
Abstract
Introduction
Neurodegeneration is the term describing the death of neurons both in the central nervous system and periphery. When affecting the central nervous system, it is responsible for diseases like Alzheimer’s disease, Parkinson’s disease, Huntington’s disorders, amyotrophic lateral sclerosis, and other less frequent pathologies. There are several common pathophysiological elements that are shared in the neurodegenerative diseases. The common denominators are oxidative stress (OS) and inflammatory responses. Unluckily, these conditions are difficult to treat. Because of the burden caused by the progression of these diseases and the simultaneous lack of efficacious treatment, therapeutic approaches that could target the interception of development of the neurodegeneration are being widely investigated. This review aims to highlight the most recent proposed novelties, as most of the previous approaches have failed. Therefore, older approaches may currently be used by healthcare professionals and are not being presented.
Methods
This review was based on an electronic search of existing literature, using PubMed as primary source for important review articles, and important randomized clinical trials, published in the last 5 years. Reference lists from the most recent reviews, as well as additional sources of primary literature and references cited by relevant articles, were used.
Results
Eighteen natural pharmaceutical substances and 24 extracted or recombinant products, and artificial agents that can be used against OS, inflammation, and neurodegeneration were identified. After presenting the most common neurodegenerative diseases and mentioning some of the basic mechanisms that lead to neuronal loss, this paper presents up to date information that could encourage the development of better therapeutic strategies.
Conclusions
This review shares the new potential pharmaceutical and not pharmaceutical options that have been recently introduced regarding OS and inflammatory responses in neurodegenerative diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
This study has made clear most of the reasons why oxidative stress definitely may represent an important reason for neuroinflammation and neurodegeneration |
The recent literature is rich of proposals of natural and synthetic products that have shown some success in blocking the oxidative stress processes |
Most of the substances indicated in this paper would deserve a deeper clinical investigation. This could represent an advancement in therapy, for pathologies that till now do not have specific and successful treatments |
Introduction
The industrialized world is aging rapidly. The European Union (EU) Joint Programme–Neurodegenerative Disease Research (JPND) states that by the year 2030, a quarter (25%) of the European population will be over age 65, a significant increase over the current 16% [1]. The implications for a “graying” Europe are not yet fully understood, but surely include an increase in debilitating and challenging-to-treat neurodegenerative diseases. Included among these is the devastating condition of Alzheimer’s disease, the most prominent subpopulation of dementia patients [1]. Other forms of neurodegenerative diseases include amyotrophic lateral sclerosis (ALS), Parkinson’s disease, spinal muscular atrophy, Huntington’s disease, prion diseases, motor neuron diseases, and spinocerebellar ataxia [1]. By and large, neurodegenerative disease treatment—where options exist at all—address symptoms rather than underlying pathophysiology.
The collective term neurodegenerative disease refers to a myriad of conditions that affect the neurons in the brain. Typically, neurodegenerative diseases are irreversible, progressive, and associated with loss of function [2]. Typical physiological signs of neurodegenerative conditions are demyelination, loss of dendrites, and neuronal death [3, 4]. As the neuronal structures deteriorate, a gradual and progressive loss of cognitive skills (dementia) and/or motor skills (ataxia) ensues, which can result in mental impairment, functional loss, and debilitation [1]. Although neurodegenerative diseases are most prevalent in the elderly, they can occur in patients at all ages [5,6,7,8]. The underlying pathophysiology common to all forms of neurodegenerative disease seems to involve neuroinflammation and oxidative stress (OS) [4, 9]. This major challenge for modern medicine is to find ways to effectively manage neurodegenerative diseases in patients who are living longer and may be multimorbid [3]. The objective of this review was to examine new approaches to these neurodegenerative diseases, specifically in relation to new therapeutic strategies involving neuroinflammation and OS, with the aim of providing an overview that may give insight to clinicians caring for patients in these challenging clinical situations. This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by the authors.
Methods
The electronic database PubMed was searched on 21 February 2019. Our search terms included “Oxidative Stress,” “Inflammatory Responses,” “Neurodegenerative Diseases,” “Parkinson,” “ALS,” “Huntington,” “Alzheimer,” “Spinal muscular atrophy,” “Prion disease,” and “Spinocerebellar ataxia”. Inclusion criteria were papers published in the last 5 years, English language, full text available, and most recent. This review also examined the bibliographies from the most recent reviews and additional sources of primary literature, as well as references cited by relevant articles. For the management of neurodegenerative diseases and OS, inclusion criteria were (1) the article had to present a specific treatment of the aforementioned diseases, either natural, extracted, recombinant products, or artificial agents; (2) the treatment had to target OS and inflammation; and (3) the substance must have been investigated in the last 5 years. These inclusion criteria were selected because to date, no therapies have been able to cure or reverse neurodegenerative diseases. This is a narrative and not a systematic review.
Results
The initial search identified 147 articles from which 95 were included as relevant. Moreover, 54 additional articles were included from the secondary search of the primary literature reference lists reviews.
Neurodegenerative Diseases and Their Pathogenesis
For a better understanding of the therapeutic approaches that will be presented, a short summary of the neurodegenerative diseases and their pathogenesis is helpful.
Alzheimer’s Disease
The most frequently occurring neurodegenerative disease is Alzheimer’s disease. The prominent symptoms of Alzheimer’s disease include progressive and irreversible cognitive deficits which can be accompanied by changes in mood and behavior [9]. Memory loss is common as the disease advances. Alzheimer’s disease has been associated with tangled strands of intracellular tau known as neurofibrillary tangles and extracellular deposits of amyloid-β (Aβ) plaques in the brain [10]. The plaque deposits have been linked to OS, which may play a substantial role in AD pathogenesis [11,12,13].
Parkinson’s Disease
Parkinson’s disease is likewise an irreversible, progressive neurodegenerative disease [14] that affects about 1% of people over age 50 [15]. As Parkinson’s disease progresses, the patient loses 50% to 70% of all dopaminergic neurons in the substantia nigra. With the loss of the brain’s dopaminergic fibers, the patient develops progressively worsening motor symptoms, which are a hallmark of Parkinson’s disease [9]. Growing evidence has implicated OS as well as immunological shifts in the pathogenesis of Parkinson’s disease. As excessive amounts of reactive oxygen species along with other free radicals accumulate, they overwhelm the dopaminergic neurons and cause them to deteriorate, setting the stage for the pathology of Parkinson’s disease [16]. A number of potential contributors to OS have been implicated in the disease process of Parkinson’s disease, namely mitochondria, endoplasmic reticulum, α-synuclein, and dopamine. It appears likely that it is their interplay rather than their individual actions that contribute to progressive neurodegeneration [17]. The role of neuroinflammation may be crucial to the pathogenesis of Parkinson’s disease, in that it has destructive effects on the nigrostriatal dopaminergic pathways. This, in turn, activates the glial cells of the brain (mainly the microglia and the astrocytes) to release various soluble factors which may be pro-inflammatory and/or neurotoxic [18].
Multiple Sclerosis
Multiple sclerosis, a chronic progressive neurodegenerative disease, is characterized by inflammation and demyelination [9]. The destruction of the myelin sheaths of the nerves as well as axonal damage and glial scarring produce its classic symptoms of muscle spasms, neuropathic pain, paralysis, and optic neuritis. During this process, an inflammatory state is promoted, mainly via myelin antigen-specific TH cells [19]. As lesions form, monocytes are recruited, which in turn produce reactive oxygen species. Observations of white matter and cerebral cortex lesions suggest that demyelination and neurodegeneration might lead to the presence of oxidized lipids in the myelin membranes, in apoptotic oligodendrocytes, and in neuronal axons [20, 21].
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) involves the progressive and irreversible degeneration of the motor neurons located in the brain cortex, brain stem, and spinal cord [22]. Most patients diagnosed with this condition will die within 3–4 years after the onset of symptoms [23]. The world-famous physicist Stephen Hawking is the exception, surviving over 50 years after his ALS diagnosis. As the name suggests, ALS involves muscular atrophy (amyotrophic) and scarring of the spinal cord (lateral sclerosis) [24]. Patients with ALS suffer progressive paralysis, dysphagia, and respiratory insufficiency. ALS is thought to be caused by any or several of 150 mutations in the superoxide dismutase 1 (SOD1) antioxidant system [25] which cause injury and death of motor neurons [26] and mitochondrial dysfunction [23]. The SOD1 mutation involves misfolding of the protein.
Motoneuron injury is a characteristic of ALS but its etiology has not been thoroughly elucidated. Extracellular mutant SOD1 is associated with motoneuron injury, but it is not known if this injury is the result of mutant SOD1 acting directly on the motoneurons or if there is some sort of indirect mediation via microglia activated by mutant SOD1. Activated microglia are presents at sites of neuronal injury [25].
Duchenne Muscular Dystrophy
Duchenne muscular dystrophy is a progressive, fatal, X-linked genetic disorder of chronic inflammation involving dystrophin gene mutations. OS is a contributor to the pathogenesis of Duchenne muscular dystrophy [27]. The muscle wasting associated with Duchenne muscular dystrophy involves the nuclear factor erythroid 2 (NF-E2)-related factor 2 along with increased levels of oxidized glutathione, combined with the accumulated oxidative damage acquired with aging [27]. The enzyme heme oxygenase 1 (HO-1) and the cytokine interleukin (IL)-6 suggest that this OS is associated with chronic inflammatory response [27].
Pathogenesis of Neurodegenerative Diseases: Main Common Mechanisms
The molecular mechanisms behind neurodegenerative diseases suggest intriguing aspects of their pathology [28,29,30,31,32]. The primary reasons for neurodegeneration appear to involve abnormal processing of proteins [28], genetic disorders [5], misfolding and aggregation of various proteins [32], activating cellular apoptosis [33], triggering mitochondrial dysfunction, creation of free radicals, and OS [32].
Link Between OS and Neurodegenerative Diseases
As more is learned about neurodegenerative diseases, OS appears to be a main contributor to disease pathogenesis [32]. OS occurs when the oxidant molecules produced by the body are not adequately counterbalanced by the body’s natural antioxidant defenses. During times of OS, free radicals overwhelm the body’s endogenous antioxidant defense system, leading to potentially ill effects. Free radicals are natural, physiological by-products of the normal aerobic metabolism, but under pathological conditions, free radicals can be produced in abnormally large—and harmful—amounts [34]. A growing body of evidence supports the idea that OS leads to the development of neurodegenerative diseases [32, 35,36,37]. OS might be defined as an elevated concentration of free radicals above the threshold recognized as being toxic [38]. In other words, OS might occur because of excessive radical oxygen species production or attenuated antioxidant defenses or a combination of both [16, 38]. The way in which this might occur is the subject of a few hypotheses. For example, a mutation in SOD1 might cause oxides to be overproduced [39]. Another hypothesis states that disrupted glutathione production and homeostasis might promote OS [38]. A systematic review of neurodegenerative disease mechanisms found that the generation of free radicals triggered the activation of multiple noxious pathways associated with the development of neurodegenerative diseases via multiple different mechanisms [40]. These multiple mechanisms, possibly seven or more, interact with each other in complex and not thoroughly elucidated ways. It is the interplay of these mechanisms rather than any one mechanism in isolation that may promote neurodegeneration [40]. On the basis of these ideas, a potential target for drug development to treat neurodegenerative disease would be agents that are able to modulate free radical production [40].
