
Abstract
Introduction
To help prevent febrile neutropenia, pegfilgrastim-cbqv (UDENYCA®; Coherus BioSciences), a pegfilgrastim (NEULASTA®; Amgen) biosimilar, is administered 24–96 h after myelosuppressive chemotherapy. Delivery of pegfilgrastim-cbqv using an on-body injector (OBI) provides an alternative method of administration, affording options in drug delivery. This study aimed to establish pharmacokinetic (PK) and pharmacodynamic (PD) bioequivalence and assess the safety of pegfilgrastim-cbqv administered using an OBI compared with a prefilled syringe (PFS).
Methods
In this open-label, two-period crossover study, healthy adult male participants (N = 189) were randomly assigned 1:1 to receive pegfilgrastim-cbqv 6 mg subcutaneously using an OBI (n = 92) or a PFS (n = 95) in period 1 and then an injection via the other method in period 2. Primary PK end points were area under the concentration–time curve from time 0 to infinity, area under the concentration–time curve from time 0 to the last quantifiable concentration, and maximum plasma concentration. Secondary PD end points, safety, immunogenicity, and tolerability were also assessed.
Results
The 90% confidence intervals (CIs) of the geometric mean ratios for the PK and PD end points fell within the predetermined range (80–125%), indicating PK and PD bioequivalence between pegfilgrastim-cbqv OBI and pegfilgrastim-cbqv PFS. Treatment-emergent adverse events (TEAEs) occurred in 87.8% and 75.8% of participants in the OBI and PFS groups, respectively. Most TEAEs were musculoskeletal effects. The most common OBI-related TEAE was injection site erythema (31.7%), which was mild, transient, and self-limiting. The incidence of treatment-emergent antidrug antibodies (ADAs) was similar between the OBI and PFS. ADAs had no apparent impact on PK, PD, or safety. Neutralizing antibodies were not detected in any participant.
Conclusions
Results of the study showed PK and PD bioequivalence of pegfilgrastim-cbqv administered using OBI compared with PFS. OBI and PFS administration had similar safety, tolerability, and immunogenicity profiles. No unexpected safety signals were identified.
Graphical Abstract available for this article.
Graphical Abstract

Plain Language Summary
Febrile neutropenia is when a patient has a fever and a lower-than-normal number of white blood cells. When white blood cell counts are low, patients are more susceptible to opportunistic infections as a result of their weakened immune systems. Severe febrile neutropenia can lead to the stopping or delaying of chemotherapy. The drug pegfilgrastim-cbqv is used 24–96 h after chemotherapy to stimulate the growth of white blood cells. Pegfilgrastim-cbqv is available in a single-dose prefilled syringe and in a prefilled autoinjector. If a patient cannot inject themselves with the drug, they must go to a clinic for the injection. Using an on-body injector applied to the skin that automatically injects the drug at a specific time could eliminate the need to go to the clinic. During this study, healthy adult male participants were given pegfilgrastim-cbqv through an on-body injector or a prefilled syringe to investigate if the movement of the drug into, through, and out of the body (pharmacokinetics) and the physiological action of the drug in the body (pharmacodynamics) were similar between the two injection methods. Side effects were also studied. The researchers found that the pharmacokinetics and pharmacodynamics for pegfilgrastim-cbqv given by on-body-injector or prefilled syringe were similar. The number and types of side effects were also similar. The most common side effect for the on-body injector was mild erythema at the injection site. This side effect resolved by itself. The treatment benefit and safety of pegfilgrastim-cbqv were very similar regardless of how the drug was administered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
The US Food and Drug Administration-licensed biosimilar pegfilgrastim-cbqv (UDENYCA®, Coherus BioSciences) was initially approved for administration using a prefilled syringe (PFS). |
The on-body injector (OBI) presentation was developed to offer a new administration option for pegfilgrastim-cbqv. |
The investigator's goals of this study were to establish the pharmacokinetic (PK) and pharmacodynamic (PD) bioequivalence of pegfilgrastim-cbqv by OBI and pegfilgrastim-cbqv by PFS and to assess the safety and tolerability of the OBI. |
What was learned from the study? |
The PK, PD, and safety assessments following administration of pegfilgrastim-cbqv by OBI were similar to those of administration by PFS. |
The use of a pegfilgrastim-cbqv OBI may be a beneficial alternative to administration of pegfilgrastim-cbqv by PFS, providing an additional option for dose delivery, which reduces the need for patient visits to health care facilities to receive the recommended next-day administration of pegfilgrastim-cbqv at a clinic or hospital. |
Digital Features
This article is published with digital features, including a Graphical Abstract, to facilitate understanding of the article. To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.24517105.
