
Abstract
Background and Objective
The combination of rosuvastatin and ezetimibe has promising clinical benefits with a significant safety and tolerability profile. However, there is a lack of clinical data supporting the drug–drug interaction (DDI) in Chinese population. Thus, the aim of this study is to assess the potential pharmacokinetic DDI between rosuvastatin and ezetimibe in a Chinese population.
Methods
In this randomized, open-label, phase 1 study, 12 healthy volunteers were randomized to three treatment groups: 10 mg rosuvastatin plus 10 mg ezetimibe, 10 mg rosuvastatin alone, and 10 mg ezetimibe alone under fasting conditions. The plasma concentrations of rosuvastatin and ezetimibe were determined, and the pharmacokinetic parameters were calculated. Primary endpoints were peak plasma concentration (Cmax), area under the curve from zero to last measurement (AUC0–t), and area under the curve from zero to infinity (AUC0–∞) that were log-transformed, and co-administration was compared with monotherapy to evaluate the DDI.
Results
The geometric mean ratios (GMRs) of rosuvastatin with 90% confidence intervals (CIs) were 0.94 (0.80–1.12) for Cmax, 0.96 (0.85–1.08) for AUC0–t, and 0.96 (0.86–1.07) for AUC0–∞ when administered in combination with ezetimibe versus administered alone. The GMRs of unconjugated ezetimibe and total ezetimibe with 90% CIs were 1.15 (1.00–1.32) and 0.93 (0.80–1.07) for Cmax, 0.96 (0.84–1.10) and 0.95 (0.83–1.08) for AUC0–t, and 1.06 (0.96–1.18) and 0.94 (0.80–1.11) for AUC0–∞, respectively, when administered in combination with rosuvastatin versus administered alone.
Conclusion
Co-administration of rosuvastatin and ezetimibe showed no clinically significant pharmacokinetic interactions in a healthy Chinese population.
Similar content being viewed by others


Co-administration of rosuvastatin and ezetimibe showed no clinically significant pharmacokinetic interactions in a healthy Chinese population. |
The treatment with combination of rosuvastatin and ezetimibe was well tolerated with no serious adverse effects. |
The study may contribute beneficial evidence for the development of a fixed-dose combination tablet to increase patient compliance. |
1 Introduction
Dyslipidemia is one of the major risk factors for coronary artery diseases, cerebrovascular diseases, and atherosclerosis [1]. The overall prevalence of dyslipidemia in Chinese adults is 43%, and by 2030, the increase in serum cholesterol level is expected to increase the incidence of cardiovascular events in China by approximately 9.2 million cases [2]. A cross-sectional study (DYSIS-China) involving 25,317 patients across China had demonstrated that up to 38.5% of patients with dyslipidemia in China were unable to reach their low-density lipoprotein cholesterol (LDL-C) targets, reflecting a huge unmet need of more efficient lipid-lowering agents [3].
The discovery of statin molecules from fungi has revolutionized the treatment of hypercholesterolemia. Statins reversibly inhibit 3-hydroxy-methyl glutaryl-coenzyme A (HMG-CoA) reductase, which is the rate-limiting enzyme in cholesterol biosynthesis, and are recommended by a number of international guidelines, including American Heart Association (AHA), National Lipid Association (NLA), and Preventive Cardiovascular Nurses Association (PCNA), as the first choice of treatment for reducing plasma cholesterol levels [4, 5]. Rosuvastatin is a fully synthetic statin that competitively binds to HMG-CoA reductase [6] and is metabolized to N-desmethyl metabolite, which is a less potent substrate, via cytochrome P450 (CYP) enzymes such as CYP2C9 and CYP2C19; however, rosuvastatin is a poor substrate for CYP450, with 90% of the drug excreted unchanged [7]. Previous studies have shown that, owing to limited metabolism by CYP enzymes, drug–drug interaction (DDI) is unlikely to occur with rosuvastatin [7]. Martin et al. showed that rosuvastatin is eliminated primarily in the feces (90%) compared with renal excretion (10%), with 19 h of plasma half-life (t1/2) [8]. A few studies have demonstrated that rosuvastatin has better efficacy in improving lipid profile than other molecules of the same class because of its high binding potency [9,10,11,12]. Although intensive statin therapies effectively reduce LDL-C, only one-fifth of statin users actually achieve the desired lipid goals [13]. Furthermore, high doses of statin have raised safety concerns, as they may promote myopathy and new-onset diabetes [14]. To address these issues, it is necessary to combine statins with additional lipid-modifying therapies that act via different mechanisms [15]. Ezetimibe is a first-in-class cholesterol absorption inhibitor that prevents the intestinal uptake of dietary and biliary cholesterol by inhibiting Niemann–Pick C1-like 1 protein [15]. Ezetimibe is metabolized into ezetimibe–glucuronide and unconjugated ezetimibe by uridine 5-diphosphate–glucuronosyltransferase (UGT) enzymes such as UGT1A1, UGT1A3, and UGT2B15 [16]. The metabolized parent drug and its conjugated metabolites are transported to the liver via portal vessels, leading to further glucuronidation of ezetimibe [17]. Eventually, ezetimibe–glucuronide gets secreted into the intestine, where it exerts its pharmacological activity and significantly contributes to the efficacy and safety profile of the drug [17]. A study by Patrick et al. on metabolism of ezetimibe in healthy male participants showed that around 90% of the total plasma radioactivity was accounted for by ezetimibe–glucuronide, while unconjugated ezetimibe contributed < 5% after 0.5 h of administration. After 24 h, human plasma contains only ezetimibe–glucuronide, whereas 10 days later, 11% and 78% of ezetimibe were recovered in urine and feces, respectively [18]. Although 69% of ezetimibe has been shown to be recovered in feces in the form of unconjugated ezetimibe, this indicates low absorption or hydrolysis of ezetimibe–glucuronide secreted in the bile [18]. Therefore, it is recommended by the US Food and Drug Administration that pharmacokinetic parameters of both total ezetimibe and unconjugated ezetimibe need to be considered during pharmacokinetic evaluations [19, 20]. Administration of ezetimibe as a monotherapy is recommended to treat hypercholesterolemia in patients who are intolerant to statin [1]. Previous trials have demonstrated that ezetimibe can lower LDL-C levels by 10% as monotherapy and by 25% if administered in combination with statins [13, 21].
