamine synthetase suppresses gastric cancer cell growth. J Cancer Res Clin Oncol. 2018;144:821–33. https://doi.org/10.1007/s00432-018-2605-9.
Wang W, Pan H, Ren F, Chen H, Ren P. Targeting ASCT2-mediated glutamine metabolism inhibits proliferation and promotes apoptosis of pancreatic cancer cells. Biosci Rep. 2022;42:BSR20212171. https://doi.org/10.1042/BSR20212171.
Dunphy MPS, Harding JJ, Venneti S, Zhang H, Burnazi EM, Bromberg J, Omuro AM, Hsieh JJ, Mellinghoff IK, Staton K, Pressl C, Beattie BJ, Zanzonico PB, Gerecitano JF, Kelsen DP, Weber W, Lyashchenko SK, Kung HF, Lewis JS. In vivo PET assay of tumor glutamine flux and metabolism: in-human trial of 18F-(2S,4R)-4-fluoroglutamine. Radiology. 2018;287:667–75. https://doi.org/10.1148/radiol.2017162610.
van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, Ritchie W, Feng Y, Bailey CG, Deng N, Harvey K, Beith JM, Selinger CI, O’Toole SA, Rasko JEJ, Holst J. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. 2016;35:3201–8. https://doi.org/10.1038/onc.2015.381.
Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136–52. https://doi.org/10.1056/NEJMra1406184.
Panuzzo C, Jovanovski A, Pergolizzi B, Pironi L, Stanga S, Fava C, Cilloni D. Mitochondria: a galaxy in the hematopoietic and leukemic stem cell universe. Int J Mol Sci. 2020;21:3928. https://doi.org/10.3390/ijms21113928.
Sánchez-Mendoza SE, Rego EM. Targeting the mitochondria in acute myeloid leukemia. Appl Cancer Res. 2017;37:22. https://doi.org/10.1186/s41241-017-0022-z.
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, Sharp KA, Levine RL, Thompson CB. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzymatic activity that converts α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–34. https://doi.org/10.1016/j.ccr.2010.01.020.
DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104:19345–50. https://doi.org/10.1073/pnas.0709747104.
Wang Y, Zhang L, Chen W-L, Wang J-H, Li N, Li J-M, Mi J-Q, Zhang W-N, Li Y, Wu S-F, Jin J, Wang Y-G, Huang H, Chen Z, Chen S-J, Tang H. Rapid diagnosis and prognosis of de novo acute myeloid leukemia by serum metabonomic analysis. J Proteome Res. 2013;12:4393–401. https://doi.org/10.1021/pr400403p.
Rudman D, Vogler WR, Howard CH, Gerron GG. Observations on the plasma amino acids of patients with acute leukemia. Cancer Res. 1971;31:1159–65.
Wang D, Tan G, Wang H, Chen P, Hao J, Wang Y. Identification of novel serum biomarker for the detection of acute myeloid leukemia based on liquid chromatography-mass spectrometry. J Pharm Biomed Anal. 2019;166:357–63. https://doi.org/10.1016/j.jpba.2019.01.022.
Willems L, Jacque N, Jacquel A, Neveux N, Maciel TT, Lambert M, Schmitt A, Poulain L, Green AS, Uzunov M, Kosmider O, Radford-Weiss I, Moura IC, Auberger P, Ifrah N, Bardet V, Chapuis N, Lacombe C, Mayeux P, Tamburini J, Bouscary D. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood. 2013;122:3521–32. https://doi.org/10.1182/blood-2013-03-493163.
Matre P, Velez J, Jacamo R, Qi Y, Su X, Cai T, Chan SM, Lodi A, Sweeney SR, Ma H, Davis RE, Baran N, Haferlach T, Su X, Flores ER, Gonzalez D, Konoplev S, Samudio I, DiNardo C, Majeti R, Schimmer AD, Li W, Wang T, Tiziani S, Konopleva M. Inhibiting glutaminase in acute myeloid leukemia: metabolic dependency of selected AML subtypes. Oncotarget. 2016;7:79722–35. https://doi.org/10.18632/oncotarget.12944.
Emadi A, Jun SA, Tsukamoto T, Fathi AT, Minden MD, Dang CV. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp Hematol. 2014;42:247–51. https://doi.org/10.1016/j.exphem.2013.12.001.