Inflammation
Inflammation is the normal, natural, expected, and adaptive reaction of the body to injury with the purpose of encouraging both the removal of the harmful stimulus and self-protection. Although the inflammatory response is integral to the immune system, imbalances can damage host tissue and even lead to organ dysfunction [41]. As a general rule, inflammation is only problematic when it fails to subside within the appropriate time [42]. Neuroinflammation occurs when the peripheral nervous system (PNS) and/or central nervous system (CNS) becomes inflamed, and this potentially insidious condition can set the stage for the development of chronic neurodegenerative diseases [43].
CNS Environment Alterations
As part of the healthy immune system, the CNS responds rapidly to injury and infection [42, 44]. In conjunction with pathological situations, cells from the body’s peripheral immune system, such as macrophages, are able to cross the blood–brain barrier (BBB) [42]. In fact, one of the most crucial recent advances in medicine has been the discovery and elucidation of an elaborate cross-talking communications network between the immune system and the CNS. In this network, an array of pro-inflammatory cytokines acts to cause the host to respond swiftly to stress, trauma, infection, and pathology via the inflammatory response. This complex cascade of responses involves activities from glial cells and mast cells, which act as inflammatory mediators. These inflammatory mediators may well play a role in the development and perpetuation of chronic pain, neurodegenerative disease, and neuropsychiatric disorders [41, 45]. A disturbance in the CNS environment, for example an infection or a neuronal injury, will activate the microglia and astrocytes. On the basis of the extent and degree of this disturbance, the cells may first respond by producing and releasing cytokines. Cytokines are important neurotropic factors that have only recently been elucidated, albeit incompletely. Prolonged neuronal damage may cause the microglia and astrocytes to release pro-inflammatory cytokines which, in turn, recruits the immune system to action and produces a localized inflammatory response. Furthermore, these activated microglia and astrocytes produce reactive oxygen species. On the one hand, reactive oxygen species can defend against microbial infection but, on the other hand, it can contribute to neurodegenerative processes [46,47,48]. When mitochondrial impairment causes an excessive buildup of reactive oxygen species, this may damage neurons and release cytosolic factors that serve to activate neighboring microglia and astrocytes. These nearby microglia and astrocytes will, in turn, release more pro-inflammatory cytokines along with reactive oxygen species and reactive nitrogen species. The release of these cytokines promotes the inflammatory response and exacerbates neuronal damage. Thus, the ongoing activation of glial cells creates a vicious circle, which both perpetuates and amplifies chronic neurodegenerative processes [49].
Microglia as Part of Immune System
Microglia serve an immunoprotective role by scavenging for damaged neurons, infectious agents, and other potentially injurious substances [50]. It is thought that microglia help with the neuroplasticity of brain remodeling during learning and memory, possibly through neurotrophic factors. Microglia also promote overall healthy brain physiology [51]. Thus, microglia are associated with numerous essential neuronal functions, such as, but not limited to, maintaining appropriate brain function, scavenging for neuronal debris, promoting neuroplasticity, and releasing specific neurotrophic factors [52, 53]. Under normal conditions, the microglia in the CNS exist in a physiological branched form, but when stimulated, they become pro-inflammatory [54]. When these microglia become pro-inflammatory, their cell bodies increase in size and they abandon their usual functions in order to promote inflammation. Among other things, this means they cease monitoring and maintaining the brain, which may lead to neuronal disruption or damage. In the event that inflammation is uncontrolled and/or becomes chronic, functional deficits can result [17]. When CNS homeostasis is disturbed, microglia may release various cytokines and growth factors to facilitate the repair of affected neurons. While microglial activation is an essential component of the healthy immune system, abnormal or prolonged microglial activation becomes maladaptive. During abnormal microglial activation, a host of pro-inflammatory cytokines and cytotoxic mediators may be induced, including but not limited to IL-12, IL-1β, IL-6, tumor necrosis factor (TNF) alpha, and reactive oxygen species. The onslaught of these substances can injure the brain by damaging neurons, and, in that way, promote neurodegenerative diseases [55,56,57]. Neurodegenerative diseases are associated with advancing age [1], because in younger individuals microglia activation is typically modest and resolved quickly, so that brain homeostasis is rapidly restored, but older individuals have a primed phenotype of microglia [54] which has a more powerful and prolonged response, resulting in the production of larger quantities of pro-inflammatory cytokines over a longer period of time (Fig. 1). These primed microglia are associated with persistent neuroinflammation, which may damage brain tissue and impair neuronal function [58] (Fig. 2).

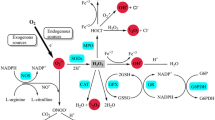
Oxidative stress and mitochondrial damage have been implicated in the pathogenesis of several neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Oxidative stress is connected with immune and glial cells crosstalk-induced inflammation. Oxidative stress and inflammation feed on each other and together contribute to the development of neurodegeneration. Oxidative stress induces activation of microglia and astrocytes with a consequent increase of pro-inflammatory mediator production and, in turn, glial activation leads to toxic radical release, exacerbating neuronal damage. Consequently, the resultant cellular damage amplifies the inflammatory response, with glial activation and leukocyte recruitment, leading to further inflammation. RNS reactive nitrogen species, ROS reactive oxygen species

Differences between resting and primed microglia. The phenotype of microglia in aging is predominantly primed. This phenotype responds to stimuli in a more intense manner, i.e., producing greater amounts of pro-inflammatory mediators and for extended periods. Primed microglia induce persistent neuroinflammation, capable of damaging tissue integrity and neuron function. Copyright, with permission from Pain Nursing Magazine
Role of TNF in OS, Inflammatory Response, and Neurodegeneration
TNFα (once known as cachectin) is part of the TNF superfamily and is a cytokine of the immune system with many functions, including initiating and promoting inflammation. While TNFα plays an important role in the healing process, uncontrolled it can bring about neurodegeneration. The activation of the pathway for nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in glial cells leads to the elevated production of pro-inflammatory cytokines, which, in turn, results in the production of the inducible form of nitric oxide (iNOS), cyclooxygenase (COX)-2, and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits, thereby activating NADPH oxidases, ultimately leading to the production of reactive oxygen species. Additionally, reactive oxygen species can activate the NF-κB pathway, thereby amplifying the TNF/reactive oxygen species/NF-κB responses. This, in turn, exacerbates neuronal damage, which further promotes neuroinflammation, resulting ultimately in a feed-forward loop that causes and perpetuates chronic neurodegeneration [49].
Key Role of Mitochondria
Reactive oxygen species are produced by the mitochondria they damage. Normally, reactive oxygen species are produced by physiological mitochondrial activity. If mitochondria are damaged, the ETC Complex I (nicotinamide adenine dinucleotide [NADH] dehydrogenase) and Complex III (cytochrome bc1) may produce an overabundance of reactive oxygen species, which can damage cells [17]. Newly discovered genes related to Parkinson’s disease, such as PINK1, PARK2 (Parkin), DJ-1, and LRRK2, encode proteins that help to regulate the balance between mitochondria and reactive oxygen species. For that reason, mitochondrial damage can dysregulate this balance and potentially lead to any number of neurodegenerative diseases, including Parkinson’s disease [59,60,61,62,63]. Mitochondria are vulnerable to damage related to OS, as they are some of the body’s main sites for the production of reactive oxygen species. Mitochondrial DNA (mtDNA) is not protected by histone proteins like nuclear DNA, meaning that mtDNA is more susceptible to oxidative damage [64]. The aging process exposes mtDNA to damage which can be 10–20-fold greater than the damage to nuclear DNA at the same age; the production of reactive oxygen species also increases with advancing age [65, 66]. The majority of the proteins encoded by mtDNA are involved in ETC, so any mutations or deletion of the mtDNA would challenge the equilibrium of the ETC and result in increased production of reactive oxygen species, thereby perpetuating the cycle of increasing mitochondrial damage [67].
Viral Insult
Neurodegeneration is known to occur following viral insult. This suggests that the microglia and astrocytes play a key role in the process of neuronal disruption. Viruses have two main prongs of attack: they can injure neurons by direct action (causing neuronal damage) or they can induce apoptosis in neuronal cells (causing neuronal death). These viral assaults may lead to neurodegenerative conditions. The neurodegenerative diseases of the elderly, such as Alzheimer’s disease, multiple sclerosis, and ALS, may occur as a result of the viral-induced neuronal apoptosis [68].
Currently Proposed Treatments
Natural Pharmaceutical Approaches (Table 1)
Pomegranate (Punica granatum)
The pomegranate fruit, which originated in India but is today cultivated around the world, is rich in antioxidants. The primary polyphenol antioxidant of the pomegranate is punicalagin (PN) or 2,3-hexahydroxydiphenoyl-gallagyl-d-glucose. As a natural antioxidant, PN offers many health benefits and has an anti-inflammatory effect. The potential neuroprotective benefit of PN in the treatment of Parkinson’s disease has been described in the literature [69].
Medicinal Plants
Traditional Persian medicine (Falij, Ra’shah, Nisyān, Khadir, Istirka) emphasizes the role of certain plants for preventing and treating neurological conditions. These botanicals use cellular and molecular mechanisms to slow or stop apoptosis, including anti-apoptotic agents (BCL-2) or by decreasing the expression and activity of pro-apoptotic proteins (BAX, caspase-3, and caspase-9). These traditional plants are thought to alleviate inflammation and reduce the production of pro-inflammatory cytokines (TNFα, various IL cytokines) while at the same time promoting antioxidation by enhanced production of superoxide dismutase and catalase. These traditional plants can modulate the transcription, transduction, and signaling of various inflammatory pathways (including ERK, p38, MAPK). By regulating these inflammatory cascades far upstream, they can help limit apoptosis and OS and, in this way, may help prevent or treat neurodegenerative conditions [70].
Vitamin D
Vitamin D reduces OS, offers both neuroprotective and anti-inflammatory actions, and may exert preventive effects on neurodegenerative diseases [71].
Vitamin C
When vitamin C (200 and 400 mg/kg body weight) was administered to rats who had colchicine-induced Alzheimer’s disease (cAD), the rats recovered some memory deficits, hippocampal neuroinflammation was reduced, and further neurodegeneration was prevented. The hippocampal neuroinflammation had had an adverse effect on the peripheral immune responses which was also recovered. However, when the rats with cAD were administered higher doses of vitamin C (600 mg/kg), memory impairments resulted along with hippocampal neuroinflammation, and neurodegeneration along with altered peripheral immune responses. These are all pro-oxidant, rather than antioxidant, effects [72].
Bakkenolide B
Bakkenolide B is a natural substance obtained from the Petasites japonicus plant which has been shown to suppress both inflammatory and allergic responses. Treatment with bakkenolide B reduced production of microglia, IL-1β, IL-6, IL-12, and TNFα [73].
Mediterranean Diet and Extra-Virgin Olive Oil
A review of scientific evidence about the Mediterranean diet and its emphasis on extra-virgin olive oil found this diet may decrease the risk for neurodegenerative diseases [74]. The health benefits of olive oil were presented in 2016 by Parkinson and Cicerale [75]. Their work came after an announcement that phenolic compounds such as oleocanthal or oleuropein found in olive oil might confer neuroprotection and possibly prevent Alzheimer’s disease [76, 77]. In fact, the Prevención con Dieta Mediterránea study (PREDIMED) found that the Mediterranean diet with extra-virgin olive oil and nuts was associated with improvements in cognition [78]. The Mediterranean diet may benefit patients at all stages of Alzheimer’s disease by slowing the rate of cognitive decline, delaying dementia, and reducing the risk of death [79]. Moreover, other benefits of extra-virgin olive oil include neuroprotective effects in the setting of depression, Parkinson’s disease, traumatic brain injury, and multiple sclerosis [80, 81].