Introduction
Febrile neutropenia (FN) is a serious complication of cancer chemotherapy. It is defined as an oral temperature of greater than 38.3 °C or two consecutive readings of greater than 38.0 °C sustained over 2 h and an absolute neutrophil count (ANC) of less than 0.5 × 109/L or an ANC expected to decrease to less than 0.5 × 109/L over a period of 48 h [1, 2]. Associated with substantial morbidity and mortality, FN is also one of the most frequent complications of chemotherapy [3,4,5,6,7,8]. As a result of FN, treatment efficacy can be compromised because of delays and dose reductions in chemotherapy [4, 8]. FN is also associated with a substantial increase in health care-related costs [3].
Granulocyte colony-stimulating factors (G-CSFs) substantially reduce the risk of infection that could result from FN and are recommended as prophylaxis by international guidelines [1, 4, 9,10,11]. Long-acting G-CSFs, such as pegfilgrastim (NEULASTA®; Amgen, Thousand Oaks, CA), are administered 24–96 h after chemotherapy and decrease the risk of FN [10,11,12,13,14,15].
Although biologic medications such as pegfilgrastim have marked clinical benefit, biologics are associated with considerable cost [16]. Biosimilars are biologic drugs proven to be highly similar in potency, safety, and efficacy to the originator biologic (reference product) but available at a lower cost [17]. Pegfilgrastim-cbqv (UDENYCA®; Coherus BioSciences, Redwood City, CA) is approved by the US Food and Drug Administration (FDA) as a biosimilar to pegfilgrastim [12, 13]. Bioequivalence between pegfilgrastim-cbqv and the reference product pegfilgrastim, along with a similar safety profile, was established during a trial of a single-dose injected using a prefilled syringe (PFS) [18].
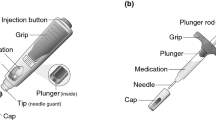
As an alternative to the pegfilgrastim-cbqv PFS, a pegfilgrastim-cbqv on-body injector (OBI) drug–device combination product was developed. It consists of a 6-mg/0.6-mL single-dose pegfilgrastim-cbqv solution contained in a PFS copackaged with an OBI. The OBI is filled by the health care provider and attached to the patient’s arm or abdomen. Patients can return home after chemotherapy wearing the OBI, which is preset to automatically deliver a dose of pegfilgrastim-cbqv 27 h after chemotherapy [14]. The dosage, route of administration, and conditions of use (i.e., indication and dosing regimen) of this new presentation are identical to those of the FDA-licensed pegfilgrastim-cbqv single-dose 6-mg PFS and the reference product pegfilgrastim [12]. The OBI provides an alternative for patients who are unable to self-administer the drug and reduces the need for a second health care visit to receive the recommended next-day administration of pegfilgrastim-cbqv [16]. The goal of the current study was to establish the pharmacokinetic (PK) bioequivalence of pegfilgrastim-cbqv OBI and pegfilgrastim-cbqv PFS and to assess the pharmacodynamic (PD) bioequivalence, the safety (including immunogenicity), and the tolerability of the OBI.
Methods
Study Design
This was a randomized, open-label, two-period, two-sequence crossover study that was conducted over a period of 14–16 weeks from November 2020 to August 2021 at two clinical sites in the USA. The study was designed to assess the PK bioequivalence of a single 6-mg subcutaneous (SC) injection of pegfilgrastim-cbqv administered using an OBI compared with a PFS. PD bioequivalence, safety (including immunogenicity), and tolerability were also assessed.
After an initial screening period (a 28-day period before dosing), participants were randomly assigned 1:1 to receive pegfilgrastim-cbqv 6 mg SC using a PFS or an OBI administered by or under health care provider supervision on day 1 of period 1. After a 6- to 8-week washout period, participants received a single injection of pegfilgrastim-cbqv 6 mg on day 1 of period 2 using the method not used in period 1 (Supplementary Material Fig. S1). A sample for hematologic testing was drawn 2 days before period 2. To be eligible for enrollment in period 2, participants were required to have an ANC of 1.7–7.2 × 103/mm3 and a white blood cell (WBC) count of 4.0–11.0 × 103/mm3. If the ANC or WBC count was outside this range, two subsequent counts were allowed, if the prescribed washout period was not exceeded and the participants’ ANC and WBC met the count criteria.
During periods 1 and 2, participants were admitted to the clinical sites 2 days before the first dose and remained at the site through the 96-h postdose time point (i.e., day 5). Blood samples were collected at specified time points for PK and PD measurements, and participants were closely monitored for safety. During the admission period, samples were collected 30 min before dosing and 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, 60, 72, 84, and 96 h after dosing. Following discharge for each period, participants returned to the clinical sites for outpatient visits for blood sample collection on days 6–11, 13, and 21 and in period 1 on day 28. Antidrug antibody (ADA) samples were collected in both periods on day 1 before dosing and on day 11 and in period 2 on day 28.