Basak NP, Banerjee S. Mitochondrial dependency in progression of acute myeloid leukemia. Mitochondrion. 2015;21:41–8. https://doi.org/10.1016/j.mito.2015.01.006.
Wagner A, Kosnacova H, Chovanec M, Jurkovicova D. Mitochondrial genetic and epigenetic regulations in cancer: therapeutic potential. Int J Mol Sci. 2022;23:7897. https://doi.org/10.3390/ijms23147897.
Sharma ND, Keewan E, Matlawska-Wasowska K. Metabolic reprogramming and cell adhesion in acute leukemia adaptation to the CNS niche. Front Cell Dev Biol. 2021;9: 767510. https://doi.org/10.3389/fcell.2021.767510.
Weng H, Huang F, Yu Z, Chen Z, Prince E, Kang Y, Zhou K, Li W, Hu J, Fu C, Aziz T, Li H, Li J, Yang Y, Han L, Zhang S, Ma Y, Sun M, Wu H, Zhang Z, Wunderlich M, Robinson S, Braas D, Hoeve JT, Zhang B, Marcucci G, Mulloy JC, Zhou K, Tao H-F, Deng X, Horne D, Wei M, Huang H, Chen J. The m6A reader IGF2BP2 regulates glutamine metabolism and represents a therapeutic target in acute myeloid leukemia. Cancer Cell. 2022;40:1566-1582.e10. https://doi.org/10.1016/j.ccell.2022.10.004.
Song M, Kim S-H, Im CY, Hwang H-J. Recent development of small molecule glutaminase inhibitors. Curr Top Med Chem. 2018;18:432–43. https://doi.org/10.2174/1568026618666180525100830.
Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, Saland E, Decroocq J, Maciel TT, Lambert M, Poulain L, Hospital MA, Sujobert P, Joseph L, Chapuis N, Lacombe C, Moura IC, Demo S, Sarry JE, Recher C, Mayeux P, Tamburini J, Bouscary D. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood. 2015;126:1346–56. https://doi.org/10.1182/blood-2015-01-621870.
Mao H, Wen Y, Yu Y, Li H, Wang J, Sun B. Bioinspired nanocatalytic tumor therapy by simultaneous reactive oxygen species generation enhancement and glutamine pathway-mediated glutathione depletion. J Mater Chem B. 2022;11:131–43. https://doi.org/10.1039/d2tb02194c.
Boysen G, Jamshidi-Parsian A, Davis MA, Siegel ER, Kore RA, Dings RPM, Griffin RJ. Glutaminase inhibitor CB-839 increases radiation sensitivity of lung tumor cells and human lung tumor xenografts in mice. Int J Radiat Biol. 2019;95:436–42. https://doi.org/10.1080/09553002.2018.1558299.
Romo-González M, Ijurko C, Hernández-Hernández Á. Reactive oxygen species and metabolism in leukemia: a dangerous liaison. Front Immunol. 2022;13: 889875. https://doi.org/10.3389/fimmu.2022.889875.
Chen Y-F, Liu H, Luo X-J, Zhao Z, Zou Z-Y, Li J, Lin X-J, Liang Y. The roles of reactive oxygen species (ROS) and autophagy in the survival and death of leukemia cells. Crit Rev Oncol Hematol. 2017;112:21–30. https://doi.org/10.1016/j.critrevonc.2017.02.004.
Kreitz J, Schönfeld C, Seibert M, Stolp V, Alshamleh I, Oellerich T, Steffen B, Schwalbe H, Schnütgen F, Kurrle N, Serve H. Metabolic plasticity of acute myeloid leukemia. Cells. 2019;8:805. https://doi.org/10.3390/cells8080805.
Gregory MA, Nemkov T, Reisz JA, Zaberezhnyy V, Hansen KC, D’Alessandro A, DeGregori J. Glutaminase inhibition improves FLT3 inhibitor therapy for acute myeloid leukemia. Exp Hematol. 2018;58:52–8. https://doi.org/10.1016/j.exphem.2017.09.007.
