
Abstract
Metabolism is a fundamental process for all cellular functions. For decades, there has been growing evidence of a relationship between metabolism and malignant cell proliferation. Unlike normal differentiated cells, cancer cells have reprogrammed metabolism in order to fulfill their energy requirements. These cells display crucial modifications in many metabolic pathways, such as glycolysis and glutaminolysis, which include the tricarboxylic acid (TCA) cycle, the electron transport chain (ETC), and the pentose phosphate pathway (PPP) [1]. Since the discovery of the Warburg effect, it has been shown that the metabolism of cancer cells plays a critical role in cancer survival and growth. More recent research suggests that the involvement of glutamine in cancer metabolism is more significant than previously thought. Glutamine, a nonessential amino acid with both amine and amide functional groups, is the most abundant amino acid circulating in the bloodstream [2]. This chapter discusses the characteristic features of glutamine metabolism in cancers and the therapeutic options to target glutamine metabolism for cancer treatment.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- Glutamine metabolism
- Glutamine addiction
- Targeting glutamine metabolism
- Transaminase upregulation
- Targeting amino acid synthesis
-
Glutamine addiction and dysregulation of the TCA cycle are characteristic features of glutamine metabolism in cancers.
-
Glutamine metabolism can be targeted by inhibiting glutaminolysis, employing combination therapies, suppressing c-MYC expression, inhibiting GDH, depleting the glutamine supply, inhibiting glutamine uptake, and exploiting glutamine analogs.
-
Transaminase upregulation and targeting of amino acid synthesis have the potential for cancer therapy.
-
The metabolic reprogramming of cancers provides them with alternative sources of glutamate: via N-acetyl-aspartyl-glutamate (NAAG) and via glutaminase II pathway.
-
Glutamine metabolism in the tumor microenvironment can impact the development of cancers.
1 Introduction
Metabolism is a fundamental process for all cellular functions. For decades, there has been growing evidence of a relationship between metabolism and malignant cell proliferation. Unlike normal differentiated cells, cancer cells have reprogrammed metabolism in order to fulfill their energy requirements. These cells display crucial modifications in many metabolic pathways, such as glycolysis and glutaminolysis, which include the tricarboxylic acid (TCA) cycle, the electron transport chain (ETC), and the pentose phosphate pathway (PPP) [1]. Since the discovery of the Warburg effect, it has been shown that the metabolism of cancer cells plays a critical role in cancer survival and growth. More recent research suggests that the involvement of glutamine in cancer metabolism is more significant than previously thought. Glutamine, a nonessential amino acid with both amine and amide functional groups, is the most abundant amino acid circulating in the bloodstream [2]. This chapter discusses the characteristic features of glutamine metabolism in cancers and the therapeutic options to target glutamine metabolism for cancer treatment.
2 Characteristic Features of Glutamine Metabolism in Cancer
2.1 Dysregulation of the TCA Cycle
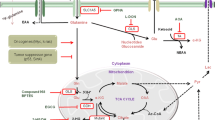
This section focuses on abnormalities within the TCA cycle, also known as the citric acid cycle or Krebs cycle (Fig. 1), that alter cancer cell metabolism. The TCA cycle is the central metabolic hub of the cell; it acts as a common pathway for the catabolism of many different sugars, fatty acids, and amino acids [3]. It also generates electrons that fuel oxidative phosphorylation, a process that produces a majority of the energy used by normoxic cells [3].

Cancer cells exhibit mutations in TCA cycle enzymes: SDH succinate dehydrogenase, FH fumarate hydratase, and IDH isocitrate dehydrogenase. The mutations result in altered activity or inhibit the activity of enzymes leading to the conversion of α-ketoglutarate (α-KG) to 2HG and the activation of HIF-1α, which, in turn, result in tumor growth. Cancer cells with these mutations rely on glutamine to feed into the TCA cycle. Glutamine can also be used to produce glutathione, an antioxidant that prevents ROS accumulation. Metabolites are shown in black. Enzymes are shown in orange. Metabolic pathways are shown as light blue arrows
Under aerobic conditions, pyruvate formed as a product of glycolysis goes through oxidative decarboxylation, a process that removes a carboxyl group as CO2 to form acetyl coenzyme A (acetyl-CoA), the typical starting molecule of the TCA cycle [4]. The TCA cycle takes place within the mitochondrial matrix [5]. The steps of the TCA cycle are as follows: (1) citrate synthase facilitates the condensation of oxaloacetate and acetyl-CoA to form citrate, (2) the enzyme aconitase then converts citrate to isocitrate, (3) isocitrate is further oxidatively decarboxylated by isocitrate dehydrogenase (IDH), (4) the resulting compound α-ketoglutarate (α-KG) is transformed into succinyl-CoA, (5) succinyl-CoA is then further converted to succinate by succinyl-CoA synthetase, (6) the succinate dehydrogenase (SDH) complex catalyzes the oxidation of succinate to fumarate, (7) fumarate is hydrated to malate by fumarate hydratase (FH), and finally (8) malate is then oxidized to oxaloacetate by malate dehydrogenase—initiating the cycle once again [6] (Fig. 1).
Mutations of TCA cycle genes have been linked to familial cancer types [6]. Recent research has found that mutations in the TCA cycle enzymes SDH, FH, and IDH resulted in a dysfunction of the TCA cycle and defects in mitochondrial metabolism in a wide range of human cancers [7, 8]. The SDH complex (also known as mitochondrial complex II) is the only membrane-bound enzyme of the TCA cycle and consists of four subunits: SDHA, SDHB, SDHC, and SDHD. SDHA and SDHB are catalytic subunits that protrude into the mitochondrial matrix, while SDHC and SDHD are anchored to the inner membrane [9]. The SDH enzyme plays an essential role in tumor suppression. Heterozygous mutations in SDH genes cause complete inactivation of the protein function and are associated with hereditary paragangliomas and pheochromocytomas [10,11,12]. Tumors exhibiting SDH mutations are more aggressive and usually proliferate at a much faster rate than normal cells [9]. In addition to these cancers, several other neoplasms have been associated with mutations in SDH subunits, including renal cell carcinoma (RCC), neuroblastoma, gastrointestinal stromal tumors, thyroid cancer, and testicular seminoma [13,14,15].
Similar to SDH, FH mutations occur throughout the genome. Research has indicated an association between heterozygous FH mutations and uterine fibroids, hereditary leiomyomatosis, and papillary renal cell cancer [16]. Additionally, loss of the wild-type allele in these cancers resulted in the absence of FH enzymatic activity. FH acts as a tumor suppressor in these cancers, and its reduced expression leads to the accumulation of the transcription factor hypoxia-inducible factor-1α (HIF-1α) [1, 17]. High levels of fumarate accumulate and act as oncometabolites, which often result in dysregulation of cellular functions in SDH- or FH-deficient cells [1]. Both accumulated succinate and fumarate are potent inhibitors of prolyl 4-hydroxylases (PHDs). PHDs are negative regulators of HIF-1α, a transcriptional factor that is upregulated under hypoxic conditions when tumor cells are deprived of adequate oxygen supplies. Impaired PHD activity leads to HIF-1α activation under normoxia, a condition known as pseudohypoxia [6]. Pseudohypoxia, in turn, facilitates tumor cell growth.
Similar to the metabolic consequences of SDH and FH mutations, mutations in the IDH enzyme also result in dysfunction of the TCA cycle. There are three isoforms of the enzyme IDH. IDH1 is found in the cytoplasm and peroxisomes; IDH2 and IDH3 are localized in the mitochondrial matrix. IDH3 is the primary form of IDH in the TCA cycle, whose function is to convert isocitrate to α-KG. Genomic analysis has identified mutations in either IDH1 or IDH2 in the vast majority of grade II and III gliomas as well as glioblastoma multiforme (GBM) patient samples [18]. The abnormal expression and activity of IDHs result in the loss of the enzyme’s ability to catalyze the conversion of isocitrate to α-KG; instead, it gains a new ability to facilitate the NADPH-dependent reduction of α-KG to D-2-hydroxyglutaric acid (D-2HG), an oncometabolite. Subsequently, excess accumulation of 2HG contributes to the formation of malignant gliomas [19]. The discovery that IDH1 and IDH2 mutations are nearly all missense mutations localized to residues R132 and R172, respectively, provides a promising biomarker for cancer diagnosis and possibly gene therapy [20, 21]. It was found that cells harboring IDH1-R132 and IDH2-R172 mutations in patients with acute myeloid leukemia gained the ability to convert α-KG to D-2HG [22].
2.2 Glutamine Addiction
In addition to glucose, proliferating cancer cells also rely on glutamine as a major source of energy and building blocks, with glutamine feeding into the TCA cycle. This condition is known as glutamine addiction. Glutamine is one of the most abundant nonessential amino acids in the bloodstream (produced by the human body and thus not an essential part of the diet) and contributes to virtually every biosynthetic pathway in proliferating cells. Moreover, it acts as a nitrogen donor in purine and pyrimidine synthesis as well as a precursor for protein and glutathione biosynthesis [23]. Many tumor cells are reliant on exogenous glutamine and have been reported to die in its absence [24].