Participants and Treatment
All participants were healthy, male, and aged 18–45 years with a body weight between 50 and 100 kg and a body mass index between 18 and 28 kg/m2. Participants who were previously given filgrastim or pegfilgrastim were not included in the study. Pegfilgrastim-cbqv was supplied either as a single 6-mg dose in a PFS or as a copackaged OBI and PFS. The OBI was a wearable drug delivery device attached to the participant’s arm or abdomen and held in place by an adhesive pad. Participants who received the OBI could not shower, bathe, or exercise until the OBI was removed. The study drug was administered at approximately the same time on day 1 of periods 1 and 2. The OBI was programmed to deliver the study drug 27 h after activation (which occurred after the OBI was filled with study drug) and over a period of 5 min.
Ethical Approval
The protocol and informed consent form were reviewed and approved by the Advarra institutional review board (IRB registration number IRB0000097).
The study was conducted in accordance with all applicable laws and regulations and complied with the International Conference for Harmonisation E6 Guideline on Good Clinical Practice. All procedures were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
PK and PD End Points
Plasma samples were analyzed for pegfilgrastim-cbqv concentrations using a validated enzyme-linked immunosorbent assay with a sensitivity of 150 pg/mL. Anti-G-CSF antibodies were used as capture and detection reagents to perform the assay. Using automated hematology analyzers that were subject to FDA regulation and underwent evaluation and approval of all analytical performance standards, Clinical Laboratory Improvement Amendments certified laboratories carried out the ANC analysis. Phoenix WinNonlin version 8.1 (Certara L.P., Princeton, NJ) was used to determine the PK parameters, while Statistical Analysis System (SAS; SAS Institute, Cary, NC) version 9.4 was used to calculate PK and PD bioequivalence.
The primary PK end points were area under the concentration–time curve (AUC) from time 0 to infinity (AUC0–inf), area under the concentration–time curve from time 0 to the last quantifiable concentration (AUC0–last), and maximum plasma concentration (Cmax). Other PK end points were time to maximum plasma concentration (Tmax), terminal elimination half-life, elimination rate constant, AUC from time 0 to 288 h, apparent extravascular clearance, volume of distribution, and percentage AUC extrapolated. The key PD parameters were ANC–time curve from time 0 to the last quantifiable absolute neutrophil count (ANC AUC0–last) and the maximum absolute neutrophil count (ANCmax). Other PD end points were time to maximum ANC and area under the ANC–time curve from time 0 to 480 h.
Safety
Measures of safety were treatment-emergent adverse events (TEAEs), AEs of special interest, serious adverse events (SAEs), local injection site reactions, laboratory tests, hematology, coagulation, urinalysis, and vital signs.
Immunogenicity
Samples were tested for the presence of ADAs using a validated electrochemiluminescence bridging assay [18,19,20]. All ADA-positive samples were characterized for titer, binding reactivity (to polyethylene glycol [PEG] or G-CSF), and neutralizing antibodies (NAbs) by use of a validated cell-based assay. Any NAb-positive samples were further tested to determine their ability to neutralize endogenous G-CSF. Details of the immunogenicity assays (ADA and NAb) are discussed in a previous publication [19].
Tolerability
Skin reactions to the OBI adhesive were evaluated using the dermal response scale [21]. The scale ranged from 0 (no evidence of irritation) to 7 (strong reaction spreading beyond the application site) [21]. Pain was evaluated using the Wong-Baker FACES pain rating scale [22]. The pain scale ranged from 0 (no hurt) to 10 (hurts worst) [22]. The number and the percentage of participants in each result category for each scale were summarized for the pegfilgrastim-cbqv OBI treatment group.
Statistical Analysis
Determination of Sample Size
The study was powered to support the primary objective of determining PK bioequivalence, and the sample size was estimated on the basis of the results from a previous pegfilgrastim-cbqv study [18]. Approximately 186 participants were randomly assigned 1:1 to each treatment sequence in the study, based on the assumptions of an intraparticipant coefficient of variation (CV) ≤ 47.5%, expected true geometric mean ratio (GMR) ≤ 1.07 for the primary PK end points, and a 35% dropout rate.
Primary PK Bioequivalence Analysis
In the PK bioequivalence assessment for the primary analysis, using the PK-evaluable population, the logarithm-transformed parameters AUC0–inf, AUC0–last, and Cmax were analyzed using an analysis of variance (ANOVA), including terms of sequence (i.e., treatment sequence), treatment (i.e., treatment group), and period as fixed effects and participant nested within sequence as a random effect. PK bioequivalence was demonstrated if the 90% confidence interval (CI) for the GMR of pegfilgrastim-cbqv OBI/PFS fell within the range of 80–125% for AUC0–inf, AUC0–last, and Cmax. The PK-evaluable population consisted of all participants who received two full doses of pegfilgrastim-cbqv and had sufficient plasma concentration–time data to permit reliable calculation of PK parameters for at least one of the primary PK end points.