Khamari R, Degand C, Fovez Q, Trinh A, Chomy A, Laine W, Dekiouk S, Ghesquiere B, Quesnel B, Marchetti P, Manier S, Kluza PJ. Key role of glutamine metabolism in persistence of leukemic cells upon exposition to FLT3 tyrosine kinase inhibitors. Exp Hematol. 2024. https://doi.org/10.1016/j.exphem.2024.104253.
Gregory MA, D’Alessandro A, Alvarez-Calderon F, Kim J, Nemkov T, Adane B, Rozhok AI, Kumar A, Kumar V, Pollyea DA, Wempe MF, Jordan CT, Serkova NJ, Tan AC, Hansen KC, DeGregori J. ATM/G6PD-driven redox metabolism promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc Natl Acad Sci U S A. 2016;113:E6669–78. https://doi.org/10.1073/pnas.1603876113.
Scheid C, Hermann K, Kremer G, Holsing A, Heck G, Fuchs M, Waldschmidt D, Herrmann H-J, Söhngen D, Diehl V, Schwenk A. Randomized, double-blind, controlled study of glycyl-glutamine-dipeptide in the parenteral nutrition of patients with acute leukemia undergoing intensive chemotherapy. Nutr Burbank Los Angel Cty Calif. 2004;20:249–54. https://doi.org/10.1016/j.nut.2003.11.018.
Timofeeva N, Ayres ML, Baran N, Santiago-O’Farrill JM, Bildik G, Lu Z, Konopleva M, Gandhi V. Preclinical investigations of the efficacy of the glutaminase inhibitor CB-839 alone and in combinations in chronic lymphocytic leukemia. Front Oncol. 2023;13:1161254. https://doi.org/10.3389/fonc.2023.1161254.
Galicia-Vázquez G, Smith S, Aloyz R. Del11q-positive CLL lymphocytes exhibit altered glutamine metabolism and differential response to GLS1 and glucose metabolism inhibition. Blood Cancer J. 2018;8:13. https://doi.org/10.1038/s41408-017-0039-2.
Jitschin R, Hofmann AD, Bruns H, Giessl A, Bricks J, Berger J, Saul D, Eckart MJ, Mackensen A, Mougiakakos D. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood. 2014;123:2663–72. https://doi.org/10.1182/blood-2013-10-532200.
Deaglio S. Glutamine and CLL: ready for prime time? Blood. 2022;140:528–9. https://doi.org/10.1182/blood.2022016696.
Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 2017;7: e577. https://doi.org/10.1038/bcj.2017.53.
Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin. 2014;64:83–103. https://doi.org/10.3322/caac.21219.
Avramis VI, Tiwari PN. Asparaginase (native ASNase or pegylated ASNase) in the treatment of acute lymphoblastic leukemia. Int J Nanomed. 2006;1:241–54.
Beard ME, Crowther D, Galton DA, Guyer RJ, Fairley GH, Kay HE, Knapton PJ, Malpas JS, Scott RB. L-asparaginase in treatment of acute leukaemia and lymphosarcoma. Br Med J. 1970;1:191–5. https://doi.org/10.1136/bmj.1.5690.191.
Patil S, Coutsouvelis J, Spencer A. Asparaginase in the management of adult acute lymphoblastic leukaemia: is it used appropriately? Cancer Treat Rev. 2011;37:202–7. https://doi.org/10.1016/j.ctrv.2010.08.002.
Nguyen TL, Nokin M-J, Terés S, Tomé M, Bodineau C, Galmar O, Pasquet J-M, Rousseau B, van Liempd S, Falcon-Perez JM, Richard E, Muzotte E, Rezvani H-R, Priault M, Bouchecareilh M, Redonnet-Vernhet I, Calvo J, Uzan B, Pflumio F, Fuentes P, Toribio ML, Khatib A-M, Soubeyran P, Murdoch PDS, Durán RV. Downregulation of glutamine synthetase, not glutaminolysis, is responsible for glutamine addiction in Notch1-driven acute lymphoblastic leukemia. Mol Oncol. 2021;15:1412–31. https://doi.org/10.1002/1878-0261.12877.
Ehsanipour EA, Sheng X, Behan JW, Wang X, Butturini A, Avramis VI, Mittelman SD. Adipocytes cause leukemia cell resistance to L-asparaginase via release of glutamine. Cancer Res. 2013;73:2998–3006. https://doi.org/10.1158/0008-5472.CAN-12-4402.