Provided that the combination of rosuvastatin and ezetimibe is both well tolerated and safe, the potential therapeutic effects of these drugs offer promising clinical benefits [22]. Previously, an in-house study assessed DDI between 10 mg Ezetrol tablet (ezetimibe) and 40 mg Crestor (rosuvastatin) tablet in Caucasians. The results showed that the 90% confidence intervals (CIs) for the geometric mean ratio (GMR) of peak plasma concentration (Cmax) and area under the curve from zero to last measurement (AUC0–t) of rosuvastatin (concomitant administration of Ezetrol versus Crestor), unconjugated ezetimibe, and total ezetimibe (concomitant administration of Crestor versus Ezetrol) were within the bioequivalence range of 80–125%, confirming that the pharmacokinetic profiles of rosuvastatin and ezetimibe were not impacted by concomitant administration in Caucasians. However, there is a lack of clinical data supporting this hypothesis in a Chinese population [22]. Hence, there is a need to establish clinical evidence on the DDI of rosuvastatin and ezetimibe in Chinese adults.
The US Food and Drug Administration recommends that rosuvastatin therapy in Asian patients be initiated at half the normal dose for non-Asians because of its increased plasma concentration [23]. Considering the pharmacokinetic profiles of these two drugs, a fixed-dose combination of rosuvastatin/ezetimibe 10 mg/10 mg in Chinese participants would be expected to result in similar average systematic exposure as compared with a 20 mg/10 mg dose in Caucasians. Therefore, in this phase 1 clinical trial, we aim to evaluate the pharmacokinetic interactions, safety, and tolerability of rosuvastatin and ezetimibe in healthy Chinese participants as per local regulatory requirements.
2 Subjects and Methods
2.1 Study Population and Inclusion Criteria
We conducted a phase 1, single-center, open-label, randomized, single-dose, three-treatment, three-period, three-sequence, crossover study to assess the DDI between ezetimibe 10 mg tablet (Ezetrol®, MSD Pharma, batch no. T000913, manufacturing date: 12 October 2019, expiry date: 11 October 2022) and rosuvastatin 10 mg tablet (Crestor®, AstraZeneca UK Limited, batch no. 500230, manufacturing date: September 2019, expiry date: August 2020) in healthy Chinese participants under fasting condition at Peking University (PKU) Care, Luzhong Hospital, China from 16 December 2020 to 18 January 2021. Healthy Chinese volunteers aged between 18 and 45 years (inclusive), with body mass index of 18.5–27.9 kg/m2 (inclusive), and certified as healthy by clinical and biological assessments were included. The serum creatinine phosphokinase levels should be no more than 2.5 times the upper limit of normal (ULN), and the aspartate aminotransferase and alanine aminotransferase levels should be no more than 1.25 times the ULN. Participants were excluded if they smoke regularly (more than five cigarettes or equivalent per week), consume alcohol heavily (more than 40 g alcohol per day on a regular basis), have any clinically significant medical histories, have abnormal physical and laboratory examination findings, or are on concomitant medication. In addition, the participants were instructed to abstain from consuming citrus fruits and other fruits (such as mango and dragon fruit) that are known to affect the activity of drug-metabolizing enzymes at least 5 days before the administration of the study drug. The study was conducted in accordance with 1964 Declaration of Helsinki and its latest amendments, good clinical practice guidelines, and other local regulatory laws and guidelines (clinical study number CTR20202332). Written informed consent was obtained from all eligible participants before induction.
2.2 Study Design and Procedure
The participants who met the inclusion criteria were randomized and assigned with a randomization number sequentially in a 1:1:1 ratio to three different sequences of treatments (sequence 1 versus sequence 2 versus sequence 3) according to a computer-generated random sequence. As the study was open label, participants, investigators, and study members had access to treatment assignment. The dose levels of ezetimibe 10 mg and rosuvastatin 10 mg for the present study were selected on the basis of the label/summary of product characteristics (SPC) of ezetimibe tablet (Ezetrol) and rosuvastatin tablet (Crestor), which is used in clinical practices in China.
The treatment regimens were as follows: treatment 1, co-administration with one rosuvastatin 10 mg film-coated tablet and one ezetimibe 10 mg tablet; treatment 2, administered with one rosuvastatin 10 mg film-coated tablet alone; and treatment 3, administered with one ezetimibe 10 mg tablet alone. The three different sequences were defined as follows: sequence 1, treatments 1, 2, and 3 in periods 1, 2, and 3, respectively; sequence 2, treatments 2, 3, and 1 in periods 1, 2, and 3, respectively; and sequence 3, treatments 3, 1, and 2 in periods 1, 2, and 3, respectively. Each treatment period was separated by a 10-day washout period. To avoid food effects, the participants were kept in fasting condition for at least 10 h before dose administration. Participants were orally administered with the assigned investigational medicinal product (IMP) with 240 mL of water. Water was not allowed for at least 1 h before and after the administration of IMP, and no food was allowed for at least 4 h after the administration. Lunch and dinner were permitted at 4 and 10 h after the administration of drug, respectively. After the administration, the participants stayed in a semi-recumbent position or stayed seated for a minimum of 2 h.