Sensitivity Analysis for PK Bioequivalence Assessment
A sensitivity analysis for PK bioequivalence was conducted to test the robustness of the primary analysis using participants in the PK-evaluable population plus PK outliers. A PK outlier was defined as any participant who had an unusually high or low PK response to either treatment that differed from the data for that participant or any of the participants. Numerically, a participant was a PK outlier if the participant had at least one intraparticipant CV that was greater than 100% in the AUC0–inf, AUC0–last, and Cmax between period 1 and period 2.
For the sensitivity analysis, the logarithm-transformed parameters AUC0–inf, AUC0–last, and Cmax were analyzed using the same ANOVA model as the primary PK bioequivalence analysis. PK bioequivalence was demonstrated if the 90% CI for the GMR of pegfilgrastim-cbqv OBI/PFS fell within the range of 80–125% for AUC0–inf, AUC0–last, and Cmax.
Secondary PD Bioequivalence Analysis
PD parameters were summarized by treatment group and study period using the PD-evaluable population. The PD-evaluable population consisted of all participants who received two full doses of pegfilgrastim-cbqv and had sufficient data to permit reliable calculation of the PD parameters for at least one of the primary PD end points. Geometric mean (GM) and geometric CV% were added to the descriptive statistics.
In the PD bioequivalence assessment, logarithmic transformations of the PD parameters were analyzed using an analysis of covariance model, including terms for sequence (i.e., treatment sequence), treatment (i.e., treatment group), and period as fixed effects; participant nested within sequence as a random effect; and baseline ANC as a covariate. PD bioequivalence was demonstrated if the 90% CI for the GMR of pegfilgrastim-cbqv OBI/PFS fell within the range of 80–125% for ANC (AUC0–last) and ANCmax in the PD-evaluable population.
Safety
Safety data were summarized and listed for the safety population. The safety population consisted of all randomly assigned participants who received at least one dose of pegfilgrastim-cbqv using a PFS or an OBI. No formal statistical analysis of the safety data was conducted; safety data were analyzed descriptively.
Immunogenicity
Incidence and time course of ADAs, ADA titer, and NAbs, if any, were summarized by treatment group and by treatment sequence group using the safety population. Additional analyses were conducted to explore the impact of ADAs or NAbs on PK, PD, and safety. The PK concentration population, comprising all participants who received any amount of pegfilgrastim-cbqv and had any measurable plasma PK data, was used to assess the impact of immunogenicity on PK. The safety population was used to assess the impact of immunogenicity on PD and safety.
Results
Participant Demographics and Treatment
A total of 189 participants were randomly assigned in a 1:1 ratio between two treatment sequences (pegfilgrastim-cbqv OBI then pegfilgrastim-cbqv PFS, n = 94; pegfilgrastim-cbqv PFS then pegfilgrastim-cbqv OBI, n = 95). Participant disposition and baseline demographics were balanced between both treatment sequences (Tables 1 and 2). Overall, 137 participants (72.5%) completed both periods, and 52 participants (27.5%) withdrew early from the study. The most common reasons for early withdrawal were AEs and failure to meet protocol-specified ANC or WBC count criteria before dosing. Overall, four participants (2.1%) from the OBI treatment group (OBI malfunction) and zero participants from the PFS group withdrew from the study because of technical problems (Table 1).
Pharmacokinetics
The mean plasma concentration versus time curves were similar between treatment groups in the PK-evaluable population. The mean pegfilgrastim-cbqv plasma concentrations peaked at approximately 16 h after dosing for both treatment groups (Fig. 1). The GMs of AUC0–last were 8790.4 h·ng/mL and 7633.3 h·ng/mL, the GMs of AUC0–inf were 8812.0 h·ng/mL and 7657.0 h·ng/mL, and the Cmax values were 229.6 ng/mL and 199.4 ng/mL for pegfilgrastim-cbqv OBI and pegfilgrastim-cbqv PFS, respectively (Supplementary Material Table S1).

Mean pegfilgrastim-cbqv concentrations by treatment group for period 1 and period 2 combined (PK-evaluable population). Concentrations collected at the follow-up visit were not included. OBI on-body injector, PFS prefilled syringe, PK pharmacokinetics
PK bioequivalence of the pegfilgrastim-cbqv OBI compared with the PFS was established on the basis of the 90% CI of GMRs for AUC0–inf, AUC0–last, and Cmax using the PK-evaluable population. For AUC0–inf and AUC0–last, the GMRs were 115.8 (90% CI 107.3–124.9) and 115.8 (90% CI 107.3–125.0), respectively. The GMR for Cmax was 115.7 (90% CI 107.2–124.8). Because the 90% CIs for the GMRs of pegfilgrastim-cbqv OBI/pegfilgrastim-cbqv PFS for the primary PK parameters fell within the predetermined range, PK bioequivalence was shown between the pegfilgrastim-cbqv OBI and PFS (Table 3).