Since glutamine-derived α-KG fuels the TCA cycle, cancer cells can employ glutaminolysis to sustain the biosynthesis of many essential molecules. In RCCs that were either ETC or TCA cycle deficient, it was found that cancer cells relied on the reductive carboxylation of glutamine-derived citrate to produce acetyl-CoA and other precursors to TCA cycle metabolites. Acetyl-CoA is a necessary intermediate for the synthesis of lipids, and without it, cancer cells are not viable. Furthermore, TCA cycle intermediates are needed to synthesize other essential cellular building blocks. Thus, cells can become utterly dependent upon glutaminolysis as a result of genetic alterations affecting oxidative mitochondrial function [25]. A study by Gameiro et al. found that the transcription factor HIF expression maintained a low level of intracellular citrate to maintain adequate lipogenesis. Therefore, the von Hippel-Lindau (VHL)-deficient RCC cells that constitutively express HIF-1α and/or HIF-2α become heavily dependent on glutamine for proliferation [26, 27].
Glutamine addiction was also found to occur in glioma cells that possess a recurrent mutation of IDH1. As mentioned in the section above, IDH1 catalyzes typically the conversion of isocitrate to α-KG, but the mutant isoform converts α-KG into D-2HG instead, which has been shown to inhibit cellular differentiation through epigenetic alterations [19]. Due to the mutant IDH1 function, glioma cells become increasingly dependent upon glutamine-derived α-KG production. Thereby cancer cells exhibiting glutamine addiction rely on glutaminase (GLS), an enzyme that converts glutamine to glutamate, which in turn is converted to α-KG for survival. The inhibition of GLS suppresses the growth of glioma cells with IDH1 mutations by decreasing the availability of glutamine-derived α-KG [28].
Further evidence suggests that the dependence of specific cancer cells on glutamine may be more profound than previously thought. Fluorodeoxyglucose-positron-emission tomography (FDG-PET) scanning, a clinical imaging technique, can detect cancers based on areas of increased glucose uptake. Specifically, 18F-FDG-PET imaging exploits increased glucose transport and hexokinase II activities in proliferating tumor tissues to locate high levels of 18F-FDG, which can be visualized by PET images. However, some cancers are invisible to PET scans and are deemed PET negative. These PET-negative cancers must rely on alternative metabolic substrates for their primary source of energy, namely glutamine [29]. Researchers have used 5-11C-(2S)-glutamine and 18F-(2S,4R)4-fluoroglutamine as glutamine-based PET imaging agents, where 18F is preferred because it has 5.5 times the half-life of 11C [30, 31]. Both tracers are useful tools for probing in vivo metabolism of glutamine and monitoring radiation effects in cancer patients.
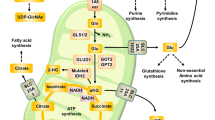
Cancer cells use precursors derived from the TCA cycle intermediates to synthesize proteins, lipids, and nucleic acids. In order to maintain mitochondrial activity, these cells must compensate for lost TCA cycle intermediates caused by their metabolic diversions [32]. This process of replenishing metabolic intermediates is known as anaplerosis [33]. Glutamine provides mitochondrial anaplerosis because of its role as a nitrogen and carbon donor to the cells [32]. It traverses the cell membrane through amino acid transporters, ASCT2 (alanine, serine, cysteine transporter 2), and system N transporter SN2 [34]. Once it enters the cytoplasm, glutamine is hydrolyzed to glutamate and ammonia (NH3) via GLS (Fig. 2) [24].

Glutamate can be catalyzed via three different pathways by the aminotransferases GPT, GOT, and PSAT1, all of which yield α-ketoglutarate (α-KG) and a different amino acid, alanine, aspartate, and phosphoserine, respectively. These enzymes are upregulated in cancer, which results in increased α-KG production. Inhibition of the enzymes results in reduced cancer cell proliferation or tumor growth. Phosphoserine can enter serine and glycine metabolism, where the enzyme SHMT is also upregulated in cancer. This results in increased production of glycine, which supports protein and purine synthesis and oxidative phosphorylation, thus leading to tumor growth. Elevated level of glycine further leads to increased level of glutathione, which mediates oxidative stress and also leads to tumor growth. Tryptophan metabolism is linked to the regulation of antitumor immune responses, where the enzymes IDO and TDO can lead to immunosuppression. Inhibition of IDO or TDO leads to reduced tumor growth. Some cancers have reduced ASS1 which leads to decreased arginine level, causing these cancers to rely on exogenous arginine supply. GPT glutamic-pyruvate transaminase, GOT glutamic-oxaloacetic transaminase, PSAT1 phosphoserine aminotransferase 1, ASS1 argininosuccinate synthetase 1, IDO indoleamine-2,3-dioxygenase, TDO tryptophan-2,3-dioxygenase, SHMT serine hydroxymethyltransferase
Glutamate can be further catabolized through the TCA cycle (via conversion to α KG) or serve as a substrate for glutathione synthesis. α-KG formation can be catalyzed by either glutamate dehydrogenase (GDH) or aminotransferases. Alternatively, glutamate can be further converted to glutathione by glutathione cysteine ligase and glutathione synthetase. Glutathione is an antioxidant vital to a cell’s immune defense, nutrient metabolism, and cellular functions [35]. It also plays an important role in the neutralization of mitochondrial reactive oxygen species (ROS). ROS are by-products of oxygen metabolism, the concentration of which plays a vital role in tumor proliferation, growth, apoptosis, and metastasis. Studies have found that ROS acts as a “double-edged sword” in some cancers; moderately elevated levels of ROS facilitate carcinogenesis while excessive levels can trigger apoptosis by causing damage to DNA and other macromolecules [36]. The dual role of ROS is also evident in the early and late stages of cancer. At precancerous and neoplastic stages, ROS tends to promote proliferation by inducing oxidative damage and base-pair mutations. In the late stages, however, increased energy need is correlated with an increase in ROS production [37]. Therefore, for a ROS-mediated therapeutic approach to be effective, this therapy must limit ROS production when ROS would induce proliferation, and it must promote ROS production when ROS would induce apoptosis.
The inhibition of glutamine metabolism has been linked to ROS overproduction, which can devastate cancer cells [38, 39]. Mitochondrial glutamine metabolism is a significant anaplerotic step in tumorigenesis. It is often enhanced in cancer cells with increased levels of TCA cycle metabolites [32]. Therefore, inhibiting GLS could effectively starve cancer cells of the glutamine essential to their survival and prevent further glutathione synthesis, thus increasing cancer cell exposure to excessive ROS levels [40].
2.3 The Metabolic Reprogramming of Cancers Provides Them with Alternative Sources of Glutamate: Via N-Acetyl-Aspartyl-Glutamate (NAAG) and via the Glutaminase II Pathway
Two recent studies have uncovered alternative sources of glutamate in cancer. The first study identified N-acetyl-aspartyl-glutamate (NAAG), which can be hydrolyzed to glutamate when needed by the oncogenic cells via glutamate carboxypeptidase II (GCP II) [41, 42], suggesting that GCPII is a viable target for cancer therapy, either alone or in combination with glutaminase inhibition. The second study demonstrated that upon the inhibition of glutaminase I pathway (the conversion of glutamine to glutamate by GLS), pancreatic cancer cells utilized the glutaminase II pathway to generate glutamate via glutamine transaminase K (GTK) [43]. Knocking down the expression of GTK inhibited the growth of pancreatic cancer cells in vitro and tumorigenesis in vivo. The uncovering of the alternative sources of glutamate in cancer opens up new strategic approaches. Specifically, these studies suggest a combination therapy of GLS1 and GCPII and/or GTK inhibition for pancreatic cancer therapy.
3 Targeting Glutamine Metabolism for Cancer Therapy
Due to its central role in many cancers, glutaminolysis is becoming an increasingly prominent target for cancer therapy. Mammalian cells express two isoforms of GLS: kidney-type GLS1 and liver-type GLS2. GLS1 is more broadly expressed in normal tissue, while GLS2 is mainly present in the liver, brain, pituitary gland, and pancreas [44]. Both encode a mitochondrial GLS, which catalyzes the hydrolysis of glutamine to glutamate [45, 46]. Studies have shown that c-MYC, a multifaceted transcriptional factor, upregulates glutamine importers and GLS1 expression and that p53, a tumor-suppressor gene/protein, upregulates GLS2 expression [46,47,48].
Using stable isotope-resolved metabolomics (SIRM) studies [49], Le et al. also reported the persistence of glutamine oxidation via the TCA cycle under hypoxia. SIRM studies track metabolic transformations using stable isotope labeling and analyze the metabolic products using nuclear magnetic resonance (NMR) and mass spectrometry (MS) at different time points. Using a human Burkitt lymphoma model P493 cell line carrying an inducible MYC vector, the group showed the coexistence of oxidative and aerobic glycolysis. Thus, inhibition of GLS induced oxidative stress and diminished ATP levels in hypoxic cells [39]. It was also found that glutamine metabolism supports cellular bioenergetics and redox homeostasis for proliferation under both aerobic and hypoxic conditions. P493 cells exhibited low glutathione levels and high ROS production under inhibition of GLS and hypoxia. Furthermore, glutamine-derived glutathione production was sustained under hypoxia as a coping method under high ROS levels [39]. These results again suggest that glutamine metabolism, specifically through GLS enzyme, is a promising target for cancer therapy.
The TP53 gene codes for a tumor-suppressor protein known to trigger cell cycle arrest, apoptosis, or senescence in response to a variety of cellular dysfunctions, including DNA damage, oncogene activation, and hypoxia [50]. It is one of the most frequently mutated genes among all cancers. However, recent studies have discovered TP53’s additional roles in regulating energy metabolism and antioxidant defense mechanisms [51, 52]. GLS2 is a p53 target gene that plays an important role in mediating the tumor-suppressant properties of p53. GLS2 increases intracellular levels of glutamate and α-KG, thus leading to enhanced mitochondrial respiration and ATP production. It also leads to increased cellular glutathione levels and thus decreased ROS levels [46]. Hu et al. demonstrated that p53 increased GLS2 expression under both stressed and non-stressed conditions—enhancing glutamate levels, mitochondrial respiration rates, and glutathione levels while decreasing ROS levels. Furthermore, the GLS2 gene promoter contains a p53 consensus DNA-binding element whose expression was induced in response to oxidative stress [1, 46]. Hu’s findings suggest that GLS2 may be a mediator to p53’s role in energy metabolism and antioxidant defense, ultimately contributing to its tumor suppression abilities.
