
Abstract
Introduction
This study evaluated the bioequivalence of ezetimibe/rosuvastatin fixed dose combination compared to the concomitant administration of individual formulations (ezetimibe and rosuvastatin) in Chinese healthy subjects under fasting conditions.
Methods
This was a phase I, randomized, open-label, two-treatment, two-period, two-sequence, crossover study conducted in healthy Chinese participants under fasting conditions. Cmax, AUC0–t, and AUC0–∞ from test and individual reference formulations were evaluated to assess bioequivalence. The safety assessments included adverse events (AEs)/treatment-emergent adverse events (TEAEs), potential clinically significant abnormalities (PCSAs) in vital signs, 12-lead electrocardiogram (12-ECG), and clinical laboratory parameters.
Results
Of the 68 subjects enrolled, 67 were treated. Systemic exposure to rosuvastatin based on Cmax, AUC0–t, and AUC0–∞ was similar in both treatments, with respective arithmetic values 12.4 ng/ml, 117 ng·h/mL, and 120 ng·h/mL for test formulation and 12.7 ng/ml, 120 ng·h/mL, and 123 ng·h/mL for reference formulations. Similarly, systemic exposure to unconjugated ezetimibe was 4.14 ng/ml, 89.7 ng·h/mL, and 102 ng·h/mL for the test formulation and 3.80 ng/ml, 89.7 ng·h/mL, and 102 ng·h/mL for reference formulations. Systemic exposure to total ezetimibe was 70.5 ng/ml, 664 ng·h/mL, and 718 ng·h/mL for test formulation and 60.2 ng/ml, 648 ng·h/mL, and 702 ng·h/mL for reference formulations. The point estimates for rosuvastatin unconjugated ezetimibe and total ezetimibe were in the acceptable range of 0.80–1.25. No deaths or serious adverse events were reported.
Conclusions
Fixed dose combination of ezetimibe/rosuvastatin (10 mg/10 mg) achieved bioequivalence with reference to commercial tablets.
Trial Registration Number
CTR20202108.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Hypercholesterolemia is a potential risk factor for cardiovascular disease (CVD). Rosuvastatin and ezetimibe are well-known lipid-lowering agents. |
This study assessed the pharmacokinetic parameters and safety of the fixed dose combination (FDC) of rosuvastatin and ezetimibe compared with their individual formulations in Chinese healthy subjects. |
What was learned from the study? |
The point estimates of formulation ratios with 90% CIs for rosuvastatin, unconjugated ezetimibe, and total ezetimibe were all within 0.80–1.25 and thus confirmed the bioequivalence of the FDC to the individual formulations. |
Overall, the FDC of rosuvastatin/ezetimibe was well tolerated without raising any safety concerns. |
Introduction
Hypercholesterolemia refers to high levels of cholesterol, triglyceride, or both and is a potential risk factor for cardiovascular disease (CVD) [1]. The World Health Organization (WHO) reported the prevalence of dyslipidemia (defined as blood levels of total cholesterol > 5 mmol/L [190 mg/dL]) in Southeast Asia and the Western Pacific to be 30.3% and 36.7%, respectively, in 2008 [2]. It is estimated that with an increase in age and prevalence of CVD in China, there will be a rise in the incidence of acute myocardial infarctions by 75 million, stroke by 118 million, and the number of cardiovascular (CV) deaths would rise by 39 million in total between 2016 to 2030 [3]. Serum cholesterol and other lipoproteins such as low-density lipoprotein (LDL), very low-density lipoprotein (VLDL), and high-density lipoprotein (HDL) are known to be related to atherosclerotic cardiovascular disease (ASCVD) [4]. LDL-cholesterol (LDL-C), when present in high amounts, leads to atherosclerosis and hence is the main target for lowering the cholesterol level [5].