The robustness of the primary PK analysis was assessed in a sensitivity analysis that included the PK-evaluable population and the PK outliers. In the sensitivity analysis, the GMRs for AUC0–inf and AUC0–last were 112.4 (90% CI 102.7–123.0) and 112.4 (90% CI 102.7–123.1), respectively. The GMR for Cmax was 113.7 (90% CI 103.4–124.9). Because the 90% CIs for the GMRs of pegfilgrastim-cbqv OBI/pegfilgrastim-cbqv PFS were within the predetermined range, PK bioequivalence was further demonstrated in the sensitivity analysis (Table 4).
Pharmacodynamics
For both treatment groups, the mean ANC peaked at approximately 72 h after dosing and returned approximately to baseline levels in the PD-evaluable population (Fig. 2). The mean and highest ANCmax and Tmax values for pegfilgrastim-cbqv OBI and pegfilgrastim-cbqv PFS were also similar (Supplementary Material Table S2). Overall, the mean ANC was similar between the two treatment groups over time.

Mean PD ANC by treatment group for period 1 and period 2 combined (PD-evaluable population). Concentrations collected at the follow-up visit were not included. ANC absolute neutrophil count, OBI on-body injector, PD pharmacodynamics, PFS prefilled syringe, SD standard deviation
The PD bioequivalence of the pegfilgrastim-cbqv OBI and PFS was established on the basis of the 90% CIs for the GMRs for ANC AUC0–last and ANCmax using the PD-evaluable population. For the ANC AUC0–last and the ANCmax, the GMRs were 100.4 (90% CI 98.4–102.4) and 102.1 (90% CI 99.8–104.5), respectively. Because the 90% CIs for the GMRs of pegfilgrastim-cbqv OBI/pegfilgrastim-cbqv PFS were within the range of 80–125%, PD bioequivalence was shown (Table 5).
Safety
Overall, 144 participants (87.8%) in the pegfilgrastim-cbqv OBI group and 122 participants (75.8%) in the pegfilgrastim-cbqv PFS group experienced at least one TEAE. Most TEAEs were considered related to the study drug and were mild. Only two participants experienced treatment-emergent serious adverse events (TESAEs) (one from each group): the participant in the pegfilgrastim-cbqv OBI group experienced moderate left nephrolithiasis on day 1 in period 2, and the participant in the pegfilgrastim-cbqv OBI group experienced a fatal asthma attack on day 41 in period 1. Neither TESAE was considered related to the study drug or the device. In addition, one participant (0.6%) in the pegfilgrastim-cbqv OBI group experienced a TEAE of special interest (perivascular dermatitis) (Table 6).
In total, eight participants (4.9%) in the pegfilgrastim-cbqv OBI group and four participants (2.5%) in the pegfilgrastim-cbqv PFS group withdrew from the study because of an AE. Only one participant (0.6%) in each group experienced a study drug-related TEAE that led to withdrawal from the study (Table 6).
The most common study drug-related TEAEs for pegfilgrastim-cbqv OBI and pegfilgrastim-cbqv PFS were musculoskeletal and connective tissue disorders (61.6% vs. 56.5%), nervous system disorders (29.9% vs. 23.0%), and general disorders and administration site conditions (6.1% vs. 4.3%). TEAEs for pegfilgrastim-cbqv OBI and pegfilgrastim-cbqv PFS were mainly those attributed to the expected musculoskeletal effects of G-CSF-related therapeutics, including myalgia (35.4% vs. 36.6%), back pain (12.8% vs. 9.3%), and spinal pain (5.5% vs. 6.8%) (Table 7).
Overall, 55 participants (33.5%) experienced TEAEs related to the OBI. The most common TEAE related to the OBI was injection site erythema (52 participants [31.7%]). The injection site erythema was considered mild and self-limiting and was deemed to be due to the adhesive of the OBI.
There were no clinically meaningful changes in the median values for the chemistry or hematology laboratory tests from baseline or any notifiable differences in the safety laboratory results for chemistry, hematology, and urinalysis parameters between treatment groups. Overall, there were no new or unexpected safety findings.
Tolerability
The pegfilgrastim-cbqv OBI performed as expected; there were no concerns with adhesion during the study. On the basis of the dermal response scale, most participants who received pegfilgrastim-cbqv OBI had minimal erythema that was barely perceptible when the OBI was removed. Five participants (3.1%) had definite erythema that was readily visible and minimal edema or minimal papular response with removal of the OBI. One participant (0.6%) had erythema and papules 48, 72, and 96 h after dosing.
Most participants who received pegfilgrastim-cbqv OBI reported having no pain at initial adhesion (169 participants [99.4%]), needle insertion (113 participants [68.9%]), or removal (117 participants [72.2%]) of OBI (i.e., rated 0 on the Wong-Baker FACES pain rating scale) [22]. Three participants (1.8%) reported having pain with needle insertion and two participants (1.2%) reported having pain during OBI removal (i.e., rated 6 on the Wong-Baker FACES pain rating scale) [22].