According to the SPC, the t1/2 of ezetimibe (Ezetrol) and rosuvastatin (Crestor) is approximately 22 and 19 h, respectively. Hence, different plasma sampling timepoints were followed for both the drugs [24, 25]. The duration for collecting pharmacokinetic blood samples for ezetimibe and rosuvastatin was considered up to 96 and 72 h, respectively, after the administration of dose with a sufficient washout period of 10 days. To measure the pharmacokinetic parameters of ezetimibe, blood samples were collected before dosing and at 0.25, 0.5, 0.75, 1, 1.25, 1.5, 1.75, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 24, 48, 72, and 96 h after dosing. In the case of rosuvastatin, as per the previous studies, the Cmax is reached at 3–5 h after the administration of the drug [26]. So, to ensure a sufficient time frame to assess the pharmacokinetic profile of rosuvastatin, extensive sampling was performed. To measure the pharmacokinetic parameters of rosuvastatin, blood samples were collected before dosing and at 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, 5, 5.5, 6, 8, 12, 24, 36, 48, and 72 h after dosing (Fig. 1).

Schematic study design of the clinical trial (three-treatment, three-period, three-sequence, crossover study)
The plasma concentrations of rosuvastatin, unconjugated ezetimibe, and total ezetimibe were measured using a validated liquid chromatography–tandem mass spectrometry (LC–MS/MS) method as discussed previously by Bhadoriya et al. (2018) [27] (Covance Pharmaceutical Research and Development, Shanghai, China) with lower limit of quantification as 0.0400, 0.0500, and 0.500 ng/mL, respectively. The samples were pretreated by liquid–liquid extraction. Di-potassium ethylenediaminetetraacetic acid (K2EDTA) was used as a plasma stabilizer in a 1:1 ratio (stabilizer: plasma). For rosuvastatin, the compounds were identified and quantified using ultra-performance liquid chromatography (UPLC)–MS/MS detection over a concentration range of 0.0400 to 40.0 ng/mL in untreated plasma and 0.0200–20.0 ng/mL in plasma treated with stabilizer. For ezetimibe, the isotopes ezetimibe-D4 and ezetimibe-D4 β-d-glucuronide were used as internal standards. The ezetimibe phenoxy β-d-glucuronide and unconjugated ezetimibe were identified and quantified over a concentration range of 0.500–200 ng/mL and 0.0500–20.0 ng/mL, respectively.
The LC–MS/MS method was followed as per Bhadoriya et al. [27]. Briefly, Shimadzu Nexera X2 UHPLC with triple quadrupole mass spectrometer detector Shimadzu LCMS-8040 (Shimadzu Corporation, Kyoto, Japan) and Waters Symmetry C18 (100 × 4.6 mm, 3.5 μm) column was used for the separation of the analytes. Acetonitrile and 10 mM ammonium formate with pH 3.5 adjusted with 0.1% formic acid in 70:30 (v/v) were used as a mobile phase with 0.8 mL/min as a flow rate. The temperature for column oven and autosampler was maintained at 30 °C and 5 °C, respectively. Isotope rosuvastatin-D6 was used as an internal standard. The MS/MS detection in the positive and negative electrospray ionization mode was used for the quantification of rosuvastatin and ezetimibe, respectively. The flow rate of 4.0 and 10.0 L/min was kept for nebulizing gas (N2) and drying gas (N2), respectively. The desolvation line and the heat block temperature were maintained at 300°C and 400°C, respectively, with interface voltage of 3.0 kV, 250 kPa of collision-induced dissociation gas (argon). The optimum values for compound-dependent parameters such as quadrupole (Q) 1 pre-bias voltage, collision energy, and Q3 pre-bias voltage were set at 25 V, 35 eV, and 25 V, respectively, for rosuvastatin; 24 V, 36 eV, and 22 V, respectively, for rosuvastatin-d6; 21 V, 17 eV, and 17 V, respectively, for ezetimibe; and 20 V, 18 eV, and 19 V, respectively, for ezetimibe d4. Q1 and Q3 were maintained at unit mass resolution, and the dwell time was set at 100 ms. Shimadzu LabSolutions software was used for data processing.
2.3 Outcomes of the Study
The primary objective of the present study was to assess the DDI between ezetimibe 10 mg tablet and rosuvastatin 10 mg tablet in healthy Chinese participants under fasting condition, whereas the secondary objective was to assess the safety and tolerability of rosuvastatin and ezetimibe.
2.3.1 Pharmacokinetics and DDI Study Endpoints
The pharmacokinetic assessment of rosuvastatin, unconjugated ezetimibe, and total ezetimibe plasma concentrations was conducted using standard noncompartmental methods. Total ezetimibe was calculated from the sum of free ezetimibe (unconjugated) and ezetimibe–glucuronide (conjugated) taking into consideration the adjustment per molecular weight, for each analyte. The pharmacokinetics and DDI endpoints of the study were Cmax, AUC0–t, and area under the curve from dosing time extrapolated to infinity (AUC0–∞), median time to reach the maximum plasma concentration (Tmax), and terminal elimination t1/2 of drug. Other parameters included in the study are summarized in Table 1. To evaluate DDI, the values of Cmax, AUC0–t, and AUC0–∞ were log-transformed and compared for co-administration (treatment 1) and administration of rosuvastatin (treatment 2) or ezetimibe alone (treatment 3).
2.3.2 Safety and Tolerability Assessments
Safety assessments were performed on safety population. Safety and tolerability were investigated through spontaneous reporting and inquiries regarding adverse events (AEs) and treatment-emergent adverse events (TEAEs) from the time of IMP administration of each treatment period to day 5 (included). Potential clinically significant abnormalities, physical examinations, vital sign measurements, 12-lead electrocardiogram, and laboratory tests, including serum chemistry, urinalysis, and hematology, were also performed to monitor tolerability. The AEs were graded on the basis of Common Terminology Criteria for Adverse Events (CTCAE v5) and were classified by System Organ Class/preferred term according the Medical Dictionary for Regulatory Activities. For AEs not included in the CTCAE, the investigator assessed the intensity of the adverse drug/biologic experience using the CTCAE general guideline.