Due to its crucial role in energy regulation and biosynthesis, targeting glutamine metabolism has the potential to affect a broad spectrum of cancers. In addition to GLS inhibition, the role of oncogenes and tumor suppressors in regulating glutamine metabolism makes it a promising venture for therapeutic strategies. However, while many drugs have been synthesized to target glutamine metabolism from its initial transport into the cell to its conversion to α-KG, most are still in preclinical stages (Table 1) [44].
3.1 Inhibition of Glutaminolysis by GLS Inhibitors
The most straightforward approach for targeting glutaminolysis is the inhibition of GLS, which catalyzes the hydrolysis of glutamine to glutamate. The kidney isoform, GLS1 (or GLS), is found in many primary tumors, while the liver isoform, GLS2, is less often expressed in cancers [53]. Inhibiting GLS can starve cancer cells by blocking the synthesis of glutamate and thus prevent α-KG from feeding the TCA cycle. After nuclear factor kappa B (NF-kB) activates glutaminase C isoform (GAC), an alternatively spliced isoform of GLS1, via phosphorylation, NF-kB itself is activated by Rho GTPases. Alteration of Rho GTPases by small-molecule inhibitors showed a significant decrease in GAC activity in human breast cancer cells [54]. The decrease in GAC activity caused breast cancer cells to stop proliferating and reduced their ability to invade surrounding cells [54]. Potent therapeutic GLS inhibitors, such as bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) and its analogs such as CB-839, are being actively investigated in GLS-targeting studies [38, 54,55,56,57,58].
BPTES allosterically inhibits GLS1 by altering the conformation of the enzyme and has been proven in many studies to inhibit cancer cell growth in vitro and slow tumor growth in vivo [39, 56, 59]. While BPTES produces formidable results in vitro, higher concentrations are needed to achieve the same effect in vivo. Due to its low solubility, BPTES tends to precipitate at high concentrations, posing a challenge to the physiological delivery of the drug in clinical trials [60]. A solution was proposed by Elgogary et al. with an emulsification method that encapsulated BPTES into biodegradable nanoparticles coated with poly(ethylene glycol) (PEG) and poly(lactic-coglycolic acid) (PLGA) to improve nanoparticle circulation time in the blood. This process enhanced the efficacy of BPTES in vivo by improving its solubility and increasing tumor drug exposure [38].
The only GLS inhibitor to have entered clinical trials is commonly referred to as CB-839 or commercially as Telaglenastat. CB-839 is a highly potent allosteric inhibitor that completed phase I clinical trials in March 2019 in patients with advanced RCC (NCT02071862) and has in 2020 moved to phase II clinical trials in patients with non-small cell lung cancer (NCT04265534). Phase I trials have been completed for hematological tumors (NCT02071888) and leukemias (NCT02071927), although no clinical data is publicized. CB-839 has shown efficacy in the treatment of triple-negative breast cancer (TNBC), showing a marked decrease in glutamine consumption, glutamate production, and levels of glutathione and other TCA cycle intermediates [55]. Elevated GLS expression is also associated with different cancer types, including glioblastoma and pancreatic cancers, thereby encouraging investigation of more targets should current clinical trials prove successful [28, 61].
Taken together, glutamine dependency may be a particular metabolic vulnerability of cancer cells, and glutaminolysis-targeting strategies could be promising approaches for glutamine-addicted cancer therapy.
3.2 Combination Therapy
The heterogeneity of cancer metabolism, even within the same tumor [62], poses many challenges for potential drug therapies. Hence, the use of combination therapies to target multiple metabolic pathways to suppress tumor growth may be most effective, especially in identifying cases that induce synthetic lethality, where two drugs induce cell death in combination, but not individually. Glutamine’s role in cellular functions makes GLS inhibition an ideal candidate for combination therapy. In their study, Elgogary et al. found that combination therapy of BPTES and metformin produced better results in pancreatic tumors than monotherapy of either drug alone. Metformin is an FDA-approved drug for the treatment of type 2 diabetes that inhibits glycolysis and glycogen synthesis. In this case, BPTES targets glutamine metabolism, and metformin targets glucose metabolism, resulting in an optimal reduction of tumor development [38]. Other treatments that are synthetically lethal with the inhibition of GLS include GLUT2q1 inhibition, mTOR inhibition, and ATF4 activation [44].
In recent years, researchers have increasingly leveraged combination therapies for the treatment of a variety of cancers (Table 2) [63]. Parlati et al. combined CB-839 with pomalidomide to target myeloma models [64]. Momcilovic et al. treated lung cancer with the synergistic combination of CB-839 and erlotinib, demonstrating significant reductions in glucose and glutamine uptake [68]. Emberley et al. explored the various combinations of CB-839 with CDK4/6 and PARP inhibitors in colorectal carcinoma (CRC), TNBC, ovarian cancer, and non-small cell lung carcinoma [67].
3.3 Knockdown of c-MYC
In a study by Wise et al., c-MYC expression was found to activate the transcription of key regulatory genes required for glutamine uptake and metabolism by selectively binding to the promoter regions of glutamine transporters ASCT2 and SN2. As a result, c-MYC induced reprogramming of mitochondrial metabolism by diverting glucose away from the TCA cycle and leaving cells susceptible to glutamine addiction to sustain anaplerosis. Moreover, c-MYC-transformed cells were found to be sensitive to GDH inhibitors. These results suggest that glutamine addiction may be a direct consequence of c-MYC activation [48]. Gao et al. found that c-MYC expression induced the expression of mitochondrial GLS in human P493 lymphoma B cells and PC3 prostate cancer cells by suppressing microRNAs miR-23a and miR-23b, which target the GLS 3′ untranslated region (UTR) seed sequence [1, 47]. Liu et al. established a direct link between glutamine and proline via MYC-induced proline biosynthesis from glutamine, suggesting novel therapeutic strategies [69]. Overall, these results may be exploited for cancer therapy using inhibitors of enzymes involved in glutamine metabolism or therapeutics that inhibit the transcriptional properties of c-MYC.
The transcriptional factor MYC is an essential growth regulator that is overexpressed in most cancers, and hence is a highly sought-after target for cancer therapies [70, 71]. Niu et al. found that suppressing c-MYC expression resulted in reduced cell growth, colony formation, and tumor formation in nasopharyngeal carcinoma cell lines both in vitro and in vivo [72]. Using RNA interference (RNAi)-mediated silencing of the c-MYC gene, Zhang et al. showed that the downregulation of c-MYC induced apoptosis in vitro and suppressed the growth of colon cancer cells in vivo [73].
Other attempts at directly or indirectly targeting MYC have included knockdown, protein/protein and DNA interaction inhibitors, and translation and expression regulation [74]. Direct inhibition of MYC expression can be achieved by using G-quadruplex stabilizers that prevent MYC transcription or antisense oligonucleotides and siRNAs that prevent MYC translation [75,76,77,78]. Small-molecule protein/protein interaction inhibitors have also been used to interfere with MYC transcriptional activation [79, 80]. Indirect inhibition methods encompass blocking transcription, hindering mRNA translation, and targeting regulators of MYC protein stability [81, 82]. The most advanced methods currently in clinical trials or commercially approved employ indirect targeting by immunotherapies, which focus on immune components required for MYC-driven tumors or checkpoints that are altered in MYC-driven tumors [83, 84].
3.4 Inhibition of Glutamate Dehydrogenase (GDH)
Inhibiting the oxidative deamination of glutamate to α-KG has devastating effects on cancer cells comparable to inhibiting glutaminolysis [61, 85]. This process is catalyzed by GDH, which can be inhibited by the preclinical compounds R162, epigallocatechin gallate (EGCG), and epicatechin gallate (ECG) [44, 86]. Using perifusion assays, Li et al. showed that EGCG and ECG blocked GDH activity by binding to the allosteric regulator ADP’s binding site [87].
Furthermore, it was demonstrated that polyphenols such as hexachlorophene and bithionol might inhibit GDH function. These inhibitors work by restricting enzyme movement, either by forming ring barriers around the enzyme or by wedging between the enzyme’s subunits. Currently, polyphenols have been shown to inhibit lung, colon, and prostate adenocarcinoma growth in xenograft models [88]. These compounds also had significant effects on glioblastoma, colon, lung, and prostate adenocarcinoma cell proliferation [89]. Additionally, it was found that GDH inhibition through siRNA resulted in a marked decrease in the proliferation of glioblastoma cells that were glutamine dependent [18, 90].
3.5 Inhibiting the TCA Cycle by Depleting Glutamine, α-Ketoglutarate, and Asparagine
One of the earlier means of suppressing glutamine metabolism arose from reducing the amount of available glutamine itself. Ollenschläger et al. found that the abundance of glutamine in the body dropped precipitously by giving L-asparaginase to patients with acute myeloid leukemia. Asparaginase catalyzes the removal of the amide nitrogen from asparagine to form aspartic acid. The administration of asparaginase also depleted stored levels of glutamine [91]. When applied in cell culture, asparaginase inhibited cell growth and induced cell death in pancreatic cancer cells. The effect of asparaginase can largely be reversed through the reintroduction of small amounts of glutamine [92].