Immunogenicity
At baseline, 18 participants were positive for ADAs. The incidence of treatment-emergent ADAs in period 1 was similar for the OBI (40.0%) and the PFS (36.6%). ADA titers were low and primarily directed to the PEG moiety of pegfilgrastim-cbqv in both treatment groups. The overall incidence of ADAs was similar in both treatment sequences (Table 8). NAbs were not detected in any participant at any time.
Impact of Immunogenicity on PK and PD
To assess the impact of ADAs on the PK of pegfilgrastim-cbqv OBI and PFS, the GMs of Cmax and AUC values were compared in ADA-positive and ADA-negative participants. On the basis of treatment, the GMs of Cmax and AUC values were similar in ADA-positive and ADA-negative participants (Supplementary Material Table S3). No impact of immunogenicity on PK was observed.
To assess the impact of ADAs on the PD of pegfilgrastim-cbqv OBI and pegfilgrastim-cbqv PFS, the GMs of ANCmax and ANC AUC0–last values were compared in ADA-positive and ADA-negative participants on the basis of treatment with the pegfilgrastim-cbqv OBI or the PFS. The GM values were similar (Supplementary Material Table S4). No impact of immunogenicity on PD was observed.
Impact of Immunogenicity on Safety
In both treatment groups, TEAEs were comparable for the ADA-positive and ADA-negative participants (Supplementary Material Table S5). No impact of immunogenicity on safety was observed.
Discussion
This study was conducted to evaluate the bioequivalence of pegfilgrastim-cbqv administered via an OBI compared with a PFS in healthy adult male participants. Because the 90% CIs of the GMRs for the PK primary end points were fully contained within the prespecified range, PK bioequivalence was demonstrated between the pegfilgrastim-cbqv OBI and PFS. Results of the additional sensitivity analysis emphasized the robustness of the PK results. PD bioequivalence was also established. The GMRs for the PK and PD parameters assessed in the current study are comparable to those found in a prior study for which the results showed PK and PD bioequivalence of pegfilgrastim-cbqv delivered via a PFS and pegfilgrastim (PFS) in healthy participants [18].
The mean plasma levels of pegfilgrastim-cbqv for the OBI and PFS treatment groups were superimposable at all time points, with both treatment groups showing a rapid increase followed by a steady decrease in the levels. The mean ANC levels for the pegfilgrastim-cbqv OBI and PFS groups were also very similar. After administration of pegfilgrastim-cbqv, the mean ANC levels decreased, followed by a return to baseline levels 2 h after administration. This fluctuation can be explained by the mechanism of action of pegfilgrastim-cbqv, which causes margination of peripheral neutrophils, followed by subsequent demargination of peripheral neutrophils and increased release of mature neutrophils from the bone marrow [23].
The majority of TEAEs with pegfilgrastim-cbqv OBI and PFS were mild and mainly attributed to the expected musculoskeletal effects of GSF-related therapeutics. The most common TEAE related to the OBI was injection site erythema, which was mild and caused by the OBI adhesive. Most participants who received pegfilgrastim-cbqv using the OBI had minimal erythema and reported no pain on adhesion, needle insertion, or removal of the OBI. Titers of ADA-positive participants were generally low and against the PEG portion of pegfilgrastim-cbqv. Overall, the safety profile of the pegfilgrastim-cbqv OBI (including immunogenicity) was similar and consistent with the known safety profile of the pegfilgrastim-cbqv PFS [12].
Failure rates of the OBI device are a key metric because a missed or partial dose due to device failure may considerably increase the risk of FN [5,6,7]. During the current study, four participants (2.1%) in the OBI group withdrew from the trial because of OBI malfunction. Results of previous studies of patients using an OBI with pegfilgrastim (the pegfilgrastim OnPro) showed that the device was subject to failure at a rate of 1–6.9% [24, 25], meaning that the failure rate of the pegfilgrastim-cbqv OBI in the current study was within the known range and lower than rates reported in studies that used a different device [24, 25].
Each method of administration has advantages and disadvantages, with specific benefits that appeal to different patients. The availability of more than one type of administration device allows the selection of the option that best fits user needs and preferences [16, 26,27,28]. The benefits of using an OBI to administer pegfilgrastim-cbqv are twofold: (1) patients who are unable to self-inject using a PFS or autoinjector have an alternative method for which no intervention from the patient is necessary and (2) the need for a second health care visit to receive the recommended next-day administration of pegfilgrastim-cbqv is reduced. Additionally, use of a pegfilgrastim-cbqv OBI could improve treatment adherence rates when used in patient groups who prefer not to self-inject [16, 28]. In a previous study of G-CSF prophylaxis, administration using an OBI compared with a PFS was associated with improved treatment adherence (94.0% [95% CI 92.9–95.2] versus 58.4% [95% CI 55.2–61.5]) [29]. The results of one study reported adherence to the prescribed regimen as 97.6% and 63.1% for patients administered treatment with the OBI and PFS, respectively [28].