Flavonoid-Rich Ethanol Extract from Leaves of Diospyros kaki
Flavonoid is a collective term for a diverse group of phytonutrients found in most fruits and vegetables. Overall, flavonoids act as antioxidants and offer neuroprotection by fighting neurotoxins. Flavonoids can reduce Aβ production. Because flavonoids in general reduce or inhibit neuroinflammation, they tend to promote proper function of the cerebrovascular system and, in that way, may improve cognition [82].
Curcumin
Recently gaining in popularity, curcumin (found in the turmeric plant) has known anti-inflammatory, antioxidative, and antitumor properties. Widely used around the world as an herbal remedy, curcumin or turmeric is increasingly recommended by Western medicine as a pain reliever. It may also confer neuroprotection [83]. Turmeric is also used as a spice and is particularly popular in Indian cuisine.
Resveratrol
Resveratrol (3,4′,5-trihydroxy-trans-stilbene) is a phytonutrient found naturally in about 70 plants, including the skin and seeds of grapes. In addition to the powerful antioxidant effect of resveratrol, its antitumor activity is currently being studied in cancer patients. Resveratrol has demonstrated neuroprotective properties in several in vivo and in vitro models because it inhibits glutathione depletion. Glutathione is an antioxidant produced by the CNS involved in numerous cellular processes. When glutathione is depleted, the neuronal system becomes vulnerable [84].
Aged Garlic Extract (AGE)
Aged garlic extract (AGE) is made when the garlic plant Allium sativum is marinated under ambient conditions in aqueous ethanol for four or more months. Through this process, much of the sharp taste and offensive odors of the garlic plant are eliminated and, more importantly, the antioxidative properties of the product are amplified. Neuroprotective benefits in Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and cerebral ischemia have been observed with AGE [85]. AGE appears to act on the signaling pathways involved in OS and neuroinflammation, although this mechanism of action remains to be more fully elucidated [85]. Two components of AGE, S-allyl-l-cysteine and N(α)-(1-deoxy-d-fructos-1-yl)-l-arginine, seem to attenuate neuroinflammation in the microglia by modulating the nuclear factor erythroid 2-like (Nrf2)-mediated pathways as well as attenuating other forms of oxidative stress signaling [85].
Phytochemicals and Botanical Extracts
NF-κB is a transcription factor involved in multiple physiological and pathological conditions. Among other things, NF-κB regulates DNA transcription, production of cytokines, and plays a role in cell survival. Thus, NF-κB is directly or indirectly involved in immunological responses, apoptosis, inflammatory processes, and carcinogenesis. The role of NF-κB and Nrf2 was investigated to better elucidate their contribution to OS and inflammation associated with neurodegenerative diseases. Botanical polyphenols have been shown to inhibit lipopolysaccharide (LPS)-induced nitric oxide responses and enhanced antioxidant responses modulated by Nrf2 [86]. Using an immortalized rat astrocyte (DI TNC1) cell line which expressed a luciferase reporter driven by the NF-κB or their Nrf2/antioxidant response element (ARE), investigators showed how these two pathways could be regulated by phytochemicals (such as quercetin, rutin, cyanidin, cyandini-3-O-glucoside) and botanical extracts from Withania somnifera (ashwagandha), Sutherlandia frutescens (sutherlandia), and Euterpe oleracea (açaí). All three of these botanical extracts inhibited LPS-induced NF-κB-reporter activity. For example, quercetin inhibited the LPS-induced NF-κB reporter activity on the one hand and induced reporter activity of Nrf2/ARE on the other hand. Cyanidin and glycoside had similar effects but only when used at much higher concentrations. Investigators stated that ashwagandha extract was more effective than sutherlandia and açaí extracts in promoting Nrf2 and HO-1 expression in DI TNC1 astrocytes, which illustrates how botanical polyphenols and extracts have different abilities in terms of downregulating LPS-induced NF-κB and upregulating NRF2/ARE [87].
Hericium erinaceus
Hericium erinaceus is a mushroom that was found to confer certain neuroprotective properties in animal studies. Rats administered the mushroom orally were found to have upregulated lipoxin A4 (LXA4) in various parts of their brains. LXA4 is thought to help reduce inflammation by halting the inflammatory processes and increase redox-sensitive proteins in response to cellular stress, such as heat shock proteins, HO-1, and thioredoxin [88]. Heat shock proteins have been implicated as a potential pathway for the prevention of protein misfolding, which has been associated with certain types of irreversible neuronal damage. It has been suggested that agents that can activate LXA4 signaling would be able to modulate the vitagene proteins that respond to stress cues; in that way, agents that activate LXA4 might be good targets for reducing the neuroinflammation and neurodegeneration associated with Alzheimer’s disease [88].
Walnut (Juglans sigillata Dode)
Peptides from the walnut have been studied for their hypothesized ability to protect against neurotoxicity. Mice were injected bilaterally in the hippocampus with amyloid-β peptide 25–35 to create an animal model of Alzheimer’s disease [89]. Mice were then treated orally with walnut peptides (200, 400, 800 mg/kg) or distilled water (placebo) for 5 weeks. The water maze and step-down avoidance tests were used to test the mice at 5 weeks for spatial learning and memory. Walnut peptides improved the cognitive abilities and memories of the animals compared to placebo. Furthermore, the two higher-dose walnut peptide groups (400 or 800 mg/kg) had restored levels of antioxidant enzymes and inflammatory mediators. This in vivo study suggests that walnut peptides might exert a neuroprotective effect in Alzheimer’s disease by reducing neuroinflammation and enhancing the endogenous antioxidant system [89].
l-3-n-Butylphthalide
One of the chemical components from Chinese celery (Apium graveolens L.), l-3-n-butylphthalide (l-NBP), has shown neuroprotective benefits in animal models of stroke and Alzheimer’s disease [90]. A synthetic version of this substance is an approved anti-ischemic agent available in China. In a preclinical study, C57BL/6 mice were administered systemic LPS that resulted in the release of pro-inflammatory cytokines and activated cerebral microglia. When these mice were treated with l-NBP, the production of pro-inflammatory cytokines such as TNFα, IL-1β, ΙL-6, and IL-10 was suppressed, and the morphological activation of microglia was decreased [91]. The phosphorylation level of the Janus kinase (JAK) mitogen-activated protein (MAP) pathways was also inhibited with the administration of l-NBP in these mice. HO-1, which helps to catalyze the degradation of heme and has anti-inflammatory and antioxidative effects, was also evaluated in this murine study and found that l-NBP was associated with an upregulation of HO-1 expression.
Gastrodia elata Blume
Highly regarded in traditional Chinese medicine, the tianma flower from the orchid family (Gastrodia elata Blume) has been used in Chinese folk medicine for centuries to treat a wide range of complaints ranging from headache to convulsions. Today it is known for its analgesic and sedative effects, and it shows promise in the treatment of neurological disorders. An extract from its rhizome has been observed to provide neuroprotective effects. In an animal model of brain injury, this extract helped brain cells to recover their function, likely by inhibition of both OS and the inflammatory response [92].
Chrysophanol
Chrysophanol is an anthraquinone well known in traditional Chinese and Korean medicine. It is widely distributed around the world in two main natural domains: Bacteria and Eukarya. It has a reputation for benefits in anticancer, hepatoprotection, neuroprotection, anti-ulcer, and antimicrobial applications. Its neuroprotective effects and its ability to reduce OS were evaluated in vitro with LPS-induced inflammatory responses in bovine BV2 cells. In this study, chrysophanol reduced the expression of iNOS and COX-2, and, in that way, nitric oxide and prostaglandin E2 production were reduced [93]. Chrysophanol inhibited DNA oxidation and was also able to reduce cell damage in these BV2 cells caused by intracellular reactive oxygen species. To measure the overall antioxidant effect, the expression levels of various antioxidative enzymes, such as superoxide dismutase, and glutathione were calculated. Chrysophanol was shown to exhibit potential anti-inflammatory and antioxidative effects in the microglia, making it a potentially valuable therapeutic product in the treatment of neurodegenerative diseases [93].
Marine Carotenoid Astaxanthin and Omega-3 Fatty Acids
Seafood is frequently recommended by dieticians for its health benefits, most of which come from omega-3 and omega-6 polyunsaturated fatty acids (PUFAs), n-3/n-6 PUFAs, and antioxidants. The ratio of omega-3 to omega-6 PUFAs is more important to health than their individual quantity. The intake of the proper ratio of omega-3 to omega-6 PUFAs can allow for effective anti-inflammatory responses that may be able to prevent or delay neurological disorders. Early in vivo and clinical studies using the marine carotenoid astaxanthin found in salmon, shrimp, and lobster show that it can protect against neuronal damage caused by free radicals that can lead to neurodegenerative processes and cognitive deficits [94]. Nutraceutical products have been created with a balanced ratio of omega-3 and omega-6 PUFAs combined with astaxanthin as protection against neurodegeneration caused by OS in the CNS. Other antioxidants such as krill oil can be combined in such products.
Palmitoylethanolamide (PEA)
Palmitoylethanolamide (PEA) is an endogenous lipid that was first brought to the attention of physicians when the anti-inflammatory effects of egg yolks were studied. (PEA occurs naturally in egg yolks.) PEA is available today as a “special food for medical purposes,” as registered by some pharmaceutical agencies. It down-modulates mast cell activation, allowing it to exert control over certain glial cell behaviors. PEA, an acylethanolamide found in diverse bodily tissues, including the nerve cells, can be synthesized by the body as needed. In fact, endogenous PEA levels vary regularly in the body, for example with stress, injury, or pain [95]. PEA is associated with reducing mast cells, down-modulating the pro-inflammatory mediators released by these mast cells, and suppressing microglial cell activation. These activities are thought to be the reason PEA is associated with anti-allodynic and anti-hyperalgesic effects. At the molecular level, PEA is a peroxisome proliferator-activated receptor alpha (PPARα) ligand with anti-inflammatory, analgesic, and neuroprotective properties [96]. Clinical trials using PEA in humans have shown promise. For example, PEA was effective in a preliminary trial of patients with failed back surgery syndrome [96] and in another study of patients with fibromyalgia [97] because of its anti-inflammatory properties. These neuroprotective and anti-neuroinflammatory properties make PEA an interesting substance to consider as a novel therapy for Alzheimer’s disease. Beggiato and colleagues found that astrocytes contribute to the neurotoxicity induced by amyloid-β42 (Aβ42); PEA could blunt the activation of Aβ42 and, in that way, induce astrocyte activation and improve neuronal cell survival in murine astrocyte–neuron co-cultures [98]. On the basis of the neuroprotective attributes exhibited in experimental models of neurodegenerative diseases, an ultra-micronized (um) form of PEA has been developed and investigated for the treatment of cognitive impairment [68]. In these studies, it was found that PEA may be a component of the homeostasis involved in the resolution of neuroinflammation. In an animal model of Parkinson’s disease, chronic PEA treatment was able to counteract behavioral impairments, motor dysfunction, loss of nigrostriatal neurons, astrocyte activation, altered iNOS expression, and proteins associated with the microtubules. In this Alzheimer’s disease model, when Aβ25–35 peptides were injected into the intracerebral ventricles of the murine brain, PEA treatment could ameliorate behavior impairments and reduce lipid peroxidation, iNOS, and caspase-3 activation [68]. In human patients with ALS, PEA improved their clinical conditions and delayed tracheotomy and death [68].