Among the cholesterol-lowering drugs, statins are widely used in lowering the LDL-C level. Rosuvastatin belongs to a class of lipid-lowering compounds which reduces the cholesterol synthesis by inhibiting the rate-limiting enzyme 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA), which represents the therapeutic target for statins by resulting in reduction of VLDL synthesis that leads to reduced delipidation of VLDL to LDL [6]. In addition, statins leads to upregulation of LDL receptors leading to an increase in clearance of both LDL and its precursors, consequently reducing LDL-C [7]. Rosuvastatin has the highest binding interactions with HMG-CoA reductase, as compared to the other statins which leads to the most powerful inhibition of cholesterol synthesis [8]. However, as a result of statin intolerance or statin resistance, many patients do not reach their target LDL-C levels. Hence, other lipid-lowering agents such as ezetimibe, fibrates, and nicotinic acid may be preferred as an add-on therapy.
Ezetimibe is a first-in class cholesterol absorption inhibitor that targets Niemann-Pick C1-Like 1 (NPC1L1) protein, which is responsible for intestinal absorption of cholesterol [9]. In the MRS-ROZE study, ezetimibe, as a monotherapy or when combined with rosuvastatin, significantly reduced total cholesterol, LDL-C, apolipoprotein B, triglycerides, and increased HDL cholesterol in patients with hypercholesterolemia [10]. A 12-week, randomized, double-blind study assessing the efficacy of the fixed dose combination (FDC) of rosuvastatin/ezetimibe in 337 Korean patients with high CV risk demonstrated that the patients on FDC achieved a higher LDL-C target of 87–95% compared to those in the monotherapy group (64–87%) [11]. Thus, FDCs provide enhanced efficacy and safety over monotherapy alone. In addition, FDCs may offer additional advantages over monotherapy such as reduced treatment cost and improved patient adherence [12]. As a result of the pill burden, the adherence to the hypercholesterolemia treatment is low; however, use of an FDC leads to better patient compliance and reduces the pill burden [13]. The effectiveness and safety of rosuvastatin/ezetimibe as FDC have been demonstrated in various studies [14]. However, the bioequivalence between the FDC and simultaneous intake of single drugs in the Chinese population under fasting conditions is unknown. This present study was therefore conducted to assess the bioequivalence between the FDC of rosuvastatin/ezetimibe (10 mg/10 mg) and the individual tablets in healthy Chinese subjects to support the substitution of rosuvastatin and ezetimibe FDC in adult patients who are adequately controlled with rosuvastatin and ezetimibe monotherapies.
Methods
Study Design
This was a phase I, randomized, open-label, two-treatment, two-period, two-sequence, crossover study conducted at Peking University (PKU) Care, Luzhong Hospital, China from 8 November 2020 to 7 December 2020 in healthy Chinese participants (CTR20202108). The investigational FDC was a test formulation [ezetimibe/rosuvastatin which contained 10 mg of both the drugs (10 mg/10 mg)] and was compared with the reference formulations [individual rosuvastatin (Crestor®, 10 mg) and ezetimibe (Ezetrol®, 10 mg]. This study was conducted in accordance with the ethical principles derived from international ethics guidelines, including the Declaration of Helsinki, and the International Council for Harmonization (ICH) guidelines for Good Clinical Practice (GCP), all applicable laws, rules, and regulations. The study received approval from the institutional ethics committee of Peking University Care, Luzhong Hospital (PKULZH-IRB-SOP-AF-013/3.0-03). Informed written consent was obtained.
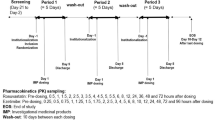
The subjects were randomized to either of the two-treatment sequences. Sequence 1 consisted of FDC administration followed by the individual formulations (test–reference), respectively in period 1 and 2. Whereas, sequence 2 consisted of administration of individual formulations followed by the FDC (reference–test), respectively, in period 1 and 2. The subjects were randomized to either sequence 1 or sequence 2 in a 1:1 ratio. The tablets were administered orally to the healthy subjects under fasting conditions. The treatment period consisted of 5 days including one treatment day in each period, followed by a washout period of 10 days between each administration.