A possible limitation of the current study could be that only healthy male participants were included. However, the FDA recognizes that healthy participants are the most sensitive population when aiming to detect differences in PK and PD, which was the objective of the study [30]. Furthermore, previous studies have shown that the PK and PD of pegfilgrastim do not vary between men and women [12, 13, 18].
Conclusions
The results of this study show that the pegfilgrastim-cbqv OBI can be used as an alternative to the PFS. The PK, PD, and safety (including immunogenicity) profiles with administration of pegfilgrastim-cbqv using an OBI are similar to those of administration using a PFS. The OBI had minimal device-specific adverse effects. Use of the pegfilgrastim-cbqv OBI may minimize the number of patient visits to the clinic and improve patient adherence.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Klastersky J, de Naurois J, Rolston K, et al. Management of febrile neutropaenia: ESMO clinical practice guidelines. Ann Oncol. 2016;27(suppl 5):v111–8.
Freifeld AG, Bow EJ, Sepkowitz KA, et al. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2011;52(4):e56-93.
Tai E, Guy GP, Dunbar A, Richardson LC. Cost of cancer-related neutropenia or fever hospitalizations, United States, 2012. J Oncol Pract. 2017;13(6):e552–61.
Kuderer NM, Dale DC, Crawford J, Lyman GH. Impact of primary prophylaxis with granulocyte colony-stimulating factor on febrile neutropenia and mortality in adult cancer patients receiving chemotherapy: a systematic review. J Clin Oncol. 2007;25(21):3158–67.
Lyman GH. Impact of chemotherapy dose intensity on cancer patient outcomes. J Natl Compr Canc Netw. 2009;7(1):99–108.
Denduluri N, Lyman GH, Wang Y, et al. Chemotherapy dose intensity and overall survival among patients with advanced breast or ovarian cancer. Clin Breast Cancer. 2018;18(5):380–6.
Denduluri N, Patt DA, Wang Y, et al. Dose delays, dose reductions, and relative dose intensity in patients with cancer who received adjuvant or neoadjuvant chemotherapy in community oncology practices. J Natl Compr Canc Netw. 2015;13(11):1383–93.
Weycker D, Li X, Edelsberg J, et al. Risk and consequences of chemotherapy-induced febrile neutropenia in patients with metastatic solid tumors. J Oncol Pract. 2015;11(1):47–54.
Aapro MS, Bohlius J, Cameron DA, et al. 2010 update of EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphoproliferative disorders and solid tumours. Eur J Cancer. 2011;47(1):8–322.
Cooper KL, Madan J, Whyte S, Stevenson MD, Akehurst RL. Granulocyte colony-stimulating factors for febrile neutropenia prophylaxis following chemotherapy: systematic review and meta-analysis. BMC Cancer. 2011;11:404.
Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Hematopoetic growth factors. Version 2.2023. © National Comprehensive Cancer Network, Inc. 2023. All rights reserved. Accessed 24 April 2023. To view the most recent and complete version of the guideline go to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way. NCCN, National Comprehensive Cancer Network® (NCCN)
Udenyca (pegfilgrastim-cbqv) injection, for subcutaneous use. Prescribing information. Coherus Biosciences Inc.; 2023.
Neulasta (pegfilgrastim) injection single-dose prefilled syringe. Prescribing information. Amgen, Inc.; 2021.
Yang BB, Morrow PK, Wu X, Moxness M, Padhi D. Comparison of pharmacokinetics and safety of pegfilgrastim administered by two delivery methods: on-body injector and manual injection with a prefilled syringe. Cancer Chemother Pharmacol. 2015;75(6):1199–206.
Arvedson T, O’Kelly J, Yang BB. Design rationale and development approach for pegfilgrastim as a long-acting granulocyte colony-stimulating factor. BioDrugs. 2015;29(3):185–98.
Humphreys SZ, Geller RB, Walden P. Pegfilgrastim biosimilars in US supportive oncology: a narrative review of administration options and economic considerations to maximize patient benefit. Oncol Ther. 2022;10(2):351–61.
Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 2018;7(4):3.
Finck B, Tang H, Civoli F, Hodge J, O’Kelly H, Vexler V. Pharmacokinetic and pharmacodynamic equivalence of pegfilgrastim-cbqv and pegfilgrastim in healthy subjects. Adv Ther. 2020;37(10):4291–307.
Civoli F, Finck B, Tang H, Hodge J, O’Kelly H, Vexler V. Biosimilar pegfilgrastim-cbqv demonstrated similar immunogenicity to pegfilgrastim in healthy subjects across three randomized clinical studies. Adv Ther. 2022;39(3):1230–46.