Extracted or Recombinant Products and Artificial Agents (Table 2)
Melatonin
As a natural substance that acts like a neurotransmitter, melatonin plays a role in a variety of bodily functions. Produced endogenously and mainly but not exclusively by the pineal gland, melatonin has known benefits for treating sleep disorders, boosting natural immunity, and offering anti-aging benefits as well. An indolamide, melatonin is perhaps best studied for its regulation of circadian rhythms, but it may be involved in a host of other functions such as oxidation, apoptosis, and mitochondrial homeostasis. When inflammatory pathologies disrupt the last three processes, melatonin appears to offer some therapeutic benefits, particularly in patients with neurodegenerative diseases or bowel disease. Among the neurodegenerative conditions in which melatonin has been studied are Alzheimer’s disease, ALS, multiple sclerosis, and Huntington’s disease; melatonin may also be effective in treating patients with ulcerative colitis [99].
Curcumin/Melatonin Hybrid 5-(4-Hydroxy-phenyl)-3-oxo-pentanoic Acid [2-(5-Methoxy-1H-indol-3-yl)-ethyl]-amide (Z-CM-I-1)
The combination of curcumin plus melatonin as an oral hybrid compound might be effective as a disease-modifying agent in the treatment of Alzheimer’s disease. In an in vivo study using a transgenic model of Alzheimer’s disease, Z-CM-I-1 administered at 50 mg/kg orally over 12 weeks decreased the accumulation of amyloid-β in the hippocampal and cortex regions of the brain and it further reduced inflammatory response and OS. In addition, with Z-CM-I-1 there was significant improvement of synaptic dysfunction (as shown by increased expression of synaptic marker proteins, PSD95, and synaptophysin which shows protective effects against synaptic degeneration). Z-CM-I-1 also increased the expression levels of complexes I, II, and IV of the mitochondrial electron transport chain in the brain tissue of mice [100].
Thioredoxin-Interacting Protein (TXNIP)
TXNIP is produced endogenously and inhibits thioredoxin, a thiol-reducing and antioxidative system in the body. In the healthy individual, the role of TXNIP is to control the processes of stress and inflammation induced by redox and glucose. During cerebrovascular accident and brain disease, TXNIP is upregulated. Recent studies suggest that pharmacological inhibition or even genetic deletion of TXNIP can serve to protect the neurological system, possibly reducing the adverse effects of neurodegenerative disease or stroke [101].
Mitochondria-Targeted Molecules
In the early stages of Alzheimer’s disease, patients are known to experience synaptic pathologies and mitochondrial oxidative damage. For this reason, mitochondria-targeted molecules were conceived as therapeutic targets that might delay or prevent the advancement of Alzheimer’s disease. Mitochondria-targeted molecules are designed to deliver treatment directly to the mitochondria, reducing reactive oxygen species and mitochondrial dysfunction and, in that way, preventing or delaying neurodegeneration. Mitochondria-targeted molecules are cell-penetrating triphenyl phosphonium lipophilic cation-based derivatives of tetrapeptide molecules or choline esters of glutathione and N-acetyl-l-cysteine [102].
Hydrogen Sulfide (H2S)
Receptors for advanced glycation end products (RAGE) are receptors expressed in numerous CNS and peripheral cells, including neurons, astrocytes, microglia, and others. These receptors bind to a select number of ligands, including non-enzymatically glycated proteins and certain lipids. The importance of RAGE is its critical role in the inflammatory process and the possible role it may play in neurodegeneration. H2S inhibits dimer formation of RAGE and, in that way, impairs the stability of its membrane. By reducing the RAGE membrane, H2S may protect cells against the adverse effects of inflammation [103, 104]. Its role in the treatment of various neurodegenerative diseases remains to be clarified.
Heat Shock Proteins
Heat shock proteins (HSPs) took their name when a family of proteins was discovered which was created during heat shock. Although heat shock proteins retain their old name, it is now known that they are produced in response to any number of external and internal stressors including thermal stress, wound healing, tissue remodeling, and others. These ubiquitous HSPs are found in nearly all living organisms and many of them have a chaperon function, whereby they help ensure the proper folding of new proteins that may be formed as a result of stress. Protein misfolding can result in permanent damage, including irreversible neurological damage. In addition to protecting cells against damage caused by inflammation, HSPs assure proper folding of new proteins and destroy misfolded proteins.
During periods of disease, HSP production may be upregulated to prevent pathology-related damage. HSP is also upregulated in response to the accumulated stress of aging. Certain HSPs can produce extracellular messengers called chaperonins to aid in modulating the immune responses of the CNS. Since HSPs are able to modulate the body’s natural immune system, they may have specific neuroprotective benefits. For these reasons, HSPs have been the subject of scientific attention as they may be able to play an important therapeutic role in the treatment of neurodegenerative diseases [105].
Intranasally Administered Erythropoietin (Neuro-EPO)
Erythropoietin (EPO) is a cytokine secreted primarily by the kidneys and liver which has certain cytoprotective actions in the brain, protecting it from inflammatory and neurodegenerative assaults. An intranasal formulation of erythropoietin has been developed and is marketed as Neuro-EPO. Neuro-EPO was evaluated for its neuroprotective properties in a transgenic mouse model of Alzheimer’s disease [106]. Low-dose Neuro-EPO was able to restore deficits in memory, learning, and the recognition of new objects. In addition, it reduced neuroinflammation, apoptosis induction, OS, and amyloid load along with alleviating altered memories.
Cyclo(His-Pro)
Thyrotropin-releasing hormone (TRH) is synthesized by the hypothalamus and confers neuroprotective benefits. Spontaneous cyclization of TRH produces His-Pro, a cyclic dipeptide. Cyclo(His-Pro), the cyclic dipeptide histidyl-proline, can be synthesized de novo endogenously and has been found throughout the body, in particular in the CNS, blood, and gastrointestinal tract. Cyclo(His-Pro) regulates glial cells and can cross the blood–brain barrier. In the brain, cyclo(His-Pro) can affect a variety of inflammatory and stress responses because of its effect on the Nrf2–NF-κB signaling axis. It has been hypothesized that cyclo(His-Pro) has the ability to interfere with many of the biological processes driving neurodegeneration and may be an important substance to study for the potential restoration of neuronal integrity. Cyclo(His-Pro) is also being studied for its possible role in the treatment of ALS [107].
Regulator of G-Protein Signaling (RGS): RGS1 and RGS10 Proteins
Guanine nucleotide-binding proteins (G proteins), part of a large family of GPPase enzymes, transmit signals of extracellular origin to the cell’s interior. A G protein is able to bind to guanosine triphosphate (GTP), which essentially opens the signaling switch; when they bind to guanosine diphosphate (GDP), the switch is turned off. Regulators of G-protein signaling (RGS) were first thought to use the proteins that accelerated the GRPase processes in order to act as negative regulators of G-protein-coupled receptor signaling. All RGS proteins contain some conserved RGS domain, but the size and regulatory functions of the domain vary. Among the many RGS proteins, RGS1 and RGS10 are relatively small and are involved in the inflammatory and autoimmunological responses in the peripheral and central nervous systems. RGS1 and RGS10 may be important targets for future drug development for anti-inflammatory interventions that might stem neurodegenerative progression [108]. RGS1 and RGS10 are of particular interest for treatment of multiple sclerosis and Parkinson’s disease.
Chitosan Oligosaccharides (COS)
Chitosan is an abundant polysaccharide with antioxidant properties and little cytotoxicity. In a rat model of Aβ1–42-induced Alzheimer’s disease, chitosan oligosaccharides inhibited OS and neuroinflammatory response and thus conferred improvements in cognitive deficits as shown by the water maze and passive-avoidance tests [109]. The antioxidative properties of COS in the hippocampus were evaluated biochemically including their effects on apoptosis (TUNEL assay) and variations in inflammatory mediators (immunohistochemistry). When COS were administered to these rats at doses of 200, 400, or 800 mg/kg, the COS effectively reduced deficits in learning and memory. Neuronal apoptosis was ameliorated at these same doses. In this study, the neuroprotection conferred by COS aligned closely with OS inhibition, as shown by the evidence of diminishing levels of malondialdehyde, 8-hydroxy-2′-deoxyguanosine and increasing levels of glutathione peroxidase along with superoxide dismutase activities. It appears that by reducing the release of pro-inflammatory cytokines, such as IL-1β and TNFα, chitosan oligosaccharides may suppress the inflammatory response and decrease quantifiable signs of inflammation [109].
Pharmacological Antagonism of IL-8 Receptor CXCR2
IL-8 is a cytokine that is produced by many types of normal cells, including neutrophils and macrophages; IL-8 is then stored in endothelial cells. IL-8 ligands bind to the CXCR1 and CXCR2 receptors and trigger various biological functions. The binding of IL-8 to CXCR2 contributes to the chemotactic responses seen in Alzheimer’s disease. A murine model of Aβ1–42-induced Alzheimer’s disease was used to evaluate the pharmacological antagonism of CXCR2 with SB332235 as a means to inhibit receptor-mediated inflammatory reactivity and, in that way, improve neuronal viability [110]. This competitive CXCR2 antagonist significantly reduced the expression of CXCR2 and microgliosis but left astrogliosis unaltered. This implies that CXCR2-dependent inflammatory responses may play a key role in induced Alzheimer’s disease in rats. Thus, the pharmacological modulation of this receptor may inhibit inflammatory reactivity, providing neuroprotection against oxidative damage [110].
Microglial Alpha-7 Nicotinic Acetylcholine Receptors (nAChRs)
The CNS is replete with nAChRs that are expressed in both neurons and non-neuronal cells. These nAChRs are receptor polypeptides that respond to endogenous acetylcholine and their activity is involved in numerous physiological functions, such as memory, learning, ambulation, attention, and others. The α7 subtype of nAChR may be involved in neuroprotection, synaptic plasticity, and neuronal survival. As such, it is a therapeutic target for numerous neurological disorders. When α7-nAChR is activated, it opens an anti-inflammatory pathway that appears to be related to the recruitment and activation of Janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) pathways. JAK2/STAT3 pathways are involved in NF-κB nuclear translocation but can also help to regulate OS via the Nrf2/HO-1 axis [111].
Stem Cells of Human Dental Pulp
Stem cells for human exfoliated deciduous teeth (SHEDs) were used to produce a serum-free conditioned medium applied in a mouse model of Alzheimer’s disease. The SHEDs were administered intranasally to mice which demonstrated substantial improvement in cognitive function. The conditioned medium made from SHED was found to contain factors involved in several neurological processes such as neuroprotection, axonal elongation, neurotransmission, inflammatory suppression, and the regulation of microglia. SHED conditioned medium attenuated pro-inflammatory effects caused by β-amyloid plaques, triggered the induction of certain anti-inflammatory microglia, and overall helped to create an anti-inflammatory environment suitable for tissue regeneration [112].