Subjects
Healthy Chinese male and female subjects of age 18 and above, body weight between 50.0 and 95.0 kg (kg) for male (inclusive), 45.0 and 90.0 kg for female (inclusive), were enrolled in this study. All the subjects were certified as healthy by a comprehensive medical assessment which included a detailed medical history and complete physical examination. Female participants were required to use at least one contraception method for 3 months after the dosing, except if the subject was menopausal or had undergone sterilization at least 3 months earlier. The subjects were excluded if they had any history or presence of any acute illness, disorder, or any drug abuse. Breastfeeding or pregnant subjects were excluded from the study. Informed written consent was obtained at the time of study enrolment.
Study Endpoints
The study aimed to evaluate the Cmax, AUC0–t, and AUC0–∞ of rosuvastatin, unconjugated and total ezetimibe from FDC and individual formulations (treatment 1 vs treatment 2) under fasting conditions. The secondary endpoints for this study were to evaluate the other pharmacokinetic (PK) parameters including t1/2, Tmax, and λz. The safety assessments included adverse events (AEs)/treatment-emergent adverse events (TEAEs), vital signs, 12-lead electrocardiogram (12-ECG), and clinical laboratory evaluations (hematology, biochemistry, urinalysis, coagulation).
PK Parameters Evaluation
Blood samples were collected at the following time points: 0 h (pre-dosing) and 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 8, 12, 24, 36, 48, and 72 h (post-dosing) for rosuvastatin and 0 h (pre-dosing) and 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 24, 48, 72, and 96 h (post-dosing) for ezetimibe. The PK parameters such as Cmax, AUC0–t, AUC0–∞, t1/2, Tmax, and λz were calculated using the noncompartmental methods from plasma rosuvastatin, unconjugated ezetimibe, and total ezetimibe concentrations obtained after single dose administration. Total ezetimibe was calculated from the sum of free ezetimibe and ezetimibe glucuronide, taking into consideration the adjustment per molecular weight for each analyte respectively.
Bioanalytical Methods
Bioanalytical methods were performed in the laboratory of Covance Pharmaceutical Research and Development (Shanghai). Liquid chromatography with tandem mass spectrometry (LC–MS/MS) was used for analysis. PK samples were used for testing analytical method performance such as comparability and incurred sample reproducibility.
Safety Evaluation
All the subjects were monitored for laboratory parameters, vital signs, ECGs, and AEs. AEs were coded according to the Medical Dictionary for Regulatory Activities (MedDRA, version 23.1). Their severity was graded according to the Common Terminology Criteria for Adverse Events (CTCAE, version 5.0). The number (%) of participants experiencing TEAEs was summarized by primary system organ class, preferred term, and treatment. For laboratory parameters, vital signs, and ECGs, incidences of potentially clinically significant abnormality (PCSA) were evaluated. The safety evaluation focused on the TEAE period, defined as the time interval from the investigational medicinal product (IMP) administration of each treatment period up to day 5 (inclusive).
Sample Size Calculation
The sample size was calculated on the basis of within-subject standard deviation (SDw) of 0.27 for rosuvastatin and unconjugated ezetimibe, and SDw of 0.22 for total ezetimibe, which was estimated from pooled SDw values of recent studies. The assumption of true difference between E10/R10 and coadministration of individual tablets on rosuvastatin and total ezetimibe is 5%, and the true difference on unconjugated ezetimibe is 7.5%, which was based on a previous Sanofi in-house bioequivalence study, ZNV-P5-545. A total of 62 subjects were required to achieve an overall power of 85% to conclude the bioequivalence of FDC to the co-administered individual tablets. But considering the potential subject dropout rate, 68 subjects were enrolled.
Statistical Analysis
PK parameters of rosuvastatin, unconjugated and total ezetimibe were summarized using descriptive statistics (such as mean, geometric mean, median, standard deviation [SD], standard error of mean [SEM], coefficient of variation [CV], minimum, and maximum) for each treatment. Listings of individual ratios (FDC versus co-administration treatment) for Cmax, AUC0–t, and area under the plasma concentration versus time curve extrapolated to infinity (AUC0–∞) were provided by subject, sequence, and summarized using descriptive statistics by treatment. The difference between FDCs and individual formulations under fasting conditions was assessed on log-transformed parameter with a linear mixed effects model with fixed term for treatment, sequence, period, and with an unstructured matrix of treatment-specific variances and covariances for subject within sequence blocks, using SAS® version 9.4.