According to the CTCAE v5, TEAE was defined as AEs that occurred, worsened, or became serious during the on-treatment phase. TEAEs were assigned to the treatment received at the time of AE onset.
2.3.3 Sample Size Calculation
On the basis of pharmacokinetic parameters (Cmax and AUC0–t) of rosuvastatin and ezetimibe, within-subject SD was calculated as 0.275 (inline studies) and was used for sample size calculation. Using a linear mixed effect model, a total number of participants N = 6, 9, 12, and 15 were considered, assuming a true within-subject SD of 0.275 for log-transformed pharmacokinetic parameters. The maximum imprecision (in terms of the 90% CI) for the ratio of analytes in co-administration of Ezetrol and Crestor versus monotherapy that was obtained with 90% assurance for different patient numbers is presented in Table 2. With nine participants, any predefined ratio mean can be estimated with maximum imprecision of 24.4% (90% CI will be 0.65 and 1/0.65 times the observed ratio), with 90% assurance. Considering the potential dropout, 12 participants were used for this study.
2.4 Statistical Analysis
All statistical analysis was conducted by SAS version 9.4. Descriptive statistics were used to analyze baseline patient demographics, pharmacokinetics, and safety profiles. The results were expressed as mean ± standard deviation (SD), except for Tmax, which was expressed as median (minimum and maximum values). To assess the associated DDI, a two-sided 90% CI for the true mean difference between treatments for pharmacokinetic parameters (Cmax, AUC0–t, and
AUC0–∞) on the log scale was computed from the following linear mixed effect model: log (parameter) = Sequence + Period + Treatment + Error, with fixed terms for sequence, period, and treatment and with an unstructured 3-by-3 matrix of treatment-specific variances and co-variance for subject within sequence. These confidence limits were then exponentiated to obtain the 90% CIs for the true GMRs for the pharmacokinetic parameters, including Cmax, AUC0–t, and AUC0–∞ (co-administration versus administered alone).
3 Results
3.1 Demographics and Baseline Characteristics
A total of 12 healthy volunteers were enrolled in this study. Among them, one volunteer discontinued the study treatment after the completion of period 1 (received only rosuvastatin, treatment 2) owing to withdrawal of consent by subject’s decision. The pharmacokinetics and the safety analysis for rosuvastatin were based on the results of 12 healthy volunteers, and the pharmacokinetics and the safety analysis for co-administration (treatment 1) and only ezetimibe monotherapy (treatment 3) were based on the results of 11 healthy volunteers. Of these 12 healthy volunteers enrolled, 8 volunteers (66.7%) were male and 4 volunteers (33.3%) were female, and the mean age was 29.9 ± 6.6 years. The demographic characteristics at baseline are presented in Table 3.
3.2 Primary Endpoints
3.2.1 Pharmacokinetic and DDI Endpoints
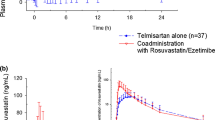
After the administration of rosuvastatin alone (treatment 2) and co-administration with ezetimibe (treatment 1), the mean Cmax ± SD of rosuvastatin was 12.6 ± 5.99 and 11.5 ± 5.33 ng/mL, respectively, whereas the mean AUC0–t ± SD was 113 ± 51.6 and 101 ± 37.1 ng·h/mL, respectively (Table 4). The mean AUC0–∞ ± SD of monotherapy of rosuvastatin and co-administration with ezetimibe was 115 ± 52.3 and 103 ± 37.6 ng·h/mL, respectively. The Tmax for both rosuvastatin administered alone and co-administered with ezetimibe was 4.5 h, with a mean t1/2 of 12.9 ± 5.10 and 12.4 ± 4.18 h, respectively (Table 4). The GMRs of Cmax, AUC0–t, and AUC0–∞ for rosuvastatin co-administered with ezetimibe versus administered alone were 0.94 (90% CI 0.80–1.12), 0.96 (90% CI 0.85–1.08), and 0.96 (90% CI 0.86–1.07), respectively (Table 5).
After the administration of ezetimibe alone (treatment 3) and co-administration with rosuvastatin (treatment 1), the mean Cmax of unconjugated ezetimibe was 4.17 ± 2.59 and 4.75 ± 2.61 ng/mL, respectively, whereas the mean AUC0–t was 102 ± 42.1 and 94.0 ± 31.0 ng·h/mL, respectively (Table 4). The mean AUC0–∞ of unconjugated ezetimibe for administration alone was 110 ± 51.9 ng·h/mL, whereas after co-administration, the mean AUC0–∞ was 98.2 ± 37.4 ng·h/mL. The median Tmax was 2 and 1.5 h for administration alone and co-administration with rosuvastatin, respectively, with a respective mean t1/2 of 21.0 ± 9.85 and 21.8 ± 7.97 h (Table 4). The GMRs of Cmax, AUC0–t, and AUC0–∞ for conjugated ezetimibe co-administered with rosuvastatin versus administered alone were 1.15 (90% CI 1.00–1.32), 0.96 (90% CI 0.84–1.10), and 1.06 (90% CI 0.96–1.18), respectively (Table 5). For total ezetimibe, the mean Cmax for monotherapy of ezetimibe and co-administration with rosuvastatin was 60 ± 35.4 and 59.2 ± 40.2 ng/mL, respectively, whereas the mean AUC0–t was 632 ± 319 and 565 ± 255 ng·h/mL, respectively. The mean AUC0–∞ of total ezetimibe for monotherapy of ezetimibe and co-administration with rosuvastatin was 688 ± 356 and 679 ± 457 ng·h/mL, respectively. The median Tmax of total ezetimibe was 0.75 h for both monotherapy of ezetimibe and co-administration with rosuvastatin, with a mean t1/2 of 22 ± 12.1 and 27.8 ± 27.7 h, respectively (Table 4). The GMRs of Cmax, AUC0–t, and AUC0–∞ for total ezetimibe co-administered with rosuvastatin versus administered alone were 0.93 (90% CI 0.80–1.07), 0.95 (90% CI 0.83–1.08), and 0.94 (90% CI 0.80–1.11), respectively (Table 5).