Piracetam
Nootropics are natural or synthetic drugs aimed at improving mental capacity. Piracetam, a synthetic derivative of gamma-aminobutyric acid (GABA), is a first-generation prototype nootropic drug. Piracetam remains enigmatic in that its mechanism of action remains unknown; it does not have an effect similar to GABA. In the cortical myoclonus, piracetam is anti-myoclonic [113]. Piracetam has been recommended for enhancing mental capacity and has been used to aid learning and sharpen memory [114]. Piracetam has been used for pediatric patients to treat breath-holding spells [115], epilepsy [116], and autism spectrum disorders where improvements were noted in a pediatric subpopulation [117]. Piracetam-induced neuroprotection remains to be elucidated; the possible role of a caspase-independent pathway has been investigated and it appears that piracetam may confer protection against LPS-induced inflammatory responses and apoptosis [118]. Piracetam appears to have antioxidative, anti-apoptotic, and neuroprotective actions against mitochondria.
Fasudil
Fasudil is a calcium channel blocker and vasodilator that works by inhibiting the Rho kinase isoforms (ROCK1 and ROCK2). Fasudil has been used to treat cerebral vasospasm, such as might occur in a subarachnoid hemorrhage, and to ameliorate the cognitive decline associated with stroke [119]. Since the ROCK pathways are involved in the dopaminergic neuronal degeneration, fasudil may be a potential therapeutic agent to treat Parkinson’s disease. In a mouse model of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), fasudil was tested for its effectiveness in treating dopaminergic neuron injury [120]. Fasudil demonstrated neuroprotective benefits against dopaminergic neurons and facilitated the recovery of motor function in the MPTP mice and suppressed inflammatory responses (IL-1β, TNFα, NF-κB-p6, and TLR-2), and reduced OS (iNOS and gp91Phox). The reduction in OS might be associated with enzymatic inhibition of ROCK or glycogen synthase kinase-3β or both. Microglial activation in the CNS can be either neuroprotective, on the one hand, or pro-inflammatory, on the other. To better describe these diverse actions, the phenotypes microglia M1 (pro-inflammatory) and M2 (neuroprotective) have been described, but have yet to be fully elucidated. It has been observed that M1 and M2 phenotypes can change with time, disease progression, and other factors. Administration of fasudil can shift microglia M1 to microglia M2, that is transition the microglial environment from a pro-inflammatory and potentially cytotoxic state to one of neuroprotection. Fasudil may also optimize the expression of antioxidative factors [119].
Intranasally Administered Plasma Rich in Growth Factors: Endoret
Growth factors are substances required to stimulate growth in living cells and may include vitamins and hormones. Growth factors may also help mediate neuroprotection. In a rodent model of Parkinson’s disease, plasma rich in growth factors (PRGF) under the brand name Endoret was administered as a moderator the inflammatory response. Endoret offered neuroprotection in this animal model of Parkinson’s disease by decreasing the inflammatory response and improving motor abilities. These benefits were associated with a marked decrease in NF-κB activation as well as reduced expression of nitric oxide, COX-2, and TNFα in the substantia nigra. It has been hypothesized that PRGF (Endoret) may be able to prevent dopaminergic degeneration by way of an NF-κB-dependent signaling process [121].
Suppression of Glia Maturation Factor Expression
Glia maturation factor (GMF) is one of several neurotrophic factors that are involved in the development and proper function of the nervous and immune systems. Altered GMF expression can have a direct impact on the production of reactive oxygen species and inflammatory mediators, which are produced by the 1-methyl-4-phenylpyridinium (MPP+) by way of NF-κB. In GMF knockout neurons and glia, the lack of GMF led to a decrease in the production of reactive oxygen species and the downregulation of NF-κB-mediated production of TNFα and IL-1β compared to wild-type neurons and glia. The overexpression of GMF induced dopamine-related neurodegeneration, while the downregulation of GMF (by GMF-specific sharp-hairpin RNA) conferred neuroprotection from the toxic effects induced by MPP. In summary, it appears that a deficiency in GMF improves the antioxidant balance, as evidenced by observed decreased lipid peroxidation levels, less reactive oxygen species, and increased levels of glutathione. Furthermore, GMF deficiency also attenuated the dopamine-associated neuronal loss by downregulating the inflammatory responses mediated by way of the NF-κB pathways [122]. GMF appears to play a role in mediating OS and the release of MPP+, both of which can result in the loss of dopamine neurons.
Preservation of Mitochondrial Neuroglobin
Conditioned medium prepared from human adipose-derived mesenchymal stem cells (CM-hMSCA) has been developed with the goal of offering therapeutic benefit in neuropathological conditions. The concept behind CM-hMSCA is to offer protection to astrocytes so that they can maintain their neuroprotective functions. In a model of human astrocytes with scratch injury, CM-hMSCA was shown to regulate cytokines IL-2, IL-6, IL-8, IL-10, granulocyte–macrophage colony-stimulating factor, and TNFα. Moreover, CM-hMSCA downregulates calcium (at the cytoplasmic level), mitochondrial dynamics, and the mitochondrial respiratory chain. CM-hMSCA can also modulate the expression of any number of proteins involved in several signaling pathways, such as the AKT/pAKT and ERK1/2/pERK. CM-hMSCA may also be able to mediate the cellular-level localization of neuroglobin, an intracellular hemoprotein expressed mainly in the central and peripheral nervous systems. These observations strongly suggest that CM-hMSCA holds promise as a potential protective agent for astrocytes in patients with brain pathology [123].
Glucagon-Like Peptide 1
Glucagon-like peptide 1 (GLP-1) is an incretin hormone produced in the gut as a product of pre-proglucagon, a polypeptide that splits to produce numerous hormones that are similar to the parent (hence the name “glucagon-like”). One role of incretins is to enhance the synthesis and secretion of insulin from the pancreas in response to food. Originally, it was thought that GLP-1 was mainly related to insulin but it is now clear that in addition to glucose control, it has multiple additional functions in the brain, heart, and kidney. Both GLP-1 and GLP-1 receptor agonists have antioxidative properties that make them effective in the treatment of certain chronic diseases, such as diabetes. The activation of GLP-1 receptor signaling may enhance neurogenesis, reduce apoptosis, and protect neuronal function, making it a promising area for further study in the treatment of neurodegenerative diseases [124].
Radix Astragali Calycosin
Calycosin belongs to the class of 7-hydroxyisoflavones and is extracted from the dry root of the Radix Astragali plant used in traditional Chinese medicine. Calycosin is the main bioactive chemical of this plant and acts like a typical phytoestrogen by binding to estrogen receptors. It has been shown to improve cognitive impairment caused by diabetes mellitus, which suggests that it might improve cognitive function in patients with Alzheimer’s disease. Using APP/PSO transgenic mice as a model for Alzheimer’s disease, investigators intraperitoneally injected the animals with three different doses of calycosin (10, 20, and 40 mg/kg) [125]. In these mice, calycosin mitigated OS and activated the protein kinase C pathway, reducing the inflammatory response in the hippocampus and improving cognition.
Therapies Targeting Metabolic Hormones
Age- and lifestyle-related changes in metabolism are often a result of increases in OS and inflammation, factors facilitating metabolic dysfunction and cognitive decline [126, 127]. Changes in insulin, leptin, and amylin resulting from poor diet, aging, or metabolic disease can result in cognitive decline and contribute to the development of AD [129,130,131]. Therefore, therapies that aim to return these metabolic systems to a homeostatic state hold the highest therapeutic potential for the subsequent treatment of both metabolic and cognitive diseases [132].
Rosiglitazone
Advanced glycation end products (AGEs) are potentially harmful compounds that accumulate with advancing age. AGEs can be produced endogenously or assumed with the diet. AGEs are associated with the pathogenesis of neuronal deficits observed in diabetes. Hyperglycemia accelerates and increases the production of AGEs, and high levels of AGE may be associated with premature aging and any number of diseases, including Alzheimer’s disease, heart disease, and kidney failure. Rosiglitazone is an antidiabetic drug that can be described as a peroxisome proliferator-activated receptor gamma (PPARγ). Rosiglitazone belongs to a family of ligand-activate nuclear receptors and its ligands are associated with a variety of physiological, pathological, and inflammatory pathways. AGEs are suspected of playing a role in the neuronal impairment that is associated with diabetes mellitus; rosiglitazone appears effective against AGE-mediated neurotoxicity on human neural stem cells. Rosiglitazone can activate PPARγ-dependent signaling, which has been shown to confer neuroprotection in AGE-treated human neural stem cells. This implies that there may be a role for PPARγ ligands in the treatment of neurodegenerative disorders [133].
Silver Nanoparticles
Silver nanoparticles (AgNPs) are actually predominantly silver oxide in the form of particles ranging in size from about 1 to 100 nm. These silver nanoparticles have antibacterial properties and are used in a variety of consumer and medical products. Neural cells can uptake nanoparticles in the size range of 3–5 nm. The potential effects of silver nanoparticles on gene expression related to inflammation and neurodegeneration was studied in murine brain ALT astrocytes, microglial BV2 cells, and neuron N2a cells [134]. After being exposed to silver nanoparticles at doses of 5, 10, and 12.5 μg/ml, the neural cells showed a marked increase in secretion of IL-1β and induced gene expression of C-X-C motif chemokine 13 (CXCL13), macrophage receptor with collagenous structure, and glutathione synthetase [134]. This study also found that after treatment with silver nanoparticles, amyloid-β plaques (a pathological feature of Alzheimer’s disease) were deposited in neural cells. Following exposure to silver nanoparticles, gene expression of the amyloid precursor protein was induced. By the same token, protein levels, neprilysin, and low-density lipoprotein receptor were decreased in neural cells. The results from this study suggest that silver nanoparticles may be able to alter gene and protein expression related to amyloid-β deposition and could potentially induce the neurodegeneration of Alzheimer’s disease [134]. Further study is warranted.
Immunomodulatory Agents
The role of immunomodulation is changing medicine and it has been suggested that immunomodulatory pharmacological agents might be an early intervention for patients with clinical signs of Parkinson’s disease or even as a prophylactic treatment in patients at elevated risk for developing Parkinson’s disease [18]. Murine studies have shown that various immunomodulators could reduce motor deficits and alleviate dopaminergic neurotoxic effects. Some, but not all, immunomodulators have an anti-inflammatory action, decrease OS, and reduce the products of lipid peroxidation. They may also decrease the activities of microglia and astrocytes and decrease the infiltration of T cells into the substantia nigra. When animals with brain lesions were pretreated with fingolimod, tanshinone I, dimethyl fumarate, thalidomide, or cocaine- and amphetamine-regulated transcript peptide, this prophylaxis ameliorated motor deficits and nigral dopaminergic neurotoxicity [18].
More specifically:
Fingolimod or FTY720 reduced functional motor deficits and reduced the loss of TH+ neurons in the substantia nigra. It also attenuated the decrease of striatal dopamine and its metabolite levels [91].
Tanshinone I ameliorated the dopaminergic neurotoxicity in the substantia nigra and striatum and protected the striatum from OS by increasing glutathione levels [135].
Tanshinone IIA reduced apomorphine-induced contralateral rotations and alleviated the loss of TH+ neurons in the substantia nigra and striatum. It also attenuated the reduction of dopamine and its metabolites [136, 137].
Dimethyl fumarate reduced motor deficits while protecting dopaminergic neurons in the substantia nigra against α-syn toxicity. It likewise decreased microgliosis and astrogliosis [138, 139].
Lenalidomide was shown to improve motor deficits and improve dopamine fiber loss in the striatum, together with a decrease in microgliosis in the striatum and hippocampus. It also reduced the expression of certain proinflammatory cytokines, namely TNFα, IL-6, IL-1β, and IFN-γ and increased the expression of the anti-inflammatory cytokines IL-10 and IL-13 [140].