Tabe Y, Lorenzi PL, Konopleva M. Amino acid metabolism in hematologic malignancies and the era of targeted therapy. Blood. 2019;134:1014–23. https://doi.org/10.1182/blood.2019001034.
Cory JG, Cory AH. Critical roles of glutamine as nitrogen donors in purine and pyrimidine nucleotide synthesis: asparaginase treatment in childhood acute lymphoblastic leukemia. Vivo Athens Greece. 2006;20:587–9.
Han Y, Zhang F, Wang J, Zhu Y, Dai J, Bu Y, Yang Q, Xiao Y, Sun X. Application of Glutamine-enriched nutrition therapy in childhood acute lymphoblastic leukemia. Nutr J. 2016;15:65. https://doi.org/10.1186/s12937-016-0187-4.
Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, Downing JR. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–80. https://doi.org/10.1126/science.1164266.
Stäubert C, Bhuiyan H, Lindahl A, Broom OJ, Zhu Y, Islam S, Linnarsson S, Lehtiö J, Nordström A. Rewired metabolism in drug-resistant leukemia cells: a metabolic switch hallmarked by reduced dependence on exogenous glutamine. J Biol Chem. 2015;290:8348–59. https://doi.org/10.1074/jbc.M114.618769.
Mannan A, Germon ZP, Chamberlain J, Sillar JR, Nixon B, Dun MD. Reactive oxygen species in acute lymphoblastic leukaemia: reducing radicals to refine responses. Antioxid Basel Switz. 2021;10:1616. https://doi.org/10.3390/antiox10101616.
Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012;5:9–19. https://doi.org/10.1097/WOX.0b013e3182439613.
Herranz D, Ambesi-Impiombato A, Sudderth J, Sánchez-Martín M, Belver L, Tosello V, Xu L, Wendorff AA, Castillo M, Haydu JE, Márquez J, Matés JM, Kung AL, Rayport S, Cordon-Cardo C, DeBerardinis RJ, Ferrando AA. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in acute lymphoblastic leukemia. Nat Med. 2015;21:1182–9. https://doi.org/10.1038/nm.3955.
Sancerni T, Renoult O, Luby A, Caradeuc C, Lenoir V, Croyal M, Ransy C, Aguilar E, Postic C, Bertho G, Dentin R, Prip-Buus C, Pecqueur C, Alves-Guerra M-C. UCP2 silencing restrains leukemia cell proliferation through glutamine metabolic remodeling. Front Immunol. 2022;13: 960226. https://doi.org/10.3389/fimmu.2022.960226.
Raho S, Capobianco L, Malivindi R, Vozza A, Piazzolla C, De Leonardis F, Gorgoglione R, Scarcia P, Pezzuto F, Agrimi G, Barile SN, Pisano I, Reshkin SJ, Greco MR, Cardone RA, Rago V, Li Y, Marobbio CMT, Sommergruber W, Riley CL, Lasorsa FM, Mills E, Vegliante MC, De Benedetto GE, Fratantonio D, Palmieri L, Dolce V, Fiermonte G. KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nat Metab. 2020;2:1373–81. https://doi.org/10.1038/s42255-020-00315-1.
Liu Y, Wan Y, Jiang Y, Zhang L, Cheng W. GPX4: The hub of lipid oxidation, ferroptosis, disease and treatment. Biochim Biophys Acta Rev Cancer. 2023;1878: 188890. https://doi.org/10.1016/j.bbcan.2023.188890.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82. https://doi.org/10.1038/s41580-020-00324-8.
Rochette L, Dogon G, Rigal E, Zeller M, Cottin Y, Vergely C. Lipid Peroxidation and iron metabolism: two corner stones in the homeostasis control of ferroptosis. Int J Mol Sci. 2022;24:449. https://doi.org/10.3390/ijms24010449.
Chen K, Albano A, Ho A, Keaney JF. Activation of p53 by oxidative stress involves platelet-derived growth factor-beta receptor-mediated ataxia telangiectasia mutated (ATM) kinase activation. J Biol Chem. 2003;278:39527–33. https://doi.org/10.1074/jbc.M304423200.
Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W, Wang J. Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cell Mol Med. 2019;23:4900–12. https://doi.org/10.1111/jcmm.14511.