For Cmax, AUC0–t, and AUC0–∞ estimates and 90% confidence intervals (CI) for geometric mean ratio of treatments (test versus individual reference formulations) were obtained by computing estimates and 90% CIs for the difference between treatment means within the mixed effects model framework, and then converting to the ratio scale by the antilog transformation. If the 90% CI of the ratio for Cmax, AUC0–t, and AUC0–∞ of rosuvastatin, unconjugated ezetimibe, and total ezetimibe all were within the range of 0.8–1.25, the bioequivalence of test formulation to co-administration of reference formulations was established. Histograms of Tmax and t1/2 values were represented by formulation. In addition, the histograms of differences in Tmax between formulations (test versus individual reference formulations) were also provided.
Results
Subject Demographics
A total of 67 healthy Chinese subjects were treated in this study. Among those 71.6% (n = 48) were male and 28.4% (n = 19) were female. The age range for the study population was 18–52 years, while the mean (SD) age was 33.2 (9.7) years. The mean body mass index (BMI) was 23.93 kg/m2. One subject discontinued the study treatment on day 3 of period 1 following the investigator’s decision because of AE (urticaria). All treated participants were evaluable for PK and safety analysis.
PK Parameters Evaluation
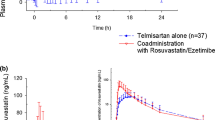
Figures 1, 2, and 3 represent the mean plasma concentrations of rosuvastatin, unconjugated ezetimibe, and total ezetimibe following single oral dose administration of the test formulation and the reference formulations in healthy Chinese subjects.

a, b Plot of mean concentrations of rosuvastatin for test (combination tablet) and reference (separate tablets) treatments

a, b Plot of mean concentrations of unconjugated ezetimibe for test (combination tablet) and reference (separate tablets) treatments

a, b Plot of mean concentrations of total ezetimibe for test (combination tablet) and reference (separate tablets) treatments
The PK parameters of rosuvastatin, unconjugated ezetimibe, and total ezetimibe following administration of test formulation and reference formulations are presented in Tables 1, 2, and 3, respectively.
PK of Rosuvastatin
The systemic exposure to rosuvastatin based on Cmax, Tmax, AUC0–t, AUC0–∞, and t1/2 was similar in both the treatments, with respective arithmetic values of 12.4 ng/ml, 4.5 h, 117 ng·h/mL, 120 ng·h/mL, and 17 h for the FDC and 12.7 ng/ml, 4.5 h, 120 ng·h/mL, 123 ng·h/mL, and 17.8 h for the individual formulations.
PK of Unconjugated Ezetimibe
The systemic exposure to unconjugated ezetimibe based on Cmax, Tmax, AUC0–t, AUC0–∞, and t1/2 was similar in both the treatments, with respective arithmetic values of 4.14 ng/mL, 1.5 h, 89.7 ng·h/mL, 102 ng·h/mL, and 25 h for the FDC and 3.80 ng/ml, 2 h, 89.7 ng·h/mL, 102 ng·h/mL, and 24.9 h for individual reference formulations.
PK of Total Ezetimibe
The systemic exposure to total ezetimibe based on Cmax, Tmax, AUC0–t, AUC0–∞, and t1/2 was similar in both the treatments, with respective arithmetic values of 70.5 ng/mL, 0.75 h, 664 ng·h/mL, 718 ng·h/mL, and 22.1 h for the FDC and 60.2 ng/ml, 1 h, 648 ng·h/mL, 702 ng·h/mL, and 24.1 h for individual reference formulations.
Bioequivalence Results
The point estimates for rosuvastatin for Cmax, AUC0–t, and AUC0–∞ were 0.98, 0.98, and 0.98, respectively. The point estimates for unconjugated ezetimibe for Cmax, AUC0–t, and AUC0–∞ were 1.07, 1.02, and 1.04, respectively. The point estimates for total ezetimibe for Cmax, AUC0–t, and AUC0–∞ were 1.17, 1.02, and 1.02, respectively. The 90% CIs for the geometric mean ratios of the primary PK parameters (Cmax, AUC0–t, and AUC0–∞) were all within the predefined equivalence range of 0.80–1.25.