Thalidomide, like lenalidomide, restored dopamine fiber loss in the striatum, and reduced the expression of TNFα, IL-6, IL-1β, and IFN-γ [140].
Ginsenoside Rg1 decreased dopaminergic neuronal loss in the substantia nigra. The regulatory T cells in the blood were increased and the serum levels of TNFα, IFN-γ, IL-1β, and IL-6 were reduced. Microgliosis was inhibited and infiltration of CD3+ T cells into the substantia nigra was also reduced [141].
CNI-1493 attenuated dopaminergic cell loss in the substantia nigra, alleviated striatal loss of dopamine content, and reduced microgliosis in the substantia nigra [142].
Pycnogenol improved behavioral motor deficits [143].
Cyclosporin enhanced motor and cognitive function, decreased the striatal level of human α-syn and partially restored the level of TH protein, exhibited an anti-inflammatory effect by lowering the expression level of NFATc3 and alleviated mitochondrial stress in the midbrain [144].
Acetoside reduced parkinsonism symptoms [145].
SA00025 was partially neuroprotective of dopaminergic neurons and fibers. It also brought about changes in microglial morphology, indicative of a resting state and a decrease in reactive microglia. The astrocyte GFAP staining intensity in the substantia nigra and IL-6 levels were also decreased [146].
C5aR antagonist DF3016A. Complement (C) membrane receptors and proteins are constitutively expressed by the CNS. Part of it is the C5a, which represents the terminal effector of the C cascade. This part is mostly involved in pain and neuroinflammation, with several clinical consequences [147]. Recently, a novel selective C5aR antagonist (DF3016A) has shown the capacity to protect the neurons against a neuroinflammation-related process, in an animal model [148]. This could represent a new therapeutic approach to controlling CNS neuroinflammatory conditions and neurodegeneration due to OS.
Other Approaches
Intermittent Fasting
Intermittent fasting is a popular practice associated with weight loss that requires the dieter to eat only for a set number of hours of the day, e.g., he or she may eat only for eight consecutive hours per day and must refrain from eating for 16 h daily. Intermittent fasting may have other health benefits in addition to weight loss, in that it appears to induce adaptive responses in the brain that suppress inflammation. In order to study whether the benefits of intermittent fasting might be lessened with age, a study found that intermittent fasting can reduce the risk for brain functional deficits and neurodegenerative disorders associated with inflammatory response, independent of age (Table 3) [149].
Conclusions
The lack of established treatments for the prevention and treatment of neurodegenerative diseases requires greater study into the underlying causes of neurodegeneration and, more specifically, the role of OS and neuroinflammatory responses. In this review, we presented both natural pharmaceutical approaches as well as treatments with extracted or recombinant products and artificial agents. Progress is being made as many new substances and treatments show considerable promise, but greater and more thorough clinical study is urgently needed particularly as populations age. We suggest more in vivo experiments and we would cautiously suggest that combination approaches may be allow more effective treatment.
References
The EU Joint Programme—Neurodegenerative Disease Research (JPND). 2017. http://www.neurodegenerationresearch.eu/. Accessed 21 Feb 2019.
Katsnelson A, De Strooper B, Zoghbi HY. Neurodegeneration: from cellular concepts to clinical applications. Sci Transl Med. 2016;8(364):364ps18.
Zis P, McHugh PC, Manca M, Sarrigiannis PG, Rao DG, Hadjivassiliou M. Increased oxidative stress as a risk factor in chronic idiopathic axonal polyneuropathy. J Mol Neurosci. 2018;66(4):547–51. https://doi.org/10.1007/s12031-018-1200-5.
Yadav S, Mansoori AN, Sisodiya JS. Current pathologic determinants of complex neurodegenerative diseases: a review. Int J Pharm Technol. 2013;5(2):2607–21.
Cravello L, Di Santo SG, Varrassi G, et al. Chronic pain in the elderly with cognitive decline: a narrative review. Pain Ther. 2019. https://doi.org/10.1007/s40122-019-0111-7.
Pearce N, Kromhout H. Neurodegenerative disease: the next occupational disease epidemic? Occup Environ Med. 2014;71(9):594–5.
Jette N, Maxwell CJ, Fiest KM, Hogan DB. Systematic reviews and meta-analyses of the incidence and prevalence of dementia and its commoner neurodegenerative causes. Can J Neurol Sci. 2016;43(Suppl 1):S1–2.
Montine TJ. Prevalence estimates for latent neurodegenerative disease. Toxicol Pathol. 2011;39(1):99–102.
González H, Pacheco R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J Neuroinflammation. 2014;11:201. https://doi.org/10.1186/s12974-014-0201-8.
Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–44. https://doi.org/10.1056/nejmra0909142.
Awasthi A, Matsunaga Y, Yamada T. Amyloid-beta causes apoptosis of neuronal cells via caspase cascade, which can be prevented by amyloid-beta-derived short peptides. Exp Neurol. 2005;196:282–9. https://doi.org/10.1016/j.expneurol.2005.08.001.
Giraldo E, Lloret A, Fuchsberger T, Viña J. A-beta and tau toxicities in Alzheimer’s are linked via oxidative stress-induced p38 activation: protective role of vitamin E. Redox Biol. 2014;2:873–7. https://doi.org/10.1016/j.redox.2014.03.002.
Liu Z, Zhou T, Ziegler AC, Dimitrion P, Zuo L Oxidative stress in neurodegenerative diseases: from molecular mechanisms to clinical applications. Oxid Med Cell Longev. 2017;2017:2525967. https://doi.org/10.1155/2017/2525967.
Lees AJ. Unresolved issues relating to the shaking palsy on the celebration of James Parkinson’s 250th birthday. Mov Disord. 2007;22(Suppl 17):S327–34.
Miller DB, O’Callaghan JP. Biomarkers of Parkinson’s disease: present and future. Metabolism. 2015;64(3 Suppl 1):S40–6.
Solleiro-Villavicencio H, Rivas-Arancibia S. Effect of chronic oxidative stress on neuroinflammatory response mediated by CD4+T cells in neurodegenerative diseases. Front Cell Neurosci. 2018;12:114. https://doi.org/10.3389/fncel.2018.00114.
Puspita L, Chung SY, Shim JW. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol Brain. 2017;10(1):53. https://doi.org/10.1186/s13041-017-0340-9.
Martinez B, Peplow PV. Neuroprotection by immunomodulatory agents in animal models of Parkinson’s disease. Neur Reg Res. 2018;13(9):1493–506.
Kobelt G, Thompson A, Berg J, Gannedahl M, Eriksson J. New insights into the burden and costs of multiple sclerosis in Europe. Mult Scler J. 2017;23:1123–36. https://doi.org/10.1177/1352458517694432.
Haider L, Fischer MT, Frischer JM, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134:1914–24. https://doi.org/10.1093/brain/awr128.
Fischer MT, Wimmer I, Höftberger R, et al. Disease-specific molecular events in cortical multiple sclerosis lesions. Brain. 2013;136:1799–815. https://doi.org/10.1093/brain/awt110.
National Institute of Neurological Disorders and Stroke. National institute of neurological disorders and stroke. Bethesda: Amyotrophic Lateral Sclerosis; 2017.
D’Amico E, Factor-Litvak P, Santella RM, Mitsumoto H. Clinical perspective on oxidative stress in sporadic amyotrophic lateral sclerosis. Free Rad Biol Med. 2013;65:509–27. https://doi.org/10.1016/j.freeradbiomed.2013.06.029.
Malaspina A, Puentes F, Amor S. Disease origin and progression in amyotrophic lateral sclerosis: an immunology perspective. Int Immunol. 2014;27:117–29. https://doi.org/10.1093/intimm/dxu099.
Hooten KG, Beers DR, Zhao W, Appel SH. Protective and toxic neuroinflammation in amyotrophic lateral sclerosis. Neurotherapeutics. 2015;12:364–75. https://doi.org/10.1007/s13311-014-0329-3.
Li Q, Spencer NY, Pantazis NJ, Engelhardt JF. Alsin and SOD1(G93A) proteins regulate endosomal reactive oxygen species production by glial cells and proinflammatory pathways responsible for neurotoxicity. J Biol Chem. 2011;286:40151–62. https://doi.org/10.1074/jbc.m111.279711.
Petrillo S, Pelosi L, Piemonte F, et al. Oxidative stress in Duchenne muscular dystrophy: focus on the NRF2 redox pathway. Hum Mol Genet. 2017;26(14):2781–90. https://doi.org/10.1093/hmg/ddx173.
Prusiner SB. Neurodegenerative diseases and prions. N Engl J Med. 2001;344(20):1516–26.
Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10:10–7.
Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296(5575):1991–5.
Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29(1):15–32.
Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004;3(3):205–14.
Vila M, Przedborski S. Targeting programmed cell death in neurodegenerative diseases. Nat Rev Neurosci. 2003;4(5):365–75.
Maritim AC, Sanders RA, Watkins JB. Diabetes, oxidative stress, and antioxidants: a review. J Biochem Mol Toxicol. 2003;17(1):24–38.
Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–95.
Islam MT. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol Res. 2017;39(1):73–82.
Santos R, Bulteau AL, Gomes CM. Neurodegeneration, neurogenesis, and oxidative stress. Oxid Med Cell Longev. 2016;2016:7632025. https://doi.org/10.1155/2016/7632025.
Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem. 2000;267(16):4904–11.
Stathopulos P, Rumfeldt J, Scholz G, et al. Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc Natl Acad. 2003;100(12):7021–6.
Yaribeygi H, Panahi Y, Javadi B, Sahebkar A. The underlying role of oxidative stress in neurodegeneration: a mechanistic review. CNS Neurol Disorders Drug Targets. 2018;17(3):207–15.
Fusco M, Skaper S, Coaccioli S, Paladini A, Varrassi G. Degenerative joint diseases and neuroinflammation. Pain Pract. 2017;17:522–32.
Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–8.
Varrassi G, Fusco M, Skaper SD, et al. A pharmacological rationale to reduce the incidence of opioid induced tolerance and hyperalgesia: a review. Pain Ther. 2018;7:59–75. https://doi.org/10.1007/s40122-018-0094-9.
Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–69.
Yong VW. Inflammation in neurological disorders: ahelpora hindrance? Neuroscientist. 2010;16:408–20.
Emerit J, Edeas M, Bricaire F. Neurodegenerative diseases and oxidative stress. Biomed Pharmacother. 2004;58:39–46.
Hsieh H-L, Yang C-M. Role of redox signaling in neuroinflammation and neurodegenerative diseases. Biomed Res Int. 2013;2013:484613. https://doi.org/10.1155/2013/484613.
Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453–62.
Fischer R, Maier O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF. Oxid Med Cell Longev. 2015;2015:610813. https://doi.org/10.1155/2015/610813.
Dröse S, Brandt U. The mechanism of mitochondrial superoxide production by the cytochrome bc1 complex. J Biol Chem. 2008;283(31):21649–54.
Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J. Neurosci. 2011;31(45):16064–9. https://doi.org/10.1523/jneurosci.4158-11.2011.
Letiembre M, Liu Y, Walter S, et al. Screening of innate immune receptors in neurodegenerative diseases: a similar pattern. Neurobiol Aging. 2009;30:759–68.
Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener. 2009;4:47.
Varrassi G, Fusco M, Coaccioli S, Paladini A. Chronic pain and neurodegenerative processes in elderly people. Pain Pract. 2015;15(1):1–3.