Yan H-F, Zou T, Tuo Q-Z, Xu S, Li H, Belaidi AA, Lei P. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6:49. https://doi.org/10.1038/s41392-020-00428-9.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308. https://doi.org/10.1016/j.molcel.2015.06.011.
Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GRS, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107:8788–93. https://doi.org/10.1073/pnas.1003428107.
Anderson NM, Mucka P, Kern JG, Feng H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell. 2018;9:216–37. https://doi.org/10.1007/s13238-017-0451-1.
Panieri E, Santoro MM. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 2016;7: e2253. https://doi.org/10.1038/cddis.2016.105.
Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, Jiang X. Role of mitochondria in ferroptosis. Mol Cell. 2019;73:354-363.e3. https://doi.org/10.1016/j.molcel.2018.10.042.
Buczkowska J, Szeliga M. Two faces of glutaminase GLS2 in carcinogenesis. Cancers. 2023;15:5566. https://doi.org/10.3390/cancers15235566.
Suzuki S, Venkatesh D, Kanda H, Nakayama A, Hosokawa H, Lee E, Miki T, Stockwell BR, Yokote K, Tanaka T, Prives C. GLS2 is a tumor suppressor and a regulator of ferroptosis in hepatocellular carcinoma. Cancer Res. 2022;82:3209–22. https://doi.org/10.1158/0008-5472.CAN-21-3914.
Suzuki S, Venkatesh D, Tanaka T, Prives C. GLS2 shapes ferroptosis in hepatocellular carcinoma. Oncotarget. 2023;14:900–3. https://doi.org/10.18632/oncotarget.28526.
Durán RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E, Hall MN. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell. 2012;47:349–58. https://doi.org/10.1016/j.molcel.2012.05.043.
Wang L, Zhu L, Wu K, Chen Y, Lee D-Y, Gucek M, Sack MN. Mitochondrial general control of amino acid synthesis 5 Like 1 regulates glutaminolysis, mTORC1 activity and murine liver regeneration. Hepatol Baltim Md. 2020;71:643–57. https://doi.org/10.1002/hep.30876.
Zhu J, Wang H, Jiang X. mTORC1 beyond anabolic metabolism: regulation of cell death. J Cell Biol. 2022;221: e202208103. https://doi.org/10.1083/jcb.202208103.
Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, Lei G, Mao C, Koppula P, Cheng W, Zhang J, Xiao Z, Zhuang L, Fang B, Chen J, Qian S-B, Gan B. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. 2021;12:1589. https://doi.org/10.1038/s41467-021-21841-w.
Jin H, Wang S, Zaal EA, Wang C, Wu H, Bosma A, Jochems F, Isima N, Jin G, Lieftink C, Beijersbergen R, Berkers CR, Qin W, Bernards R. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. Elife. 2020;9: e56749. https://doi.org/10.7554/eLife.56749.
Li Q-Q, Pan S-Y, Chen Q-Y, Zhou W, Wang S-Q. Effect of competitive antagonist of transmembrane glutamine flux V-9302 on apoptosis of acute myeloid leukemia cell lines HL-60 and KG-1. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2021;29:685–9. https://doi.org/10.19746/j.cnki.issn.1009-2137.2021.03.005.
Li Y, Shao H, Da Z, Pan J, Fu B. High expression of SLC38A1 predicts poor prognosis in patients with de novo acute myeloid leukemia. J Cell Physiol. 2019;234:20322–8. https://doi.org/10.1002/jcp.28632.
Corti A, Dominici S, Piaggi S, Belcastro E, Chiu M, Taurino G, Pacini S, Bussolati O, Pompella A. γ-Glutamyltransferase enzyme activity of cancer cells modulates L-γ-glutamyl-p-nitroanilide (GPNA) cytotoxicity. Sci Rep. 2019;9:891. https://doi.org/10.1038/s41598-018-37385-x.
Emadi A, Law JY, Strovel ET, Lapidus RG, Jeng LJB, Lee M, Blitzer MG, Carter-Cooper BA, Sewell D, Van Der Merwe I, Philip S, Imran M, Yu SL, Li H, Amrein PC, Duong VH, Sausville EA, Baer MR, Fathi AT, Singh Z, Bentzen SM. Asparaginase Erwinia chrysanthemi effectively depletes plasma glutamine in adult patients with relapsed/refractory acute myeloid leukemia. Cancer Chemother Pharmacol. 2018;81:217–22. https://doi.org/10.1007/s00280-017-3459-6.