Studies of acute lymphoblastic leukemia indicated that asparaginase activity correlated with glutamine depletion in the bloodstream and improved treatment outcomes [93, 94]. Furthermore, cancer cells, with a deficiency of asparagine synthase (ASNS), an enzyme that generates asparagine, such as acute lymphoblastic leukemia (ALL), must use asparagine from the blood [95]. In 1979, Ertel et al. treated ALL patients with asparaginase, which exhausted the asparagine supply in the blood. This treatment re-induced remission in up to 60% of cases [96]. ALL can upregulate ASNS to restore intracellular asparagine levels and satisfy their asparagine demand [97]. However, some studies show that ASNS levels may not impact the sensitivity of ALL to asparaginase treatments in all cases [98]. The diverse metabolic phenotypes of malignant cells create many challenges for therapeutic strategies. It seems that a combination of drug therapy targeting both asparagine and glutamine metabolism could be a promising treatment.
Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl), a drug previously known to inhibit cancer cell proliferation and increase vulnerability to other cytotoxic agents, has been shown to interfere with glutamine metabolism through inhibition of IDH1/2 and slowing of the TCA cycle [99]. This effect was found both in vitro and in vivo and showed that Tempol might be a powerful therapeutic in combination with other cancer drugs.
Another possible therapeutic in development is phenylbutyrate, a drug that lowers glutamine concentrations in the plasma. It is currently approved by the FDA and has shown clinical improvement in patients with hormone-refractory prostatic carcinoma and GBM [94, 100, 101].
3.6 Inhibiting Glutamine Uptake
The c-MYC-activated amino acid transporter ASCT2 (or SLC1A5) is upregulated in many cancers and is involved in controlling glutamine uptake [48, 94]. High levels of ASCT2 are correlated with aggressive tumor growth and short survival time. ASCT2 inhibitors include benzylserine, l-γ-glutamyl-p-nitroanilide (GPNA), and V-9302 [44, 102, 103]. Additionally, Colas et al. found that ASCT2 ligands chloroalanine, aminooxetane-3-carboxylate (AOC), and γ-FBP also inhibit ASCT2-mediated glutamine uptake in human melanoma cells [104]. Research shows that the inhibition of glutamine importers significantly slowed growth in human colon and lung cancer cells [105, 106].
Benzylserine and GPNA are amino acid analogs; however, while they reduce glutamine uptake, they do not exclusively inhibit the ASCT2 function [107]. Their low affinity and specificity make them unsuitable for clinical trials that specifically target glutamine addiction via ASCT2. V-9302 is a more contemporary inhibitor; however, research has indicated that its efficacy was likely unrelated to ASCT2 inhibition, yielding again to the problem of non-specificity [108].
The development of blocking antibodies and antibody-drug conjugates presents an alternative, perhaps a more viable approach to inhibiting glutamine uptake by regulating the ASCT2 transporter [109]. MEDI7247 is the only ASCT2 antibody-drug conjugate currently in phase I clinical trials (NCT03811652, NCT03106428).
3.7 Using Glutamine Mimetics
Another means of decreasing the availability of glutamine is the creation of glutamine analogs. Analogs such as 6-diazo-5-oxo-l-norleucine (DON) and acivicin did show cytotoxic effects against several tumor types, including leukemia and colorectal cancers; however, these analogs are no longer clinically available due to patient toxicity [110]. DON prodrugs have shown enhanced cerebrospinal fluid delivery, although toxicity was still observed [111].
A few glutamine analogs, namely acivicin, azaserine, and DON, have been extensively researched in an effort to inhibit glutamine metabolism. DON is a substrate analog of glutamine that binds to the active site of human kidney GLS to serve as an inhibition mechanism [112]. However, DON has had difficulty progressing into clinical trials due to concerns regarding its lack of selectivity to GLS and toxicity [110, 113, 114]. Clinical studies of DON co-administered with PEGylated glutaminase (PEG-PGA) suggested improved efficacy and it holds promise [115]. Similar to DON, acivicin and azaserine are also glutamine analogs that interrupt nucleotide synthesis by inhibiting amidotransferases [110, 116]. All three analogs exhibit excessive side effects and toxicity that have prevented them from reaching or advancing in clinical trials.
4 Transaminase Upregulation and Targeting Amino Acid Synthesis for Cancer Therapy
Another means of inhibiting glutaminolysis is to target alanine transaminase through L-cycloserine or aspartate transaminase through the inhibitor amino oxyacetate, which could almost completely halt the growth of breast cancer in xenograft mice [117, 118]. What is truly promising is that there appears to be little to no toxicity in non-neoplastic cells. The effectiveness of the inhibitor, combined with the lack of toxicity, makes inhibition of aspartate aminotransferase a potentially successful chemotherapeutic target.
Transaminases, also known as aminotransferases, are enzymes that catalyze reactions between amino acids and α-keto acids. Specifically, aminotransferases can convert glutamate to α-KG without producing ammonia. Glutamate acts as a nitrogen donor in these transaminations. Alanine aminotransferase, also known as glutamic-pyruvate transaminase (GPT), and aspartate aminotransferase, also known as glutamic-oxaloacetic transaminase (GOT), are abundantly present in the liver and often serve as markers for liver toxicity. There are three aminotransferase pathways through which glutamate can be transformed to α-KG. These key enzymes in these pathways are GPT, GOT, and phosphoserine aminotransferase 1 (PSAT1)—each of which produces a different amino acid by-product in addition to α-KG. As illustrated in Fig. 2, GPT transfers nitrogen from glutamate to pyruvate to produce alanine and α-KG. GOT transfers nitrogen from glutamate to oxaloacetate to produce aspartate and α-KG. PSAT1 transfers nitrogen from glutamate to 3-phosphohydroxypyruvate to produce phosphoserine and α-KG [44]. PSAT1 is also involved in the serine synthesis pathway, which is essential for many breast cancers [119]. Serine is essential for the synthesis of proteins necessary for cell proliferation. PSAT1 expression has recently been demonstrated to be upregulated in cancers in many studies [44]. Possemato et al. found that serine pathway flux is augmented in some breast cancer cell lines and that suppression of PSAT1 inhibited proliferation of these cells in addition to causing a significant reduction of α-KG [120]. In a study by Son et al., aspartate aminotransferases were demonstrated to be vital in maintaining redox homeostasis in pancreatic ductal adenocarcinoma (PDAC) cells. Furthermore, oncogenic mutant KRAS activity was found to upregulate the expression of aminotransferases, hence yielding high ROS levels and slowing tumor growth in vivo [85, 116, 121]. Taken together, these works suggest that targeting the amino acid synthesis pathway may be another effective strategy for cancer therapy.
Apart from glutamine, many other amino acids play important roles in tumorigenesis, namely arginine, tryptophan, serine, and glycine. Arginine is a precursor for the synthesis of proteins, urea, and various signaling molecules [122]. Although arginine is considered a nonessential amino acid, many cancer cells are dependent upon arginine for proliferation. Argininosuccinate synthetase 1 (ASS1) catalyzes the conversion of citrulline into argininosuccinate in the arginine synthesis pathway. Loss or suppression of ASS1 in osteosarcoma cells resulted in the depletion of arginine. Studies have shown that ASS1 acts as a tumor suppressor because cells with low ASS1 expression could not grow in an environment without arginine [123, 124].
Tryptophan is linked to the regulation of antitumor immune responses [125]. Figure 2 shows that it can be degraded to kynurenine via two enzymes: indoleamine-2,3-dioxygenase (IDO) and tryptophan-2,3-dioxygenase (TDO). IDO activity commonly leads to the suppression of the immune system [126]. Dendritic cells (immune system cells that present antigens to T cells) expressing IDO can limit tryptophan supply to T cells in the extracellular matrix, thus limiting T-cell response to tumor growth [127]. Studies have shown that mice transfected with IDO-induced cells developed large tumors and exhibited poor survival, while mice transfected with IDO-negative cells showed no signs of tumor development [128]. To further support this finding, immunohistochemical staining for IDO expression revealed a correlation between high IDO expression and low levels of immune cells CD3+, CD8+, and CD57+ [129]. This, in turn, can be correlated with aggressive tumor progression and poor survival in cancer patients with high IDO expression [129].
There are currently four IDO inhibitors under clinical development and more in preclinical testing [130]. In 2013, Beatty et al. studied the effects of the small-molecule IDO inhibitor INCB024360 treatment in 52 cancer patients. The drug was well tolerated by patients and successfully inhibited more than 90% of IDO activity when administered twice a day. Results showed stable disease conditions in 30% of patients [131]. Because INCB024360 was well tolerated, it has the potential to be potent as either a monotherapy or part of combination therapy. Phase II clinical trials of this inhibitor have been completed for patients with ovarian cancer and myelodysplastic syndrome (NCT01822691, NCT01685255). Combinatorial therapies with IDO inhibitors and cancer vaccines have also shown progress. A phase I study of indoximod, another IDO inhibitor, in combination with docetaxel, an antimitotic chemotherapy drug, showed stable or partially stable disease conditions in more than 50% of patients [132]. Other combinatorial therapies being tested in the clinic include INCB024360 and MK3475, an immune checkpoint inhibitor [130].