Civoli F, Kasinath A, Cai XY, et al. Recommendations for the development and validation of immunogenicity assays in support of biosimilar programs. AAPS J. 2019;22(1):7.
US Food and Drug Administration. Assessing the irritation and sensitization potential of transdermal and topical delivery systems for ANDAs. Draft guidance for industry. 2023. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/assessing-irritation-and-sensitization-potential-transdermal-and-topical-delivery-systems-andas. Accessed May 23, 2023.
Stuppy DJ. The faces pain scale: reliability and validity with mature adults. Appl Nurs Res. 1998;11(2):84–9.
Yang BB, Kido A. Pharmacokinetics and pharmacodynamics of pegfilgrastim. Clin Pharmacokinet. 2011;50(5):295–306.
Stuessy P, Sanchez FA, Schober M. Retrospective review of pegfilgrastim on-body injector delivery rates in a large health system. J Clin Oncol. 2017;35(15_suppl):e18273.
Townley C, Porter C, McMullen N. Comparing grade 4 neutropenia associated with pegfilgrastim administered via the Onpro device versus manual injection with a prefilled syringe. JHOP. 2018;8(3):119–25.
Schwarzenbach F, Dao Trong M, Grange L, et al. Results of a human factors experiment of the usability and patient acceptance of a new autoinjector in patients with rheumatoid arthritis. Patient Pref Adher. 2014;8:199–209.
Vermeire S, D’Heygere F, Nakad A, et al. Preference for a prefilled syringe or an auto-injection device for delivering golimumab in patients with moderate-to-severe ulcerative colitis: a randomized crossover study. Patient Prefer Adher. 2018;12:1193–202.
Metz M, Semsek D, Rogmans G, et al. Patient, nurse and physician preferences: final results of the CONVENIENCE study evaluating pegfilgrastim prophylaxis via pre-filled syringe or On-body injector (OBI) in cancer patients. Ann Oncol. 2020;31(suppl 4):S1071.
Rifkin RM, Crawford J, Mahtani RL, et al. A prospective study to evaluate febrile neutropenia incidence in patients receiving pegfilgrastim on-body injector vs other choices. Support Care Cancer. 2022;30(10):7913–22.
US Department for Health and Human Services. Guidance for industry. Clinical pharmacology data to support a demonstration of biosimilarity to a reference product. Updated May 5, 2020. Silverspring; 2016. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-pharmacology-data-support-demonstration-biosimilarity-reference-product. Accessed August 10, 2023.
Acknowledgements
Medical Writing Assistance.
Medical writing and editorial assistance were provided by Chantell Hayward, PharmD; Cindi A. Hoover, PhD; and Chantel Kowalchuk, PhD (ApotheCom, San Francisco, CA), and funded by Coherus BioSciences (Redwood City, CA).
Authorship.
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Funding
Funding for this study was provided by Coherus BioSciences (Redwood City, CA). All authors had full access to all of the data in the study and take complete responsibility for the integrity of the data and accuracy of the data analysis. Journal Rapid Service and Open Access Fees were also funded by Coherus BioSciences (Redwood City, CA).
Author information
Authors and Affiliations
Contributions
Hong Tang, Francesca Civoli, Suzanna Tatarewicz contributed to the curation, formal analysis, and validation of the data. Barbara Finck assisted with the formal analysis of the data. Francesca Civoli and Suzanna Tatarewicz contributed to project administration. Hong Tang conceptualized the study. Hong Tang, Francesca Civoli, Suzanna Tatarewicz, Nathalie Vandenkoornhuyse, and Barbara Finck contributed to the study design of the manuscript and reviewed and approved the final content of this manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Hong Tang, Suzanna Tatarewicz, Nathalie Vandenkoornhuyse, and Barbara Finck are employees of and own stock in Coherus BioSciences (Redwood City, CA). Francesca Civoli is a former employee and owns stock in Coherus BioSciences (Redwood City, CA); she is currently affiliated with Francesca Civoli Consulting LLC.
Ethical Approval
The protocol and informed consent form were reviewed and approved by the Advarra institutional review board (IRB registration number IRB0000097). The study was conducted in accordance with all applicable laws and regulations and complied with the International Conference for Harmonisation E6 Guideline on Good Clinical Practice. All procedures were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Additional information
Francesca Civoli was affiliated with Coherus BioSciences at the time of the study.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Tang, H., Civoli, F., Tatarewicz, S. et al. A Randomized, Open-Label Study Conducted to Evaluate the Bioequivalence of Pegfilgrastim-cbqv On-Body Injector Versus Prefilled Syringe in Healthy Male Participants. Adv Ther 41, 991–1009 (2024). https://doi.org/10.1007/s12325-023-02735-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-023-02735-3