Zavorka Thomas ME, Lu X, Talebi Z, Jeon JY, Buelow DR, Gibson AA, Uddin ME, Brinton LT, Nguyen J, Collins M, Lodi A, Sweeney SR, Campbell MJ, Sweet DH, Sparreboom A, Lapalombella R, Tiziani S, Baker SD. Gilteritinib inhibits glutamine uptake and utilization in FLT3-ITD-positive AML. Mol Cancer Ther. 2021;20:2207–17. https://doi.org/10.1158/1535-7163.MCT-21-0071.
Konopleva M, DiNardo C, Bhagat T, Baran N, Lodi A, Saxena K, Cai T, Su X, Skwarska A, Guerra V, Kuruvilla V, Konoplev S, Gordon-Mitchell S, Pradhan K, Aluri S, Collins M, Sweeney S, Busquet J, Rathore A, Deng Q, Green M, Grant S, Demo S, Choudhary G, Sahu S, Agarwal B, Spodek M, Thiruthuvanathan V, Will B, Steidl U, Tippett G, Burger J, Borthakur G, Jabbour E, Pemmaraju N, Kadia T, Kornblau S, Daver N, Naqvi K, Short N, Garcia-Manero G, Tiziani S, Verma A. Glutaminase inhibition in combination with azacytidine in myelodysplastic syndromes: clinical efficacy and correlative analyses. Res Sq. 2023. https://doi.org/10.21203/rs.3.rs-2518774/v1.
Lemberg KM, Vornov JJ, Rais R, Slusher BS. We’re not “DON” yet: optimal dosing and prodrug delivery of 6-Diazo-5-oxo-L-norleucine. Mol Cancer Ther. 2018;17:1824–32. https://doi.org/10.1158/1535-7163.MCT-17-1148.
Hanaford AR, Alt J, Rais R, Wang SZ, Kaur H, Thorek DLJ, Eberhart CG, Slusher BS, Martin AM, Raabe EH. Orally bioavailable glutamine antagonist prodrug JHU-083 penetrates mouse brain and suppresses the growth of MYC-driven medulloblastoma. Transl Oncol. 2019;12:1314–22. https://doi.org/10.1016/j.tranon.2019.05.013.
Emadi A, Kapadia B, Bollino D, Bhandary B, Baer MR, Niyongere S, Strovel ET, Kaizer H, Chang E, Choi EY, Ma X, Tighe KM, Carter-Cooper B, Moses BS, Civin CI, Mahurkar A, Shetty AC, Gartenhaus RB, Kamangar F, Lapidus RG. Venetoclax and pegcrisantaspase for complex karyotype acute myeloid leukemia. Leukemia. 2021;35:1907–24. https://doi.org/10.1038/s41375-020-01080-6.
Michelozzi IM, Granata V, De Ponti G, Alberti G, Tomasoni C, Antolini L, Gambacorti-Passerini C, Gentner B, Dazzi F, Biondi A, Coliva T, Rizzari C, Pievani A, Serafini M. Acute myeloid leukaemia niche regulates response to L-asparaginase. Br J Haematol. 2019;186:420–30. https://doi.org/10.1111/bjh.15920.
Ardalan B, Arakawa M, Villacorte D, Jayaram H, Cooney DA. Effect of L-glutamine antagonists on 5-phosphoribosyl 1-pyrophosphate levels in P388 leukemia and in murine colon adenocarcinomas in vivo. Biochem Pharmacol. 1982;31:1509–13. https://doi.org/10.1016/0006-2952(82)90373-2.
Zuo F, Yu J, He X. Single-cell metabolomics in hematopoiesis and hematological malignancies. Front Oncol. 2022;12: 931393. https://doi.org/10.3389/fonc.2022.931393.
Polet F, Corbet C, Pinto A, Rubio LI, Martherus R, Bol V, Drozak X, Grégoire V, Riant O, Feron O. Reducing the serine availability complements the inhibition of the glutamine metabolism to block leukemia cell growth. Oncotarget. 2016;7:1765–76. https://doi.org/10.18632/oncotarget.6426.