Other than IDO, cancer cells can also use TDO, an immunosuppressive enzyme, to avoid immune destruction. TDO is abundantly present in melanomas, bladder carcinomas, and hepatocarcinomas. Similar to IDO, the use of TDO inhibitors prevents the growth of TDO-expressing tumor cells [133]. There are several other enzymes that cancer cells exploit for immune tolerance; hence, targeting tryptophan metabolism with combinatorial approaches may yield optimal therapies [123].
The serine and glycine biosynthesis pathways are interconnected. They both provide methyl groups for the one-carbon pool that supports purine and pyrimidine synthesis in proliferating cancer cells [134]. Research has shown that phosphoglycerate dehydrogenase (PHGDH), the enzyme that catalyzes the first reaction in the serine synthesis pathway, was highly upregulated in metastatic breast cancer and was correlated to short patient survival times [135, 136]. The gene encoding PHGDH is also amplified in melanoma and breast cancer types [120]. In addition to PHGDH, serine hydroxymethyltransferase (SHMT) is also implicated in tumorigenesis. SHMT catalyzes the conversion of serine to glycine and is regulated by c-MYC, an oncogene that controls the transcription of 15% of human genes [123, 137, 138]. Glycine is a component of glutathione and is required for regulating cellular redox balance. It also fuels nucleotide biosynthesis and sustains oxidative phosphorylation in mitochondria. Thus, glycine metabolism has been shown to promote rapid tumor proliferation [134, 139, 140]. In an attempt to block glycine biosynthesis, researchers are using antimetabolites (drugs that interfere with the effects of metabolites), methotrexate, and pemetrexed to inhibit SHMT [134, 141]. Since serine and glycine are considered nonessential, their depletion can be tolerated in vivo. Maddocks et al. found that mice fed diets lacking serine and glycine showed a reduction in tumor sizes and survived longer than those fed diets containing the amino acids, indicating that diet regulation may be a potential therapy for investigation [142].
Many cancers become dependent on exogenous amino acid supplies to increase de novo synthesis of other amino acids. This characteristic can be exploited for cancer therapy by depleting amino acid supplies, blocking uptake by transporters, and inhibiting biosynthetic enzymes. The identification of novel therapeutic strategies targeting amino acid pathways could allow for the emergence of new drugs and enhance the current therapeutic efficacy.
5 Glutamine Metabolism in the Tumor Microenvironment
In addition to its effects on metabolism in cancer cells, glutamine can have relevant effects on the metabolic, and subsequently functional, profiles of cells in a tumor microenvironment [143]. Immune cells are present throughout the tumor microenvironment, and they can heavily influence tumor progression [144]. Natural killer (NK) cells and T cells are regularly involved in early detection, control, and clearance of tumors [144, 145]. Macrophages, on the other hand, can contribute to a microenvironment conducive to tumor growth, invasion into nearby tissues, and metastasis [146, 147]. In addition, other cell types, such as fibroblasts, play important roles in promoting tumor growth and metastasis, providing growth factors and contributing to extracellular matrix remodeling with matrix metalloproteinases [148, 149]. These cell types elicit unique responses to different metabolites, each of which could enhance or impede a cancer therapy’s effectiveness in clearing cancers.
The responses to glutamine that these cell types exhibit add to the vast heterogeneity of possible metabolic schemes that cancers can adopt. Studying these metabolic processes builds upon a well-mapped understanding of cancer metabolism, which, in concert with existing and newly developed therapies, can make an immense difference in a cancer patient’s course of treatment.
5.1 The Role of Glutamine Metabolism in T Cells and NK Cells
T cells and NK cells are the cytotoxic effector cells of productive immune response to cancer, but they can be as influenced by a metabolic landscape as cancer cells are. Presnell et al. showed that human CD8+ cytotoxic T cells, when stimulated in vitro, produce significantly less interferon-γ and tumor necrosis factor-α (TNFα) in a glutamine-deprived environment, compared to a baseline glutamine-supplemented environment [150]. This decrease in cytokine production correlates with a decrease in CD8+ T-cell effector function when in a low-glutamine environment. Thioredoxin-interacting protein (TXNIP), a suppressor of glucose uptake known to be active in low-glutamine conditions as a glycolytic sensor, was found to be strongly expressed in the glutamine-deprived setting in activated CD8+ T cells [151]. These findings were corroborated in the work presented by Song et al., who showed that ovarian cancer could induce endoplasmic reticulum stress in cytotoxic T cells, which depleted glutamine transporters in these cells and subsequently led to T-cell dysfunction [152]. This phenotype was rescued by repairing glutamine uptake in these T cells, further suggesting that glutamine deprivation in the tumor microenvironment serves as a powerful tool for tumor immune evasion.
However, Presnell et al. also showed that NK cells are more resilient against glutamine depletion and still produced interferon-γ in a glutamine-limited setting [150]. Additionally, activated NK cells did not show upregulation of TXNIP expression in a low-glutamine environment. This was investigated further, and, when faced with a low-glucose/low-glutamine environment, these cells were shown to rely on fatty acid oxidation or acetate for energy to mount a cytokine response, further highlighting the metabolic resilience of NK cells.
Considering these findings in the greater scheme of cancer treatments and given the explosive development of cancer immunotherapy technologies to kick-start T-cell activity, it will be important for clinicians to consider the metabolic implications that different combinations of drugs can have.
5.2 The Role of Glutamine Metabolism in Tumor-Associated Macrophages
Macrophages in the tumor microenvironment can define the immune landscape of a tumor, depending on the cytokines that they produce. Macrophages polarize to either M1 macrophages or M2 macrophages, and these phenotypes are dependent on the broader environment that the cells are in [146]. In a cancer setting, M1 macrophages generally promote inflammation and active antitumor immune responses. In contrast, M2 macrophages produce cytokine signals like IL-6 and IL-10 that help cancer cells evade immune surveillance, recruit endothelial cells for angiogenesis, and avoid apoptosis, thus allowing them to proliferate more freely [146, 153].
Fu et al. showed that, in a microenvironment deprived of glutamine, due to glutamine addiction in clear cell renal cell carcinoma (ccRCC), tumor-associated macrophages (TAMs) promote an immunosuppressive microenvironment [154]. Glutamine-addicted ccRCC tumors deprive the tumor microenvironment of glutamine. This study showed that, in glutamine-deprived microenvironments, TAMs began producing interleukin-23 (IL-23) via HIF1α activation. IL-23 subsequently activated the immune-suppressive regulatory T cells (Treg) in the tumor microenvironment. Through the production of IL-10 and transforming growth factor-beta (TGF-β), Tregs suppressed nearby cytotoxic cells, leading to immune evasion for the tumor [155, 156]. This phenotype was explored in both mouse models and in vitro with an anti-IL-23 antibody to block IL-23 signaling where mice experienced prolonged survival and decreased tumor burden, and CD8+ cells in mice and in vitro showed enhanced cytotoxicity in the IL-23-depleted state [154] (Fig. 3).
This example highlights a direct link between glutamine addiction and immune function in the tumor microenvironment. These findings show that an understanding of cancer metabolism can be used to not only shape metabolically targeted therapies but also hint at therapies against other factors impacting the landscape of the cancer treatment.
5.3 The Role of Glutamine Metabolism in Cancer-Associated Fibroblasts
Fibroblasts play a key role in solid tumors, fulfilling important functions like secreting growth factors, remodeling the extracellular matrix, and promoting metastasis [148]. They may also influence the metabolic behaviors of cancer cells. Zhao et al. reported that cancer-associated fibroblasts (CAFs) secreted exosomes that contain metabolites, which are taken up by cancer cells and can alter the metabolic patterns within those cancer cells [157, 158]. Using an in vitro system of prostate cancer cell lines and patient-derived CAFs, this study visualized exosome uptake and noted that the uptake of these exosomes was followed by a decrease in cancer cell mitochondrial oxidative phosphorylation, concomitantly leading to an increase in glycolysis. Through 13C5-glutamine labeling experiments, they found that these prostate cancer cells also exhibited a greater reliance on glutamine following exosome uptake, with increased levels of 13C-labeled m + 5 α-KG and m + 5 glutamate indicative of reductive glutamine metabolism for anaplerosis into the TCA cycle. This data suggests that CAFs are capable of shifting the metabolism of cancer cells from mitochondrial dependent to glycolytic dependent and upregulating their glutamine metabolism. These findings open the opportunities for therapy targeting metabolic cross talk between cancer cells and cancer-associated fibroblasts [159].
6 Conclusion
Glutaminolysis is a metabolic process that has been shown to play a critical part in a wide variety of cancers. As a result, glutamine metabolism is an important potential target for cancer therapy. Cancer metabolism is heterogeneous. Just as only some cancers are dependent upon glucose for the TCA cycle, only some cancers will exhibit aberrant glutaminolysis. Even within a single patient, the cancer cells may exhibit vast differences in their dependence on metabolic fuel supplies. This implies that not all cancers will respond in the same manner, or to the same extent, to the inhibition of glutaminolysis. It is important to note that inhibiting glutaminolysis will be more effective on cancers that display glutamine addiction. That being said, there is a huge potential for inhibition of glutaminolysis in cancers [160, 161]. As stated before, genetic alterations, as well as the tumor microenvironment, can influence cancer cells’ use of glutaminolysis. Developing and exploring glutaminolysis inhibitors present a strategic course of action toward the goal of finding an effective treatment for the many glutamine-dependent cancers. Inhibitors of GLS, GDH, GCPII, GTK, or other key enzymes could be used in combination with standard chemotherapy treatments to increase their overall effectiveness (Fig. 3).