Mueller AM, Yoon BH, Sadiq SA. Inhibition of hyaluronan synthesis protects against central nervous system (CNS) autoimmunity and increases CXCL12 expression in the inflamed CNS. J Biol Chem. 2014;289:22888–99.
Slusarczyk J, Trojan E, Glombik K, et al. Anti-inflammatory properties of tianeptine on lipopolysaccharide-induced changes in microglial cells involve toll-like receptor-related pathways. J Neurochem. 2016;136:958–70.
Tao L, Zhang F, Hao L, et al. 1-O-Tigloyl-1-O-deacetyl-nimbolinin B inhibits LPS-stimulated inflammatory responses by suppressing NF-kappaB and JNK activation in microglia cells. J Pharmacol Sci. 2014;125:364–74.
Paladini A, Fusco M, Coaccioli S, Skaper SD, Varrassi G. Chronic pain in the elderly: the case for new therapeutic strategies. Pain Physician. 2015;18(5):E863–76.
Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60. https://doi.org/10.1126/science.10962844.
Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–7.
Nichols WC, Pankratz N, Hernandez D, et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet. 2005;365:410–2.
Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–9. https://doi.org/10.1126/science.1077209.
Gilks Abou-Sleiman PM, Gandhi S, et al. A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet. 2005;365:415–41.
Richter Park JW, Ames BNC. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci. 1988;85:6465–7. https://doi.org/10.1073/pnas.85.17.6465.
Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med. 2000;29:222–30. https://doi.org/10.1016/s0891-5849(00)00317-8.
Lee HC, Chang CM, Chi CW. Somatic mutations of mitochondrial DNA in aging and cancer progression. Ageing Res Rev. 2010;9(Suppl 1):S47–58. https://doi.org/10.1016/j.arr.2010.08.009.
Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007;100:460–73. https://doi.org/10.1161/01.res.0000258450.44413.96.
Palazzo E, Luongo L, Guida F, et al. Role of N-acylethanolamines in the neuroinflammation: ultramicronized palmitoylethanolamide in the relief of chronic pain and neurodegenerative diseases. Neuropsychiatry. 2019;9:2035–46.
Chu J, Han W. Punicalagin exerts beneficial functions in 6-hydroxydopamine-treated SH-SY5Y cells by attenuating mitochondrial dysfunction and inflammatory responses. Med Sci Monit. 2018;24:5905–13. https://doi.org/10.12659/msm.909969.
Farzaei MH, Shahpiri Z, Mehri MR, et al. Medicinal plants in neurodegenerative diseases: perspective of traditional Persian medicine. Curr Drug Metab. 2018;19(5):429–42. https://doi.org/10.2174/1389200219666180305150256.
Gil Á, Plaza-Diaz J, Mesa MD. Vitamin D: classic and novel actions. Ann Nutr Metab. 2018;72(2):87–95.
Sil S, Ghosh T, Gupta P, Ghosh R, Kabir SN, Roy A. Dual role of vitamin C on the neuroinflammation mediated neurodegeneration and memory impairments in colchicine induced rat model of Alzheimer disease. J Mol Neurosci. 2016;60(4):421–35.
Park SY, Choi MH, Park G, Choi YW. Petasites japonicus bakkenolide B inhibits lipopolysaccharide-induced pro-inflammatory cytokines via AMPK/Nrf2 induction in microglia. Int J Mol Med. 2018;41(3):1683–92.
Casas R, Estruch R, Sacanella E. The protective effects of extra virgin olive oil on immune-mediated inflammatory responses. Endocr Metab Immune Disord Drug Targets. 2018;18(1):23–35.
Parkinson L, Cicerale S. The health benefiting mechanisms of virgin olive oil phenolic compounds. Molecules. 2016;16:21. https://doi.org/10.3390/molecules21121734.
Casamenti F, Grossi C, Rigacci S, Pantano D, Luccarini I, Stefani M. Oleuropein aglycone: a possible drug against degenerative conditions in vivo evidence of its effectiveness against Alzheimer’s disease. J Alzheimers Dis. 2015;45:679–88.
Li W, Sperry JB, Crowe A, Trojanowski JQ, Smith AB, Lee VMY. Inhibition of tau fibrillization by oleocanthal via reaction with the amino groups of tau. J Neurochem. 2009;110:1339–51.
Valls-Pedret C, Sala-Vila A, Serra-Mir M, et al. Mediterranean diet and age-related cognitive decline: a randomized clinical trial. JAMA Intern Med. 2015;175:1094–103.
Panza F, Solfrizzi V, Tortelli R, Resta F, Sabbà C, Logroscino G. Prevention of late-life cognitive disorders: diet related factors, dietary patterns, and frailty models. Curr Nutr Rep. 2014;3:110–29.
Achour I, Arel-Dubeau AM, Renaud J, et al. Oleuropein prevents neuronal death, mitigates mitochondrial superoxide production and modulates autophagy in a dopaminergic cellular model. Int J Mol Sci. 2016;17:1293. https://doi.org/10.3390/ijms17081293.
Khalatbary AR. Olive oil phenols and neuroprotection. Nutr Neurosci. 2013;16:243–9.
Ma Y, Ma B, Shang Y, et al. Flavonoid-rich ethanol extract from the leaves of Diospyros kaki attenuates cognitive deficits, amyloid-beta production, oxidative stress, and neuroinflammation in APP/PS1 transgenic mice. Brain Res. 2018;1678:85–93.
Dhouib IB, Annabi A, Doghri R, et al. Neuroprotective effects of curcumin against acetamiprid-induced neurotoxicity and oxidative stress in the developing male rat cerebellum: biochemical, histological, and behavioral changes. Env Sci Poll Res. 2017;24(35):27515–24.
Arús BA, Souza DG, Bellaver B, et al. Resveratrol modulates GSH system in C6 astroglial cells through heme oxygenase 1 pathway. Mol Cell Biochem. 2017;428(1–2):67–77.
Qu Z, Mossine VV, Cui J, Sun GY, Gu Z. Protective effects of AGE and its components on neuroinflammation and neurodegeneration. Neuromol Med. 2016;18(3):474–82.
Sun Y, Moore MJ, Zhang S, et al. Phylogenomic and structural analyses of 18 complete plastomes across nearly all families of early-diverging eudicots, including an angiosperm-wide analysis of IR gene content evolution. Mol Phylogen Evolut. 2016;96:93–101.
Ajit D, Simonyi A, Li R, et al. Phytochemicals and botanical extracts regulate NF-κB and Nrf2/ARE reporter activities in DI TNC1 astrocytes. Neurochem Intern. 2016;97:49–56.
Trovato A, Siracusa R, Di Paola R, et al. Redox modulation of cellular stress response and lipoxin A4 expression by Hericium erinaceus in rat brain: relevance to Alzheimer’s disease pathogenesis. Immun Ageing. 2016;13(1):23. https://doi.org/10.1186/s12979-016-0078-8.
Zou J, Cai PS, Xiong CM, Ruan JL. Neuroprotective effect of peptides extracted from walnut (Juglans Sigilata Dode) proteins on Aβ25-35-induced memory impairment in mice. J Huazhong Univ Sci Technol Med Sci. 2016;36(1):21–30.
Peng Y, Hu Y, Xu S, et al. l-3-n-Butylphthalide reduces tau phosphorylation and improves cognitive deficits in AβPP/PS1-Alzheimer’s transgenic mice. J Alzheimer Dis. 2012;29(2):379–91.
Zhao CY, Lei H, Zhang Y, et al. l-3-n-Butylphthalide attenuates neuroinflammatory responses by downregulating JNK activation and upregulating Heme oxygenase-1 in lipopolysaccharide-treated mice. J Asian Nat Prod Res. 2016;18(3):289–302.
Jang JH, Son Y, Kang SS, et al. Neuropharmacological potential of Gastrodia elata Blume and its components. Evid Based Complement Alternat Med. 2015;2015:309261. https://doi.org/10.1155/2015/309261.
Lin F, Zhang C, Chen X, et al. Chrysophanol affords neuroprotection against microglial activation and free radical-mediated oxidative damage in BV2 murine microglia. Int J Clin Exp Med. 2015;8(3):3447–55.
Barros M, Poppe S, Bondan E. Neuroprotective properties of the marine carotenoid astaxanthin and omega-3 fatty acids, and perspectives for the natural combination of both in krill oil. Nutrients. 2014;6(3):1293–317.
Paladini A, Fusco M, Cenacchi T, Schievano C, Piroli A, Varrassi G. Palmitoylethanolamide, a special food for medical purposes, in the treatment of chronic pain: a pooled data meta-analysis. Pain Physician. 2016;19(2):11–24.
Paladini A, Varrassi G, Bentivegna G, Carletti S, Piroli A, Coaccioli S. Palmitoylethanolamide in the treatment of failed back surgery syndrome. Pain Res Treat. 2017;2017:1486010. https://doi.org/10.1155/2017/1486010.
Del Giorno R, Skaper S, Paladini A, Varrassi G, Coaccioli S. Palmitoylethanolamide in fibromyalgia: results from prospective and retrospective observational studies. Pain Ther. 2015;4(2):169–78.
Beggiato S, Borelli AC, Ferraro L, Tanganelli S, Antonelli T, Tomasini MC. Palmitoylethanolamide blunts amyloid-β42-induced astrocyte activation and improves neuronal survival in primary mouse cortical astrocyte-neuron co-cultures. J Alzheimer Dis. 2018;61(1):389–99. https://doi.org/10.3233/jad-170699.
Carrascal L, Nunez-Abades P, Ayala A, Cano M. Role of melatonin in the inflammatory process and its therapeutic potential. Curr Pharm Design. 2018;24(14):1563–88.
Gerenu G, Liu K, Chojnacki JE, Saathoff JM, et al. Curcumin/melatonin hybrid 5-(4-hydroxy-phenyl)-3-oxo-pentanoic acid [2-(5-methoxy-1H-indol-3-yl)-ethyl]-amide ameliorates AD-Like pathology in the APP/PS1 mouse model. ACS Chem Neurosci. 2015;6(8):1393–9.
Nasoohi S, Ismael S, Ishrat T. Thioredoxin-interacting protein (TXNIP) in cerebrovascular and neurodegenerative diseases: regulation and implication. Mol Neurobiol. 2018;55(10):7900–20.
Reddy AP, Reddy PH. Mitochondria-targeted molecules as potential drugs to treat patients with Alzheimer’s disease. Progr Mol Biol Transl Sci. 2017;146:173–201.
Zhou H, Ding L, Wu Z, et al. Hydrogen sulfide reduces RAGE toxicity through inhibition of its dimer formation. Free Rad Biol Med. 2017;104:262–71.
Derk J, MacLean M, Juranek J, Schmidt AM. The receptor for advanced glycation endproducts (RAGE) and mediation of inflammatory neurodegeneration. J Alzheimers Dis Parkinsonism. 2018;8(1):pii:421. https://doi.org/10.4172/2161-0460.1000421.
van Noort MJ, Bugiani M, Amor S. Heat shock proteins: old and novel roles in neurodegenerative diseases in the central nervous system. CNS Neurol Disord Drug Targets. 2017;16(3):244–56.
Rodriguez Cruz Y, Strehaiano M, Rodriguez Obaya T, Garcia Rodriguez JC, Maurice T. An intranasal formulation of erythropoietin (Neuro-EPO) prevents memory deficits and amyloid toxicity in the APP Swe transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis. 2017;55(1):231–48.