Tables 4, 5, and 6 present the point estimates of formulation ratios with 90% CIs for rosuvastatin, unconjugated ezetimibe, and total ezetimibe.
Safety Results
All 67 subjects were administered the test formulation and reference individual formulations. One subject receiving test formulation withdrew from the study as a result of AE (urticaria). There were no deaths, serious adverse events (SAE), or adverse events of special interest (AESI) reported in this study. A total of 15 subjects reported at least one treatment TEAE in the study (9 out of 66 subjects in the individual reference treatment group and 6 out of 67 subjects in the test formulation treatment group). One subject in the test formulation treatment group reported one TEAE leading to permanent treatment discontinuation. Most of the TEAE were of grade 1 or 2. Only one subject reported grade 3 (blood triglycerides increased). There were no AESIs or serious TEAEs reported during the study (Table 7). All TEAEs were resolved by the end of the study without any sequelae. There were four PCSAs observed in laboratory tests or 12-lead ECG parameters. Only one PCSA in laboratory test (blood triglycerides increased) detected on the ambulatory visit on day 5 of period 2 was reported as a TEAE.
Discussion
This study assessed the PK parameters and safety of the FDC of rosuvastatin and ezetimibe compared with their individual formulations in Chinese healthy subjects. The point estimates of formulation ratios with 90% CIs for rosuvastatin, unconjugated ezetimibe, and total ezetimibe were all within 0.80–1.25 and thus confirmed the bioequivalence of the FDC to the individual formulations. The mean concentration–time profile was also similar for rosuvastatin, unconjugated ezetimibe, and total ezetimibe.
Previously published studies on rosuvastatin/ezetimibe FDC have established the benefit of FDC over the individual formulations. A phase III study, I-ROSETTE (NCT02749994), stated that an FDC significantly improved the lipid profiles when compared to rosuvastatin monotherapy (92.3% vs 79.9%), with a mean decrease of at least 50% in the LDL-C levels [15]. A 6-week ACTE study also stated that a significant reduction in LDL-C level was observed when ezetimibe was added to rosuvastatin [16]. In another study where patients with hypercholesterolemia were randomized to receive rosuvastatin 10 mg plus ezetimibe 10 mg, rosuvastatin 10 mg plus placebo, ezetimibe 10 mg plus placebo, or two placebo tablets, greater reductions in LDL-C levels were achieved with co-administration of rosuvastatin and ezetimibe than placebo or either monotherapy [17]. Similarly, in a study by Kim et al., a higher proportion of patients receiving the combination of statin with ezetimibe achieved LDL-C concentrations of less than 70 mg/dL and lower intolerance-related drug discontinuation or dose reduction than those receiving high-intensity statin monotherapy [18]. Based on the 2019 European Society of Cardiology/European Atherosclerosis Society (ESC/EAS) dyslipidemia guidelines, a simulation model with a 5-year horizon was developed and showed that treatment with statin or statin plus ezetimibe FDC compared with statin and ezetimibe as multiple pills can result in better LDL-C control and population-level cardiovascular events averted [19].
Co-administration of rosuvastatin and ezetimibe does not appear to produce any clinically significant PK interactions in healthy adults [20]. An open-label, single-dose, crossover study with rosuvastatin/ezetimibe FDC and individual drugs reported a geometric mean ratio and 90% CI for the rosuvastatin Cmax and AUC0–t of 106.20 (96.62–116.74) and 102.88 (96.32–109.90), respectively, and for ezetimibe Cmax and AUC0–t were 108.96 (98.56–120.51) and 98.13 (92.01–104.66), respectively. The mean Cmax and AUC0–t values of rosuvastatin were 12.5 ng/mL and 115.6 ng.h/mL for the FDC, and 12.2 ng/mL and 115.1 ng·h/mL for the individual drugs, respectively. All treatments were well tolerated during this study, with no SAEs reported [21]. A previous Sanofi in-house bioequivalence study (ZNV-P5-545) compared an FDC (10 mg/40 mg of ezetimibe and rosuvastatin) with individual formulations (10 mg of ezetimibe and 40 mg of rosuvastatin). The results showed that the geometric mean ratio of Cmax and AUC0–t of rosuvastatin (FDC vs coadministration of individual formulations) and that of Cmax and AUC0–72 of unconjugated ezetimibe were within the standard acceptance range of 0.80–1.25 [22]. Similar results were observed in another study conducted on healthy Korean subjects [23].