Cancer cells with glutamine addiction result in a glutamine-deprived tumor microenvironment. Decreased glutamine level results in decreased interferon-γ and TNF-α production in T cells leading to tumor immune evasion. However, NK cells are not affected by decreased glutamine level. Decreased glutamine level also results in HIF-1α activation in tumor-associated macrophages which leads to immunosuppression through IL-23 signaling. Inhibition of IL-23 results in prolonged survival and reduced tumor growth. Cancer-associated fibroblasts in the tumor microenvironment can release exosomes containing metabolites, which when absorbed by the cancer cells can promote glycolysis and glutamine metabolism
Currently, the use of SIRM with NMR has been very useful in tracking and examining metabolite usage within certain cancer lines [56]. Increased efforts should be made in the future to use metabolomic technologies for the analysis of different cancers.
Abbreviations
- 2HG:
-
2-Hydroxyglutaric acid
- Acetyl-CoA:
-
Acetyl coenzyme A
- ASS:
-
Argininosuccinate synthetase
- CAF:
-
Cancer-associated fibroblast
- ccRCC:
-
Clear cell renal cell carcinoma
- ECG:
-
Epicatechin gallate
- EGCG:
-
Epigallocatechin gallate
- ETC:
-
Electron transport chain
- FDG-PET:
-
Fluorodeoxyglucose-positron-emission tomography
- FH:
-
Fumarate hydratase
- GBM:
-
Glioblastoma multiforme
- GDH:
-
Glutamate dehydrogenase
- GLS:
-
Glutaminase
- GOT:
-
Glutamic-oxaloacetic transaminase
- GPNA:
-
l-γ-Glutamyl-p-nitroanilide
- GPT:
-
Glutamic-pyruvate transaminase
- HIF:
-
Hypoxia-inducible factor
- IDH:
-
Isocitrate dehydrogenase
- IDO:
-
Indoleamine-2,3-dioxygenase
- IL:
-
Interleukin
- NK:
-
Natural killer (cell)
- NMR:
-
Nuclear magnetic resonance
- PEG:
-
Poly(ethylene glycol)
- PHD:
-
Prolyl 4-hydroxylases
- PLGA:
-
Poly(lactic-co-glycolic acid)
- PPP:
-
Pentose phosphate pathway
- PSAT:
-
Phosphoserine aminotransferase
- RCC:
-
Renal cell carcinomas
- SDH:
-
Succinate dehydrogenase
- SHMT:
-
Serine hydroxymethyltransferase
- SIRM:
-
Stable isotope-resolved metabolomics
- TAM:
-
Tumor-associated macrophage
- TCA:
-
Tricarboxylic acid
- TDO:
-
Tryptophan-2,3-dioxygenase
- TGFβ:
-
Transforming growth factor beta
- TNBC:
-
Triple-negative breast cancer
- TNFα:
-
Tumor necrosis factor alpha
- Treg:
-
Regulatory T cell
- TXNIP:
-
Thioredoxin-interacting protein
- UTR:
-
Untranslated region
- α-KG:
-
α-Ketoglutarate
References
Chen, J. Q., & Russo, J. (2012). Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochimica et Biophysica Acta, 1826(2), 370–384.
Scriver, C. R., & Rosenberg, L. E. (1973). Amino acid metabolism and its disorders. Major Problems in Clinical Pediatrics, 10, 1–478.
Berg, J. M., Tymoczko, J. L., & Stryer, L. (2012). Biochemistry (7th ed.). New York: W.H. Freeman. xxxii, 1054, 43, 41, 48 p.
Vander Heiden, M. G., Cantley, L. C., & Thompson, C. B. (2009). Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science, 324(5930), 1029–1033.
Akram, M. (2014). Citric acid cycle and role of its intermediates in metabolism. Cell Biochemistry and Biophysics, 68(3), 475–478.
Cardaci, S., & Ciriolo, M. R. (2012). TCA cycle defects and cancer: When metabolism tunes redox state. International Journal of Cell Biology, 2012, 161837.
Cairns, R. A., Harris, I. S., & Mak, T. W. (2011). Regulation of cancer cell metabolism. Nature Reviews. Cancer, 11(2), 85–95.
Laurenti, G., & Tennant, D. A. (2016). Isocitrate dehydrogenase (IDH), succinate dehydrogenase (SDH), fumarate hydratase (FH): Three players for one phenotype in cancer? Biochemical Society Transactions, 44(4), 1111–1116.
Bardella, C., Pollard, P. J., & Tomlinson, I. (2011). SDH mutations in cancer. Biochimica et Biophysica Acta, 1807(11), 1432–1443.
Baysal, B. E., et al. (2000). Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science, 287(5454), 848–851.
Baysal, B. E., et al. (2002). Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. Journal of Medical Genetics, 39(3), 178–183.
Burnichon, N., et al. (2010). SDHA is a tumor suppressor gene causing paraganglioma. Human Molecular Genetics, 19(15), 3011–3020.
Cascon, A., et al. (2008). Molecular characterisation of a common SDHB deletion in paraganglioma patients. Journal of Medical Genetics, 45(4), 233–238.
Ricketts, C., et al. (2008). Germline SDHB mutations and familial renal cell carcinoma. Journal of the National Cancer Institute, 100(17), 1260–1262.
Zantour, B., et al. (2004). A thyroid nodule revealing a paraganglioma in a patient with a new germline mutation in the succinate dehydrogenase B gene. European Journal of Endocrinology, 151(4), 433–438.
Tomlinson, I. P., et al. (2002). Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nature Genetics, 30(4), 406–410.
Shanmugasundaram, K., et al. (2014). The oncometabolite fumarate promotes pseudohypoxia through noncanonical activation of NF-kappaB signaling. The Journal of Biological Chemistry, 289(35), 24691–24699.
Quinones, A., & Le, A. (2021). The multifaceted glioblastoma: From genomic alterations to metabolic adaptations. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_4
Dang, L., et al. (2010). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature, 465(7300), 966.
Parsons, D. W., et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science, 321(5897), 1807–1812.
Yan, H., et al. (2009). IDH1 and IDH2 mutations in gliomas. The New England Journal of Medicine, 360(8), 765–773.
Ward, P. S., et al. (2010). The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell, 17(3), 225–234.
DeBerardinis, R. J., & Cheng, T. (2010). Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene, 29(3), 313–324.
Still, E. R., & Yuneva, M. O. (2017). Hopefully devoted to Q: Targeting glutamine addiction in cancer. British Journal of Cancer, 116(11), 1375–1381.
Mullen, A. R., et al. (2011). Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature, 481(7381), 385–388.
Gameiro, P. A., et al. (2013). In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metabolism, 17(3), 372–385.
Zarisfi, M., et al. (2021). The heterogeneity metabolism of renal cell carcinomas. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_8
Seltzer, M. J., et al. (2010). Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Research, 70(22), 8981–8987.
Ma, W. W., et al. (2009). [18F]fluorodeoxyglucose positron emission tomography correlates with Akt pathway activity but is not predictive of clinical outcome during mTOR inhibitor therapy. Journal of Clinical Oncology, 27(16), 2697–2704.
Qu, W., et al. (2012). Preparation and characterization of L-[5-11C]-glutamine for metabolic imaging of tumors. Journal of Nuclear Medicine, 53(1), 98–105.
Zhu, L., et al. (2017). Metabolic imaging of glutamine in cancer. Journal of Nuclear Medicine, 58(4), 533–537.
Jeong, S. M., et al. (2016). Enhanced mitochondrial glutamine anaplerosis suppresses pancreatic cancer growth through autophagy inhibition. Scientific Reports, 6, 30767.
Owen, O. E., Kalhan, S. C., & Hanson, R. W. (2002). The key role of anaplerosis and cataplerosis for citric acid cycle function. The Journal of Biological Chemistry, 277(34), 30409–30412.
Umapathy, N. S., et al. (2008). Expression and function of system N glutamine transporters (SN1/SN2 or SNAT3/SNAT5) in retinal ganglion cells. Investigative Ophthalmology & Visual Science, 49(11), 5151–5160.
Wu, G., et al. (2004). Glutathione metabolism and its implications for health. The Journal of Nutrition, 134(3), 489–492.
Zhang, L., et al. (2016). Reactive oxygen species and targeted therapy for pancreatic cancer. Oxidative Medicine and Cellular Longevity, 2016, 1616781.
Assi, M. (2017). The differential role of reactive oxygen species in early and late stages of cancer. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 313(6), R646–R653.
Elgogary, A., et al. (2016). Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America, 113(36), E5328–E5336.
Le, A., et al. (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metabolism, 15(1), 110–121.
Erickson, J. W., & Cerione, R. A. (2010). Glutaminase: A hot spot for regulation of cancer cell metabolism? Oncotarget, 1(8), 734–740.
Nguyen, T., et al. (2019). Uncovering the role of N-acetyl-aspartyl-glutamate as a glutamate reservoir in cancer. Cell Reports, 27(2), 491–501. e6.
Asaka, R., & Le, A. (2019). Dual role of N-acetyl-aspartyl-glutamate metabolism in cancer monitor and therapy. Molecular & Cellular Oncology, 6(5), e1627273.
Udupa, S., et al. (2019). Upregulation of the glutaminase II pathway contributes to glutamate production upon glutaminase 1 inhibition in pancreatic cancer. Proteomics, 19(21-22), e1800451.
Altman, B. J., Stine, Z. E., & Dang, C. V. (2016). From Krebs to clinic: Glutamine metabolism to cancer therapy. Nature Reviews. Cancer, 16(11), 749.
Colombo, S. L., et al. (2011). Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. Proceedings of the National Academy of Sciences of the United States of America, 108(52), 21069–21074.