Grottelli S, Ferrari I, Pietrini G, Peirce M, Minelli A, Bellezza I. The role of cyclo(His-Pro) in neurodegeneration. Int J Mol Sci. 2016;17(8):1332. https://doi.org/10.3390/ijms17081332.
Lee JK, Dagher JB. Regulator of G-protein signaling (RGS) 1 and RGS10 proteins as potential drug targets for neuroinflammatory and neurodegenerative diseases. AAPS J. 2016;18(3):545–9.
Jia S, Lu Z, Gao Z, et al. Chitosan oligosaccharides alleviate cognitive deficits in an amyloid-β1–42-induced rat model of Alzheimer’s disease. Int J Biol Macromol. 2016;83:416–25.
Ryu JK, Cho T, Choi HB, Jantaratnotai N, McLarnon JG. Pharmacological antagonism of interleukin-8 receptor CXCR2 inhibits inflammatory reactivity and is neuroprotective in an animal model of Alzheimer’s disease. J Neuroinflammation. 2015;12(1):144. https://doi.org/10.1186/s12974-015-0339-z.
Egea J, Buendia I, Parada E, Navarro E, León R, Lopez MG. Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem Pharmacol. 2015;97(4):463–72.
Mita T, Furukawa-Hibi Y, Takeuchi H, et al. Conditioned medium from the stem cells of human dental pulp improves cognitive function in a mouse model of Alzheimer’s disease. Behav Brain Res. 2015;293:189–97.
Gouliaev AH, Senning A. Piracetam and other structurally related nootropics. Brain Res Rev. 1994;19(2):180–222.
Lee AW, Hillis AE. The pharmacological treatment of aphasia. In: Stemmer B, Whitaker HA, editors. Handbook of the neuroscience of language. Amsterdam: Elsevier, 2008. p. 407–416
Fenichel GM. Clinical pediatric neurology: a signs and symptoms approach. Amsterdam: Elsevier Health Sciences; 2009.
Porter RJ, Dhir A, Macdonald RL, Rogawski MA. Mechanisms of action of antiseizure drugs. Handb Clin Neurol. 2012;108:663–81.
Farmer CA, Aman MG. Pharmacological intervention for disruptive behaviors in intellectual and developmental disabilities: the glass is half full. Int Rev Res Dev Disabil. 2013;2013(44):281–325. https://doi.org/10.1016/b978-0-12-401662-0.00009-9.
Verma DK, Gupta S, Biswas J, et al. New therapeutic activity of metabolic enhancer piracetam in treatment of neurodegenerative disease: participation of caspase independent death factors, oxidative stress, inflammatory responses and apoptosis. Biochim Biophys Acta Mol Basis Dis. 2018;1864:2078–96. https://doi.org/10.1016/j.bbadis.2018.03.
Shibuya M, Suzuki Y. Treatment of cerebral vasospasm by a protein kinase inhibitor AT 877. Nō To Shinkei. 1993;45(9):819–24. (In Japanese).
Zhao YF, Zhang Q, Xi JY, Li YH, Ma CG, Xiao BG. Multitarget intervention of fasudil in the neuroprotection of dopaminergic neurons in MPTP-mouse model of Parkinson’s disease. J Neurol Sci. 2015;353(1–2):28–37.
Anitua E, Pascual C, Pérez-Gonzalez R, Orive G, Carro E. Intranasal PRGF-Endoret enhances neuronal survival and attenuates NF-κB-dependent inflammation process in a mouse model of Parkinson’s disease. J Control Release. 2015;203:170–80.
Khan MM, Zaheer S, Nehman J, Zaheer A. Suppression of glia maturation factor expression prevents 1-methyl-4-phenylpyridinium (MPP+)-induced loss of mesencephalic dopaminergic neurons. Neuroscience. 2014;277:196–205.
Baez-Jurado E, Guio-Vega G, Hidalgo-Lanussa O, et al. Mitochondrial neuroglobin is necessary for protection induced by conditioned medium from human adipose-derived mesenchymal stem cells in astrocytic cells subjected to scratch and metabolic injury. Mol Neurobiol. 2019;56(7):5167–87. https://doi.org/10.1007/s12035-018-1442-9.
Oh Y, Jun HS. Effects of glucagon-like peptide-1 on oxidative stress and Nrf2 signaling. Int J Mol Sci. 2017;19(1):26. https://doi.org/10.3390/ijms19010026.
Song L, Li X, Bai XX, Gao J, Wang CY. Calycosin improves cognitive function in a transgenic mouse model of Alzheimer’s disease by activating the protein kinase C pathway. Neural Regen Res. 2017;12(11):1870–6.
Floyd RA, Hensley K. Oxidative stress in brain aging: implications for therapeutics of neurodegenerative diseases. Neurobiol Aging. 2002;23(5):795–807.
Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–9.
Posey KA, Clegg DJ, Printz RL, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab. 2009;296(5):E1003–12.
Talbot K, Wang H-Y, Kazi H, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122(4):1316–38.
Bonda DJ, Stone JG, Torres SL, et al. Dysregulation of leptin signaling in Alzheimer disease: evidence for neuronal leptin resistance. J Neurochem. 2014;128(1):162–72.
Adler BL, Yarchoan M, Hwang HM, et al. Neuroprotective effects of the amylin analogue pramlintide on Alzheimer’s disease pathogenesis and cognition. Neurobiol Aging. 2014;35(4):793–801.
Grizzanti J, Lee HG, Camins A, Pallas M, Casadesus G. The therapeutic potential of metabolic hormones in the treatment of age-related cognitive decline and Alzheimer’s disease. Nutr Res. 2016;36(12):1305–15.
Chiang MC, Cheng YC, Nicol CJ, Lin CH. The neuroprotective role of rosiglitazone in advanced glycation end product treated human neural stem cells is PPARgamma-dependent. Int J Biochem Cell Biol. 2017;92:121–33.
Huang CL, Hsiao IL, Lin HC, Wang CF, Huang YJ, Chuang CY. Silver nanoparticles effect on gene expression of inflammatory and neurodegenerative responses in mouse brain neural cells. Environ Res. 2015;136:253–63.
Wang S, Jing H, Yang H, et al. Tanshinone I selectively suppress pro-inflammatory genes expression in activated microglia and prevents nigrostriatal dopaminergic neurodegeneration in a mouse model of Parkinson’s disease. J Ethnopharmacol. 2015;164:247–55.
Zhang XS, Ha S, Wang XL, Shi YL, Duan SS, Li ZA. Tanshinone IIA protects dopaminergic neurons against 6-hydroxydopamine-induced neurotoxicity through miR-153/NF-E2-related factor 2/antioxidant response element signaling pathway. Neuroscience. 2015;303:489–502.
Ren B, Zhang YX, Zhou HX, et al. Tanshinone IIA prevents the loss of nigrostriatal dopaminergic neurons by inhibiting NADPH oxidase and iNOS in the MPTP model of Parkinson’s disease. J Neurol Sci. 2015;348:142–52.
Lestes-Becker I, García-Yagüe AJ, Scannevin RH, et al. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinpathy in Parkinson’s disease. Antioxid Redox Signal. 2016;25:61–77.
Jing X, Shi H, Zhang C, et al. Dimethyl fumarate attenuates 6-OHDA-induced neurotoxicity in SH-SY5Y cells and in animal model of Parkinson’s disease by enhancing Nrf2 activity. Neuroscience. 2015;286:131–40.
Valera E, Mante M, Anderson S, Rockenstein E, Masliah E. Lenalidomide reduces microglial activation and behavioral deficits in a transgenic model of Parkinson’s disease. J Neuroinflammation. 2015;12:93. https://doi.org/10.1186/s12974-015-0320-x.
Zhou TT, Zu G, Wang X, et al. Immunomodulatory and neuroprotective effects of ginsenoside Rg1 in the MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine)-induced mouse model of Parkinson’s disease. Int Immunopharmacol. 2015;29:334–43.
Noelker C, Stuckenholz V, Reese JP, et al. CNI-1493 attenuates neuroinflammation and dopaminergic neurodegeneration in the acute MPTP mouse model of Parkinson’s disease. Neurodegener Dis. 2013;12:103–10.
Khan MM, Kempuraj D, Thangavel R, Zaheer A. Protection of MPTP-induced neuroinflammation and neurodegeneration by pycnogenol. Neurochem Int. 2013;62:379–88.
Tamburino A, Churchill MJ, Wan OW, et al. Cyclosporin promotes neurorestoration and cell replacement therapy in pre-clinical models of Parkinson’s disease. Acta Neuropathol Commun. 2015;3:84. https://doi.org/10.1186/s40478-015-0263-6.
Yuan J, Ren J, Wang Y, He X, Zhao Y. Acetoside binds to caspase-3 and exerts neuroprotection in the rotenone rat model of Parkinson’s disease. PLoS One. 2016;11:e0162696.
Smith GA, Rocha EM, Rooney T, et al. A Nurr1 agonist causes neuroprotection in a Parkinson’s disease lesion model primed with the toll-like receptor 3 dsRNA inflammatory stimulant poly(I:C). PLoS One. 2015;10:121072.
Quadros AU, Cunha TM. C5a and pain development: an old molecule, a new target. Pharmacol Res. 2016;112:58–67. https://doi.org/10.1016/j.phrs.2016.02.004.
Brandolini L, Grannonico M, Bianchini G, et al. The novel C5aR antagonist DF3016A protects neurons against ischemic neuroinflammatory injury. Neurotox Res. 2019;36(1):163–74. https://doi.org/10.1007/s12640-019-00026-w.
Vasconcelos AR, Kinoshita PF, Yshii LM, et al. Effects of intermittent fasting on age-related changes on Na,K-ATPase activity and oxidative status induced by lipopolysaccharide in rat hippocampus. Neurobiol Aging. 2015;36(5):1914–23. https://doi.org/10.1016/k.neurobiolaging.2015.02.020.
Acknowledgements
The authors are very grateful to Mariella Fusco for her scientific feedback and Luca Di Giacomo for his continuous technical assistance with the graphic design.
Funding
This paper has been possible thanks to the grant provided by the Paolo Procacci Foundation, Via Tacito 7, 00,193 Roma, Italy. It was also partially funded by the grant received by “Fondazione Internazionale Salvatore Maugeri”, Research Project “Modello di Unità di Terapia del Dolore e di Cure Palliative, Integrate con Associazioni di Volontariato”. No Rapid Service Fee was received by the journal for the publication of this article.
Medical Writing Assistance
The authors acknowledge medical editing services by Jo Ann LeQuang of LeQ Medical whose activity has been supported by Paolo Procacci Foundation and “Fondazione Internazionale Salvatore Maugeri” funds.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
M Rekatsina carried out most of the research and drafted the initial manuscript. All the other authors have contributed to review and ameliorate the quality of the paper. All the authors have reviewed and approved the final draft of the manuscript.
Disclosures
Martina Rekatsina, Alba Piroli, Panagiotis Zis have nothing to disclose. Antonella Paladini is a member of the journal’s Editorial Board. Joseph V. Pergolizzi is a member of the journal’s Editorial Board. Giustino Varrassi is a member of the journal’s Editorial Board.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.10247852.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rekatsina, M., Paladini, A., Piroli, A. et al. Pathophysiology and Therapeutic Perspectives of Oxidative Stress and Neurodegenerative Diseases: A Narrative Review. Adv Ther 37, 113–139 (2020). https://doi.org/10.1007/s12325-019-01148-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-019-01148-5