The PK parameters of ezetimibe in our study are similar to those in a previous study in Korean patients where it was found that for the total ezetimibe Tmax was 1 h and t1/2 was 17.3, thus supporting that the exposure of ezetimibe is similar in the two Asian populations [20]. Similarly, the PK parameters of rosuvastatin 10 mg reported in this study are comparable to those in a previous study in various ethnic populations, which found that the ratios for rosuvastatin AUC0–t were 2.31, 1.91, and 1.63 and the ratios of maximum plasma concentration were 2.36, 2.00, and 1.68 in Chinese, Malay, and Asian–Indian subjects, respectively, compared with White subjects, providing evidence that exposure to rosuvastatin is higher in Asians compared with Caucasians [24].
There were no AESIs or serious TEAEs reported during the study. Therefore, the safety of the FDC was comparable to the co-administration of individual rosuvastatin and ezetimibe drugs in healthy patients. Overall, the FDC of rosuvastatin/ezetimibe was well tolerated without raising any safety concerns. There are few limitations that warranted mention. First, we enrolled healthy subjects in the study as this decreases the potential for concomitant medications and the presence of underlying disease, which may introduce study bias. Nevertheless, in real-world clinical practice, the PK might be different in other targeted populations, especially in elderly patients, or in various dosage regimens. Second, this study analyzed the bioequivalence under fasting conditions only. The effect of food on the PK of rosuvastatin and ezetimibe as FDC has not been studied. However, the effect of food on individual drugs is available. Administration of rosuvastatin with food did not affect the AUC of rosuvastatin and, hence, it can be given with or without food. Concomitant food administration (high fat or non-fat meals) had no effect on the oral bioavailability of ezetimibe 10 mg tablets. Ezetimibe can be administered with or without food [25]. Third, the combination of proposed doses (10 mg of ezetimibe and 10 mg of rosuvastatin) was considered because of the known differences in PK profiles between Asian and Caucasian, resulting in prescription of lower dose of statin in Asian population [26, 27].
Conclusion
The combination tablet containing 10 mg of ezetimibe and 10 mg of rosuvastatin was bioequivalent to the simultaneous administration of the separate commercial tablets in healthy Chinese subjects under fasting conditions. Ezetimibe and rosuvastatin, administered either as a combination tablet or as separate tablets, were safe and well tolerated in Chinese subjects.
References
Nelson RH. Hyperlipidemia as a risk factor for cardiovascular disease. Prim Care. 2013;40:195–211.
Lin C-F, Chang Y-H, Chien S-C, Lin Y-H, Yeh H-Y. Epidemiology of dyslipidemia in the Asia Pacific region. Int J Gerontol. 2018;12:2–6.
Stevens W, Peneva D, Li JZ, et al. Estimating the future burden of cardiovascular disease and the value of lipid and blood pressure control therapies in China. BMC Health Serv Res. 2016;16:175.
Borén J, Chapman MJ, Krauss RM, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41:2313–30.
Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139:e1082–143.
Grigore L, Norata GD, Catapano AL. Combination therapy in cholesterol reduction: focus on ezetimibe and statins. Vasc Health Risk Manag. 2008;4:267–78.
Young SG, Fong LG. Lowering plasma cholesterol by raising LDL receptors—revisited. N Engl J Med. 2012;366:1154–5.
Rosenson RS. Rosuvastatin: a new inhibitor of HMG-CoA reductase for the treatment of dyslipidemia: expert review of cardiovascular therapy. 2003;1(4):495–505. https://doi.org/10.1586/14779072.1.4.495.