The mean (SD) plasma concentration–time profiles for rosuvastatin, unconjugated ezetimibe, and total ezetimibe are illustrated in Figs. 2, 3, and 4, respectively. Each figure is displayed on the linear and semi-logarithmic scales.

Mean (SD) plasma concentration–time profiles for rosuvastatin alone following co-administration with ezetimibe. a Linear scale, b Semi-logarithmic scale. LLOQ lower limit of quantification, SD standard deviation

Mean (SD) plasma concentration–time profiles for unconjugated ezetimibe: administration of ezetimibe alone following co-administration with rosuvastatin. a Linear scale, b semi-logarithmic scale. LLOQ lower limit of quantification, SD standard deviation

Mean (SD) plasma concentration–time profiles for total ezetimibe: administration of ezetimibe alone following co-administration with rosuvastatin. a Linear scale, b semi-logarithmic scale. SD standard deviation
3.3 Secondary Endpoints
3.3.1 Safety Analysis
Only 1 (9.1%) of 11 healthy volunteers from ezetimibe monotherapy and 1 (8.3%) of 12 healthy volunteers of rosuvastatin monotherapy were reported grade 1 TEAEs. None of the volunteers reported TEAEs with co-administration of rosuvastatin and ezetimibe. For the ezetimibe and rosuvastatin monotherapy groups, the AEs were reported as increased blood creatinine and triglyceride levels, respectively. No other abnormal findings on clinical laboratory tests, physical examinations, or vital signs were reported. None of the volunteers withdrew from the study owing to AEs, and no deaths were reported.
4 Discussion
The current study evaluated the potential pharmacokinetic interaction between rosuvastatin and ezetimibe. These two drugs have been widely used as a co-therapy in many parts of the world in different ethnic groups to generate additive effects for controlling the levels of LDL-C and have been recommended by various international guidelines such as the European Society of Cardiology/European Atherosclerotic Society (ESC/EAS) and American College of Cardiology and American Heart Association (ACC/AHA) guidelines [28, 29]. Furthermore, several well-designed clinical studies (EXPLORER, ACTE, and GRAVITY) conducted in Caucasian populations reported that significantly more patients administered with co-therapy achieved target LDL-C goal compared with those on rosuvastatin monotherapy [30,31,32]. Kouvelos et al. conducted a randomized clinical trial in Greece where patients were administered 10 mg rosuvastatin as monotherapy or co-therapy of 10 mg rosuvastatin and 10 mg ezetimibe [33]. The study reported intensified lipid-lowering results in terms of reduced LDL-C levels with co-therapy compared with rosuvastatin monotherapy [33]. In a real-world analysis by Foody et al., in the USA, the reported odds of attaining LDL-C increased by 3.1-fold in patients who were administered with ezetimibe on top of rosuvastatin [34]. Other clinical trials in Asian (Korean and Japanese) populations also demonstrated that co-therapy of rosuvastatin and ezetimibe resulted in additive therapeutic effects, resulting in better LDL-C levels [35, 36].
According to the Center for Drug Evaluation regulations in China, any drug marketed abroad that has to be premarketed in China needs pharmacokinetic, pharmacodynamic, pharmacokinetic/ pharmacodynamic, and DDI data of the Chinese population, unless there is evidence that no racial difference exists between Chinese and other populations [37]. Hence, the present study was conducted to study pharmacokinetics and DDI of rosuvastatin and ezetimibe in a Chinese population. Contextually, two studies reported rosuvastatin to have an at least twofold higher exposure in Asians compared with whites in the same conditions after controlling for only solute carrier organic anion transporter family member 1B1 (SLCO1B1) wild type [38, 39]. Another study reported that reduced-function single-nucleotide polymorphism frequency for SLCO1B1 *type15 is more widespread in Eastern Asians than in whites (14% versus 2.7%) [23]. Reduced-function polymorphisms and minor allele frequency of SLCO1B1 (gene encoding OATP1B1) are suggested as the key reasons for interethnic disparity in rosuvastatin pharmacokinetics and DDIs. Interestingly, a recent study reported differences in SLCO1B1 haplotype frequencies even within the Asian population, where SLCO1B1 gene haplotype frequency demonstrated a prevalence of 1.2% in Chinese, whereas it was absent among the Korean and Vietnamese populations [40]. Hence, it is crucial to explicitly rule out any possibility of pharmacokinetic interactions of rosuvastatin with ezetimibe in the Chinese population.
The mean Cmax and AUC0–t of rosuvastatin administered as co-administration with ezetimibe (Cmax: 11.5 ng/mL, AUC0–t: 101 ng·h/mL) versus administered alone (Cmax: 12.6 ng/mL, AUC0–t: 113 ng·h/mL) in the present study were comparable to those reported by Kang et al. The study reported 12.5 ng/mL and 115.5 ng·h/mL of mean Cmax and AUC0–t values with rosuvastatin co-administration (10 mg rosuvastatin + 10 mg ezetimibe) and 12.2 ng/mL and 115.1 ng·h/mL when administered alone, respectively [41]. As demonstrated in previous studies [1, 42], the mean Cmax, AUC0–t, and AUC0–∞ values in our study were also similar between administered alone and co-administration, indicating that both rosuvastatin and ezetimibe were almost unaffected by their respective co-administration treatments. Moreover, the Tmax and the t1/2 values in our study were comparable between concomitantly and individually administered rosuvastatin and ezetimibe, further confirming that the pharmacokinetic properties of these two drugs are not affected by co-administration of each other in healthy Chinese volunteers under fasting conditions. As demonstrated previously, rosuvastatin was observed to have relatively shorter terminal t1/2 compared with total ezetimibe and unconjugated ezetimibe [42]. The time–concentration curve of ezetimibe exhibited multiple peaks due to enterohepatic circulation, which were consistent with the findings from previous reports [19, 42, 43].