Hu, W., et al. (2010). Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proceedings of the National Academy of Sciences of the United States of America, 107(16), 7455–7460.
Gao, P., et al. (2009). c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature, 458(7239), 762–765.
Wise, D. R., et al. (2008). Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences of the United States of America, 105(48), 18782–18787.
Hoang, G., Udupa, S., & Le, A. (2019). Application of metabolomics technologies toward cancer prognosis and therapy. International Review of Cell and Molecular Biology, 347, 191–223.
Zilfou, J. T., & Lowe, S. W. (2009). Tumor suppressive functions of p53. Cold Spring Harbor Perspectives in Biology, 1(5), a001883.
Matoba, S., et al. (2006). p53 regulates mitochondrial respiration. Science, 312(5780), 1650–1653.
Sablina, A. A., et al. (2005). The antioxidant function of the p53 tumor suppressor. Nature Medicine, 11(12), 1306–1313.
Xiang, Y., et al. (2015). Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. The Journal of Clinical Investigation, 125(6), 2293–2306.
Wang, J. B., et al. (2010). Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell, 18(3), 207–219.
Gross, M. I., et al. (2014). Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Molecular Cancer Therapeutics, 13(4), 890–901.
Robinson, M. M., et al. (2007). Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochemical Journal, 406(3), 407–414.
Willis, R. C., & Seegmiller, J. E. (1977). The inhibition by 6-diazo-5-oxo-l-norleucine of glutamine catabolism of the cultured human lymphoblast. Journal of Cellular Physiology, 93(3), 375–382.
Zimmermann, S. C., et al. (2016). Allosteric glutaminase inhibitors based on a 1,4-di(5-amino-1,3,4-thiadiazol-2-yl)butane scaffold. ACS Medicinal Chemistry Letters, 7(5), 520–524.
Elgadi, K. M., et al. (1999). Cloning and analysis of unique human glutaminase isoforms generated by tissue-specific alternative splicing. Physiological Genomics, 1(2), 51–62.
Shukla, K., et al. (2012). Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. Journal of Medicinal Chemistry, 55(23), 10551–10563.
Son, J., et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature, 496(7443), 101–105.
Nabi, K., & Le, A. (2021). The intratumoral heterogeneity of cancer metabolism. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_11
Xu, X., et al. (2019). Overview of the development of glutaminase inhibitors: Achievements and future directions. Journal of Medicinal Chemistry, 62(3), 1096–1115.
Parlati, F., et al. (2014). Glutaminase inhibitor CB-839 synergizes with pomalidomide in preclinical multiple myeloma models. Blood, 124(21), 4720–4720.
Parlati, F. (2015). CB-839, a selective glutaminase inhibitor, synergizes with signaling pathway inhibitors to produce an anti-tumor effect in cell lines and tumor xenografts. Cancer Research, 75, 4711.
Emberley, E., et al. (2017). CB-839, a selective glutaminase inhibitor, has anti-tumor activity in renal cell carcinoma and synergizes with cabozantinib and everolimus, in keystone symposia, tumor metabolism: Mechanisms and targets. Canada: Whistler.
Emberley, E. D., et al. (2018). The glutaminase inhibitor CB-839 synergizes with CDK4/6 and PARP inhibitors in pre-clinical tumor models. Cancer Research, 78(13), 3509–3509.
Momcilovic, M., et al. (2017). Targeted inhibition of EGFR and glutaminase induces metabolic crisis in EGFR mutant lung cancer. Cell Reports, 18(3), 601–610.
Liu, W., et al. (2012). Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proceedings of the National Academy of Sciences of the United States of America, 109(23), 8983–8988.
Dang, C. V., Le, A., & Gao, P. (2009). MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clinical Cancer Research, 15(21), 6479–6483.
Le, A., & Dang, C. V. (2013). Studying Myc’s role in metabolism regulation. Methods in Molecular Biology, 1012, 213–219.
Niu, Z., et al. (2015). Knockdown of c-Myc inhibits cell proliferation by negatively regulating the Cdk/Rb/E2F pathway in nasopharyngeal carcinoma cells. Acta Biochimica et Biophysica Sinica Shanghai, 47(3), 183–191.
Zhang, X., Ge, Y. L., & Tian, R. H. (2009). The knockdown of c-myc expression by RNAi inhibits cell proliferation in human colon cancer HT-29 cells in vitro and in vivo. Cellular & Molecular Biology Letters, 14(2), 305–318.
Whitfield, J. R., Beaulieu, M. E., & Soucek, L. (2017). Strategies to inhibit myc and their clinical applicability. Frontiers in Cell and Development Biology, 5, 10.
Brooks, T. A., & Hurley, L. H. (2010). Targeting MYC expression through G-quadruplexes. Genes & Cancer, 1(6), 641–649.
Brown, R. V., et al. (2011). Demonstration that drug-targeted down-regulation of MYC in non-Hodgkin’s lymphoma is directly mediated through the promoter G-quadruplex. Journal of Biological Chemistry, 286(47), 41018–41027.
Devi, G. R., et al. (2005). In vivo bioavailability and pharmacokinetics of a c-MYC antisense phosphorodiamidate morpholino oligomer, AVI-4126, in solid tumors. Clinical Cancer Research, 11(10), 3930–3938.
Tolcher, A. W., et al. (2015). Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. Journal of Clinical Oncology, 33(15_suppl), 11006–11006.
Stellas, D., et al. (2014). Therapeutic effects of an anti-Myc drug on mouse pancreatic cancer. Journal of the National Cancer Institute, 106, 12.
Yin, X., et al. (2003). Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene, 22(40), 6151–6159.
Farrell, A. S., et al. (2014). Targeting inhibitors of the tumor suppressor PP2A for the treatment of pancreatic cancer. Molecular Cancer Research, 12(6), 924–939.
Polivka, J., Jr., & Janku, F. (2014). Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacology & Therapeutics, 142(2), 164–175.
Casey, S. C., et al. (2016). MYC regulates the antitumor immune response through CD47 and PD-L1. Science, 352(6282), 227–231.
Masso-Valles, D., Jauset, T., & Soucek, L. (2016). Ibrutinib repurposing: From B-cell malignancies to solid tumors. Oncoscience, 3(5-6), 147–148.
Jin, L., Alesi, G. N., & Kang, S. (2016). Glutaminolysis as a target for cancer therapy. Oncogene, 35(28), 3619–3625.
Li, C., et al. (2006). Green tea polyphenols modulate insulin secretion by inhibiting glutamate dehydrogenase. The Journal of Biological Chemistry, 281(15), 10214–10221.
Li, C., et al. (2011). Green tea polyphenols control dysregulated glutamate dehydrogenase in transgenic mice by hijacking the ADP activation site. The Journal of Biological Chemistry, 286(39), 34164–34174.
Yang, C. S., et al. (2009). Cancer prevention by tea: Animal studies, molecular mechanisms and human relevance. Nature Reviews. Cancer, 9(6), 429–439.
Li, M., et al. (2009). Novel inhibitors complexed with glutamate dehydrogenase: Allosteric regulation by control of protein dynamics. The Journal of Biological Chemistry, 284(34), 22988–23000.
Yang, C., et al. (2009). Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Research, 69(20), 7986–7993.
Ollenschlager, G., et al. (1988). Asparaginase-induced derangements of glutamine metabolism: The pathogenetic basis for some drug-related side-effects. European Journal of Clinical Investigation, 18(5), 512–516.
Wu, M. C., Arimura, G. K., & Yunis, A. A. (1978). Mechanism of sensitivity of cultured pancreatic carcinoma to asparaginase. International Journal of Cancer, 22(6), 728–733.
Grigoryan, R. S., et al. (2004). Changes of amino acid serum levels in pediatric patients with higher-risk acute lymphoblastic leukemia (CCG-1961). In Vivo, 18(2), 107–112.
Lukey, M. J., Wilson, K. F., & Cerione, R. A. (2013). Therapeutic strategies impacting cancer cell glutamine metabolism. Future Medicinal Chemistry, 5(14), 1685–1700.
Nguyen, H. A., Su, Y., & Lavie, A. (2016). Structural insight into substrate selectivity of Erwinia chrysanthemi L-asparaginase. Biochemistry, 55(8), 1246–1253.
Ertel, I. J., et al. (1979). Effective dose of L-asparaginase for induction of remission in previously treated children with acute lymphocytic leukemia: A report from Children’s Cancer Study Group. Cancer Research, 39(10), 3893–3896.
Panosyan, E. H., et al. (2014). Asparagine depletion potentiates the cytotoxic effect of chemotherapy against brain tumors. Molecular Cancer Research, 12(5), 694–702.
Stams, W. A., et al. (2003). Sensitivity to L-asparaginase is not associated with expression levels of asparagine synthetase in t(12;21)+ pediatric ALL. Blood, 101(7), 2743–2747.
Ye, S., et al. (2020). The heterocyclic compound Tempol inhibits the growth of cancer cells by interfering with glutamine metabolism. Cell Death & Disease, 11(5), 312.
Darmaun, D., et al. (1998). Phenylbutyrate-induced glutamine depletion in humans: Effect on leucine metabolism. The American Journal of Physiology, 274(5 Pt 1), E801–E807.
Thibault, A., et al. (1994). A phase I and pharmacokinetic study of intravenous phenylacetate in patients with cancer. Cancer Research, 54(7), 1690–1694.
Schulte, M. L., et al. (2018). Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nature Medicine, 24(2), 194–202.
Wahi, K., & Holst, J. (2019). ASCT2: A potential cancer drug target. Expert Opinion on Therapeutic Targets, 23(7), 555–558.