Phan BAP, Dayspring TD, Toth PP. Ezetimibe therapy: mechanism of action and clinical update. Vasc Health Risk Manag. 2012;8:415–27.
Kim K-J, Kim S-H, Yoon YW, et al. Effect of fixed-dose combinations of ezetimibe plus rosuvastatin in patients with primary hypercholesterolemia: MRS-ROZE (Multicenter Randomized Study of ROsuvastatin and eZEtimibe). Cardiovasc Ther. 2016;34(5):371–82.2021.
Yang Y-J, Lee S-H, Kim BS, et al. Combination therapy of rosuvastatin and ezetimibe in patients with high cardiovascular risk. Clin Ther. 2017;39:107–17.
Bell DSH. Combine and conquer: advantages and disadvantages of fixed-dose combination therapy. Diabetes Obes Metab. 2013;15:291–300.
Iellamo F, Werdan K, Narkiewicz K, et al. Practical applications for single pill combinations in the cardiovascular continuum. Card Fail Rev. 2017;03:40.
Mikhailidis DP, Lawson RW, McCormick AL, et al. Comparative efficacy of the addition of ezetimibe to statin vs statin titration in patients with hypercholesterolaemia: systematic review and meta-analysis. Database Abstr. Rev. Eff. DARE Qual.-Assess. Rev. Centre for Reviews and Dissemination (UK); 2011. https://www.ncbi.nlm.nih.gov/books/NBK80801/. Accessed 14 Jul 2021.
Hong SJ, Jeong HS, Ahn JC, et al. A phase III, multicenter, randomized, double-blind, active comparator clinical trial to compare the efficacy and safety of combination therapy with ezetimibe and rosuvastatin versus rosuvastatin monotherapy in patients with hypercholesterolemia: I-ROSETTE (Ildong rosuvastatin and ezetimibe for hypercholesterolemia) randomized controlled trial. Clin Ther. 2018;40:226–241.e4.
Bays HE, Davidson MH, Massaad R, et al. Safety and efficacy of ezetimibe added on to rosuvastatin 5 or 10 mg versus up-titration of rosuvastatin in patients with hypercholesterolemia (the ACTE study). Am J Cardiol. 2011;108:523–30.
Kosoglou T, Statkevich P, Yang B, et al. Pharmacodynamic interaction between ezetimibe and rosuvastatin. Curr Med Res Opin. 2004;20:1185–95.
Kim B-K, Hong S-J, Lee Y-J, et al. Long-term efficacy and safety of moderate-intensity statin with ezetimibe combination therapy versus high-intensity statin monotherapy in patients with atherosclerotic cardiovascular disease (RACING): a randomised, open-label, non-inferiority trial. Lancet. 2022;400:380–90.
Farnier M, Santos RD, Cosin-Sales J, et al. Projected impact of treatment intensification with statin, ezetimibe, and statin plus ezetimibe fixed-dose combination on MACE across six countries. Eur J Prev Cardiol. 2022;29:2264–71.
Kim CH, An H, Kim SH, Shin D. Pharmacokinetic and pharmacodynamic interaction between ezetimibe and rosuvastatin in healthy male subjects. Drug Des Devel Ther. 2017;11:3461–9.
Kang WY, Seong SJ, Ohk B, et al. Pharmacokinetics and bioequivalence of a rosuvastatin/ezetimibe fixed-dose combination tablet versus single agents in healthy male subjects. Int J Clin Pharmacol Ther. 2018;56:43–52.
CZ_H_0816_002_PAR.pdf. https://file.wuxuwang.com/hma/CZ_H_0816_002_PAR.pdf. Accessed 27 May 2021.
Min KL, Park MS, Jung J, Chang MJ, Kim CO. Comparison of pharmacokinetics and safety of a fixed-dose combination of rosuvastatin and ezetimibe versus separate tablets in healthy subjects. Clin Ther. 2017;39:1799–810.
Lee E, Ryan S, Birmingham B, et al. Rosuvastatin pharmacokinetics and pharmacogenetics in white and Asian subjects residing in the same environment. Clin Pharmacol Ther. 2005;78. https://pubmed.ncbi.nlm.nih.gov/16198652/. Accessed 10 Jul 2021.