Culp-Hill R, D’Alessandro A, Pietras EM. Extinguishing the embers: targeting AML metabolism. Trends Mol Med. 2021;27:332–44. https://doi.org/10.1016/j.molmed.2020.10.001.
Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’Dwyer KM, Liesveld JL, Brookes PS, Becker MW, Jordan CT. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–41. https://doi.org/10.1016/j.stem.2012.12.013.
Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, Bosc C, Sugita M, Stuani L, Fraisse M, Scotland S, Larrue C, Boutzen H, Féliu V, Nicolau-Travers M-L, Cassant-Sourdy S, Broin N, David M, Serhan N, Sarry A, Tavitian S, Kaoma T, Vallar L, Iacovoni J, Linares LK, Montersino C, Castellano R, Griessinger E, Collette Y, Duchamp O, Barreira Y, Hirsch P, Palama T, Gales L, Delhommeau F, Garmy-Susini BH, Portais J-C, Vergez F, Selak M, Danet-Desnoyers G, Carroll M, Récher C, Sarry J-E. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017;7:716–35. https://doi.org/10.1158/2159-8290.CD-16-0441.
Tabe Y, Konopleva M, Andreeff M. Fatty acid metabolism, bone marrow adipocytes, and AML. Front Oncol. 2020;10:155. https://doi.org/10.3389/fonc.2020.00155.
Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. 2016;16:732–49. https://doi.org/10.1038/nrc.2016.89.
Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis. 2016;5: e189. https://doi.org/10.1038/oncsis.2015.49.
Chen Q, Kirk K, Shurubor YI, Zhao D, Arreguin AJ, Shahi I, Valsecchi F, Primiano G, Calder EL, Carelli V, Denton TT, Beal FM, Gross SS, Manfredi G, D’Aurelio M. Rewiring of glutamine metabolism is a bioenergetic adaptation of human cells with mitochondrial DNA mutations. Cell Metab. 2018;27:1007-1025.e5. https://doi.org/10.1016/j.cmet.2018.03.002.
Polet F, Martherus R, Corbet C, Pinto A, Feron O. Inhibition of glucose metabolism prevents glycosylation of the glutamine transporter ASCT2 and promotes compensatory LAT1 upregulation in leukemia cells. Oncotarget. 2016;7:46371–83. https://doi.org/10.18632/oncotarget.10131.
van Gastel N, Spinelli JB, Sharda A, Schajnovitz A, Baryawno N, Rhee C, Oki T, Grace E, Soled HJ, Milosevic J, Sykes DB, Hsu PP, Vander Heiden MG, Vidoudez C, Trauger SA, Haigis MC, Scadden DT. Induction of a timed metabolic collapse to overcome cancer chemoresistance. Cell Metab. 2020;32:391-403.e6. https://doi.org/10.1016/j.cmet.2020.07.009.
Ma G, Zhang Z, Li P, Zhang Z, Zeng M, Liang Z, Li D, Wang L, Chen Y, Liang Y, Niu H. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun Signal CCS. 2022;20:114. https://doi.org/10.1186/s12964-022-00909-0.
Sornsuvit C, Komindr S, Chuncharunee S, Wanikiat P, Archararit N, Santanirand P. Pilot Study: effects of parenteral glutamine dipeptide supplementation on neutrophil functions and prevention of chemotherapy-induced side-effects in acute myeloid leukaemia patients. J Int Med Res. 2008;36:1383–91. https://doi.org/10.1177/147323000803600628.
Liu P-S, Wang H, Li X, Chao T, Teav T, Christen S, Di Conza G, Cheng W-C, Chou C-H, Vavakova M, Muret C, Debackere K, Mazzone M, Huang H-D, Fendt S-M, Ivanisevic J, Ho P-C. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18:985–94. https://doi.org/10.1038/ni.3796.
Acknowledgements
We thank Editage (www.editage.com) for their editing support on this manuscript.
Funding
No funding was received to assist with the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
W.Z. wrote and structured the paper; W.Z. analyzed literature data and wrote the paper; Y.Q. and L.M. reviewed the literature; all authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Z., Liu, M. & Yang, Q. Glutamine and leukemia research: progress and clinical prospects. Discov Onc 15, 391 (2024). https://doi.org/10.1007/s12672-024-01245-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-024-01245-0