In the current study, DDI assessment indicated no clinically significant interactions between the two drugs in healthy Chinese volunteers. The results were in accordance with reported clinical studies that have demonstrated a lack of clinically significant pharmacokinetic DDI in both Asian and non-Asian populations [1, 22, 42]. In a South Korean population, the GMR of Cmax for rosuvastatin, total ezetimibe, and free ezetimibe was 1.099, 0.996, and 1.182, respectively, which was comparable to the results of our study [1]. Similarly, Kang et al. also reported a lack of clinically significant DDI with GMR for ezetimibe Cmax and AUC0–t as 108.96% and 98.13% and for rosuvastatin as 106.20% and 102.88%, respectively [41].
The insignificant interaction between the two drugs could be due to several reasons. Firstly, neither drug is reported to affect any of the major CYP enzymes and thus does not disturb each other’s pharmacokinetic properties [44]. Secondly, transporter-mediated DDI is also not a concern because, although transporter inhibitors may affect the exposure of rosuvastatin, ezetimibe is neither an inhibitor nor an inducer of these transporters and does not alter rosuvastatin exposure. Thirdly, the pathways by which these two drugs are metabolized are quite different. Although rosuvastatin is a hepato-selective hydrophilic drug that is metabolized by CYP2C19, CYP3A4, and CYP2D6, ezetimibe is majorly metabolized presystemically by glucuronidation in the intestinal wall and further metabolized by UGT1A1, UGT1A3, and UGT2B15 enzymes [7, 16].
The treatments in our study were well tolerated with no serious AEs. Both of the AEs reported in rosuvastatin and ezetimibe administered alone were mild and resolved without any complications. This study may contribute important evidence for developing fixed-dose combination tablet in China to increase patient compliance. Limitations of our study include that only healthy volunteers were eligible for inclusion as this reduces the potential impact of underlying diseases and concomitant medications. Nevertheless, it should be noted that, in real-world clinical practice, the pharmacokinetics of rosuvastatin and ezetimibe still might be influenced by concomitant medications or underlying diseases. Moreover, we determined only the pharmacokinetic characteristics but not pharmacological effects, and the LDL-C-lowering effects were not evaluated in patients with dyslipidemia.
5 Conclusions
In conclusion, co-administration of rosuvastatin and ezetimibe showed no clinically significant pharmacokinetic interactions, and the combined administration of these two drugs was well tolerated in healthy Chinese participants.
References
Kim H, Choi HY, Kim Y-H, et al. Pharmacokinetic interactions and tolerability of rosuvastatin and ezetimibe: an open-label, randomized, multiple-dose, crossover study in healthy male volunteers. Drug Des Dev Ther. 2018;12:815–21. https://doi.org/10.2147/DDDT.S158408.
Moran A, Gu D, Zhao D, et al. Future cardiovascular disease in China: Markov model and risk factor scenario projections from the Coronary Heart Disease Policy Model-China. Circ Cardiovasc Qual Outcomes. 2010;3:243–52. https://doi.org/10.1161/CIRCOUTCOMES.109.910711.
Wang F, Ye P, Hu D, et al. Lipid-lowering therapy and lipid goal attainment in patients with metabolic syndrome in China: subgroup analysis of the Dyslipidemia International Study-China (DYSIS-China). Atherosclerosis. 2014;237:99–105. https://doi.org/10.1016/j.atherosclerosis.2014.08.023.
Sirtori CR. The pharmacology of statins. Pharmacol Res. 2014;88:3–11. https://doi.org/10.1016/j.phrs.2014.03.002.
Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol. J Am Coll Cardiol. 2019;73:e285–350. https://doi.org/10.1016/j.jacc.2018.11.003.
Cortese F, Gesualdo M, Cortese A, et al. Rosuvastatin: beyond the cholesterol-lowering effect. Pharmacol Res. 2016;107:1–18. https://doi.org/10.1016/j.phrs.2016.02.012.
Luvai A, Mbagaya W, Hall AS, Barth JH. Rosuvastatin: a review of the pharmacology and clinical effectiveness in cardiovascular disease. Clin Med Insights Cardiol. 2012;6:17–33. https://doi.org/10.4137/CMC.S4324.
Martin PD, Warwick MJ, Dane AL, et al. Metabolism, excretion, and pharmacokinetics of rosuvastatin in healthy adult male volunteers. Clin Ther. 2003;25:2822–35. https://doi.org/10.1016/s0149-2918(03)80336-3.
Jones PH, Davidson MH, Stein EA, et al. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial). Am J Cardiol. 2003;92:152–60. https://doi.org/10.1016/s0002-9149(03)00530-7.
Davidson MH. Rosuvastatin safety: lessons from the FDA review and post-approval surveillance. Expert Opin Drug Saf. 2004;3:547–57. https://doi.org/10.1517/14740338.3.6.547.
Schuster H, Barter PJ, Stender S, et al. Effects of switching statins on achievement of lipid goals: Measuring Effective Reductions in Cholesterol Using Rosuvastatin Therapy (MERCURY I) study. Am Heart J. 2004;147:705–13. https://doi.org/10.1016/j.ahj.2003.10.004.
Ballantyne CM, Bertolami M, Hernandez Garcia HR, et al. Achieving LDL cholesterol, non-HDL cholesterol, and apolipoprotein B target levels in high-risk patients: Measuring Effective Reductions in Cholesterol Using Rosuvastatin Therapy (MERCURY) II. Am Heart J. 2006;151:975.e1-9. https://doi.org/10.1016/j.ahj.2005.12.013.