Ezetimibe 10mg Tablets—Summary of Product Characteristics (SmPC)—(emc). https://www.medicines.org.uk/emc/product/8618/smpc#gref. Accessed 11 Jan 2023.
Tzeng T-B, Schneck DW, Birmingham BK, et al. Population pharmacokinetics of rosuvastatin: implications of renal impairment, race, and dyslipidaemia. Curr Med Res Opin. 2008;24:2575–85.
Lee E, Ryan S, Birmingham B, et al. Rosuvastatin pharmacokinetics and pharmacogenetics in white and Asian subjects residing in the same environment. Clin Pharmacol Ther. 2005;78:330–41.
Acknowledgements
Funding
This study and Journal’s rapid service fee was funded by Sanofi.
Medical Writing and/or Editorial Assistance
The authors acknowledge Dan Zhang, Medical Communication, Sanofi, China, for publication process coordination, Ya Li, Man Xia, Qiang Ma, Xuan Zhao and Chuang Zhao, CSU, Sanofi, China for their contribution to the implementation of clinical trials, Haibiao Jiang, TMCP, Sanofi, China for the protocol design, Jing He and Ran Hu, R&D, Sanofi, China for study conduct, and Na Chen, CSO, Sanofi, China for CSR writing support. In addition, the authors would like to thank all the participants of the study. Medical writing support was provided by Dr Amit Bhat from Indegene which was funded by Sanofi.
Author Contributions
Conceptualization: Jie Hou, Yujing Di, Zhaojun Wang, Xiaochuan Xie, Chanyan Hu, Fang Xie. Data acquisition: Chuandong Jia, Xin Xie, Shanshan Yang, Wenhua Wang. Data analysis or interpretation: Jie Hou, Yujing Di, Zhaojun Wang, Xiaochuan Xie, Qian Wang, Chanyan Hu, Fang Xie, Mohamed Abdel-Moneim, Lionel Hovsepian, Yanzhen Wu, Na Yang. Drafting: Xiaochuan Xie, Jie Hou, Yujing Di, Zhaojun Wang. Critical Revision: Jie Hou, Yujing Di, Zhaojun Wang, Chuandong Jia, Xin Xie, Shanshan Yang, Wenhua Wang, Xiaochuan Xie, Qian Wang, Chanyan Hu, Fang Xie, Mohamed Abdel-Moneim, Lionel Hovsepian, Yanzhen Wu, Na Yang. Final Approval: Jie Hou, Yujing Di, Zhaojun Wang, Chuandong Jia, Xin Xie, Shanshan Yang, Wenhua Wang, Xiaochuan Xie, Qian Wang, Chanyan Hu, Fang Xie, Mohamed Abdel-Moneim, Lionel Hovsepian, Yanzhen Wu, Na Yang.
Disclosures
Xiaochuan Xie, Qian Wang, Chanyan Hu, Fang Xie, Mohamed Abdel-Moneim, Lionel Hovsepian, Yanzhen Wu and Na Yang are employees of Sanofi and may hold shares and/or stock options in the company for the Sanofi employees. Yujing Di, Zhaojun Wang, Chuandong Jia, Xin Xie, Shanshan Yang, Wenhua Wang, and Jie Hou have no conflict of interests.
Compliance with Ethics Guidelines
This study was conducted in accordance with the ethical principles derived from international ethics guidelines, including the Declaration of Helsinki, and the International Council for Harmonization (ICH) guidelines for Good Clinical Practice (GCP), all applicable laws, rules, and regulations. The study received approval from the institutional ethics committee of Peking University Care, Luzhong Hospital (PKULZH-IRB-SOP-AF-013/3.0-03). Informed written consent was obtained at the time of study enrolment.
Data Availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Di, Y., Wang, Z., Jia, C. et al. A Bioequivalence Study of Ezetimibe/Rosuvastatin Fixed Dose Combination (10 mg/10 mg) Versus the Individual Formulations Taken Concomitantly. Adv Ther 40, 2205–2216 (2023). https://doi.org/10.1007/s12325-023-02439-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-023-02439-8