Colas, C., et al. (2015). Ligand discovery for the alanine-serine-cysteine transporter (ASCT2, SLC1A5) from homology modeling and virtual screening. PLoS Computational Biology, 11(10), e1004477.
Fuchs, B. C., & Bode, B. P. (2005). Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Seminars in Cancer Biology, 15(4), 254–266.
Hassanein, M., et al. (2013). SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clinical Cancer Research, 19(3), 560–570.
Chiu, M., et al. (2017). GPNA inhibits the sodium-independent transport system L for neutral amino acids. Amino Acids, 49(8), 1365–1372.
Broer, A., Fairweather, S., & Broer, S. (2018). Disruption of amino acid homeostasis by novel ASCT2 inhibitors involves multiple targets. Frontiers in Pharmacology, 9, 785.
Osanai-Sasakawa, A., et al. (2018). An anti-ASCT2 monoclonal antibody suppresses gastric cancer growth by inducing oxidative stress and antibody dependent cellular toxicity in preclinical models. American Journal of Cancer Research, 8(8), 1499–1513.
Ahluwalia, G. S., et al. (1990). Metabolism and action of amino acid analog anti-cancer agents. Pharmacology & Therapeutics, 46(2), 243–271.
Rais, R., et al. (2016). Discovery of 6-Diazo-5-oxo-l-norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: A potential treatment for glioblastoma. Journal of Medicinal Chemistry, 59(18), 8621–8633.
Thangavelu, K., et al. (2014). Structural basis for the active site inhibition mechanism of human kidney-type glutaminase (KGA). Scientific Reports, 4, 3827.
Ortlund, E., et al. (2000). Reactions of pseudomonas 7A glutaminase-asparaginase with diazo analogues of glutamine and asparagine result in unexpected covalent inhibitions and suggests an unusual catalytic triad Thr-Tyr-Glu. Biochemistry, 39(6), 1199–1204.
Ovejera, A. A., et al. (1979). Efficacy of 6-diazo-5-oxo-L-norleucine and N-[N-gamma-glutamyl-6-diazo-5-oxo-norleucinyl]-6-diazo-5-oxo-norleucine against experimental tumors in conventional and nude mice. Cancer Research, 39(8), 3220–3224.
Mueller, C., et al. (2008). A phase IIa study of PEGylated glutaminase (PEG-PGA) plus 6-diazo-5-oxo-L-norleucine (DON) in patients with advanced refractory solid tumors. Journal of Clinical Oncology, 26(15_suppl), 2533–2533.
Lukey, M. J., Katt, W. P., & Cerione, R. A. (2017). Targeting amino acid metabolism for cancer therapy. Drug Discovery Today, 22(5), 796–804.
Beuster, G., et al. (2011). Inhibition of alanine aminotransferase in silico and in vivo promotes mitochondrial metabolism to impair malignant growth. The Journal of Biological Chemistry, 286(25), 22323–22330.
Thornburg, J. M., et al. (2008). Targeting aspartate aminotransferase in breast cancer. Breast Cancer Research, 10(5), R84.
Tan, J., & Le, A. (2021). The heterogeneity of breast cancer metabolism. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_6
Possemato, R., et al. (2011). Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature, 476(7360), 346–350.
Camelo, F., & Le, A. (2021). The intricate metabolism of pancreatic cancers. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_5
Wu, G., & Morris, S. M., Jr. (1998). Arginine metabolism: Nitric oxide and beyond. Biochemical Journal, 336(Pt 1), 1–17.
Ananieva, E. (2015). Targeting amino acid metabolism in cancer growth and anti-tumor immune response. World Journal of Biological Chemistry, 6(4), 281–289.
Kobayashi, E., et al. (2010). Reduced argininosuccinate synthetase is a predictive biomarker for the development of pulmonary metastasis in patients with osteosarcoma. Molecular Cancer Therapeutics, 9(3), 535–544.
Grohmann, U., & Bronte, V. (2010). Control of immune response by amino acid metabolism. Immunological Reviews, 236, 243–264.
Godin-Ethier, J., et al. (2011). Indoleamine 2,3-dioxygenase expression in human cancers: Clinical and immunologic perspectives. Clinical Cancer Research, 17(22), 6985–6991.
Mellor, A. L., & Munn, D. H. (1999). Tryptophan catabolism and T-cell tolerance: Immunosuppression by starvation? Immunology Today, 20(10), 469–473.
Uyttenhove, C., et al. (2003). Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nature Medicine, 9(10), 1269–1274.
Ino, K., et al. (2008). Inverse correlation between tumoral indoleamine 2,3-dioxygenase expression and tumor-infiltrating lymphocytes in endometrial cancer: Its association with disease progression and survival. Clinical Cancer Research, 14(8), 2310–2317.
Moon, Y. W., et al. (2015). Targeting the indoleamine 2,3-dioxygenase pathway in cancer. Journal for ImmunoTherapy of Cancer, 3, 51.
Beatty, G. L., et al. (2013). A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clinical Cancer Research, 19(22), 6286–6295.
Soliman, H. H., et al. (2016). A phase I study of indoximod in patients with advanced malignancies. Oncotarget, 7(16), 22928–22938.
Pilotte, L., et al. (2012). Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proceedings of the National Academy of Sciences of the United States of America, 109(7), 2497–2502.
Amelio, I., et al. (2014). Serine and glycine metabolism in cancer. Trends in Biochemical Sciences, 39(4), 191–198.
DeBerardinis, R. J. (2011). Serine metabolism: Some tumors take the road less traveled. Cell Metabolism, 14(3), 285–286.
Pollari, S., et al. (2011). Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Research and Treatment, 125(2), 421–430.
Dang, C. V. (2012). MYC on the path to cancer. Cell, 149(1), 22–35.
Nikiforov, M. A., et al. (2002). A functional screen for Myc-responsive genes reveals serine hydroxymethyltransferase, a major source of the one-carbon unit for cell metabolism. Molecular and Cellular Biology, 22(16), 5793–5800.
di Salvo, M. L., et al. (2013). Glycine consumption and mitochondrial serine hydroxymethyltransferase in cancer cells: The heme connection. Medical Hypotheses, 80(5), 633–636.
Jain, M., et al. (2012). Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science, 336(6084), 1040–1044.
Daidone, F., et al. (2011). In silico and in vitro validation of serine hydroxymethyltransferase as a chemotherapeutic target of the antifolate drug pemetrexed. European Journal of Medicinal Chemistry, 46(5), 1616–1621.
Maddocks, O. D., et al. (2013). Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature, 493(7433), 542–546.
Antonio, M. J., Zhang, C., & Le, A. (2021). Different tumor microenvironments lead to different metabolic phenotypes. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_10.
Binnewies, M., et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature Medicine, 24(5), 541–550.
Larsen, S. K., Gao, Y., & Basse, P. H. (2014). NK cells in the tumor microenvironment. Critical Reviews in Oncogenesis, 19(1-2), 91–105.
Wang, J., et al. (2019). Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment. Cancer Medicine, 8(10), 4709–4721.
Wu, L., et al. (2019). Tumor-associated neutrophils in cancer: Going pro. Cancers (Basel), 11, 4.
Xing, F., Saidou, J., & Watabe, K. (2010). Cancer associated fibroblasts (CAFs) in tumor microenvironment. Frontiers in Bioscience (Landmark Ed), 15, 166–179.
Weinberg, R. A. (2014). The biology of cancer. New York, NY: Garland Science, Taylor & Francis Group LLC.
Presnell, S. R., et al. (2020). Differential fuel requirements of human NK cells and human CD8 T cells: Glutamine regulates glucose uptake in strongly activated CD8 T cells. Immunohorizons, 4(5), 231–244.
Parikh, H., et al. (2007). TXNIP regulates peripheral glucose metabolism in humans. PLoS Medicine, 4(5), e158.
Song, M., et al. (2018). IRE1α–XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature, 562(7727), 423–428.
Owen, J. A., et al. (2013). Kuby Immunology. New York: W.H. Freeman.
Fu, Q., et al. (2019). Tumor-associated macrophage-derived interleukin-23 interlinks kidney cancer glutamine addiction with immune evasion. European Urology, 75(5), 752–763.
Viguier, M., et al. (2004). Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. Journal of Immunology, 173(2), 1444–1453.
Bradley, C. A. (2018). IL-23 links glutamine addiction and immune function. Nature Reviews. Urology, 15(12), 725.
Zhao, H., et al. (2016). Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife, 5, e10250.
Sazeides, C., & Le, A. (2021). Metabolic relationship between cancerassociated fibroblasts and cancer cells. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_14
Jung, J. G., & Le, A. (2021). Targeting metabolic cross talk between cancer cells and cancer-associated fibroblasts. Advances in Experimental Medicine and Biology, 1311, https://doi.org/10.1007/978-3-030-65768-0_15.
Dang, C. V., et al. (2011). Therapeutic targeting of cancer cell metabolism. Journal of Molecular Medicine (Berlin), 89(3), 205–212.
Hirschey, M. D., et al. (2015). Dysregulated metabolism contributes to oncogenesis. Seminars in Cancer Biology, 35(Suppl), S129–S150.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Li, T., Copeland, C., Le, A. (2021). Glutamine Metabolism in Cancer. In: Le, A. (eds) The Heterogeneity of Cancer Metabolism. Advances in Experimental Medicine and Biology, vol 1311. Springer, Cham. https://doi.org/10.1007/978-3-030-65768-0_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-65768-0_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-65767-3
Online ISBN: 978-3-030-65768-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)