Strilchuk L, Tocci G, Fogacci F, Cicero AFG. An overview of rosuvastatin/ezetimibe association for the treatment of hypercholesterolemia and mixed dyslipidemia. Expert Opin Pharmacother. 2020;21:531–9. https://doi.org/10.1080/14656566.2020.1714028.
Hu M, Cheung BMY, Tomlinson B. Safety of statins: an update. Ther Adv Drug Saf. 2012;3:133–44. https://doi.org/10.1177/2042098612439884.
Savarese G, De Ferrari GM, Rosano GMC, Perrone-Filardi P. Safety and efficacy of ezetimibe: a meta-analysis. Int J Cardiol. 2015;201:247–52. https://doi.org/10.1016/j.ijcard.2015.08.103.
Ghosal A, Hapangama N, Yuan Y, et al. Identification of human UDP-glucuronosyltransferase enzyme(s) responsible for the glucuronidation of ezetimibe (Zetia). Drug Metab Dispos. 2004;32:314–20. https://doi.org/10.1124/dmd.32.3.314.
Kosoglou T, Statkevich P, Johnson-Levonas AO, et al. Ezetimibe: a review of its metabolism, pharmacokinetics and drug interactions. Clin Pharmacokinet. 2005;44:467–94. https://doi.org/10.2165/00003088-200544050-00002.
Patrick JE, Kosoglou T, Stauber KL, et al. Disposition of the selective cholesterol absorption inhibitor ezetimibe in healthy male subjects. Drug Metab Dispos. 2002;30:430–7. https://doi.org/10.1124/dmd.30.4.430.
Min KL, Park MS, Jung J, et al. Comparison of pharmacokinetics and safety of a fixed-dose combination of rosuvastatin and ezetimibe versus separate tablets in healthy subjects. Clin Ther. 2017;39:1799–810. https://doi.org/10.1016/j.clinthera.2017.07.038.
Bioequivalence Studies With Pharmacokinetic Endpoints for Drugs Submitted Under an Abbreviated New Drug Application. 2021. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioequivalence-studies-pharmacokinetic-endpoints-drugs-submitted-under-abbreviated-new-drug. Accessed 25 Oct 2022.
Jellinger PS, Handelsman Y, Rosenblit PD, et al. American Association of Clinical Endocrinologists and American College of Endocrinology Guidelines for management of dyslipidemia and prevention of cardiovascular disease. Endocr Pract. 2017;23:1–87. https://doi.org/10.4158/EP171764.APPGL.
Kosoglou T, Statkevich P, Yang B, et al. Pharmacodynamic interaction between ezetimibe and rosuvastatin. Curr Med Res Opin. 2004;20:1185–95. https://doi.org/10.1185/030079904125004213.
Wu H-F, Hristeva N, Chang J, et al. Rosuvastatin pharmacokinetics in Asian and white subjects wild type for both OATP1B1 and BCRP under control and inhibited conditions. J Pharm Sci. 2017;106:2751–7. https://doi.org/10.1016/j.xphs.2017.03.027.
Crestor 10mg film-coated tablets. https://www.medicines.org.uk/emc/product/7559/smpc#gref. Accessed 25 Oct 2022.
Ezetrol 10mg Tablets. https://www.medicines.org.uk/emc/product/6792/smpc#gref. Accessed 25 Oct 2022.
Li Y, Jiang X, Lan K, et al. Pharmacokinetic properties of rosuvastatin after single-dose, oral administration in Chinese volunteers: a randomized, open-label, three-way crossover study. Clin Ther. 2007;29:2194–203. https://doi.org/10.1016/j.clinthera.2007.10.005.
Bhadoriya A, Sanyal M, Shah PA, Shrivastav PS. Simultaneous quantitation of rosuvastatin and ezetimibe in human plasma by LC–MS/MS: pharmacokinetic study of fixed-dose formulation and separate tablets. Biomed Chromatogr. 2018;32: e4291. https://doi.org/10.1002/bmc.4291.
Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111–88. https://doi.org/10.1093/eurheartj/ehz455.
Reiter-Brennan C, Osei AD, Iftekhar Uddin SM, et al. ACC/AHA lipid guidelines: personalized care to prevent cardiovascular disease. CCJM. 2020;87:231–9. https://doi.org/10.3949/ccjm.87a.19078.
Ballantyne CM, Hoogeveen RC, Raya JL, et al. Efficacy, safety and effect on biomarkers related to cholesterol and lipoprotein metabolism of rosuvastatin 10 or 20 mg plus ezetimibe 10 mg vs. simvastatin 40 or 80 mg plus ezetimibe 10 mg in high-risk patients: results of the GRAVITY randomized study. Atherosclerosis. 2014;232:86–93. https://doi.org/10.1016/j.atherosclerosis.2013.10.022.
Ballantyne CM, Weiss R, Moccetti T, et al. Efficacy and safety of rosuvastatin 40 mg alone or in combination with ezetimibe in patients at high risk of cardiovascular disease (results from the EXPLORER study). Am J Cardiol. 2007;99:673–80. https://doi.org/10.1016/j.amjcard.2006.10.022.
Bays HE, Davidson MH, Massaad R, et al. Safety and efficacy of ezetimibe added on to rosuvastatin 5 or 10 mg versus up-titration of rosuvastatin in patients with hypercholesterolemia (the ACTE Study). Am J Cardiol. 2011;108:523–30. https://doi.org/10.1016/j.amjcard.2011.03.079.
Kouvelos GN, Arnaoutoglou EM, Matsagkas MI, et al. Effects of rosuvastatin with or without ezetimibe on clinical outcomes in patients undergoing elective vascular surgery: results of a pilot study. J Cardiovasc Pharmacol Ther. 2013;18:5–12. https://doi.org/10.1177/1074248412445506.
Foody JM, Toth PP, Tomassini JE, et al. Changes in LDL-C levels and goal attainment associated with addition of ezetimibe to simvastatin, atorvastatin, or rosuvastatin compared with titrating statin monotherapy. Vasc Health Risk Manag. 2013;9:719–27. https://doi.org/10.2147/VHRM.S49840.
Verschuren L, Radonjic M, Wielinga PY, et al. Systems biology analysis unravels the complementary action of combined rosuvastatin and ezetimibe therapy. Pharmacogenet Genom. 2012;22:837–45. https://doi.org/10.1097/FPC.0b013e328359d274.
Masuda J, Tanigawa T, Yamada T, et al. Effect of combination therapy of ezetimibe and rosuvastatin on regression of coronary atherosclerosis in patients with coronary artery disease. Int Heart J. 2015;56:278–85. https://doi.org/10.1536/ihj.14-311.
Circular of the Center for Drug Evaluation of the State Food and Drug Administration on Issuing the “Clinical Technical Requirements for Overseas Marketed and Domestic Unmarketed Drugs” (2020 No. 29). 2022. https://www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20201016145016178.html. Accessed 24 Feb 2022.
Lee E, Ryan S, Birmingham B, et al. Rosuvastatin pharmacokinetics and pharmacogenetics in white and Asian subjects residing in the same environment. Clin Pharmacol Ther. 2005;78:330–41. https://doi.org/10.1016/j.clpt.2005.06.013.
Birmingham BK, Bujac SR, Elsby R, et al. Impact of ABCG2 and SLCO1B1 polymorphisms on pharmacokinetics of rosuvastatin, atorvastatin and simvastatin acid in Caucasian and Asian subjects: a class effect? Eur J Clin Pharmacol. 2015;71:341–55. https://doi.org/10.1007/s00228-014-1801-z.
Na Nakorn C, Waisayarat J, Dejthevaporn C, et al. Genetic variations and frequencies of the two functional single nucleotide polymorphisms of SLCO1B1 in the Thai population. Front Pharmacol. 2020;11:728. https://doi.org/10.3389/fphar.2020.00728.
Kang WY, Seong SJ, Ohk B, et al. Pharmacokinetics and bioequivalence of a rosuvastatin/ezetimibe fixed-dose combination tablet versus single agents in healthy male subjects. Int J Clin Pharmacol Ther. 2018;56:43–52. https://doi.org/10.5414/CP203026.
Kim CH, An H, Kim SH, Shin D. Pharmacokinetic and pharmacodynamic interaction between ezetimibe and rosuvastatin in healthy male subjects. Drug Des Devel Ther. 2017;11:3461–9. https://doi.org/10.2147/DDDT.S146863.
Ezzet F, Krishna G, Wexler DB, et al. A population pharmacokinetic model that describes multiple peaks due to enterohepatic recirculation of ezetimibe. Clin Ther. 2001;23:871–85. https://doi.org/10.1016/s0149-2918(01)80075-8.
Genest J. Combination of statin and ezetimibe for the treatment of dyslipidemias and the prevention of coronary artery disease. Can J Cardiol. 2006;22:863–8. https://doi.org/10.1016/s0828-282x(06)70305-1.
Acknowledgements
The authors acknowledge Dan Zhang, Medical Communication, Sanofi, China for publication process coordination; Ya Li, Sanofi, China for her contribution to the implementation of clinical trials; Haibiao Jiang, Sanofi, China for the protocol design; Jing He and Ran Hu, R&D, Sanofi, China for study conduct; and Boya Feng, Sanofi, China for CSR (Clinical Study Report) writing support. The authors would like to thank all the participants of the study. In addition, the authors would like to thank Dr. Amit Bhat, Fatemah Bensaheb, and Dr. Satya Lavanya Jakki from Indegene Pvt Ltd, India for their medical writing support with inputs from all the authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Sanofi.
Conflict of Interest
Xiaochuan Xie, Qian Wang, Chanyan Hu, Fang Xie, Mohamed Abdel-Moneim, Lionel Hovsepian, Zhihong Zhao, and Na Yang are employees of Sanofi, and they may hold shares and/or stock options in the company. The other authors have no conflicts of interest.
Author Contributions
Conceptualization: J.H., L.W., Y.L., X.X., C.H., and F.X. Data acquisition: C.J., X.X., and Z.Z.. Data analysis or interpretation: J.H., L.W., Y.L., X.X., Q.W., C.H., F.X., M.A.-M., L.H., Z.Z., and N.Y. Drafting: X.X., J.H., L.W., and Y.L. Critical revision: J.H., L.W., Y.L., C.J., X.X., Z.Z., X.X., Q.W., C.H., F.X., M.A.-M., L.H., Z.Z., and N.Y. Final approval: J.H., L.W., Y.L., C.J., X.X., Z.Z., X.X., Q.W., C.H., F.X., M.A.-M., L.H., Z.Z., and N.Y.
Ethical Considerations
The study was conducted in accordance with 1964 Declaration of Helsinki and its latest amendments, good clinical practice guidelines, and other local regulatory laws and guidelines (clinical study number CTR20202332). The study was approved by the Clinical Trial Ethics Committee of Peking University.
Consent to Participate
Written informed consent was obtained from all eligible participants before induction.
Consent for Publication
Not applicable.
Code Availability
Not applicable.
Availability of Data and Materials
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Wang, L., Luan, Y., Jia, C. et al. Pharmacokinetic Interactions and Tolerability of Rosuvastatin and Ezetimibe: A Randomized, Phase 1, Crossover Study in Healthy Chinese Participants. Eur J Drug Metab Pharmacokinet 48, 51–62 (2023). https://doi.org/10.1007/s13318-022-00798-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-022-00798-1